 Jérémie Martinet1
Jérémie Martinet1 Gwladys Bourdenet1
Gwladys Bourdenet1 Amine Meliani2,3
Amine Meliani2,3 Laetitia Jean1
Laetitia Jean1 Sahil Adriouch1
Sahil Adriouch1 Jose L. Cohen4
Jose L. Cohen4 Federico Mingozzi2,3
Federico Mingozzi2,3 Olivier Boyer1*
Olivier Boyer1*
- 1Normandie University, UNIROUEN, Pathophysiology and Biotherapy of Inflammatory and Autoimmune Diseases, INSERM, CHU Rouen, Rouen, France
- 2U974, INSERM, University Pierre and Marie Curie, Paris, France
- 3Genethon, Evry, France
- 4U955 Team 21, Inserm, Créteil, France
Background: Gene therapy is a promising treatment option for hemophilia and other protein deficiencies. However, immune responses against the transgene product represent an obstacle to safe and effective gene therapy, urging for the implementation of tolerization strategies. Induction of a hematopoietic chimerism via bone marrow transplantation (BMT) is a potent means for inducing immunological tolerance in solid organ transplantation.
Objectives: We reasoned, here, that the same viral vector could be used, first, to transduce BM cells for inducing chimerism-associated transgene-specific immune tolerance and, second, for correcting protein deficiencies by vector-mediated systemic production of the deficient coagulation factor.
Methods: Evaluation of strategies to induce B and T cell tolerance was performed using ex vivo gene transfer with lentiviral (LV) vectors encoding coagulation factor IX (FIX) or the SIINFEKL epitope of ovalbumin. Following induction of microchimerism via BMT, animals were challenged with in vivo gene transfer with LV vectors.
Results: The experimental approach prevented humoral immune response against FIX, resulting in persistence of therapeutic levels of circulating FIX, after LV-mediated gene transfer in vivo. In an ovalbumin model, we also demonstrated that this approach effectively tolerized the CD8+ T cell compartment in an antigen-specific manner.
Conclusion: These results provide the proof-of-concept that inducing a microchimerism by gene-modified BMT is a powerful tool to provide transgene-specific B and T cell tolerance in a gene therapy setting.
Introduction
A major complication of enzyme replacement therapy in hemophilia and other genetic defects is the development of immune responses toward the recombinant therapeutic protein (1–3). Similarly, a potential important concern for gene therapy is represented by the risk of immunization against the therapeutic transgene product, as shown in animal models of hemophilia B (4–8). In particular, while factor IX (FIX) gene therapy using adeno-associated or lentiviral (LV) vectors in vivo is a promising treatment option for hemophilia B (9, 10), humoral and cell-mediated immune responses triggered by the transgene may result in lack of therapeutic efficacy (11, 12). Only few tolerizing strategies have been investigated to tackle this issue, mostly by targeting FIX expression to the liver (5, 13) or detargeting transgene expression from antigen-presenting cells (11). Some other approaches have also been proposed to induce tolerance to factor VIII (FVIII) in hemophilia A, i.e., administration of B cell blasts transduced by FVIII-immunodominant domains using a retrovirus-mediated gene transfer (14) or intraosseous infusion of LV encoding FVIII under the control of platelet-specific promoters (15).
Induction of a hematopoietic chimerism is a potent means for inducing immunological tolerance in solid organ transplantation. For instance, transplantation of alloantigen-expressing BM cells results in a strong state of tolerance in allogeneic (16) or syngeneic gene-modified settings (17). In hemophilia, expression of coagulation factors at therapeutic levels by transduced BM cells has been shown to provide FVIII- or FIX-specific tolerance in settings where transgene expression is restricted to the hematopoietic compartment (18–22). Here, we evaluated the hypothesis that induction of a microchimerism (<0.5%) by grafting LV-modified BM may be sufficient to elicit transgene-specific tolerance and to sustain transgene expression after subsequent systemic LV administration or LV injection to an extra-hematopoietic tissue.
Materials and Methods
Lentiviral Vectors and Gene Transfer
We developed LV-OVA and LV-FIX vectors by replacing the GFP gene of the PGK promoter-driven LV-GFP vector (23) by a SIINFEKL/β2-microglobulin/H-2Kb fusion construct (24) or human FIX cDNA (25), respectively (Figure 1). LV titers (expressed as transducing units, TU/mL) were determined by flow cytometry for LV-OVA and LV-GFP (26) and by qPCR for LV-FIX (10). BM cells from female Ly5.1 C57BL/6 (Ly5.1 B6) mice (Charles River Laboratories) were transduced with LV at a multiplicity of infection of 1 (17).
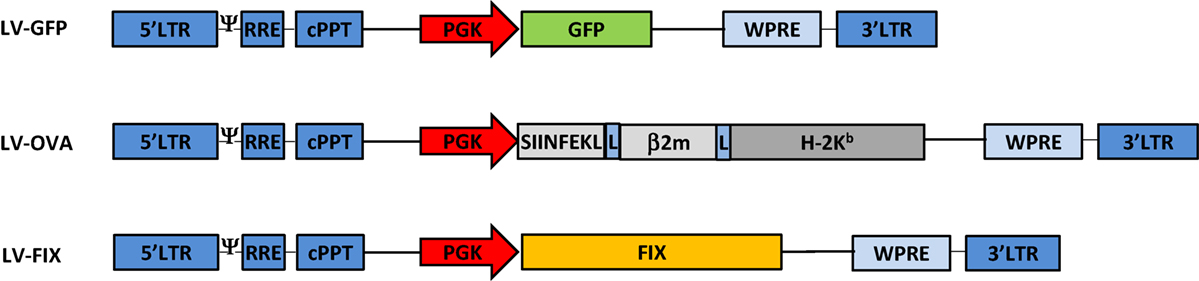
Figure 1. Lentiviral vectors design. Schematic representation of the LV-GFP, LV-OVA, and LV-FIX lentiviral vectors.
Induction of BM Chimerism
Animal experiments were approved by an ethics committee according to French legislation (authorization N/35-11-12/58/11-15).
Ly5.2 C57BL/6 (B6) mice (Charles River Laboratories) were sub-lethally irradiated (5 Gy) using an X-ray Faxitron apparatus. BM cells from Ly5.1 B6 mice, transduced by LV and 107 cells, were injected IV into irradiated recipients. Two months after injection, BM cells from transplanted mice were stained with APC-labeled anti-CD45.1 and PerCP-Cy5.5-labeled anti-CD45.2 monoclonal antibodies (eBioscience). The percentage of CD45.1+ donor-type among CD45.2+ recipient-type BM cells was determined by flow cytometry (FACS CantoII, Becton Dickinson).
In LV-OVA experiments, transduction efficacy was determined by flow cytometry after staining with the 25-D1.16 monoclonal antibody recognizing the H-2Kb-OVA complex (eBioscience).
Transgene Persistence
Mice received intramuscular injection of LV-OVA (4 × 109 TU/mouse) or IV injection of LV-FIX (109 TU/mouse). LV-OVA mRNA was quantified from injected muscles by qPCR with SYBR green (Roche) using a LightCycler480 Roche, as described (27, 28). Relative amounts of LV-Ova mRNA were determined using a standard curve (serial dilutions of plasmid) and normalized by the amount of Eef2. Alternatively, FIX production was measured in plasma by ELISA (29).
Immune Response toward the Transgene Product
Subcutaneous injection of 20 μg human FIX (LFB, Les Ulis, France) emulsified in complete Freund’s adjuvant (Sigma) was carried out on the day of IV injection of LV-FIX to provoke immunization, as classically performed (10). The level of FIX-specific antibodies was measured in plasma by ELISA (30).
CD8+ T cells recognizing the OVA-specific SIINFEKL peptide were numerated by flow cytometry after staining with PE-conjugated H-2Kb/SIINFEKL dextramers (Immudex).
Results
We, first, evaluated whether inducing microchimerism by transplantation of gene-modified BM could be sufficient for inhibiting the production of transgene-specific antibodies in a gene therapy setting. For this purpose, we attempted to tolerize non-lethally irradiated Ly5.2 B6 mice against FIX by injecting BM cells from Ly5.1 congenic animals that had been transduced ex vivo with LV-FIX or LV-GFP as control (Figure 2A). One month after BM graft, mice were tested for human FIX expression in plasma. No circulating human FIX was found in both groups, demonstrating that the low frequency of LV-FIX expressing BM cells after non-lethal conditioning is not enough to produce circulating human FIX. Then, mice were challenged using a strong immunogenic regimen (FIX in complete Freund’s adjuvant) to induce both cell-mediated and humoral responses to FIX antigens, as described (5). At the same time, mice were injected IV with 109 TU of LV-FIX to promote the endogenous expression of human FIX. One month later, we observed a mixed BM chimerism where donor Ly5.1+ cells represented 21 ± 2.1% of the total BM cells. In mice that had received LV-FIX-modified BM cells, there was expression of human FIX at therapeutic level in plasma (612 ± 591 ng/mL, Figure 2B). In contrast, no circulating human FIX (<1.5 ng/mL) was found in controls that had received LV-GFP BM. These results indicate that the tolerization regimen had prevented mice from mounting an anti-FIX humoral immune response. Indeed, no FIX-specific antibodies were found in tolerized mice, whereas there was a strong humoral response in controls (Figure 2C).
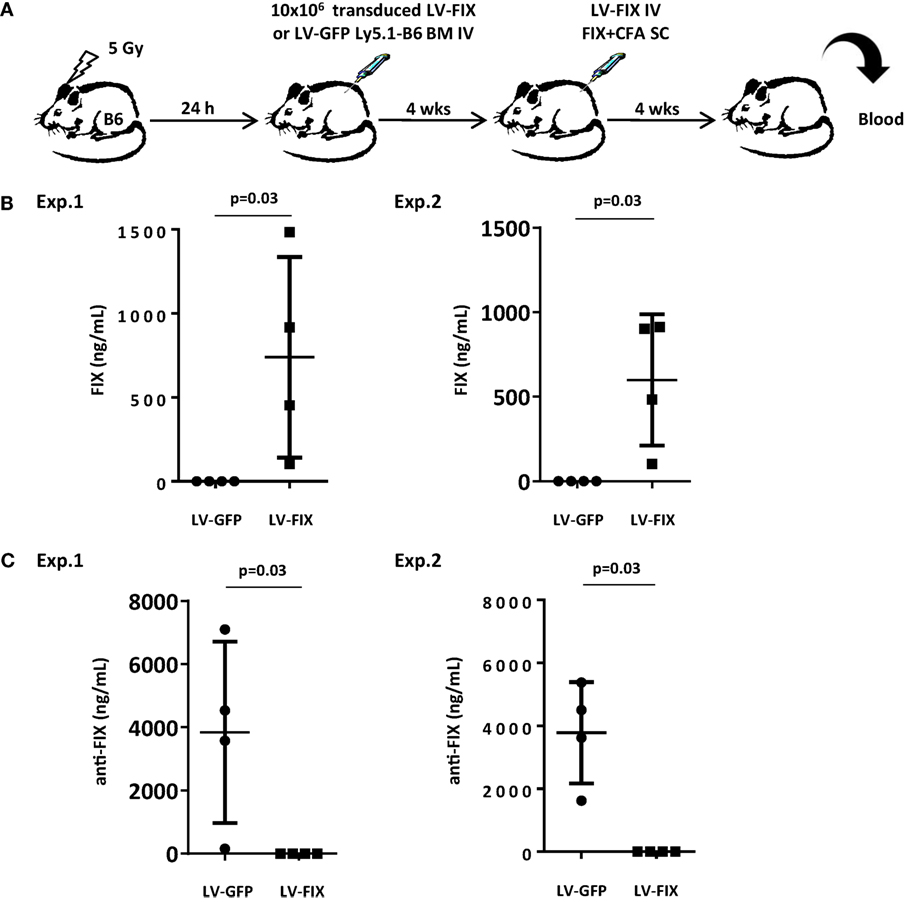
Figure 2. Non-lethally irradiated mice grafted with BM cells transduced by LV-FIX are tolerized toward human FIX. (A) Non-lethally irradiated (5 Gy) B6 mice (Ly5.2) were transplanted with BM cells transduced ex vivo with LV-FIX (or control LV-GFP) from Ly5.1 congenic B6 mice. One month after transplantation, chimeric mice were immunized by subcutaneous (SC) injection of recombinant FIX in complete Freund’s adjuvant and injected IV with the FIX-LV. (B) One month later, production of FIX in blood was evaluated by ELISA. Results are from two independent experiments using N = 4 mice per group. (C) Anti-FIX antibodies were titrated by ELISA.
We, next, evaluated the effect of this strategy on the cytotoxic cellular immune response in another model where the transgene product is membrane-bound and expressed from muscle. 5-Gy-conditioned B6 recipient mice were tolerized by grafting BM cells derived from Ly5.1 congenic donor mice transduced with a LV expressing the SIINFEKL immunodominant peptide of ovalbumin (OVA257–264) fused to H-2Kb (LV-OVA) or a control LV-GFP (Figure 3A). One month post-grafting, mice from the LV-OVA and LV-GFP tolerized groups both received LV-OVA intramuscularly for studying the OVA-specific cellular response and transgene persistence.
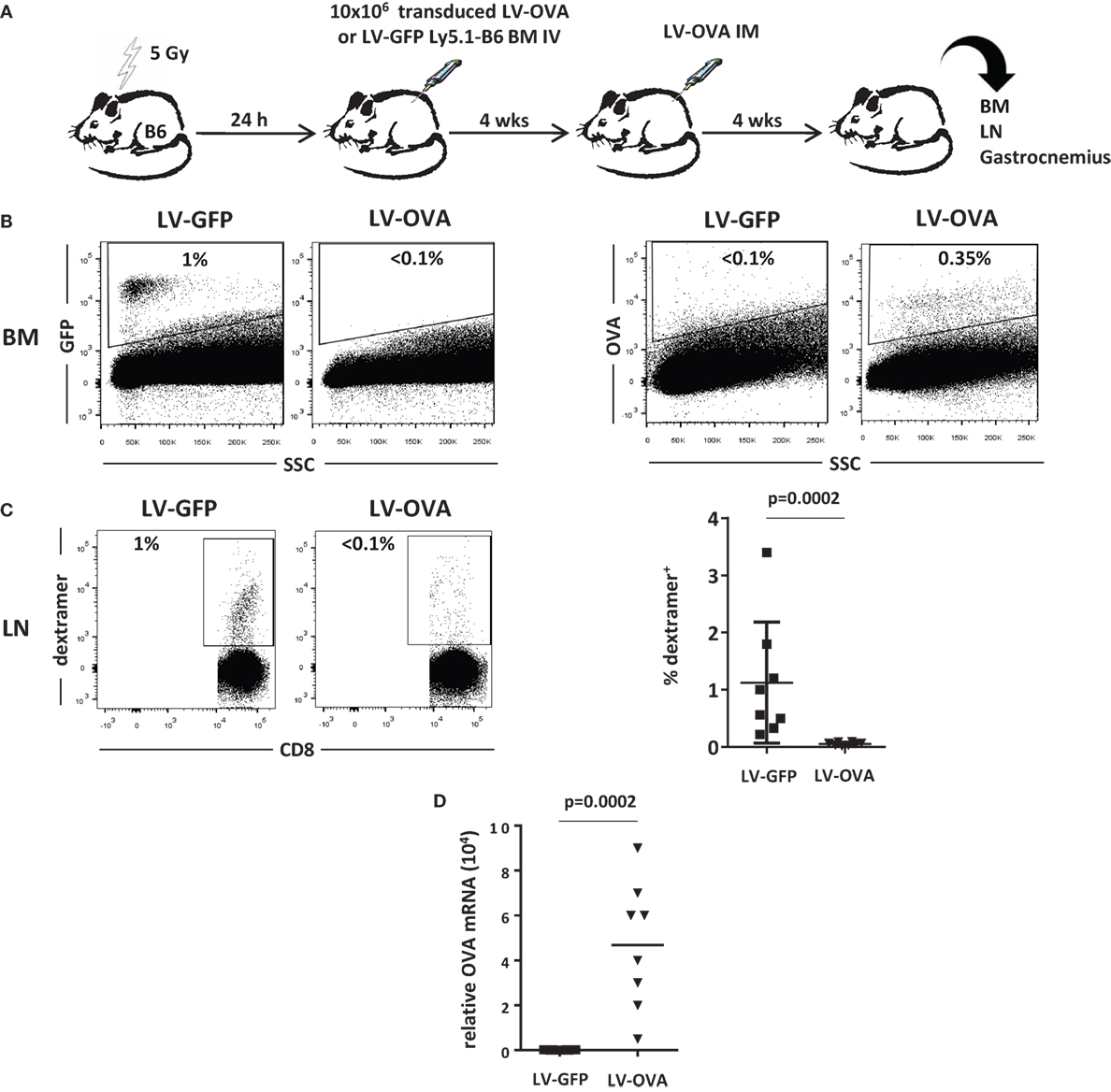
Figure 3. Non-lethally irradiated mice grafted with LV-OVA transduced BM cells are tolerized toward a membrane-bound transgene product. (A) Irradiated (5 Gy) B6 mice were transplanted with Ly5.1 B6 BM cells that had been transduced ex vivo with a LV expressing the SIINFEKL immunodominant peptide of ovalbumin (OVA257-264) covalently linked to H-2Kb so that to be expressed at the cell surface (LV-OVA) or a LV-GFP control. Then, chimeric mice were injected intramuscularly (IM) in one gastrocnemius with LV-OVA. (B) One month later, BM transgene expression was evaluated by flow cytometry for H-2Kb-OVA and GFP expression. (C) Immunization against transgenic OVA was evaluated in draining lymph nodes by staining specific CD8+ T cells with H-2Kb/SIINFEKL dextramers. Results are from two independents experiments with eight mice per group. (D) Expression of OVA mRNA was evaluated by RT-qPCR in injected gastrocnemius.
One month after LV-OVA challenge, the mixed chimerism could be confirmed and Ly5.1 donor cells represented 20 ± 3% of BM cells. Gene-modified cells only represented a minority of BM cells after conditioning, i.e., 1.0 ± 0.6% (GFP) or 0.3 ± 0.1% (OVA) (Figure 3B).
The cellular immune response was quantified by evaluating the percentage of SIINFEKL-specific CD8+ T cells in lymph nodes. In control mice that had received LV-GFP BM cells, specific CD8+ T cells expanded to reach 1.2 ± 0.5%, whereas they remained below the level of detection in tolerized mice that had received LV-OVA BM cells (Figure 3C). Hence, induction of microchimerism using gene-modified BM cells also prevented mice from mounting a transgene-specific cellular immune response.
To evaluate whether this state of immunological unresponsiveness also supported transgene persistence, we determined the level of transgene expression in the injected muscle. In control LV-GFP-tolerized mice, there was no detectable expression, indicating that transduced cells had been rejected by the CD8+ cytotoxic response (Figure 3D). In contrast, mice that had been tolerized by LV-OVA BM cells still expressed significant levels of transgenic mRNA 1 month after the intramuscular LV-OVA challenge.
Discussion
One of the major causes of FIX replacement therapy failure in hemophilia B is the development of inhibitors, i.e., anti-FIX antibodies (3). Similarly, development of antibodies in protein- and gene-replacement therapy represents an important potential complication in the treatment of many diseases (1–3). Furthermore, transgene-specific T CD8+ lymphocytes can destroy transduced cells and provoke failure of gene therapy as seen in preclinical animal models of hemophilia B (31, 32) and in clinical trials of gene therapy for muscular dystrophy (33).
Here, we demonstrated that expression of a transgene in a minority of BM cells after a non-lethal conditioning regimen is able to tolerize mice in an antigen-specific manner. With this intervention, we were able to prevent both humoral and CD8+ T cells responses, allowing for sustained transgene expression after subsequent systemic or tissue-specific in vivo gene transfer.
Induction of tolerance by transplantation of syngeneic BM cells that had been transduced by an alloantigen-specific transgene was previously explored (17). The underlying mechanisms involved T cell negative selection in the thymus, leading to a robust and lasting central T cell tolerance (34). Another study showed that grafting alloantigen-expressing BM cells is able to induce regulatory T cells, leading to peripheral tolerance (35). It is presumable that such mechanisms are also involved in the approach described, herein.
Oral tolerance or liver gene transfer has been used to tolerize mice to transgene products. Oral tolerance can be efficient in the context of gene therapy, but requires repeated oral administration of high doses of the tolerogen (28). Liver gene transfer is able to reduce the level of FIX-specific inhibitors and sustain long-term transgene expression (10, 13, 31, 36), but cannot be applied for other organs gene therapy. Notably, the approach proposed herein could be proposed for liver, muscle, or other organ gene transfer to improve transgene tolerance and long-term expression after a unique administration of a low number of gene-modified BM cells.
The present results are consistent with that of other studies in which FIX was expressed from transduced BM cells for both tolerization and therapeutic purposes. However, a high level of transduction was required to achieve tolerance and to also produce therapeutic levels of FIX (18–22). Here, we show that, with the current strategy, only a low frequency of LV-modified BM cells (less than 0.5%) is sufficient to tolerize mice. Importantly, this approach is compatible with different gene therapy settings, i.e., injecting the vector in blood, muscle, or potentially other extra-hematopoietic tissues. Therefore, it may prove useful for providing transgene-specific tolerance in the context of gene therapy of monogenic diseases beyond hemophilia.
In conclusion, the present results provide proof-of-concept of induction of tolerance via syngeneic microchimerism in a clinically translatable gene therapy setting. The use of LV-transduced BM cells after reduced intensity conditioning (37, 38) may represent a feasible candidate approach for augmenting the probability of success of different indications of gene therapy, by preventing detrimental humoral and cellular immune responses to the therapeutic transgene whatever its mode of expression.
Author Contributions
JM, GB, AM, LJ, and SA performed experiments; JM, JC, FM, and OB designed the research, analyzed data, and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We kindly thank Ted H. Hansen for providing the plasmid coding for the trimeric SIINFEKL/β2-microglobulin/H-2Kb fusion protein, Reynald Publier for his help in mice irradiation, Laurent Drouot for his contribution to qRT-PCR analysis, and Serge Jacquot and Sophie Candon for critical reading of the manuscript.
Funding
This work was funded in part by an Immunology-Hematology-Pneumology (IHP) ITMO collaborative grant from Inserm.
References
1. Astermark J, Lacroix-Desmazes S, Reding MT. Inhibitor development. Haemophilia (2008) 14(Suppl 3):36–42. doi:10.1111/j.1365-2516.2008.01711.x
2. van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet (2008) 372:1342–53. doi:10.1016/S0140-6736(08)61555-X
3. DiMichele DM. Immune tolerance in haemophilia: the long journey to the fork in the road. Br J Haematol (2012) 159:123–34. doi:10.1111/bjh.12028
4. Fields PA, Arruda VR, Armstrong E, Chu K, Mingozzi F, Hagstrom JN, et al. Risk and prevention of anti-factor IX formation in AAV-mediated gene transfer in the context of a large deletion of F9. Mol Ther (2001) 4:201–10. doi:10.1006/mthe.2001.0441
5. Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest (2003) 111:1347–56. doi:10.1172/JCI16887
6. Wang L, Cao O, Swalm B, Dobrzynski E, Mingozzi F, Herzog RW. Major role of local immune responses in antibody formation to factor IX in AAV gene transfer. Gene Ther (2005) 12:1453–64. doi:10.1038/sj.gt.3302539
7. Chen J, Wu Q, Yang P, Hsu HC, Mountz JD. Determination of specific CD4 and CD8 T cell epitopes after AAV2- and AAV8-hF.IX gene therapy. Mol Ther (2006) 13:260–9. doi:10.1016/j.ymthe.2005.10.006
8. Vandendriessche T, Thorrez L, Acosta-Sanchez A, Petrus I, Wang L, Ma L, et al. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost (2007) 5:16–24. doi:10.1111/j.1538-7836.2006.02220.x
9. High KA. The gene therapy journey for hemophilia: are we there yet? Blood (2012) 120:4482–7. doi:10.1182/blood-2012-05-423210
10. Annoni A, Cantore A, Della Valle P, Goudy K, Akbarpour M, Russo F, et al. Liver gene therapy by lentiviral vectors reverses anti-factor IX pre-existing immunity in haemophilic mice. EMBO Mol Med (2013) 5:1684–97. doi:10.1002/emmm.201302857
11. Brown BD, Sitia G, Annoni A, Hauben E, Sergi LS, Zingale A, et al. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood (2007) 109:2797–805. doi:10.1182/blood-2006-10-049312
12. Rogers GL, Martino AT, Zolotukhin I, Ertl HC, Herzog RW. Role of the vector genome and underlying factor IX mutation in immune responses to AAV gene therapy for hemophilia B. J Transl Med (2014) 12:25. doi:10.1186/1479-5876-12-25
13. Markusic DM, Hoffman BE, Perrin GQ, Nayak S, Wang X, LoDuca PA, et al. Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol Med (2013) 5:1698–709. doi:10.1002/emmm.201302859
14. Lei TC, Scott DW. Induction of tolerance to factor VIII inhibitors by gene therapy with immunodominant A2 and C2 domains presented by B cells as Ig fusion proteins. Blood (2005) 105:4865–70. doi:10.1182/blood-2004-11-4274
15. Wang X, Shin SC, Chiang AF, Khan I, Pan D, Rawlings DJ, et al. Intraosseous delivery of lentiviral vectors targeting factor VIII expression in platelets corrects murine hemophilia A. Mol Ther (2015) 23:617–26. doi:10.1038/mt.2015.20
16. Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med (2008) 358:353–61. doi:10.1056/NEJMoa071074
17. Jindra PT, Tripathi S, Tian C, Iacomini J, Bagley J. Tolerance to MHC class II disparate allografts through genetic modification of bone marrow. Gene Ther (2013) 20(5):478–86. doi:10.1038/gt.2012.57
18. Moayeri M, Hawley TS, Hawley RG. Correction of murine hemophilia A by hematopoietic stem cell gene therapy. Mol Ther (2005) 12:1034–42. doi:10.1016/j.ymthe.2005.09.007
19. Bigger BW, Siapati EK, Mistry A, Waddington SN, Nivsarkar MS, Jacobs L, et al. Permanent partial phenotypic correction and tolerance in a mouse model of hemophilia B by stem cell gene delivery of human factor IX. Gene Ther (2006) 13:117–26. doi:10.1038/sj.gt.3302638
20. Ide LM, Gangadharan B, Chiang KY, Doering CB, Spencer HT. Hematopoietic stem-cell gene therapy of hemophilia A incorporating a porcine factor VIII transgene and nonmyeloablative conditioning regimens. Blood (2007) 110:2855–63. doi:10.1182/blood-2007-04-082602
21. Ramezani A, Zweier-Renn LA, Hawley RG. Factor VIII delivered by haematopoietic stem cell-derived B cells corrects the phenotype of haemophilia A mice. Thromb Haemost (2011) 105:676–87. doi:10.1160/TH10-11-0725
22. Verhoeyen E, Relouzat F, Cambot M, Costa C, Negre D, Legrand F, et al. Stem cell factor-displaying simian immunodeficiency viral vectors together with a low conditioning regimen allow for long-term engraftment of gene-marked autologous hematopoietic stem cells in macaques. Hum Gene Ther (2012) 23:754–68. doi:10.1089/hum.2012.020
23. Follenzi A, Ailles LE, Bakovic S, Geuna M, Naldini L. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat Genet (2000) 25:217–22. doi:10.1038/76095
24. Yu YY, Netuschil N, Lybarger L, Connolly JM, Hansen TH. Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. J Immunol (2002) 168:3145–9. doi:10.4049/jimmunol.168.7.3145
25. Grimm D, Zhou S, Nakai H, Thomas CE, Storm TA, Fuess S, et al. Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood (2003) 102:2412–9. doi:10.1182/blood-2003-02-0495
26. Gay V, Moreau K, Hong SS, Ronfort C. Quantification of HIV-based lentiviral vectors: influence of several cell type parameters on vector infectivity. Arch Virol (2012) 157:217–23. doi:10.1007/s00705-011-1150-5
27. Adriouch S, Franck E, Drouot L, Bonneau C, Jolinon N, Salvetti A, et al. Improved immunological tolerance following combination therapy with CTLA-4/Ig and AAV-mediated PD-L1/2 muscle gene transfer. Front Microbiol (2011) 2:199. doi:10.3389/fmicb.2011.00199
28. Hardet R, Chevalier B, Dupaty L, Naimi Y, Riou G, Drouot L, et al. Oral-tolerization prevents immune responses and improves transgene persistence following gene transfer mediated by adeno-associated viral vector. Mol Ther (2016) 24(1):87–95. doi:10.1038/mt.2015.146
29. Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood (2006) 108:3321–8. doi:10.1182/blood-2006-04-017913
30. Crudele JM, Finn JD, Siner JI, Martin NB, Niemeyer GP, Zhou S, et al. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood (2015) 125:1553–61. doi:10.1182/blood-2014-07-588194
31. Brown BD, Cantore A, Annoni A, Sergi LS, Lombardo A, Della Valle P, et al. A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice. Blood (2007) 110:4144–52. doi:10.1182/blood-2007-03-078493
32. Nayak S, Herzog RW. Progress and prospects: immune responses to viral vectors. Gene Ther (2010) 17:295–304. doi:10.1038/gt.2009.148
33. Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med (2010) 363:1429–37. doi:10.1056/NEJMoa1000228
34. Kang ES, Iacomini J. Induction of central deletional T cell tolerance by gene therapy. J Immunol (2002) 169:1930–5. doi:10.4049/jimmunol.169.4.1930
35. Forman D, Kang ES, Tian C, Paez-Cortez J, Iacomini J. Induction of alloreactive CD4 T cell tolerance in molecular chimeras: a possible role for regulatory T cells. J Immunol (2006) 176:3410–6. doi:10.4049/jimmunol.176.6.3410
36. Annoni A, Goudy K, Akbarpour M, Naldini L, Roncarolo MG. Immune responses in liver-directed lentiviral gene therapy. Transl Res (2013) 161:230–40. doi:10.1016/j.trsl.2012.12.018
37. Childs R, Clave E, Contentin N, Jayasekera D, Hensel N, Leitman S, et al. Engraftment kinetics after nonmyeloablative allogeneic peripheral blood stem cell transplantation: full donor T-cell chimerism precedes alloimmune responses. Blood (1999) 94:3234–41.
Keywords: gene therapy, hemophilia B, tolerance, microchimerism, animal model
Citation: Martinet J, Bourdenet G, Meliani A, Jean L, Adriouch S, Cohen JL, Mingozzi F and Boyer O (2016) Induction of Hematopoietic Microchimerism by Gene-Modified BMT Elicits Antigen-Specific B and T Cell Unresponsiveness toward Gene Therapy Products. Front. Immunol. 7:360. doi: 10.3389/fimmu.2016.00360
Received: 18 July 2016; Accepted: 02 September 2016;
Published: 15 September 2016
Edited by:
Duncan Howie, University of Oxford, UKReviewed by:
Bruce Milne Hall, University of New South Wales, AustraliaDavid William Scott, Unversity of the Health Sciences, USA
Copyright: © 2016 Martinet, Bourdenet, Meliani, Jean, Adriouch, Cohen, Mingozzi and Boyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Boyer, olivier.boyer@chu-rouen.fr