 Hang Yang
Hang Yang Mona Helena Biermann
Mona Helena Biermann Jan Markus Brauner
Jan Markus Brauner Yi Liu
Yi Liu Yi Zhao
Yi Zhao Martin Herrmann
Martin Herrmann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 August 2016
Sec. Molecular Innate Immunity
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00302
This article is part of the Research Topic NETosis 2: the excitement continues View all 39 articles
Recent data suggest that NETosis plays a crucial role in the innate immune response and disturbs the homeostasis of the immune system. NETosis is a form of neutrophil-specific cell death characterized by the release of large web-like structures referred to as neutrophil extracellular traps (NETs). NETs are composed of DNA strands associated with histones and decorated with about 20 different proteins, including neutrophil elastase, myeloperoxidase, cathepsin G, proteinase 3, high mobility group protein B1, and LL37. Reportedly, NETosis can be induced by several microbes, and particulate matter including sterile stimuli, via distinct cellular mechanisms. Meanwhile, suicidal NETosis and vital NETosis are controversial. As we enter the second decade of research on NETosis, we have partly understood NETs as double-edged swords of innate immunity. In this review, we will discuss the mechanisms of NETosis, its antimicrobial action, and role in autoimmune diseases, as well as the relatively new field of NET-associated mitochondrial DNA.
Neutrophil granulocytes are the most abundant type of white blood cells in humans and play a vital role in innate immunity by defending the host against invading pathogens. The immune regulatory functions of neutrophils include phagocytosis, generation of reactive oxygen species (ROS), degranulation, and the formation of neutrophil extracellular traps (NETs), a process referred to as NETosis. NETosis is accepted as a specific form of cell death subroutine performed by granulocytes, differing from apoptosis and necrosis (1, 2). When neutrophils undergo NETosis, nuclear and granular membranes disintegrate, the chromatin decondenses, and it diffuses into the cytoplasm, mixing with cytoplasmic proteins. This is followed by plasma membrane rupture and the release of chromatin, decorated with granular proteins, into the extracellular space (2, 3). NETs consist of chromatin fibers with diameters of 15–17 nm that contain DNA and the histones H1, H2A, H2B, H3, and H4. Moreover, the DNA fibers are decorated with several proteins like neutrophil elastase (NE), myeloperoxidase (MPO), cathepsin G, proteinase 3 (PR3), high mobility group protein B1 (HMGB1), and LL37, thus displaying proinflammatory characteristics (1). In the past decade, new aspects of neutrophil functions have emerged unveiling their significance not only in defending the host against microbes but also in contributing to many autoimmune pathological conditions. Therefore, the purpose of this review is to present and discuss the current knowledge about the mechanisms of NETosis and its role in the pathogenesis of autoimmune diseases.
Neutrophil suicide, distinct from either necrosis or apoptosis, was first described following chemical stimulation with phorbol 12-myristate 13-acetate (PMA) in 1996 (4). This form of cell death was characterized by the disintegration of nuclear and granular membranes and by the release of decondensed chromatin into the cytoplasm. In 2004, Zychlinsky and colleagues reported that neutrophil suicide resulted in the release of large web-like structures composed of decondensed chromatin and neutrophil antimicrobial factors, and coined the name neutrophil extracellular traps (1). In their studies, they used PMA and interleukin-8 (IL-8) to elicit NETs in vitro. In 2007, it was reported that, going along with chromatin decondensation, neutrophils undergo an NADPH oxidase-dependent death process that includes nuclear envelope disintegration and the mixing of nucleic acids and granule proteins within a large intracellular vacuole (3). After the association of nucleic acids and granule proteins, NETs are released via plasma membrane perforation and cell lysis. This process is completed1–4 h after the inciting stimulus. The released chromatin structures are prone to bind particular matter, e.g., bacteria. The authors concluded that PMA-induced NETosis is a form of a beneficial suicide (3). Apart from PMA and IL-8, bacteria, fungi, protozoa, antibody–antigen complexes (5), autoantibodies (6), tumor necrosis factor (TNF), interferon (IFN) (7), and further stimuli also trigger NETosis.
Conventional suicidal NETosis has long been recognized as a distinct form of active cell death. In addition, some researchers have described a different mechanism by which NETs are formed, termed vital NETosis. This non-suicidal pathway allows NET release from neutrophils staying viable (8–12).
Conventional suicidal NETosis is frequently initiated by ligand binding to neutrophil toll-like receptors and receptors for IgG–Fc, complement, or cytokines (1, 5, 13). Upon activation of these receptors, calcium storages of the endoplasmic reticulum release calcium ions into the cytoplasm. Elevated cytoplasmic calcium levels increase protein kinase C (PKC) activity and phosphorylation of gp91phox (14). This induces the assembly of the cytosolic and membrane-bound subunits of NADPH oxidase into functional complexes at cytoplasmic or phagosomal membranes (also called phagocytic oxidase, PHOX) and the subsequent generation of ROS (15). Under the influence of ROS, granules and the nuclear envelope rupture. Subsequently, the released nuclear, granular, and cytoplasmic contents blend. NE and MPO, usually stored in azurophilic granules, migrate to the nucleus. Here, NE degrades the linker histone H1 and processes the core histones, and MPO enhances chromatin decondensation (15). Histone deimination by peptidyl arginine deiminase 4 (PAD4) and proteolytic cleavage of histones initiated before nuclear breakdown additionally contribute to chromatin decondensation (16, 17). The rupture of the plasma membrane allows the release of NETs and leads to cell death and the loss of viable cell functions of like migration and phagocytosis (Figure 1) (15).
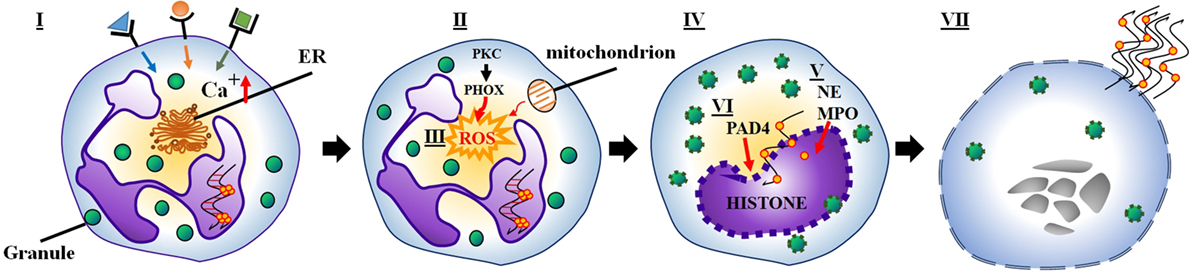
Figure 1. (I) Several stimuli (e.g., bacteria, viruses, fungi) initiate NETosis by binding to neutrophil receptors (e.g., Fc receptors, TLRs), which activate the endoplasmic reticulum to release stored calcium ions. (II) Elevated cytoplasmic calcium levels increase PKC activity, which induces NADPH oxidase to assemble into a functional complex (PHOX). (III) Subsequently, PHOX (or alternatively the mitochondrial respiratory chain) generate ROS. (IV) ROS generation leads to the rupture of granules and the nuclear envelope. (V) Meanwhile, NE and MPO translocate to the nucleus. (VI) As a result, histone deimination and chromatin decondensation contribute to the formation of NETs. (VII) Finally, the rupture of the plasma membrane causes neutrophil lysis and allows the release of NETs.
The generally accepted notion that ROS play a crucial role in the classical suicidal NETosis pathway is based on two important observations: (1) Neutrophils from patients with chronic granulomatous disease (CGD), not capable of performing the oxidative burst, show strongly reduced abilities to form NETs. This is independent of the type of mutation leading to a defective PHOX complex. CGD patients suffer from severe and often chronic infections (3, 18). Moreover, treatment with H2O2 rescued the production of NETs in neutrophils from CGD patients, downstream of the PHOX complex (3). (2) ROS scavengers, such as N-acetylcysteine, or trolox reportedly inhibit NETosis (3, 19). In fact, it remains unclear how ROS participate in the dismantling of the nuclear envelope or the mixing of the NET components. Some studies suggest that ROS directly promote the morphologic changes observed during NETosis (14). ROS may alternatively inactivate caspases, thereby inhibiting apoptosis and favoring autophagy. This leads to dissolution of cellular membranes (20). These two alternatives are not mutually exclusive: under certain experimental conditions, each of them can also act independently. There is now growing evidence that some stimuli induce NETosis independent of NADPH oxidase. Oxidant-independent release of NETs was studied in detail by Winterbourn and colleagues (21).
Peptidyl arginine deiminase 4 catalyzes the conversion of arginine residues to citrullinein polypeptides, thereby eliminating a positive charge of the protein. Thus, citrullination of histones weakens the stability of nucleosomes (22, 23). Loss of positive charges causes the opening of the compact structure of chromatin and allows decondensation and dispersion of chromatin in the form of NETs. Consistently, neutrophils from mice with a PAD4 deficiency display impaired capacities to form NETs and are highly susceptible to severe skin infections in vivo (16, 17). However, PAD4 deficiency does not contribute to lung infections caused by influenza virus (16).
Contrary to previous studies describing the canonical pathways of NETosis as a process requiring several hours, Clark et al. reported in 2007 that lipopolysaccharide (LPS)-stimulated NETosis occurred within just 30 min involving TLR4 on platelets (8). It was demonstrated that neutrophils that released NETs remained impermeable for SYTOX Green, indicating that they remained structurally intact. Therefore, the authors later coined the term vital NETosis (12). Electron microscopy revealed that NET release induced by Staphylococcus aureus occurs via blebbing of the nuclear envelope and vesicular exportation in vitro and in vivo (9). As a result, this pathway preserved the integrity of the neutrophils’ plasma membranes (Figure 2). NETting neutrophils became anuclear cytoplasts capable of chasing and imprisoning live Staphylococci (10). Candida albicans was reported a further stimulus of vital NETosis (11). It still remains controversial whether and how suicidal and vital NETosis coexist. Furthermore, it is not clear if a neutrophil that has ejected (parts of) its DNA should be termed “viable.”
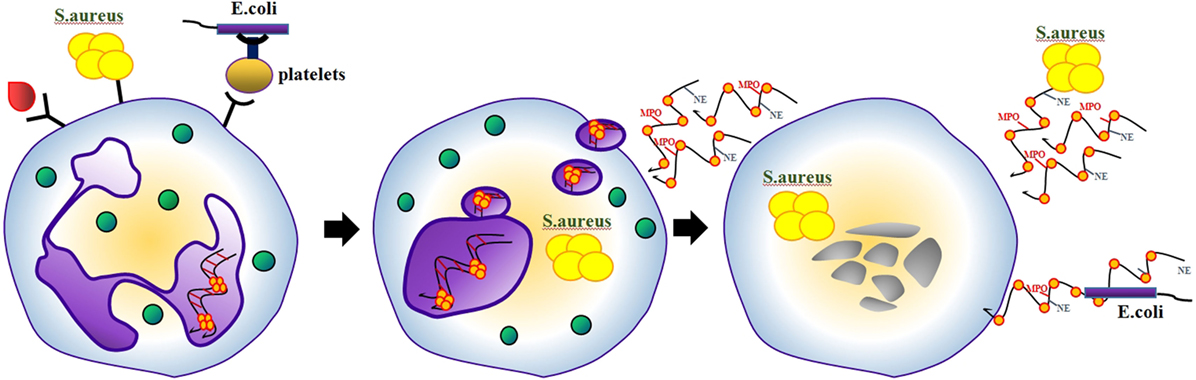
Figure 2. Some NETosis-inducing stimuli involve TLR4 on platelets. Under these conditions, neutrophils release NETs via blebbing of the nuclear envelope and vesicular exportation. As a result, neutrophils become nuclear cytoplasts, which are still able to migrate and retain several conventional functions of viable neutrophil.
As mentioned above, ROS are indispensable for several kinds of NETosis (24). In mammals, both the mitochondrial respiration chain and the NADPH oxidase independently contribute to the production of ROS (25). Recently, it has been observed that in vivo inhibition of mitochondrial ROS production reduced intracellular ROS levels and NETosis (26). Ribonucleoprotein immune complexes (RNP ICs) were used to stimulate neutrophils and mitochondrial ROS generation. Mitochondria became hypopolarized, translocated to the cell surface, and were observed within the expelled NETs. Concomitantly, mitochondrial ROS oxidized mitochondrial DNA (mtDNA), which is proinflammatory in vitro. When injected into mice, oxidized mtDNA triggered inflammation and type I IFN production via a pathway dependent on the DNA sensor STING (7, 26). Mitochondria have evolved from bacteria and contain unmethylated CpG motifs (27) as well as N-formylated peptides (28). Similar to bacteria, extracellular mitochondria are stimulators of proinflammatory signaling. Several reports attribute this effect to the unmethylated CpG DNA repeats within the mtDNA (29), others highlight the effect of DNA oxidation (28).
In patients with systemic lupus erythematosus (SLE), abnormal NETosis and defects in the clearance of NETs were found to promote the production and release of type I IFN (30). In contrast, patients with CGD carry an increased risk to suffer from SLE, despite lacking functional NADPH oxidase activity (18), the major source of ROS in activated healthy neutrophils. Based on this observation, one might question whether increased NETosis is a factor contributing to the etiopathogenesis of SLE. Instead, deficiency of the clearance of NETs is likely to foster the antinuclear autoimmunity in patients with SLE (30, 31). However, Kaplan and colleagues reexamined the importance of ROS in low-density granulocytes from patients with CGD and observed that this granulocyte subpopulation undergoes spontaneous NETosis and that their mitochondrial respiration produces sufficient amounts of ROS to execute NETosis. The levels of ROS derived from low-density granulocytes correlated with the levels of type I IFN in the corresponding patients. Accordingly, the authors not only confirmed that mitochondria drove NETosis but also concluded that NETosis is a pathological factor able to foster SLE (26, 32). Consistently, anti-mtDNA antibodies were elevated in the sera of patients with SLE, and antibody levels correlated with IFN scores and disease activity. Immune complexes containing mtDNA induced more IFN-α than those with nuclear dsDNA. Thus, anti-mtDNA antibodies can be considered as typical for driving both SLE and lupus nephritis (33). As a drug that selectivity inhibits mitochondrial respiratory chain complex I and decreases NADPH oxidase activity (34), metformin may be a new option to treat SLE (33).
Numerous microbes reportedly induce formation of NETs (Table 1). NET-inducing molecules include the bacterial cell surface components LPS, lipoteichoic acid, and their breakdown products. Several bacteria and fungi were reported to potently induce NET formation, such as Staphylococcus aureus (9, 35), Streptococcus sp. (36), Haemophilus influenzae (37), Klebsiella pneumonia (15), Listeria monocytogenes (38), Mycobacterium tuberculosis (39), Shigella flexneri (1), Aspergillus nidulans, Aspergillus fumigatus, and Candida albicans (40–42). Further examples are pathogens, such as Yersinia (1) and members of the oral microbiome, including Porphyromonas gingivalis (43). NETs can immobilize and kill a broad range of microbes, including bacteria, fungi, and protozoa (1, 9, 15, 35–42), and thus prevent the dissemination of microbial pathogens (37). Some studies have questioned the killing capabilities of NETs since viable Staphylococcus aureus and Candida albicans blastospores were released from NETs by incubation with DNases (44). Branzk et al. found that in response to large pathogens, like filamentous Candida albicans, neutrophils selectively released NETs (45). Intriguingly, NETosis did not occur in response to the yeast form of Candida albicans or single bacteria. Phagocytosis via dectin-1 acted as a sensor of microbial size and prevented NET release by downregulating translocation of NE to the nucleus. Apart from directly killing microbes, NETs inactivate microbial “virulence factors” that alter the function of host cells. NET-associated NE specifically cleaved virulence factors of Shigella flexneri, Salmonella typhimurium, and Yersinia enterocolitica (1). The serine proteases cathepsin G and PR3 may also destroy virulence factors of further classes of microbes (46). NETs contain several proteins that inhibit microbes, including enzymes, antimicrobial peptides, calgranulin, and histones. The microbicidal activity of NETs results from the combined action of several components being enhanced by the high local concentrations of mediators on the NETs’ surfaces (15).

Table 1. Pathogens that induce NETs.
Various components of NETs contribute to different aspects of microbicidal activity. It was shown that the activity of MPO on NETs is essential to eliminate S. aureus (47). The antifungal activity of NETs has been assigned to calgranulin (48), which chelates zinc, a cation required for fungal growth (15). Also, histones restrict microbial growth very efficiently, and antibodies against histones prevent NET-mediated microbicidal activity (1). Microbes are suggested to be entrapped due to electrostatic interactions between the positively charged bacterial surface and the negatively charged chromatin fibers based on electrostatic interactions (49). Encapsulated pathogens or those that can change their surface charge may escape entrapment (50). Importantly, several bacteria are able to degrade NETs by nucleases and thus escape NET-mediated entrapment (Table 2). These include the Gram-negative pathogen Vibrium cholera (51) and the Gram-positive bacteria Streptococcus pneumoniae (52), Streptococcus pyogenes (53), Yersinia ssp. (54), Streptococcus agalactiae (55), Streptococcus suis (56), Staphylococcus aureus (57), and Aeromonas hydrophila (58). This emphasizes the importance of nucleases as pathogenic factors.

Table 2. Pathogens which evade entrapments via degrading NETs.
Vasculitis manifests in vessel wall inflammation and can affect any organ system of the body. ANCA-associated vasculitis (AAV), a subgroup of the vasculitides, is characterized by involvement of the small vessels, a neutrophil-rich necrotizing inflammation, and the presence of anti-neutrophil cytoplasmic antibodies (ANCAs) (59). AAV comprises granulomatosis with polyangiitis (formerly Wegener’s granulomatosis), microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis (formerly Churg–Strauss syndrome). Many ANCAs are directed against PR3 or MPO, enzymes typically found in the azurophilic granules of neutrophils and on the surfaces of NETs (60). NETs are reportedly released by ANCA-stimulated neutrophils and in turn contain the autoantigens PR3 and MPO (39). This suggests that NET formation triggers vasculitis and promotes the autoimmune response against neutrophil components in individuals with small-vessel vasculitis (61). Consistently, increased levels of NET remnants containing complexes of nucleosomes and MPO have been detected in the circulation of patients with active vasculitis (39) and in patients with active AAV (60). Neutrophils of patients with AAV exhibited an increased tendency for spontaneous cell death. The levels of NET remnants were positively correlated with disease activity and neutrophil count, but inversely with ANCA at least during remission.
Systemic lupus erythematosus is a complex multifactorial autoimmune disease associated with severe organ damage. NETs are considered a potential source of autoantigens. Polymorphonuclear leukocytes (PMNs) of patients with SLE display an increased propensity to execute NETosis in conjunction with impaired degradation of NETs by circulating DNase1. The aberrant NETs induce type I IFN, which is associated with vascular complications and tissue damage (30, 62).
High numbers of low-density granulocytes have been identified as a particular subset of neutrophils in SLE patients. Low-density granulocytes persistently produced TNF and type 1 IFN, and spontaneously underwent NETosis (24). Furthermore, increased IFN-α in SLE patients is an important driving force that primes neutrophils for the execution of NETosis (63).
Not only production but also degradation of NETs is altered in SLE patients. Sera of a subgroup of SLE patients degrade NETs less efficiently than those of healthy controls (30). The deficient clearance of NETs in patients with SLE correlates with high titers of anti-NET antibodies and renal involvement (30). In healthy individuals, mononuclear phagocytes clear NETs in cooperation with DNase1 and C1q both synergizing in predigesting the chromatin part (64). The activities of serum DNase1 in patients with SLE are lower than that of healthy controls (65). Increased serum levels of DNase1 inhibitors, rare mutations in the gene of DNase1, and anti-DNase 1 antibodies may explain the decreased activity of DNase 1 (66, 67). Circulating chromatin in the form of immune complexes in individuals with SLE contains LL37, which triggers TLR9 in plasmacytoid dendritic cells, induces IFN-α synthesis, and protects nucleic acids from degradation by nucleases (68, 69). A study found that the individual NET degradation activity in the circulation of a given patient changed with disease activity. Sera of patients with SLE, which were not able to degrade NETs, showed increased complement consumption, since NETs activate the classical complement pathway due to their interaction with C1q (70). Thus, strategies that eliminate NETs and their components from the circulation pose a promising therapeutic approach for the treatment of patients with SLE (70).
Neutrophil extracellular traps promote thrombosis by providing a scaffold and stimulus for platelet and red blood cell adhesion and aggregation (71), thus enhancing coagulation (72). Neutrophils in thrombi are required for propagation of deep venous thromboses by binding factor XII and supporting its activation through NETosis (73). The major components of NETs (DNA, histones, and proteases) all display procoagulant properties. DNA induces thrombin generation in plasma and increases the protease activity of coagulation factors (74, 75). Histones may directly induce epithelial and endothelial cell death (76), and can mediate thrombosis in vivo (77). Histones were found to inhibit anticoagulation of plasma by promoting thrombin generation and hamper thrombomodulin function (78, 79). Elastase inactivated the tissue factor pathway inhibitor; thus, further increasing coagulation and fibrin deposition in vivo (72). Release of NETs in the vascular compartment triggered a procoagulant state and promoted binding and activation of platelets leading to thrombosis (80).
In the autoimmune disease, rheumatoid arthritis (RA), the formation of autoantibodies to citrullinated proteins (ACPA) is thought to be a key pathogenic factor. Given that histone citrullination is implicated in NET formation, NETosis may play a critical role in RA (81). In 2013, Kaplan and colleagues found that neutrophils from patients with RA had a greater tendency to release NETs than neutrophils from healthy controls. RA serum and synovial fluid was a strong inducer of NETosis (82). Furthermore, NETosis resulted in the externalization of citrullinated protein antigens and immune-stimulatory molecules that may promote aberrant adaptive and innate immune responses in the joint.
Diabetes mellitus (or diabetes) is a chronic, lifelong condition, in which impaired insulin secretion and variable degrees of peripheral insulin resistance lead to hyperglycemia and affect the body’s ability to use food energy. Under conditions of hyperglycemia, neutrophils reportedly produce more superoxide and cytokines, like TNF-α, which triggers NETosis (83, 84). Based on these studies, we speculated that hyperglycemia may facilitate NETosis. Recently, Wong et al. (85) isolated neutrophils from type 1 and type 2 diabetic humans and mice. Nearly twice as many neutrophils derived from patients released NETs in comparison to cells from healthy controls. The authors attributed this to PAD4 and revealed a fourfold upregulation of PAD4 protein expression in the neutrophils from individuals with diabetes as compared to healthy controls. It is well established that delayed wound healing is a hallmark of patients with diabetes. The authors reported that large quantities of NETs were found in excisional skin wounds of diabetic mice and that DNase1, which dismantled NETs, accelerated wound healing. Despite the triggers of NETosis in wounds remaining elusive, it has been confirmed that inhibiting NETosis or degrading NETs improved wound healing and reduced NET-driven chronic inflammation in diabetes (85). However, the exact role of NETosis in wound healing remains to be revealed.
NETosis may influence tumor development during many stages, including growth, angiogenesis, and metastasis. It has been observed that there is a large necrotic area of dead neutrophils and NET-like structures in Lewis lung carcinoma and Ewing sarcoma (86, 87). It remains to be clarified whether these NETs are responsible for the generation of the necrotic areas. Alternatively, NETs may serve to shield healthy tissues from necrotic areas. A study observed NET deposition on the microvasculature and subsequent local trapping of circulating cancer cells. The tumor cells, immobilized by NETs, survived and proliferated to form nodules. This suggests a role for NETs in enhancing tumor metastasis (88). However, whether NETs just protect or anchor cancer cells physically or whether they promote tumor growth is still elusive.
The pathology of sepsis results from infection, hyperinflammatory host response, and immune paralysis. During sepsis, NETs are released in the vascular system, where they trap bacteria (8, 12). Trapped bacteria can be killed, protecting patients from bacterial overflow (1, 44). In contrast, NET deposition in organs and their pro-thrombotic activities may also contribute to organ failure (89, 90). When researchers subjected mice to polymicrobial sepsis following cecal ligation and puncture, PAD4-deficient mice showed a similar survival rate when compared to wild-type controls (91). However, PAD4-deficient mice were partially protected from LPS-induced shock, indicating that NETs may contribute to the toxic inflammatory and procoagulant host response to bacteria in sepsis. The authors proposed that preventing NET formation by PAD4 inhibition in inflammatory or thrombotic diseases is not likely to increase host vulnerability to bacterial infections (91).
Progress in the research on NETosis has greatly increased our understanding of its role in immunological processes and autoimmune disorders. Recent studies described how autoantigens, released during NETosis, activate immune cells and that cytokines in turn give rise to further NETosis. Aggregated NETs finally sequester and degrade proinflammatory mediators to avoid excessive inflammation (8, 14). The published data also revealed that blocking the process of NETosis or inhibiting the activity of components in NETs might be effective in the treatment of autoimmune diseases. Future work investigating the exact process of NETosis and the interplay of NET components and the immune system will contribute to a deeper understanding of the role of neutrophils in the induction and resolution of inflammation.
HY and YZ wrote the first draft of this article. HY and YL designed the figures. YL, MB, JB, and MH critically revised the manuscript for important intellectual content. All authors approved the final version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The present work was supported by the National Natural Science Foundation of China (81273286) to YZ, the international collaborative project in Science and Technology of the Sichuan province (2015HH0050) to YZ and MH, the international collaborative project in Science and Technology of the Sichuan province (2014HH0027) to YL and MH. MH was supported by the German Research Council CRC-643-B5 and B8, CRC-1181-C03, KFO257, SPP 1468 Osteoimmunology IMMUNOBONE, and the doctoral training programs GK1660 and SFB643.
1. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):12–5. doi:10.1126/science.1092385
2. Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE (2007) 27(379):pe11. doi:10.1126/stke.3792007pe11
3. Tobias AF, Ulrike A, Christian G, Robert H, Ilka S, Volker W, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176(2):231–41. doi:10.1083/jcb.200606027
4. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol (1996) 59(2):229–40.
5. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi:10.1126/scitranslmed.3001201
6. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15(6):623–5. doi:10.1038/nm.1959
7. Sibylla M, Mirjana U, Arezoo D, Patrick AO, Christa B, Martin FF, et al. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem (2004) 279(42):44123–32. doi:10.1074/jbc.M405883200
8. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13(4):463–9. doi:10.1038/nm1565
9. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185(12):7413–25. doi:10.4049/jimmunol.1000675
10. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18(9):1386–93. doi:10.1038/nm.2847
11. Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol (2013) 190(8):4136–48. doi:10.4049/jimmunol.1202671
12. Yipp BG, Kubes P. NETosis: how vital is it? Blood (2013) 122(16):2784–94. doi:10.1182/blood-2013-04-457671
13. Munks MW, McKee AS, Macleod MK, Powell RL, Degen JL, Reisdorph NA, et al. Aluminum adjuvants elicit fibrin-dependent extracellular traps in vivo. Blood (2010) 116(24):5191–9. doi:10.1182/blood-2010-03-275529
14. Kaplan MJ, Marko R. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol (2012) 189(6):2689–95. doi:10.4049/jimmunol.1201719
15. Papayannopoulos V, Metzler MD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191(3):677–91. doi:10.1083/jcb.201006052
16. Saskia H, John RT, Sanja A, Kerra AM. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One (2011) 6(7):e22043. doi:10.1371/journal.pone.0022043
17. Pingxin L, Ming L, Lindberg MR, Kennett MJ, Na X, Yanming W. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207(9):1853–62. doi:10.1084/jem.20100239
18. Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood (2009) 114(13):2619–22. doi:10.1182/blood-2009-05-221606
19. Lim MB, Kuiper JW, Katchky A, Goldberg H, Glogauer M. Rac2 is required for the formation of neutrophil extracellular traps. J Leukoc Biol (2011) 90(4):771–6. doi:10.1189/jlb.1010549
20. Remijsen Q, Vanden BT, Wirawan E, Asselbergh E, Parthoens E, De RR, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res (2011) 21(2):290–304. doi:10.1038/cr.2010.150
21. Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol (2012) 92(4):841–9. doi:10.1189/jlb.1211601
22. Wang Y, Li M, Sonja S, Sarah C, Pingxin L, Wang DC, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol (2009) 184(2):205–13. doi:10.1083/jcb.200806072
23. Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science (2004) 306(5694):279–83. doi:10.1126/science.1101400
24. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol (2011) 7(12):691–9. doi:10.1038/nrrheum.2011.132
25. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell (2014) 159(7):1549–62. doi:10.1016/j.cell.2014.11.036
26. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22(2):146–53. doi:10.1038/nm.4027
27. Cardon LR, Burge C, Clayton DA, Karlin S. Pervasive CpG suppression in animal mitochondrial genomes. Proc Natl Acad Sci U S A (1994) 91(9):3799–803. doi:10.1073/pnas.91.9.3799
28. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med (2011) 17(11):1381–90. doi:10.1038/nm.2514
29. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
30. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A (2010) 107(21):9813–8. doi:10.1073/pnas.0909927107
31. Janko C, Schorn C, Grossmayer GE, Frey B, Herrmann M, Gaipl US, et al. Inflammatory clearance of apoptotic remnants in systemic lupus erythematosus (SLE). Autoimmun Rev (2008) 8(1):9–12. doi:10.1016/j.autrev.2008.07.015
32. Eric B, Paul RF. Connective tissue diseases: mitochondria drive NETosis and inflammation in SLE. Nat Rev Rheumatol (2016) 12(4):195–6. doi:10.1038/nrrheum.2016.24
33. Haiting W, Ting L, Sheng C, Yueying G, Shuang Y. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol (2015) 67(12):3190–200. doi:10.1002/art.39296
34. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) (2012) 122(6):253–70. doi:10.1042/CS20110386
35. von Kökritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood (2008) 111(6):3070–80. doi:10.1182/blood-2007-07-104018
36. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol (2006) 16(4):401–7. doi:10.1016/j.cub.2006.01.056
37. Juneau RA, Pang B, Weimer KE, Armbruster CE, Swords WE. Non typeable Haemophilus influenzae initiates formation of neutrophil extracellular traps. Infect Immun (2011) 79(1):431–8. doi:10.1128/IAI.00660-10
38. Munafo DB, Johnson JL, Brzezinska AA, Ellis BA, Wood MR, Catz SD. DNase I inhibits a late phase of reactive oxygen species production in neutrophils. J Innate Immun (2009) 1(6):527–42. doi:10.1159/000235860
39. Ramos KV, Mondragón F, Mondragón CM, Gonzalez PS, Muñiz HS, Rojas EO, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb) (2009) 89(1):29–37. doi:10.1016/j.tube.2008.09.009
40. McCormick A, Heesemann L, Wagener J, Marcos V, Hartl D, Loeffler J, et al. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect (2010) 12(12–13):928–36. doi:10.1016/j.micinf.2010.06.009
41. Bianchi M, Niemiec MJ, Siler U, Urban CF, Reichenbach J. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J Allergy Clin Immunol (2011) 127(5):1243.e–52.e. doi:10.1016/j.jaci.2011.01.021
42. Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol (2006) 8(4):668–76. doi:10.1111/j.1462-5822.2005.00659.x
43. Delbosc S, Alsac JM, Journe C, Louedec L, Castier Y, Bonnaure-Mallet M, et al. Porphyromonas gingivalis participates in pathogenesis of human abdominal aortic aneurysm by neutrophil activation: proof of concept in rats. PLoS One (2011) 6(4):e18679. doi:10.1371/journal.pone.0018679
44. Renzo M, Eva D, Pietro D. Killing by neutrophil extracellular traps: fact or folklore? Blood (2012) 119(5):1214–6. doi:10.1182/blood-2011-07-364604
45. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol (2014) 15(11):1017–25. doi:10.1038/ni.2987
46. Averhoff P, Kolbe M, Zychlinsky A, Weinrauch Y. Single residue determines the specificity of neutrophil elastase for Shigella virulence factors. J Mol Biol (2008) 377(4):1053–66. doi:10.1016/j.jmb.2007.12.034
47. Parker H, Albrett AM, Kettle AJ, Winterbourn CC. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol (2012) 91(3):369–76. doi:10.1189/jlb.0711387
48. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog (2009) 5(10):e1000639. doi:10.1371/journal.ppat.1000639
49. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol (2007) 5(8):577–82. doi:10.1038/nrmicro1710
50. Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, et al. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol (2007) 9(5):1162–71. doi:10.1111/j.1462-5822.2006.00857.x
51. Seper A, Hosseinzadeh A, Gorkiewicz G, Lichtenegger S, Roier S, Leitner DR, et al. Vibrio cholerae evades neutrophil extracellular traps by the activity of two extracellular nucleases. PLoS Pathog (2013) 9(9):e1003614. doi:10.1371/journal.ppat.1003614
52. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques NB. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol (2006) 16(4):401–7. doi:10.1016/j.cub.2006.01.056
53. Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol (2006) 16(4):396–400. doi:10.1016/j.cub.2005.12.039
54. Möllerherm H, Neumann A, Schilcher K, Blodkamp S, Zeitouni NE, Dersch P, et al. Yersinia enterocolitica-mediated degradation of neutrophil extracellular traps. FEMS Microbiol Lett (2015) 362(23):fnv192. doi:10.1093/femsle/fnv192
55. Derré-Bobillot A, Cortes-Perez NG, Yamamoto Y, Kharrat P, Couvé E, Da Cunha V. Nuclease A (Gbs0661), an extracellular nuclease of Streptococcus agalactiae, attacks the neutrophil extracellular traps and is needed for full virulence. Mol Microbiol (2013) 89(3):518–31. doi:10.1111/mmi.12295
56. de Buhr N, Neumann A, Jerjomiceva N, von Köckritz-Blickwede M, Baums CG. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology (2014) 160(Pt2):385–95. doi:10.1099/mic.0.072199-0
57. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun (2010) 2(6):576–86. doi:10.1159/000319909
58. Brogden G, von Köckritz-Blickwede M, Adamek M, Reuner F, Jung SV, Naim HY, et al. β-Glucan protects neutrophil extracellular traps against degradation by Aeromonas hydrophila in carp (Cyprinuscarpio). Fish Shellfish Immunol (2012) 33(4):1060–4. doi:10.1016/j.fsi.2012.08.009
59. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med (1997) 337(21):1512–23. doi:10.1056/NEJM199711203372106
60. Söderberg D, Kurz T, Motamedi A, Hellmark T, Eriksson P, Segelmark M. Increased levels of neutrophil extracellular trap remnants in the circulation of patients with small vessel vasculitis, but an inverse correlation to anti-neutrophil cytoplasmic antibodies during remission. Rheumatology (Oxford) (2015) 54(11):2085–94. doi:10.1093/rheumatology/kev217
61. Lögters T, Margraf S, Altrichter J, Cinatl J, Mitzner S, Windolf J, et al. The clinical value of neutrophil extracellular traps. Med Microbiol Immunol (2009) 198(4):211–9. doi:10.1007/s00430-009-0121-x
62. Aparna M, Martin H, Luis EM. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol (2016) 8(7):35. doi:10.3389/fimmu.2016.00035
63. Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in systemic lupus erythematosus. J Biomed Biotechnol (2010) 2010:948364. doi:10.1155/2010/948364
64. Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol (2013) 191(5):2647–56. doi:10.4049/jimmunol.1300436
65. Martinez VF, Balada E, Ordi RJ, Bujan RS, Sellas FA, Vilardell TM. DNase 1 activity in patients with systemic lupus erythematosus: relationship with epidemiological, clinical, immunological and therapeutical features. Lupus (2009) 18(5):418–23. doi:10.1177/0961203308098189
66. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet (2001) 28(4):313–4. doi:10.1038/91070
67. Frost PG, Lachmann PJ. The relationship of desoxyribonuclease inhibitor levels in human sera to the occurrence of antinuclear antibodies. Clin Exp Immunol (1968) 3(5):447–445.
68. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187(1):538–52. doi:10.4049/jimmunol.1100450
69. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra19. doi:10.1126/scitranslmed.3001180
70. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol (2012) 188(7):3522–31. doi:10.4049/jimmunol.1102404
71. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A (2010) 107(36):15880–5. doi:10.1073/pnas.1005743107
72. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med (2010) 16(8):887–96. doi:10.1038/nm.2184
73. von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi:10.1084/jem.20112322
74. Swystun LL, Mukherjee S, Liaw PC. Breast cancer chemotherapy induces the release of cell-free DNA, a novel procoagulant stimulus. J Thromb Haemost (2011) 9(11):2313–21. doi:10.1111/j.1538-7836.2011.04465.x
75. Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A (2007) 104(15):6388–93. doi:10.1073/pnas.0608647104
76. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One (2012) 7(2):e32366. doi:10.1371/journal.pone.0032366
77. Jun X, Xiaomei Z, Rosana P, Marc M, Concetta TA, Fabrizio S, et al. Extracellular histones are major mediators of death in sepsis. Nat Med (2009) 15(11):1318–21. doi:10.1038/nm.2053
78. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood (2011) 118(7):1952–61. doi:10.1182/blood-2011-03-343061
79. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost (2011) 9(9):1795–803. doi:10.1111/j.1538-7836.2011.04422.x
80. Demers M, Wagner DD. NETosis: a new factor in tumor progression and cancer-associated thrombosis. Semin Thromb Hemost (2014) 40(3):277–83. doi:10.1055/s-0034-1370765
81. Dwivedi N, Upadhyay J, Neeli I, Khan S, Pattanaik D, Myers L, et al. Felty’s syndrome autoantibodies bind to deiminated histones and neutrophil extracellular traps. Arthritis Rheum (2011) 64(4):982–92. doi:10.1002/art.33432
82. Ritika K, Carmelo CR, Anuradha VG, Alison G, Srilakshmi Y, Jason SK, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med (2013) 5(178):178ra140. doi:10.1126/scitranslmed.3005580
83. Hanses F, Park S, Rich J, Lee JC. Reduced neutrophil apoptosis in diabetic mice during staphylococcal infection leads to prolonged TNFα production and reduced neutrophil clearance. PLoS One (2011) 6(8):e23633. doi:10.1371/journal.pone.0023633
84. Krystallenia IA, Christina P, Panayiotis DZ, Nikolaos VA, Konstantinos M, Vassiliki S, et al. Cytokine secretion in long-standing diabetes mellitus type 1 and 2: associations with low-grade systemic inflammation. J Clin Immunol (2008) 28(4):314–21. doi:10.1007/s10875-007-9164-1
85. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med (2015) 21(7):815–9. doi:10.1038/nm.3887
86. Ho-Tin-Noé B, Carbo C, Demers M, Cifuni SM, Goerge T, Wagner DD. Innate immune cells induce hemorrhage in tumors during thrombocytopenia. Am J Pathol (2009) 175(4):1699–708. doi:10.2353/ajpath.2009.090460
87. Berger-Achituv S, Brinkmann V, Abed UA, Kühn LI, Ben-Ezra J, Elhasid R, et al. A proposed role for neutrophil extracellular traps in cancer immune editing. Front Immunol (2013) 4:48. doi:10.3389/fimmu.2013.00048
88. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest (2013) 123(8):3446–58. doi:10.1172/JCI67484
89. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med (2009) 15(11):1318–21. doi:10.1038/nm.2053
90. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2013) 13(1):34–45. doi:10.1038/nri3345
Keywords: NETosis, NETs, antimicrobial activity, autoimmune diseases, mitochondrial DNA
Citation: Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y and Herrmann M (2016) New Insights into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Front. Immunol. 7:302. doi: 10.3389/fimmu.2016.00302
Received: 29 April 2016; Accepted: 26 July 2016;
Published: 12 August 2016
Edited by:
Ivan Zanoni, University of Milano-Bicocca, ItalyReviewed by:
Ben Croker, Boston Children’s Hospital, USACopyright: © 2016 Yang, Biermann, Brauner, Liu, Zhao and Herrmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Zhao, emhhby55MTk3N0AxNjMuY29t
†Hang Yang, Mona Helena Biermann, Jan Markus Brauner, Yi Liu, Yi Zhao, and Martin Herrmann contributed equally.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.