
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 July 2016
Sec. Vaccines and Molecular Therapeutics
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00285
This article is part of the Research Topic HSPs - ambiguous mediators of immunity View all 10 articles
 Zarah Batulan1
Zarah Batulan1 Vivek Krishna Pulakazhi Venu1
Vivek Krishna Pulakazhi Venu1 Yumei Li1
Yumei Li1 Geremy Koumbadinga1
Geremy Koumbadinga1 Daiana Gisela Alvarez-Olmedo2
Daiana Gisela Alvarez-Olmedo2 Chunhua Shi1
Chunhua Shi1 Edward R. O’Brien1*
Edward R. O’Brien1*
Heat shock protein 27 (HSP27) is traditionally viewed as an intracellular chaperone protein with anti-apoptotic properties. However, recent data indicate that a number of heat shock proteins, including HSP27, are also found in the extracellular space where they may signal via membrane receptors to alter gene transcription and cellular function. Therefore, there is increasing interest in better understanding how HSP27 is released from cells, its levels and composition in the extracellular space, and the cognate cell membrane receptors involved in effecting cell signaling. In this paper, the knowledge to date, as well as some emerging paradigms about the extracellular function of HSP27 is presented. Of particular interest is the role of HSP27 in attenuating atherogenesis by modifying lipid uptake and inflammation in the plaque. Moreover, the abundance of HSP27 in serum is an emerging new biomarker for ischemic events. Finally, HSP27 replacement therapy may represent a novel therapeutic opportunity for chronic inflammatory disorders, such as atherosclerosis.
Since its first discovery in 1962 by Ritossa, the heat shock response has been established as the classical molecular mechanism underlying the cellular response to heat stress, involving the upregulation of a spectrum of protein families designated as “heat shock proteins” (HSPs) (1, 2). These proteins range in molecular weight from 10 to 110 kDa and primarily function as “chaperones” that facilitate proper protein folding (3, 4). Exposed hydrophobic residues in nascent or misfolded proteins act as the signal to recruit and activate HSPs, making this class of proteins powerful sensors of environmental (e.g., heat, oxidative) and physiological (e.g., infection, inflammation) stress (5, 6). Although initially characterized as intracellular chaperones, subsequent studies pointed to the presence of HSPs in the extracellular space (7, 8). Extracellular HSPs (eHSPs) have consequently been ascribed novel functions (such as “outside–in” signaling leading to cellular proliferation and immune response modulation), expanding their canonical role of maintaining homeostasis in the cell to that of the whole organism.
Unlike the larger molecular weight HSPs, HSP27 was first identified as an estrogen-responsive protein (9–11), which was later confirmed to be heat-inducible and sharing sequence homology with other HSPs (12–16). It is a member of the small heat shock protein beta (HSPB) family, an HSP subgroup characterized by a lower molecular weight range (12–43 kDa) and a shared α-crystallin domain flanked by variable N- and C-terminal sequences (17, 18). The encoding gene, HSPB1, is located on chromosome 7 (7q11.23) (19), and its associated mutations have been linked to Charcot–Marie–Tooth disease type 2 (20, 21) and possibly Williams syndrome (22, 23). The intracellular chaperoning function of HSP27 is largely regulated by the phosphorylation/dephosphorylation of three key N-terminal serine residues at positions 15, 78, and 82 (24–26), which influences the assembly of HSP27 monomers into large oligomeric complexes (200–800 kDa). Such complexes create an ATP-independent chaperone network that effectively traps misfolded protein substrates, preventing their aggregation through a series of controlled binding and release reactions that, in conjunction with other larger HSPs (HSP70, HSP90), facilitate their correct folding (27–29). Thus, in concert with other chaperones, HSP27 maintains cellular homeostasis by holding misfolded proteins in a soluble, refolding-competent form. Intracellular HSP27 has also been implicated in (i) cytoskeletal architecture and dynamics (30–32), (ii) mRNA stabilization (33), (iii) antioxidant responses (34–36), and (iv) anti-apoptosis (37–40). Not surprisingly, its multiple, cytoprotective effects are associated with the amelioration of a range of pathological conditions, including degenerative, ischemic, neurological, and cardiovascular disease (41). For more comprehensive reviews on the biology of intracellular HSP27, several references are available (17, 42–44).
Heat shock proteins were first observed in the extracellular space nearly 30 years ago as proteins that were transferred from glia to neurons in the squid giant axon (45) and later confirmed to be released after heat shock in mammalian cells (46). Like other HSPs, HSP27 has been found extracellularly – initial evidence indicated its secretion from tumor cells (47–51). The presence of HSP27 has been detected in human serum, with elevations observed after extreme exercise (52, 53). Increases in serum HSP27 have also been measured in patients with chronic pancreatitis (54), gastric adenocarcinoma (55), insulin resistance (56), and during acute attacks in multiple sclerosis (57), while decreases have been associated with type 1 diabetes (58). HSP27 has also been found in the cerebrospinal fluid during brain and spinal cord ischemia (59).
In the context of cardiovascular disease, two independent groups utilizing a proteomic analysis of human atherogenic arterial tissue have shown that HSP27 is differentially secreted into the extracellular milieu (60, 61). Moreover, serum levels of HSP27 have been detected in patients with atherosclerosis (60, 62, 63), acute coronary syndromes (61), and reperfusion after ischemic clamping during heart bypass surgery (64). The presence of serum HSP27 was confirmed in an atherosclerosis mouse model (ApoE−/−), overexpressing HSP27 (HSP27o/e) using commercially available ELISA kits (65, 66). However, these experiments revealed the presence of HSP27-reactive autoantibodies (AAB) in the serum that effectively shield the detection of HSP27, thereby resulting in erroneously low levels of HSP27. Consequently, our laboratory has developed a mass spectrometric-based method that can more sensitively detect serum HSP27 at levels higher than those quantified using commercial ELISA kits (67). Even though the detection of serum HSP27 levels can only improve, results to date firmly establish the extracellular presence of HSP27. However, key questions remain (1) how is HSP27 released from cells; (2) what is its function outside the cell; and (3) how does it signal from outside to inside the cell?
Secreted proteins typically exit eukaryotic cells via the endoplasmic reticulum–Golgi network (known as the “classical secretory pathway”). Such proteins contain an N-terminal signal peptide that marks them for secretion – which interestingly is absent in other types of secreted proteins, including HSPs. Several groups have shown that despite this, HSPs can still be released – for example, early experiments that pharmacologically blocked the classical pathway still resulted in HSP secretion (in this case, HSP70) (46). Although HSP release was proposed to be a consequence of passive transport, i.e., necrosis (68), secretion can also occur independent of cell death, implying an active transport process that involves alternative, “non-classical” secretory mechanisms (46, 69–71). Indeed, some secretory proteins lacking the signal peptide for the classical pathway have since been identified, including interleukin (IL)-1a, IL-1b, and fibroblast growth factor (FGF)-2, and increasing evidence indicates that these proteins along with some HSPs are released by non-classical secretory pathways (7, 72, 73). Although there is still much mechanistic information to uncover regarding how HSP27 is secreted, this review will discuss a few key findings indicating that it may leave the cell via lysosomes and/or exosomes and bring up the possibility of direct protein translocation (Figure 1).
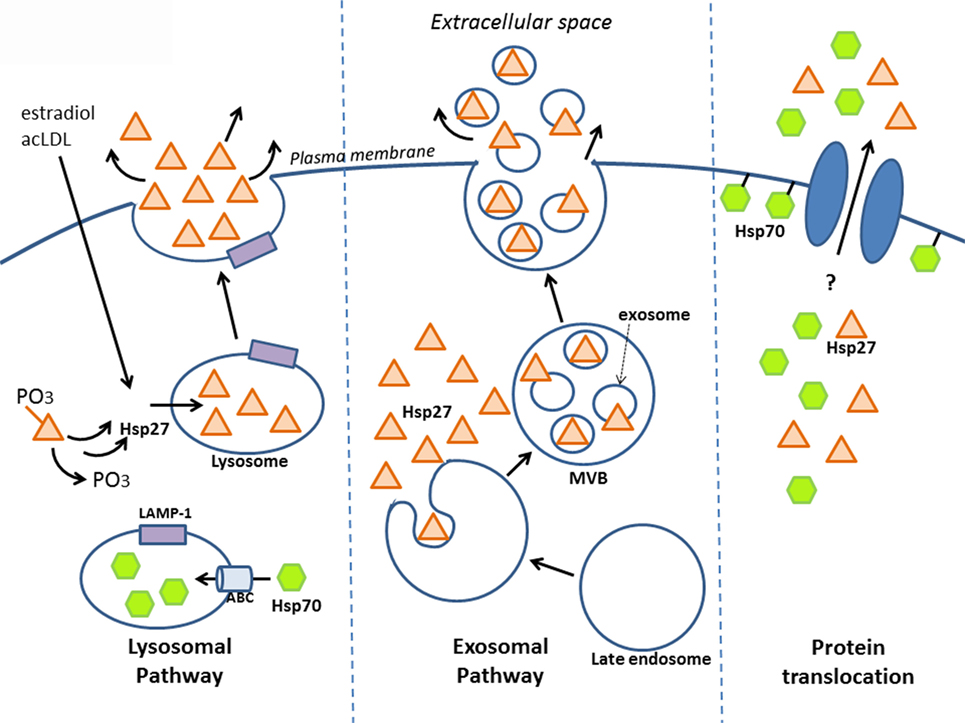
Figure 1. Release of HSP27 involves non-classical secretory mechanisms. Experimental evidence suggests that HSP27 exits cells via the endolysosomal pathway and through exosomes. Localization of HSP27 to lysosomes (65) may involve the dephosphorylation of two serine residues at amino acids 15 and 82 (74); meanwhile, HSP70 appears to enter lysosomes with the aid of ABC transporters spanning the lysosomal membrane (75). Although direct protein translocation accounts for the unconventional secretion of FGF-2 (76, 77), it remains to be determined whether this mechanism plays a part in HSP27 extracellular release. Like FGF-2, HSP70 can interact with lipids as well as integrate into lipid membranes (7, 46, 78–81), and it also appears that HSP70 can integrate into artificial lipid bilayers forming channels.
Known as the recycling units of the cell, lysosomes are vesicular organelles containing enzymes that break down biomolecules (proteins, lipids, nucleic acids, carbohydrates) into their basic components. In addition to their role in the degradation of cellular waste, lysosomes can also serve as storage compartments for proteins targeted for secretion. These “secretory lysosomes” have been found in blood cells, among other cell types, and share similarities with conventional lysosomes, including an acidic pH interior (82). Utilization of this alternative secretion pathway has been observed in the case of HSP70. In one study using prostate carcinoma cell lines, lysosomal fractions contained more HSP70 following heat shock, and conversely, treatment with the lysosomal inhibitors, methylamine, and ammonium chloride prevented heat shock-mediated release of HSP70 into the culture media – which together suggest that the lysosome is involved in secretion (75). It is likely that HSP70 enters the lysosomal interior via ATP-binding cassette (ABC) transporters, since inhibition with glibenclamide, a general ABC transporter inhibitor, and DIDS, a specific ABCA1 inhibitor, reduced HSP70 release (75). As well, its release was associated with the relocalization of lysosomal markers, typically found in the lysosomal interior to the exterior cell membrane leaflet (75, 83). Taken together, these findings indicate that HSP70 may exit cells via secretory lysosomes, resulting in their fusion to the cell membrane. Similar findings were seen in cultured primary human peripheral blood mononuclear cells (PBMCs), whereby HSP70 secretion was significantly inhibited by the lysosomal inhibitor, methylamine, but not by brefeldin A (a blocker of transport from the ER to Golgi) (70). Furthermore, the presence of HSP70 in the lumen of lysosomes has been reported by another group (84).
What is the evidence that HSP27 uses the endolysosomal pathway? Findings from our laboratory have shown that upon treatment of macrophages with estradiol or acetylated low-density lipoprotein (ac-LDL) – conditions that stimulate HSP27 secretion – there was increased co-localization of HSP27 with the lysosomal markers, LAMP-1, and lysotracker (65). A later study yielded new insight into how HSP27 could be released through lysosomes (74). Their experiments utilized two types of HSP27 mutations, a constitutively phosphorylated mimic (S15D/S82D) and a non-phosphorylatable form (S15A/S82A), which were linked to GFP in order to visualize cellular localization in endothelial (HUVEC) cells. Interestingly, it was observed that the S15A/S82A mutation co-localized more frequently with the lysosomal marker, LAMP-1, using fluorescence microscopy. Furthermore, using His-tagged HSP27 mutants expressed in HUVEC cells, the non-phosphorylatable HSP27 was once more found at higher levels in cell culture media compared to S15D/S82D, as assessed by Western blotting for the histidine tag. These findings suggest that in order for HSP27 to localize to the lysosome (for subsequent secretion), these two serine residues must first be dephosphorylated (Figure 1).
Exosomes are the most commonly accepted vehicle for HSP release (69). This type of secretory vesicle is derived from a multistep process that first involves the internalization of the plasma membrane to form endosomes, which then develop into late endosomes. Late endosomal membranes are invaginated within, forming smaller vesicles (exosomes) that measure 40–100 nm in diameter and contain cytoplasmic components. These complex structures, referred to as “multivesicular bodies” (MVBs), fuse to the cell membrane, releasing exosomes into the extracellular space (Figure 1) (69, 85). Numerous findings have indicated the presence of several HSPs (HSP70, HSP60, HSP90) in exosomes, with localization observed at both the exosomal membrane (conceivably, facilitating interactions with cell surface receptors on target cells) and lumen (implying that for HSPs to have an effect on target cells, exosomes will have to burst to release their contents) (7, 86–90).
There is mounting evidence indicating that HSP27 is found in exosomes (Figure 1). HSP27 has been detected in exosomes derived from a variety of cell and tissue types, including keratinocytes, platelets, cancer cells, saliva, thymus, and urine, making the exosomal pathway a compelling system for HSP27 export (http://exocarta.org/gene_summary?gene_id=3315). Experiments in B-lymphoblastoid cells also demonstrate the presence of HSP27 in exosomes (90). Here, basal levels of HSP27, HSP70, and HSP90 localized to exosome fractions as assessed by Western blotting – an effect that increased with duration of heat shock treatment. When isolated exosomes were coupled to beads for flow cytometry analysis, positive staining for exosomal surface markers (CD81 and MHC class I), but not HSPs, were noted, implying that these HSPs were not expressed on the exosomal surface. However, when the exosome–bead complexes were solubilized by boiling in SDS (disrupting the exosomes) and total protein analyzed by Western blotting, elevated levels of HSP70 were detected in exosomes from heat-shocked cells compared to controls. These findings indicate that disruption of the exosomal membrane was required in order to detect HSP70, implying its localization within the exosome. Unfortunately, experiments detecting HSP27 after exosomal disruption were not presented in this study, thus it could not be confirmed whether the absence of HSP27 in the exosomal membrane points to a luminal presence. Results from our laboratory also support that exosomes may be one system involved in HSP27 release, since the treatment of human macrophages with an exosomal inhibitor, dimethyl amiloride (DMA), substantially decreased extracellular HSP27 levels (66). More recently, HSP27 was detected in exosomal fractions originating from primary cultures of rat astrocytes (91). Consensus has yet to be achieved with regards to the precise location of HSPs in exosomes. The suggestion of a luminal localization for HSP70 (90) is in contrast to results in macrophages infected with two different viruses (88, 92) and in blood cells after ischemic preconditioning (87), which indicate the presence of HSP70 in the exosomal membrane. Differences in exosomal localization is likely determined by the type of HSP, the cell from which exosomes originate, and the kind of stimuli triggering exosomal release (e.g., heat shock vs. viral infection). Another aspect of the exosomal pathway that is worth further exploration is determining the sorting signal that directs HSPs to populate exosomes. In the case of HSP60, for example, it appears to be ubiquitinated in exosomal fractions (86), suggesting that ubiquitinylation may signal sorting to exosomes. Whether this applies to HSP27 is currently unknown.
A final, intriguing possibility for HSP27 export may involve direct protein translocation across the plasma membrane, as has been proposed for FGF-2 (73, 93). Exit of HSPs from cells could be mediated by transmembrane proteins, like ABC transporters (also located on cell membranes), which, as already mentioned, are instrumental in transporting HSP70 in the lysosomes for secretion (75, 83). Another hypothetical way for HSPs to leave the cell is by directly associating with the cell membrane, which could then somehow facilitate their extracellular release. In fact, such lipid interactions have been observed with HSP70 (Figure 1) (46, 78–80), and it also appears that HSP70 can integrate into artificial lipid bilayers forming channels (7, 81). Despite the lack of evidence supporting the direct release of HSP70 (and other HSPs) after insertion or association into lipid membranes, it is still possible to consider direct membrane translocation of HSPs as a mechanism for exit from cells by looking to the cellular export paradigm utilized by FGF-2.
Over the years, the Nickel research group has systematically delineated the various steps involved in the export mechanism for FGF-2 (76, 77). According to their findings, FGF-2 localizes to the inner leaflet of the cell membrane via interactions with lipids, and after phosphorylation by Tec kinase, is then primed to oligomerize and integrate into the membrane, forming a transient pore structure, which itself is speculated to disassemble into FGF-2 monomers during extracellular release (76). Subsequent results highlight the importance of two surface cysteines unique to FGF-2 (notably absent in other FGF family members) that mediate disulfide bond formation with another FGF-2 monomer, forming dimers that then assemble into hexamers – the final FGF-2 structural complex that is inserted into the membrane (93). Since mutations of these two cysteines abrogate FGF-2 secretion, it appears that disulfide formation between cysteines facilitates the insertion of FGF-2 oligomers into the cell membrane and its subsequent release (93). Perhaps, similar mechanisms are involved with HSP export. FGF-2 and HSP70 have both been observed to interact with lipids and integrate into membranes. In addition, HSP27 also shares a key feature with FGF-2 – both contain unique cysteine (C) residues that happen to be missing in members of their respective protein families. In HSP27, this unique cysteine residue, C137, located in the α-crystallin domain, also mediates dimerization with other HSP27 monomers, however the consequence of dimerization differs in that disulfide formation between opposing cysteine residues modulates HSP27 function during oxidative stress (94). In reducing conditions, cysteines are directly opposite each other at the dimer interface, while under oxidative conditions, a disulfide bond forms, altering the structure of HSP27’s α-crystallin domain, leading to increased exposure of hydrophobic residues and promoting chaperoning activity (95–97). Although C137 is thought to be critical in mediating HSP27’s chaperoning activity during oxidative stress (i.e., disulfide bond can more easily bind to misfolded proteins that arise from oxidative stress), it will be interesting to see if mutation of this cysteine can alter secretion of HSP27, as has been observed with FGF-2. Another issue to address is whether HSP27 can interact with membrane lipids, like HSP70 (98–100). There is much to be discovered before ruling out that HSP27 could, in part, be exported from cells via direct protein transport (Figure 1).
Once outside the cell, what does extracellular HSP27 do? Most evidence suggests that, like other HSPs, HSP27 serves as a signaling molecule released by cells, which bind to surface receptors on distant cellular targets, such as endothelial and immune cells (7, 101, 102). Thus far, it appears that HSP27 can function outside the cell as (i) an uncomplexed, extracellular protein, (ii) part of a larger protein complex, for example, via interactions with AAB in the serum, and (iii) molecular cargoes located on the surface or inside exosomes. This review will focus on how recombinant extracellular HSP27 modifies receptor-mediated signaling in target cells and will only briefly comment on its effects as part of antibody complexes and exosomes.
Several cell membrane receptors have been identified for eHSPs, including CD91, CD40, CD36, CD14, toll-like receptors (TLRs), and scavenger receptor-A (SR-A) (8, 102). HSP27 has also been shown to interact with TLRs, as well as other types of receptors, altering signaling mechanisms in a variety of cell types and conditions (Figure 2). For example, HSP27 treatment of human macrophages promoted NF-κB transcriptional activation as well as protein expression and secretion of the NF-κB-dependent cytokines, GM-CSF and IL-10 – the latter important in anti-inflammatory responses (103). NF-κB activation by HSP27 was also observed in mouse coronary endothelial cells, occurring via interactions with TLR-2 and TLR-4 and resulting in the upregulation of intercellular adhesion molecule-1 (ICAM-1) and monocyte chemoattractant protein-1 (MCP-1) (64). In human coronary endothelial cells, HSP27 also upregulated ICAM-1, MCP-1, as well as IL-6 and IL-8, although the receptors mediating the response in human cells were not identified (64). In contrast, TLR-3 was implicated as the receptor utilized by HSP27 in human microvascular endothelial cells, leading to NF-κB activation and secretion of IL-8 and vascular endothelial growth factor (VEGF) (104). The effect of HSP27 on VEGF was corroborated in endothelial progenitor cells (EPCs), although this study did not address which receptors play a part in VEGF secretion (105). HSP27 increased secretion of IL-6 in bone marrow-derived dendritic cells, as well as TNF-α, IL-1β, IL-12p35, and IL-12p40, which partly occurred through TLR-4 signaling (106). In primary human monocytes, release of IL-10 by HSP27 involved phosphorylation of p38 and MAPKAPK-2, while increased TNF-α levels were attributed to the activation of both p38 and ERK1/2 signaling pathways (107). HSP27 also interacts with estrogen receptor-β (ER-β) (108, 109). Altogether, these findings implicate extracellular HSP27 in (i) immune signaling (via activation of TLR signaling pathways), leading to cytokine production and modulation of the immune response, (ii) cellular migration (due to the upregulation of ICAM-1 and VEGF), and (iii) cellular proliferation (through its interactions with ER-β).
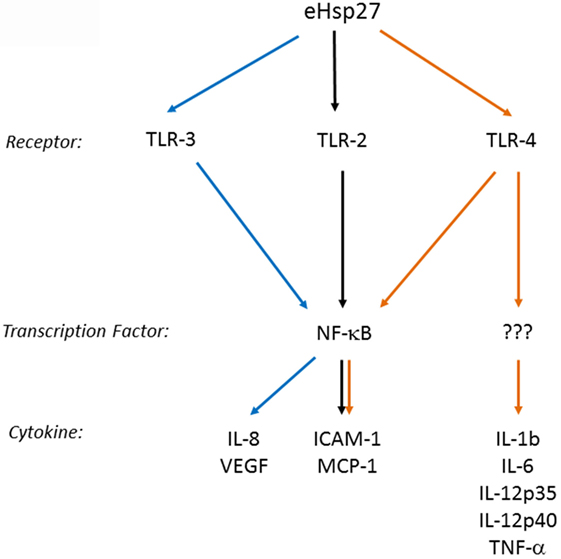
Figure 2. Extracellular HSP27 (eHSP27) can trigger TLR signaling, leading to NF-κB activation and cytokine upregulation (64, 103–106).
Extracellular HSP27 has also been found as part of larger macromolecular complexes. For example, our laboratory’s attempts to develop a more sensitive method for measuring HSP27 in serum samples led to the serendipitous discovery that this protein is complexed to AAB, which effectively shields the detection of serum HSP27. In vitro experiments later showed that AAB could potentiate the signaling effects of HSP27 (67). Another example of HSP27 being part of a larger complex is its presence in exosomes. Exosomes carrying HSP27 (either on the surface or in its interior) could then interact with target cells through receptor interactions leading to endocytosis, or by simple membrane fusion that is independent of receptors (69). Precisely how exosomes cargoing HSP27 affect target cells is unknown; however, it may instigate a variety of effects depending on the exosomal source and downstream cellular target. This has been observed with HSP70, whose expression on exosomal surfaces has been found to (i) trigger TLR-4 signaling leading to myocardial protection (87) and (ii) stimulate natural killer (NK) cells, increasing its cytolytic activity against HSP70-membrane positive tumors (110). Thus, by interacting with receptors on both immune and endothelial cells, leading to the differential release of cytokines and growth factors, extracellular HSP27 is a potentially important modulator of the immune response – an important process that contributes to atherosclerosis.
Atherosclerosis is a chronic cardiovascular condition that is marked by the progressive accumulation of inflammatory scar tissue containing lipid deposits, calcification, and necrotic debris within the walls of arteries that can lead to the formation of unstable plaques at risk of rupture. Plaque rupture is unpredictable and is the final phase that leads to a reduction in blood flow, ischemia in the distal vascular bed, and clinical symptoms or events (e.g., heart attack and/or death). Although the etiology of atherosclerosis was primarily thought to originate from a lipid/cholesterol imbalance (be it genetic or environmental), it appears that atheroprogression is a complex, multifactorial process that is, in part, due to coordinated immune responses. Recently, several lines of evidence support the emerging role of HSP27 in modulating atheroprogression, by altering the inflammatory components of atherosclerotic plaques.
Although evidence of cardiovascular disease has been traced back to thousands of years, with various prehistoric populations displaying signs of arterial plaque (111), it is only within the past 100 years that this condition has emerged as the leading cause of mortality in the world. Epidemiological studies from the twentieth century have indicated that women are at lower risk for developing cardiovascular diseases compared to men; however, this bias toward the female sex disappears after menopause, implicating female hormones as protective factors against CVD (112). Consequently, various clinical trials conducted in the 1970s–2000s sought to test whether the replacement of ovarian hormones could protect menopausal women from CVD. Unfortunately, the conclusions of these trials indicate that hormone replacement therapy not only failed to protect women from CVD but also increased side effects as well as CVD risk (113, 114). Although hormone replacement therapies were ineffective against CVD, the question of how the presence of female hormones protect against CVD in premenopausal women remains. Given that HSP27 was initially identified as an estrogen-inducible gene (10, 11), it was hypothesized that HSP27 is one important downstream “foot soldier” of estrogen signaling that mediates cardiovascular protection (108).
There is a precedent in establishing a link between other HSPs and cardiovascular disease. For example, some studies correlate HSP60 with atherosclerosis severity and suggest that it could serve as a biomarker (115–117), while others associate HSP70 serum levels with low CVD risk (118, 119). HSP27 was first implicated in atherosclerosis more than 10 years ago, when a proteomic screen comparing levels of released proteins noted lower levels of HSP27 secretion from human atherosclerotic arteries compared to normal arteries (60). Indeed, HSP27 secretion and expression in atherosclerotic human coronary arteries (108, 120) decreases with increasing plaque progression, complexity, and instability. Conversely, areas of normal-appearing atherosclerotic arteries display increased HSP27 expression compared to plaque-filled areas from the same vessel (61). Three independent studies confirm the reduction in secreted HSP27 levels in atherosclerosis patients. The first study involving small patient cohorts (28 patients vs. 12 controls) noted significantly lower HSP27 serum levels in CVD patients compared to healthy controls (60). Two later studies in larger patient cohorts (76 patients vs. 53 controls; 80 patients vs. 80 controls) validated this finding, with CVD patients again displaying lower levels of serum HSP27 (62, 63). Moreover, our laboratory also demonstrated that in patients diagnosed with coronary atherosclerosis (>50% stenosis by angiography), lower levels of HSP27 was predictive of subsequent major clinical events (i.e., heart attack, stroke, cardiovascular death) over a 5-year period (63). However, in a prospective, nested, case–controlled study involving initially healthy female participants who later developed cardiovascular disease, baseline HSP27 plasma levels are inversely associated with age but neither significantly associated with other established cardiovascular risk factors nor with future cardiovascular events (121). These seemingly disparate findings regarding HSP27’s potential as a marker in predicting cardiovascular events can be reconciled if one considers that all of the female subjects studied in the nested case–control series were, on average, 61 years of age at baseline and beyond menopause. Therefore, one would expect relatively low serum levels of HSP27 in both the controls and later CVD event patients at baseline. Moreover, at this age, coronary atherogenesis may have already reached a moderately advanced stage.
Hence, the results to date reveal that plasma HSP27 levels may indeed be a potential marker for atherosclerosis and predictive of adverse cardiovascular events. With further refinements in the HSP27 serum assay that will remove the confounding presence of HSP27 AAB, we are confident that this conclusion will be further validated. Additionally, diminished expression of HSP27 in atherosclerotic plaques compared to vessels free of disease further highlights the association between augmented HSP27 levels and protection against atherosclerosis.
Some investigators suggest that extracellular HSP27 may be subject to protease degradation that is more abundant in atherosclerotic compared to normal arteries. Indeed, it has been shown that plasmin is upregulated in atherosclerotic plaques and can cause the degradation and aggregation of recombinant HSP27 (rHSP27) as well as HSP27 endogenously secreted from arterial explants – findings reversed by aprotinin, a specific plasmin inhibitor (122). Other enzymes that are more abundant in vulnerable atherosclerotic plaques due to a proteolytic imbalance, like matrix metalloproteases (MMPs) (123), can also interact with (124, 125) and possibly degrade HSPs (126). Another idea to consider is that HSP27 (as well as other HSPs) could function as “danger signals” – highly abundant self-proteins normally found intracellularly in healthy tissues, which are released into the extracellular space when tissues are damaged, infected, or stressed. Such danger signals are capable of activating antigen-presenting cells (APCs) to initiate the adaptive immune response (127). Stress and damage associated with atheroprogression could thus release more HSP27 from cells into the circulation, resulting in the following possibilities: (i) less HSP27 expression in cells as plaque accumulates, (ii) proteolytic cleavage and degradation of extracellular HSP27, (iii) increased susceptibility to apoptosis of cells that have secreted HSP27, and (iv) activation/modulation of the immune response. Although the link between reduced HSP27 levels in the serum and atherosclerotic vessel walls is strong, it remains unclear whether HSP27 levels are a secondary feature of atherosclerosis or the stress response during atherogenesis, or if HSP27 plays a causal role in directly modulating vessel wall homeostasis.
In order to understand how HSP27 is involved in atheroprogression and whether it could protect against development of CVD, the various stages and key cellular players involved in atherosclerosis must first be highlighted. Atherosclerosis is characterized by the development in arteries of lesions or plaques containing lipids, inflammatory markers, and dead cells surrounded by a fibrotic cap structure. Some areas of the vasculature are more susceptible to plaque accumulation. For example, branch points in the circulatory tree experience altered laminar blood flow that could lead to atherosclerosis-promoting changes (128), including endothelial dysfunction, accumulation of oxidized low-density lipoproteins (ox-LDL), alterations in matrix protein composition, and accumulation of proteoglycans, which increase ox-LDL accumulation at the cellular level (129). Plaque accumulation can impair blood flow, causing health complications, and if left alone, can escalate to the grave consequence of plaque “rupture,” exposing pro-thrombotic (pro-clotting) material from the plaque to the blood and causing occlusion of the artery. These local arterial blood clots deprive nearby cells of oxygen, leading to cell death and various adverse outcomes (depending on the rupture site), including heart failure, stroke, renal impairment, and/or hypertension (129).
The first stage of plaque formation involves the development of “fatty streaks.” This gradual process originates in endothelial cells lining the inner arterial wall (“intima”) and involves (1) the uptake and accumulation of circulating oxidized lipids and low-density lipoprotein (LDL), (2) expression of adhesion molecules, and (3) chemokine release, resulting in the recruitment and deposition of immune cells (mainly lipid-containing macrophages also known as foam cells and T-cells) along the intima (130). Fatty streaks do not give rise to overt symptoms and are reversible (129). It is interesting to note that the partial attenuation of HSP27 expression starts in this stage of atherogenesis (Figure 3) (108, 131).
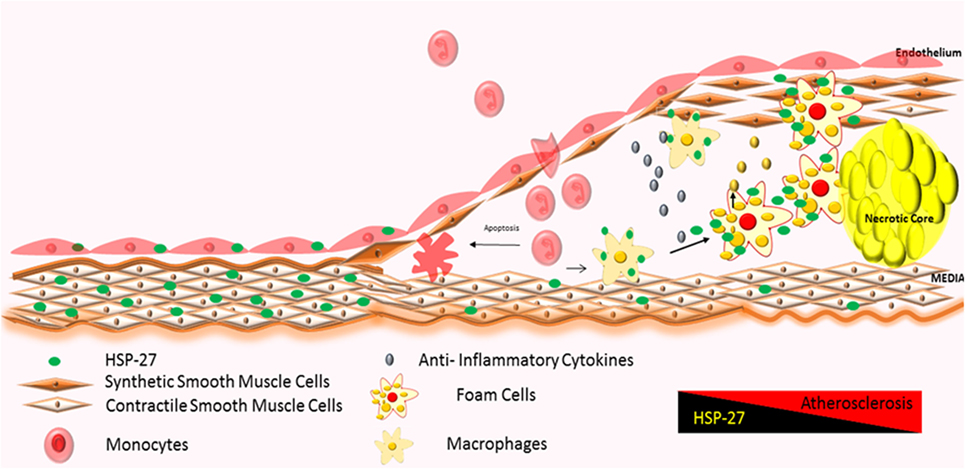
Figure 3. Atherosclerotic plaque progression: severity of atherosclerosis correlates with increased infiltration of monocytes and their local activation into macrophages and foam cells, further promoting the inflammatory process (128, 129). HSP27 is expressed in normal areas of arteries, with reduction in expression in areas of advanced plaque (60, 61, 108, 120). Increases in HSP27 expression have also been noted in the fibrous cap and media (122).
Over time, fatty streaks become mature atherosclerotic plaques (“atheromas”) distinguished by the following regions: (1) a core region, consisting of foam cells, lipids, immune cells, such as dendritic cells, mast cells, and B-cells (129), apoptotic cells, debris, cholesterol crystals (132–134), and proteases like plasmin (122); (2) a fibrous cap region surrounding the core, made up of smooth muscle cells (122) and a collagen-rich matrix; and (3) an interface between core and fibrous cap regions (“shoulder” region), housing mainly T-cells and macrophages (129) (Figure 3). Immunohistochemical analysis of atherosclerotic arteries obtained from patients undergoing cardiac surgery show HSP27 localization mainly in the fibrous cap and media (122); however, its abundance diminishes as the complexity and volume of the plaque increases (108).
The presence of immune cells early in plaque formation indicates the involvement of the immune response; however, it is still unclear whether this promotes or attenuates atherosclerosis. The canonical idea that atherosclerosis is primarily caused by dietary and lifestyle factors still carries weight in that the rise of cardiovascular diseases came about within the last century, coinciding with increasingly sedentary lifestyles and increased caloric and fat intake. Given that fatty streaks are found in young people, it appears to be a normal adaptive process to have an immune response generated against lipids in the arteries. However, for this response to promote atheroprogression likely involves an interplay between the presence of cholesterol (in plaques) derived from the diet and circulating immune cells that become resident in the arterial wall. Hence, the human body can be tipped into developing atherosclerosis if the source of cholesterol becomes dysregulated (dietary or genetic predisposition to the development of hypercholesterolemia) or if the immune system becomes abnormally sensitized to normal levels of lipid in the blood vessels, or both.
The immune cells primarily responsible for propagating the inflammatory response against lipids in atherosclerotic plaque are macrophages. Upon encountering lipids, particularly ox-LDL, macrophages engulf ox-LDL by binding to LDL membrane receptors. Internalization of ox-LDL then triggers a cascade of events that transform macrophages into foam cells. Foam cells also secrete pro-inflammatory cytokines that result in the recruitment and activation of additional immune cells to the lesion site – further amplifying the immune response and promoting the development of complex, advanced plaques. Our previous results have shown that HSP27 competes with LDL binding to SR-A, known to take up atherogenic lipids, as well as downregulating SR-A expression (65, 135), thus attenuating foam cell formation. Furthermore, administration of recombinant HSP27 has been shown to block the differentiation of monocytes to dendritic cells, a cell type that can also promote the pro-inflammatory process (136). Another way that HSP27 can regulate the inflammatory response involved in atherosclerotic plaque progression is through the release of GM-CSF, which we have seen in cultured human macrophages, following rHSP27 treatment (103). Depending on the existing cytokine milieu, GM-CSF appears to promote the development of the M1 macrophage lineage (137), considered to play a role in the pro-inflammatory immune response (138). HSP27 may thus favor the preponderance of one macrophage phenotype over another, thereby affecting plaque progression.
Finally, another immune cell type involved in atheroprogression (albeit to a lesser degree) are T-regulatory cells, which mediate atheroprotection by amplifying the release of IL-10, thereby resulting in immune suppression (139–142). However, the role of HSP27 in T-regulatory function during atheroprogression is uncertain (143). Mast cells, a key cell type that populates atherosclerotic plaque, are classically known to react immediately to allergens via cross-linking of cell surface IgE resulting in degranulation, histamine release, and synthesis of arachidonic acid metabolites, which can activate stress-signaling and pro-apoptotic pathways (144). Mast cells have been shown to release proteases that can degrade HSP27 (145, 146).
The key findings from our research group and others showing that (i) the loss of HSP27 serum levels correlates with plaque and disease progression and (ii) HSP27 is absent at sites of mature, complex plaque (while present in normal intima from the same patient with atherosclerosis), suggest that HSP27 could be atheroprotective. To address this possibility, mice prone to atherosclerosis (ApoE−/−) (147) were crossed to transgenic mice overexpressing (human) HSP27 (HSP27o/e) (65). Circulating HSP27 serum levels were higher in HSP27o/e ApoE−/− mice compared to ApoE−/− control mice and were inversely correlated with lesion area in both male and female mice (65, 148). More importantly, HSP27 overexpression protected mice from atherosclerosis, particularly in female mice (65) – a process that was abrogated by ovariectomy but rescued by administration of exogenous estrogens (66). Taken together, these data indicate that secretion of HSP27 into the circulation requires estrogens (66). Compared to ApoE−/− mice, HSP27o/e ApoE−/− mice exhibited reductions in cholesterol cleft areas, macrophage infiltration, apoptotic cell number in the plaques, and free serum cholesterol. Features indicative of favorable plaque remodeling (e.g., increased smooth muscle cell and collagen content and greater vessel stiffness) were also evident (148). Administering HSP27 to atheroprone mice via bone marrow transplant from HSP27o/e mice or recombinant HSP27 injections also reduced atherosclerotic lesions and increased plaque stability (63). These in vivo findings yielded several mechanistic possibilities underlying HSP27-mediated atheroprotection. First, the presence of estrogen appears to be important for HSP27’s extracellular release. Second, HSP27 could modify various processes involved in atherosclerosis, including cholesterol homeostasis/trafficking, inflammation, immune cell infiltration into and migration out of plaques, activation of macrophages to foam cells, and plaque remodeling (Figure 4).
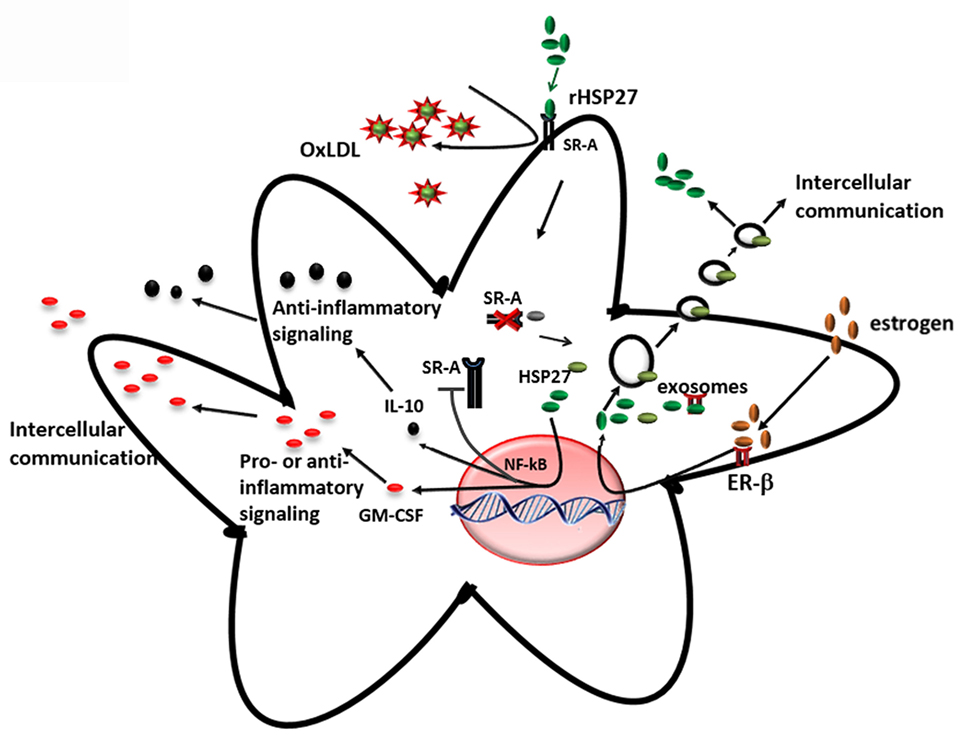
Figure 4. Extracellular HSP27, whose release is stimulated by estrogen, affects macrophages by (i) reducing ox-LDL uptake and (ii) activating NF-κB, which results in increased secretion of IL-10 and GM-CSF (65, 103, 135).
An interesting finding from the HSP27 overexpressing atherogenic mouse model is the reduction in cholesterol, in both plaque and serum (148), suggesting that HSP27 is involved in lipid homeostasis. One class of receptors known to bind to modified lipoproteins and facilitate their internalization in macrophages is SR-A (149, 150). It has been suggested that unregulated uptake of modified LDL in macrophages promotes the differentiation of monocyte-derived macrophages into pro-inflammatory foam cells, resulting in their accumulation in the lumen and, consequently, the narrowing and hardening of blood vessels (151, 152). In vitro experiments from our laboratory have shown that extracellular HSP27 affects SR-A activity via protein interactions on the surface of macrophages (65). This interaction reduces the binding of LDL to SR-A, and consequently, the formation of cholesterol-laden macrophages (65, 135). However, recent experimental data also support the role of HSP27 as a downregulator of SR-A protein expression (135). It has been shown that treatment of macrophages with extracellular recombinant HSP27 (rHSP27) is associated with reductions in both SR-A mRNA levels (−39%) and cell surface expression (−58%) as well as less foam cell formation – effects that occur via NF-κB signaling (135). Knockout of SR-A in ApoE−/− atheroprone mice abrogated the protective effects (reductions in atherosclerotic lesions) of HSP27, indicating HSP27 atheroprotection appears to depend, in part, on the presence of SR-A (135). Recent experiments in our laboratory indicate that HSP27 modulates cholesterol efflux in macrophages via ABCA1 and ABCG1 transporters (153). Thus, HSP27 has numerous, pleiotropic effects on cholesterol trafficking, modulating both import and export of cholesterol in macrophages (Figure 4).
Less atherosclerotic lesions in HSP27o/e ApoE−/− suggested that HSP27 can modulate the immune processes involved in atherosclerosis. One possibility lies in HSP27-mediated regulation of NF-κB activity in macrophages. NF-κB transcription factors regulate a vast number and diversity of gene targets, including those involved in cell proliferation, apoptosis, the cell stress response, inflammation, and both innate and adaptive immune responses (154, 155). Normally found in the cytoplasm as inactive dimers associated with IκB, NF-κB translocates to the nucleus once IκB is phosphorylated by the upstream IκB kinase complex (IKK), leading to the transactivation of numerous gene targets (154, 156). NF-κB-dependent pathways may influence the inflammatory process in atherosclerosis. Various experimental models indicate that intracellular HSP27 attenuates NF-κB activation and signaling. In skeletal muscle disuse-mediated atrophy, the activation of NF-κB is reversed by HSP27 (157). Cardiac-specific overexpression of HSP27 was found to be protective against LPS-induced cardiac dysfunction in mice via inhibition of the NF-κB pathway (158). Also, heat shock treatment, which upregulated HSP27, was thought to protect against angiotensin II-induced inflammation via an inhibitory effect of HSP27 on the NF-κB pathway by reducing both phosphorylated and non-phosphorylated IKK-α (159, 160). However, several studies show the opposite effect of HSP27 on NF-κB. HSP27 overexpression enhances the 26S proteasome and augments the degradation of phosphorylated IκBα. As described previously, this leads to the activation and nuclear translocation of NF-κB, which the authors suggest is responsible for HSP27’s downstream anti-apoptotic properties (161). More recently, Park and colleagues found that some pro-inflammatory cytokines, such as TNF-α, can enhance the binding of HSP27 to other NF-κB protein partners, such as IκB kinases (IKKs), and this interaction is promoted by the phosphorylation of HSP27 (162). While these studies all focused on intracellular actions of HSP27, whether extracellular HSP27 has an effect on the NF-κB pathway is starting to be explored.
Within atherosclerotic plaque, NF-κB is activated mainly in endothelial cells, smooth muscle cells, and macrophages. Based on the evidence supporting the role of HSP27 in the activation of NF-κB, and also the release of HSP27 from macrophages by estrogen (66, 135), it is apparent that extracellular HSP27 may also contribute to NF-κB signaling during atherosclerosis (Figure 4). Indeed, macrophages exposed to an extracellular recombinant HSP27 (rHSP27) consistently activated NF-κB through the degradation of IκBα. This effect was followed by a change in the transcriptional profile of some target genes coding for pro- and anti-inflammatory cytokines, such as IL-6, GM-CSF, TNF, or IL-10 (103), confirming the evidence that cytokines are implicated at various stages of atherosclerosis. Moreover, it was also shown that HSP27 attenuated ac-LDL-induced release of the pro-inflammatory cytokine, IL-1β, while increasing secretion of the anti-inflammatory cytokine, IL-10, in macrophages (65). Previous findings in primary human monocytes support this anti-inflammatory role, as extracellular rHSP27 significantly induced IL-10 production and only minimally upregulated TNF-α (107).
Extracellular HSP27 may also protect the endothelium from various stress factors inherent in the vasculature. The vessel wall and subendothelial space are continually exposed to stress from hemodynamic strain, as well as damaging factors within the blood itself (i.e., pro-inflammatory molecules, pathogens, carcinogens, and products of oxidative stress). Reactive oxygen species (ROS) can modify LDLs in the blood, turning it into ox-LDL, which has been shown to activate NADPH oxidase and xanthine oxidase, thereby inducing oxidative stress (163). Additionally, exposure of endothelial cells to LDL and ox-LDL increases the levels of the ROS species, O2−, and NO (164, 165). During oxidative stress, HSP27 functions as an antioxidant in cells, lowering the levels of ROS by reducing the levels of intracellular iron and raising intracellular levels of glutathione (166). HSP27’s presence in the circulation may therefore affect ox-LDL’s contribution to atherogenesis on two levels – by competing with its uptake into macrophages (Figure 4) (65) and by lowering intracellular ROS production in endothelial cells, which would then curb the subsequent oxidative modification of LDL in the blood. An additional cardioprotective effect stems from the ability of HSP27 to protect the endothelium from ischemic insult by maintaining the integrity of cytoskeletal proteins (167–171). HSP27 can also preserve the endothelial barrier by functioning as a general anti-apoptotic protein, since downregulation of HSP27 in retinal capillary endothelial cells by both cytokines and siRNA increased apoptosis (172). Thus, HSP27-based treatment strategies – which could potentially prevent LDL oxidative modification as well as protect the endothelium/accelerate its repair – may be effective in reducing lesion formation. Although the majority of the above findings related to endothelium protection involve intracellular HSP27, recent results from our laboratory implicate extracellular HSP27 in the regeneration of the endothelial barrier (105). Using primary human EPCs treated with rHSP27 and mice overexpressing HSP27 (HSP27o/e mice), it was found that after vascular injury, re-endothelialization improved in both the experimental models, and neointima formation was decreased in HSP27o/e mice compared to wild type – effects partly mediated through the secretion of VEGF and its paracrine effect on EPCs (105).
By mediating the immunological process of atherosclerosis (Figures 3 and 4) and attenuating disease symptoms and progression, HSP27 has considerable therapeutic potential in the treatment of cardiovascular diseases. Currently, our laboratory is considering several strategies to capitalize on the atheroprotective effect of HSP27 via its emerging role as an immune modulator.
The first and simplest approach is to administer HSP27 to patients. Indeed, as described above, in mice prone to developing atherosclerosis, subcutaneous injections of rHSP27 reduced lesion formation and total cholesterol levels (63). Although these preclinical findings in animal models are encouraging, several questions remain in the context of its application in the clinical setting – for example, what is the best route of administration (subcutaneous vs. intravenous injection vs. oral intake)? Also, what is the most effective formulation (the complete HSP27 protein vs. an HSP27 protein fragment)? An important caveat to keep in mind as well is that the administration of HSP27 may pose risks in the case of cancer, since extracellular HSP27 has been shown to promote (i) the transendothelial migration of primary tumor cells (49) and (ii) the differentiation of macrophages to phenotypes that tolerate human breast cancer cell progression (48). One must therefore carefully weigh the advantages and disadvantages concerning exogenous administration of HSP27 as a therapeutic strategy in atherosclerosis.
Another approach to consider is to harness the organism’s inherent immunity when exposed to rHSP27. The discovery in our laboratory of HSP27 being complexed with AAB in the blood (thus confounding its detection through conventional ELISA kits) led to the development of a more sensitive, mass spectrometric-based detection assay, as well as mechanistic insight into how HSP27 exists/functions outside the cell. Our preliminary studies suggest that HSP27 AAB may, in fact, have salutary effects on HSP27 signaling and atheroprotection (67). The existence of AAB against HSPs in the context of atherosclerosis is not new – for example, antibodies generated against microbial HSP60 may cross-react with endogenous HSP60 expressed in the vessel lumen (molecular mimicry), enhancing inflammation and promoting atherosclerosis (173). HSP27 AAB are present in other disease states like cancer (174) and acute heart disease (175–177), although it is difficult to gage how levels correlate with disease. Higher antibody titers are associated with improved survival of breast cancer patients, but elevations also correlate with adverse conditions, such as metabolic syndrome (177) and the early phase of acute coronary syndromes (e.g., myocardial infarction and unstable angina) – although in acute coronary syndromes, HSP27 antibody levels rapidly fall after a brief spike (175). It is still unclear how HSP27 AAB are linked to CVD – one study correlated elevations with CVD (176), while other findings indicate the opposite, that HSP27 autoantibody titers (67, 178) are higher in healthy control subjects compared to CVD patients. Indeed, if the presence of HSP27 antibodies complexed to HSP27 promotes atheroprotection, immunization strategies, either active (vis-à-vis injection with an HSP27-derived antigenic peptide with or without adjuvant) or passive (administration of the antigenic peptide combined with already generated AAB), could provide exciting, new therapeutic possibilities.
Finally, there are alternative methods for boosting HSP27 function. For example, some microRNAs (“miRs,” which are 20–22 nt-long non-coding RNA molecules that regulate gene expression by binding to nascent mRNA transcripts, targeting them for degradation), which downregulate HSP27 mRNA transcripts, can be targeted for degradation by its complimentary sequence, known as “miR inhibitor” or “anti-miR” (179, 180). Proof of this principle has been demonstrated in an insulin-resistance rat model, where decreased expression of miR-1 and miR-133 increased the level of HSP27, contributing to the loss of muscle adaptability (181). Our laboratory is currently investigating which miR species are specifically upregulated in atherosclerosis and whether these alter HSP27 levels; if so, there is a potential to target such miRs with anti-miR therapy in atherosclerosis patients. There are also endogenous methods of increasing levels of circulating HSPs – for example, a huge literature already exists correlating exercise with increased serum HSP levels (182). In light of a recent Finnish study linking frequency of sauna use with reduced risk of CVD (183), it appears that subjecting the body to periods of mild to moderate heat stress may be very beneficial for the health. These sentiments – extolling the virtues of heat – are best expressed by a quote attributed to the ancient sage and physician Hippocrates: “That which drugs fail to cure, the scalpel can cure. That which the scalpel fails to cure, heat can cure. If the heat cannot cure, it must be determined to be incurable” (184, 185). By modulating atheroprogression through cross talk with the immune system, strategies that increase HSP27 levels are promising therapeutic approaches to consider.
ZB – planning and organizing structure of the review; contributed to the sections (writing): introduction, background, extracellular HSP27, HSP27 in CVD, immunological contribution, mechanisms of atheroprotection, future directions; and planning and creation of figures. VV – contributed to the sections (writing): immunological contribution; planning and creation of figures. YL – planning and organizing structure of the review; contributed to the sections (writing): background, HSP27 in CVD, mechanisms of atheroprotection; planning and creation of figures. GK – planning and organizing structure of the review; contributed to the sections (writing): HSP27 in CVD, immunological contribution, mechanisms of atheroprotection; and planning and creation of figures. DA-O – contributed to the sections (writing): mechanisms of atheroprotection. CS – planning and organizing structure of the review; contributed to the sections: background, extracellular HSP27, HSP27 in CVD. EO – planning and organizing structure of the review; contributed to the sections writing/critically reviewing manuscript, planning the structure/outline; critical review of the manuscripts; and contributed to all sections (writing/editing).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by operating grants jointly funded by the Canadian Institute for Health Research (CIHR) and Medtronic Canada (ISO 110836) as well as the Heart and Stroke Foundation of Canada (G-13-0001599). VV is the recipient of a University of Calgary Cumming School of Medicine Eyes High Postdoctoral Fellowship Award. CIHR and Medtronic also collectively fund a peer-reviewed research chair [grant IRC 57093] to EO.
1. Ritossa F. A new puffing pattern induced by temperature shock and DNP in Drosophila. Experientia (1962) 18(12):571–3. doi:10.1007/BF02172188
2. Tissieres A, Mitchell HK, Tracy UM. Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J Mol Biol (1974) 84(3):389–98. doi:10.1016/0022-2836(74)90447-1
3. Houry WA. Chaperone-assisted protein folding in the cell cytoplasm. Curr Protein Pept Sci (2001) 2(3):227–44. doi:10.2174/1389203013381134
4. Lindquist S. The heat-shock response. Annu Rev Biochem (1986) 55:1151–91. doi:10.1146/annurev.bi.55.070186.005443
5. Morimoto RI. Heat shock: the role of transient inducible responses in cell damage, transformation, and differentiation. Cancer Cells (1991) 3(8):295–301.
6. Mathew A, Morimoto RI. Role of the heat-shock response in the life and death of proteins. Ann N Y Acad Sci (1998) 851:99–111. doi:10.1111/j.1749-6632.1998.tb08982.x
7. De Maio A, Vazquez D. Extracellular heat shock proteins: a new location, a new function. Shock (2013) 40(4):239–46. doi:10.1097/SHK.0b013e3182a185ab
8. Calderwood SK. Heat shock proteins in extracellular signaling. Methods (2007) 43(3):167. doi:10.1016/j.ymeth.2007.09.004
9. Ciocca DR, Adams DJ, Edwards DP, Bjercke RJ, McGuire WL. Distribution of an estrogen-induced protein with a molecular weight of 24,000 in normal and malignant human tissues and cells. Cancer Res (1983) 43(3):1204–10.
10. Ciocca DR, Oesterreich S, Chamness GC, McGuire WL, Fuqua SA. Biological and clinical implications of heat shock protein 27,000 (Hsp27): a review. J Natl Cancer Inst (1993) 85(19):1558–70. doi:10.1093/jnci/85.19.1558
11. Ciocca DR, Dufau ML. Estrogen-dependent Leydig cell protein recognized by monoclonal antibody to MCF-7 cell line. Science (1984) 226(4673):445–6. doi:10.1126/science.6387908
12. Gaestel M, Gross B, Benndorf R, Strauss M, Schunk WH, Kraft R, et al. Molecular cloning, sequencing and expression in Escherichia coli of the 25-kDa growth-related protein of Ehrlich ascites tumor and its homology to mammalian stress proteins. Eur J Biochem (1989) 179(1):209–13. doi:10.1111/j.1432-1033.1989.tb14542.x
13. Fuqua SA, Blum-Salingaros M, McGuire WL. Induction of the estrogen-regulated “24K” protein by heat shock. Cancer Res (1989) 49(15):4126–9.
14. Strahler JR, Kuick R, Eckerskorn C, Lottspeich F, Richardson BC, Fox DA, et al. Identification of two related markers for common acute lymphoblastic leukemia as heat shock proteins. J Clin Invest (1990) 85(1):200–7. doi:10.1172/JCI114413
15. Ciocca DR, Luque EH. Immunological evidence for the identity between the hsp27 estrogen-regulated heat shock protein and the p29 estrogen receptor-associated protein in breast and endometrial cancer. Breast Cancer Res Treat (1991) 20(1):33–42. doi:10.1007/BF01833355
16. Mendelsohn ME, Zhu Y, O’Neill S. The 29-kDa proteins phosphorylated in thrombin-activated human platelets are forms of the estrogen receptor-related 27-kDa heat shock protein. Proc Natl Acad Sci U S A (1991) 88(24):11212–6. doi:10.1073/pnas.88.24.11212
17. Bakthisaran R, Tangirala R, Rao ChM. Small heat shock proteins: role in cellular functions and pathology. Biochim Biophys Acta (2015) 1854(4):291–319. doi:10.1016/j.bbapap.2014.12.019
18. Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones (2009) 14(1):105–11. doi:10.1007/s12192-008-0068-7
19. Stock AD, Spallone PA, Dennis TR, Netski D, Morris CA, Mervis CB, et al. Heat shock protein 27 gene: chromosomal and molecular location and relationship to Williams syndrome. Am J Med Genet A (2003) 120A(3):320–5. doi:10.1002/ajmg.a.20055
20. Zuchner S, Vance JM. Molecular genetics of autosomal-dominant axonal Charcot-Marie-Tooth disease. Neuromolecular Med (2006) 8(1–2):63–74. doi:10.1385/NMM:8:1:63
21. Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet (2004) 36(6):602–6. doi:10.1038/ng1354
22. Boggio PS, Asthana MK, Costa TL, Valasek CA, Osorio AA. Promoting social plasticity in developmental disorders with non-invasive brain stimulation techniques. Front Neurosci (2015) 9:294. doi:10.3389/fnins.2015.00294
23. Korenberg JR, Chen XN, Hirota H, Lai Z, Bellugi U, Burian D, et al. VI. Genome structure and cognitive map of Williams syndrome. J Cogn Neurosci (2000) 12(Suppl 1):89–107. doi:10.1162/089892900562002
24. Arrigo AP. Structure-functions of HspB1 (Hsp27). Methods Mol Biol (2011) 787:105–19. doi:10.1007/978-1-61779-295-3_9
25. Lambert H, Charette SJ, Bernier AF, Guimond A, Landry J. HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J Biol Chem (1999) 274(14):9378–85. doi:10.1074/jbc.274.14.9378
26. Landry J, Lambert H, Zhou M, Lavoie JN, Hickey E, Weber LA, et al. Human HSP27 is phosphorylated at serines 78 and 82 by heat shock and mitogen-activated kinases that recognize the same amino acid motif as S6 kinase II. J Biol Chem (1992) 267(2):794–803.
27. Ellis RJ, van der Vies SM. Molecular chaperones. Annu Rev Biochem (1991) 60:321–47. doi:10.1146/annurev.bi.60.070191.001541
28. Basha E, O’Neill H, Vierling E. Small heat shock proteins and alpha-crystallins: dynamic proteins with flexible functions. Trends Biochem Sci (2012) 37(3):106–17. doi:10.1016/j.tibs.2011.11.005
29. Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem (1993) 268(3):1517–20.
30. Huot J, Houle F, Spitz DR, Landry J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res (1996) 56(2):273–9.
31. Mounier N, Arrigo AP. Actin cytoskeleton and small heat shock proteins: how do they interact? Cell Stress Chaperones (2002) 7(2):167–76. doi:10.1379/1466-1268(2002)007<0167:ACASHS>2.0.CO;2
32. Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol (1995) 15(1):505–16. doi:10.1128/MCB.15.1.505
33. Knapinska AM, Gratacos FM, Krause CD, Hernandez K, Jensen AG, Bradley JJ, et al. Chaperone Hsp27 modulates AUF1 proteolysis and AU-rich element-mediated mRNA degradation. Mol Cell Biol (2011) 31(7):1419–31. doi:10.1128/MCB.00907-10
34. Mehlen P, Kretz-Remy C, Preville X, Arrigo AP. Human hsp27, Drosophila hsp27 and human alphaB-crystallin expression-mediated increase in glutathione is essential for the protective activity of these proteins against TNFalpha-induced cell death. EMBO J (1996) 15(11):2695–706.
35. Preville X, Salvemini F, Giraud S, Chaufour S, Paul C, Stepien G, et al. Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp Cell Res (1999) 247(1):61–78. doi:10.1006/excr.1998.4347
36. Parcellier A, Schmitt E, Brunet M, Hammann A, Solary E, Garrido C. Small heat shock proteins HSP27 and alphaB-crystallin: cytoprotective and oncogenic functions. Antioxid Redox Signal (2005) 7(3–4):404–13. doi:10.1089/ars.2005.7.404
37. Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol (2000) 2(9):645–52. doi:10.1038/35023595
38. Gibert B, Eckel B, Gonin V, Goldschneider D, Fombonne J, Deux B, et al. Targeting heat shock protein 27 (HspB1) interferes with bone metastasis and tumour formation in vivo. Br J Cancer (2012) 107(1):63–70. doi:10.1038/bjc.2012.188
39. Gao C, Zou Z, Xu L, Moul J, Seth P, Srivastava S. p53-dependent induction of heat shock protein 27 (HSP27) expression. Int J Cancer (2000) 88(2):191–4. doi:10.1002/1097-0215(20001015)88:2<191::AID-IJC7>3.0.CO;2-A
40. O’Callaghan-Sunol C, Gabai VL, Sherman MY. Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res (2007) 67(24):11779–88. doi:10.1158/0008-5472.CAN-07-2441
41. Vidyasagar A, Wilson NA, Djamali A. Heat shock protein 27 (HSP27): biomarker of disease and therapeutic target. Fibrogenesis Tissue Repair (2012) 5(1):7. doi:10.1186/1755-1536-5-7
42. Katsogiannou M, Andrieu C, Baylot V, Baudot A, Dusetti NJ, Gayet O, et al. The functional landscape of Hsp27 reveals new cellular processes such as DNA repair and alternative splicing and proposes novel anticancer targets. Mol Cell Proteomics (2014) 13(12):3585–601. doi:10.1074/mcp.M114.041228
43. Arrigo AP, Gibert B. Protein interactomes of three stress inducible small heat shock proteins: HspB1, HspB5 and HspB8. Int J Hyperthermia (2013) 29(5):409–22. doi:10.3109/02656736.2013.792956
44. Mymrikov EV, Seit-Nebi AS, Gusev NB. Large potentials of small heat shock proteins. Physiol Rev (2011) 91(4):1123–59. doi:10.1152/physrev.00023.2010
45. Tytell M, Greenberg SG, Lasek RJ. Heat shock-like protein is transferred from glia to axon. Brain Res (1986) 363(1):161–4. doi:10.1016/0006-8993(86)90671-2
46. Hightower LE, Guidon PT Jr. Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J Cell Physiol (1989) 138(2):257–66. doi:10.1002/jcp.1041380206
47. Feng JT, Liu YK, Song HY, Dai Z, Qin LX, Almofti MR, et al. Heat-shock protein 27: a potential biomarker for hepatocellular carcinoma identified by serum proteome analysis. Proteomics (2005) 5(17):4581–8. doi:10.1002/pmic.200401309
48. Banerjee S, Lin CF, Skinner KA, Schiffhauer LM, Peacock J, Hicks DG, et al. Heat shock protein 27 differentiates tolerogenic macrophages that may support human breast cancer progression. Cancer Res (2011) 71(2):318–27. doi:10.1158/0008-5472.CAN-10-1778
49. Thuringer D, Berthenet K, Cronier L, Solary E, Garrido C. Primary tumor- and metastasis-derived colon cancer cells differently modulate connexin expression and function in human capillary endothelial cells. Oncotarget (2015) 6(30):28800–15. doi:10.18632/oncotarget.4894
50. Ciocca DR, Adams DJ, Edwards DP, Bjercke RJ, McGuire WL. Estrogen-induced 24K protein in MCF-7 breast cancer cells is localized in granules. Breast Cancer Res Treat (1984) 4(4):261–8. doi:10.1007/BF01806037
51. Fanelli MA, Cuello Carrion FD, Dekker J, Schoemaker J, Ciocca DR. Serological detection of heat shock protein hsp27 in normal and breast cancer patients. Cancer Epidemiol Biomarkers Prev (1998) 7(9):791–5.
52. Lancaster GI, Febbraio MA. Mechanisms of stress-induced cellular HSP72 release: implications for exercise-induced increases in extracellular HSP72. Exerc Immunol Rev (2005) 11:46–52.
53. Lancaster GI, Moller K, Nielsen B, Secher NH, Febbraio MA, Nybo L. Exercise induces the release of heat shock protein 72 from the human brain in vivo. Cell Stress Chaperones (2004) 9(3):276–80. doi:10.1379/CSC-18R.1
54. Liao WC, Wu MS, Wang HP, Tien YW, Lin JT. Serum heat shock protein 27 is increased in chronic pancreatitis and pancreatic carcinoma. Pancreas (2009) 38(4):422–6. doi:10.1097/MPA.0b013e318198281d
55. Huang Q, Ye J, Huang Q, Chen W, Wang L, Lin W, et al. Heat shock protein 27 is over-expressed in tumor tissues and increased in sera of patients with gastric adenocarcinoma. Clin Chem Lab Med (2010) 48(2):263–9. doi:10.1515/CCLM.2010.043
56. Burut DF, Borai A, Livingstone C, Ferns G. Serum heat shock protein 27 antigen and antibody levels appear to be related to the macrovascular complications associated with insulin resistance: a pilot study. Cell Stress Chaperones (2010) 15(4):379–86. doi:10.1007/s12192-009-0152-7
57. Ce P, Erkizan O, Gedizlioglu M. Elevated HSP27 levels during attacks in patients with multiple sclerosis. Acta Neurol Scand (2011) 124(5):317–20. doi:10.1111/j.1600-0404.2010.01475.x
58. Pourhamidi K, Skarstrand H, Dahlin LB, Rolandsson O. HSP27 concentrations are lower in patients with type 1 diabetes and correlate with large nerve fiber dysfunction. Diabetes Care (2014) 37(3):e49–50. doi:10.2337/dc13-1780
59. Hecker JG, McGarvey M. Heat shock proteins as biomarkers for the rapid detection of brain and spinal cord ischemia: a review and comparison to other methods of detection in thoracic aneurysm repair. Cell Stress Chaperones (2011) 16(2):119–31. doi:10.1007/s12192-010-0224-8
60. Martin-Ventura JL, Duran MC, Blanco-Colio LM, Meilhac O, Leclercq A, Michel JB, et al. Identification by a differential proteomic approach of heat shock protein 27 as a potential marker of atherosclerosis. Circulation (2004) 110(15):2216–9. doi:10.1161/01.CIR.0000136814.87170.B1
61. Park HK, Park EC, Bae SW, Park MY, Kim SW, Yoo HS, et al. Expression of heat shock protein 27 in human atherosclerotic plaques and increased plasma level of heat shock protein 27 in patients with acute coronary syndrome. Circulation (2006) 114(9):886–93. doi:10.1161/CIRCULATIONAHA.105.541219
62. Jin C, Phillips VL, Williams MJ, van Rij AM, Jones GT. Plasma heat shock protein 27 is associated with coronary artery disease, abdominal aortic aneurysm and peripheral artery disease. Springerplus (2014) 3:635. doi:10.1186/2193-1801-3-635
63. Seibert TA, Hibbert B, Chen YX, Rayner K, Simard T, Hu T, et al. Serum heat shock protein 27 levels represent a potential therapeutic target for atherosclerosis: observations from a human cohort and treatment of female mice. J Am Coll Cardiol (2013) 62(16):1446–54. doi:10.1016/j.jacc.2013.05.041
64. Jin C, Cleveland JC, Ao L, Li J, Zeng Q, Fullerton DA, et al. Human myocardium releases heat shock protein 27 (HSP27) after global ischemia: the proinflammatory effect of extracellular HSP27 through toll-like receptor (TLR)-2 and TLR4. Mol Med (2014) 20:280–9. doi:10.2119/molmed.2014.00058
65. Rayner K, Chen YX, McNulty M, Simard T, Zhao X, Wells DJ, et al. Extracellular release of the atheroprotective heat shock protein 27 is mediated by estrogen and competitively inhibits acLDL binding to scavenger receptor-A. Circ Res (2008) 103(2):133–41. doi:10.1161/CIRCRESAHA.108.172155
66. Rayner K, Sun J, Chen YX, McNulty M, Simard T, Zhao X, et al. Heat shock protein 27 protects against atherogenesis via an estrogen-dependent mechanism: role of selective estrogen receptor beta modulation. Arterioscler Thromb Vasc Biol (2009) 29(11):1751–6. doi:10.1161/ATVBAHA.109.193656
67. Shi C, Chen Y, Li Y, O’Brien ER. When auto-antibodies potentiate: the paradoxical signalling role of anti-Hsp27 auto-antibody immune complexes improves athero-protection. Circulation (2014) 130(Suppl 2):A12771.
68. Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol (2000) 12(11):1539–46. doi:10.1093/intimm/12.11.1539
69. De Maio A. Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones (2011) 16(3):235–49. doi:10.1007/s12192-010-0236-4
70. Hunter-Lavin C, Davies EL, Bacelar MM, Marshall MJ, Andrew SM, Williams JH. Hsp70 release from peripheral blood mononuclear cells. Biochem Biophys Res Commun (2004) 324(2):511–7. doi:10.1016/j.bbrc.2004.09.075
71. Mambula SS, Calderwood SK. Heat induced release of Hsp70 from prostate carcinoma cells involves both active secretion and passive release from necrotic cells. Int J Hyperthermia (2006) 22(7):575–85. doi:10.1080/02656730600976042
72. Nickel W, Seedorf M. Unconventional mechanisms of protein transport to the cell surface of eukaryotic cells. Annu Rev Cell Dev Biol (2008) 24:287–308. doi:10.1146/annurev.cellbio.24.110707.175320
73. Steringer JP, Bleicken S, Andreas H, Zacherl S, Laussmann M, Temmerman K, et al. Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J Biol Chem (2012) 287(33):27659–69. doi:10.1074/jbc.M112.381939
74. Lee YJ, Lee HJ, Choi SH, Jin YB, An HJ, Kang JH, et al. Soluble HSPB1 regulates VEGF-mediated angiogenesis through their direct interaction. Angiogenesis (2012) 15(2):229–42. doi:10.1007/s10456-012-9255-3
75. Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol (2006) 177(11):7849–57. doi:10.4049/jimmunol.177.11.7849
76. Rabouille C, Malhotra V, Nickel W. Diversity in unconventional protein secretion. J Cell Sci (2012) 125(Pt 22):5251–5. doi:10.1242/jcs.103630
77. La Venuta G, Zeitler M, Steringer JP, Muller HM, Nickel W. The startling properties of fibroblast growth factor 2: how to exit mammalian cells without a signal peptide at hand. J Biol Chem (2015) 290(45):27015–20. doi:10.1074/jbc.R115.689257
78. Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature (2010) 463(7280):549–53. doi:10.1038/nature08710
79. Arispe N, Doh M, Simakova O, Kurganov B, De Maio A. Hsc70 and Hsp70 interact with phosphatidylserine on the surface of PC12 cells resulting in a decrease of viability. FASEB J (2004) 18(14):1636–45. doi:10.1096/fj.04-2088com
80. Schilling D, Gehrmann M, Steinem C, De Maio A, Pockley AG, Abend M, et al. Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J (2009) 23(8):2467–77. doi:10.1096/fj.08-125229
81. Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, et al. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol (2008) 180(6):4299–307. doi:10.4049/jimmunol.180.6.4299
82. Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol (2002) 3(2):122–31. doi:10.1038/nrm732
83. Mambula SS, Stevenson MA, Ogawa K, Calderwood SK. Mechanisms for Hsp70 secretion: crossing membranes without a leader. Methods (2007) 43(3):168–75. doi:10.1016/j.ymeth.2007.06.009
84. Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med (2004) 200(4):425–35. doi:10.1084/jem.20040531
85. Johnstone RM. Exosomes biological significance: a concise review. Blood Cells Mol Dis (2006) 36(2):315–21. doi:10.1016/j.bcmd.2005.12.001
86. Gupta S, Knowlton AA. HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Heart Circ Physiol (2007) 292(6):H3052–6. doi:10.1152/ajpheart.01355.2006
87. Vicencio JM, Yellon DM, Sivaraman V, Das D, Boi-Doku C, Arjun S, et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J Am Coll Cardiol (2015) 65(15):1525–36. doi:10.1016/j.jacc.2015.02.026
88. Anand PK. Exosomal membrane molecules are potent immune response modulators. Commun Integr Biol (2010) 3(5):405–8. doi:10.4161/cib.3.5.12474
89. Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem (2005) 280(24):23349–55. doi:10.1074/jbc.M502017200
90. Clayton A, Turkes A, Navabi H, Mason MD, Tabi Z. Induction of heat shock proteins in B-cell exosomes. J Cell Sci (2005) 118(Pt 16):3631–8. doi:10.1242/jcs.02494
91. Nafar F, Williams JB, Mearow KM. Astrocytes release HspB1 in response to amyloid-beta exposure in vitro. J Alzheimers Dis (2015) 49(1):251–63. doi:10.3233/JAD-150317
92. Anand PK, Anand E, Bleck CK, Anes E, Griffiths G. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS One (2010) 5(4):e10136. doi:10.1371/journal.pone.0010136
93. Muller HM, Steringer JP, Wegehingel S, Bleicken S, Munster M, Dimou E, et al. Formation of disulfide bridges drives oligomerization, membrane pore formation, and translocation of fibroblast growth factor 2 to cell surfaces. J Biol Chem (2015) 290(14):8925–37. doi:10.1074/jbc.M114.622456
94. Pasupuleti N, Gangadhariah M, Padmanabha S, Santhoshkumar P, Nagaraj RH. The role of the cysteine residue in the chaperone and anti-apoptotic functions of human Hsp27. J Cell Biochem (2010) 110(2):408–19. doi:10.1002/jcb.22552
95. Hochberg GK, Ecroyd H, Liu C, Cox D, Cascio D, Sawaya MR, et al. The structured core domain of alphaB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc Natl Acad Sci U S A (2014) 111(16):E1562–70. doi:10.1073/pnas.1322673111
96. Rajagopal P, Liu Y, Shi L, Clouser AF, Klevit RE. Structure of the alpha-crystallin domain from the redox-sensitive chaperone, HSPB1. J Biomol NMR (2015) 63(2):223–8. doi:10.1007/s10858-015-9973-0
97. Peschek J, Braun N, Rohrberg J, Back KC, Kriehuber T, Kastenmuller A, et al. Regulated structural transitions unleash the chaperone activity of alphaB-crystallin. Proc Natl Acad Sci U S A (2013) 110(40):E3780–9. doi:10.1073/pnas.1308898110
98. Vigh L, Nakamoto H, Landry J, Gomez-Munoz A, Harwood JL, Horvath I. Membrane regulation of the stress response from prokaryotic models to mammalian cells. Ann N Y Acad Sci (2007) 1113:40–51. doi:10.1196/annals.1391.027
99. Vigh L, Horvath I, Maresca B, Harwood JL. Can the stress protein response be controlled by ‘membrane-lipid therapy’? Trends Biochem Sci (2007) 32(8):357–63. doi:10.1016/j.tibs.2007.06.009
100. Gehrmann M, Liebisch G, Schmitz G, Anderson R, Steinem C, De Maio A, et al. Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS One (2008) 3(4):e1925. doi:10.1371/journal.pone.0001925
101. Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C. Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. J Leukoc Biol (2007) 81(1):15–27. doi:10.1189/jlb.0306167
102. Binder RJ, Vatner R, Srivastava P. The heat-shock protein receptors: some answers and more questions. Tissue Antigens (2004) 64(4):442–51. doi:10.1111/j.1399-0039.2004.00299.x
103. Salari S, Seibert T, Chen YX, Hu T, Shi C, Zhao X, et al. Extracellular HSP27 acts as a signaling molecule to activate NF-kappaB in macrophages. Cell Stress Chaperones (2013) 18(1):53–63. doi:10.1007/s12192-012-0356-0
104. Thuringer D, Jego G, Wettstein G, Terrier O, Cronier L, Yousfi N, et al. Extracellular HSP27 mediates angiogenesis through toll-like receptor 3. FASEB J (2013) 27(10):4169–83. doi:10.1096/fj.12-226977
105. Ma X, Hibbert B, McNulty M, Hu T, Zhao X, Ramirez FD, et al. Heat shock protein 27 attenuates neointima formation and accelerates reendothelialization after arterial injury and stent implantation: importance of vascular endothelial growth factor up-regulation. FASEB J (2014) 28(2):594–602. doi:10.1096/fj.13-230417
106. Yusuf N, Nasti TH, Huang CM, Huber BS, Jaleel T, Lin HY, et al. Heat shock proteins HSP27 and HSP70 are present in the skin and are important mediators of allergic contact hypersensitivity. J Immunol (2009) 182(1):675–83. doi:10.4049/jimmunol.182.1.675
107. De AK, Kodys KM, Yeh BS, Miller-Graziano C. Exaggerated human monocyte IL-10 concomitant to minimal TNF-alpha induction by heat-shock protein 27 (Hsp27) suggests Hsp27 is primarily an antiinflammatory stimulus. J Immunol (2000) 165(7):3951–8. doi:10.4049/jimmunol.165.7.3951
108. Miller H, Poon S, Hibbert B, Rayner K, Chen YX, O’Brien ER. Modulation of estrogen signaling by the novel interaction of heat shock protein 27, a biomarker for atherosclerosis, and estrogen receptor beta: mechanistic insight into the vascular effects of estrogens. Arterioscler Thromb Vasc Biol (2005) 25(3):e10–4. doi:10.1161/01.ATV.0000156536.89752.8e
109. Al-Madhoun AS, Chen YX, Haidari L, Rayner K, Gerthoffer W, McBride H, et al. The interaction and cellular localization of HSP27 and ERbeta are modulated by 17beta-estradiol and HSP27 phosphorylation. Mol Cell Endocrinol (2007) 270(1–2):33–42. doi:10.1016/j.mce.2007.02.002
110. Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res (2005) 65(12):5238–47. doi:10.1158/0008-5472.CAN-04-3804
111. Thompson RC, Allam AH, Lombardi GP, Wann LS, Sutherland ML, Sutherland JD, et al. Atherosclerosis across 4000 years of human history: the Horus study of four ancient populations. Lancet (2013) 381(9873):1211–22. doi:10.1016/S0140-6736(13)60598-X
112. Rossouw JE. Hormones, genetic factors, and gender differences in cardiovascular disease. Cardiovasc Res (2002) 53(3):550–7. doi:10.1016/S0008-6363(01)00478-3
113. Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women’s health initiative randomized controlled trial. JAMA (2002) 288(3):321–33. doi:10.1001/jama.288.3.321
114. Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, et al. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med (2003) 349(6):523–34. doi:10.1056/NEJMoa030808
115. Xu Q, Kleindienst R, Waitz W, Dietrich H, Wick G. Increased expression of heat shock protein 65 coincides with a population of infiltrating T lymphocytes in atherosclerotic lesions of rabbits specifically responding to heat shock protein 65. J Clin Invest (1993) 91(6):2693–702. doi:10.1172/JCI116508
116. Pockley AG, Wu R, Lemne C, Kiessling R, de Faire U, Frostegard J. Circulating heat shock protein 60 is associated with early cardiovascular disease. Hypertension (2000) 36(2):303–7. doi:10.1161/01.HYP.36.2.303
117. Xu Q, Schett G, Perschinka H, Mayr M, Egger G, Oberhollenzer F, et al. Serum soluble heat shock protein 60 is elevated in subjects with atherosclerosis in a general population. Circulation (2000) 102(1):14–20. doi:10.1161/01.CIR.102.1.14
118. Pockley AG, Georgiades A, Thulin T, de Faire U, Frostegard J. Serum heat shock protein 70 levels predict the development of atherosclerosis in subjects with established hypertension. Hypertension (2003) 42(3):235–8. doi:10.1161/01.HYP.0000086522.13672.23
119. Zhu J, Quyyumi AA, Wu H, Csako G, Rott D, Zalles-Ganley A, et al. Increased serum levels of heat shock protein 70 are associated with low risk of coronary artery disease. Arterioscler Thromb Vasc Biol (2003) 23(6):1055–9. doi:10.1161/01.ATV.0000074899.60898.FD
120. Lepedda AJ, Cigliano A, Cherchi GM, Spirito R, Maggioni M, Carta F, et al. A proteomic approach to differentiate histologically classified stable and unstable plaques from human carotid arteries. Atherosclerosis (2009) 203(1):112–8. doi:10.1016/j.atherosclerosis.2008.07.001
121. Kardys I, Rifai N, Meilhac O, Michel JB, Martin-Ventura JL, Buring JE, et al. Plasma concentration of heat shock protein 27 and risk of cardiovascular disease: a prospective, nested case-control study. Clin Chem (2008) 54(1):139–46. doi:10.1373/clinchem.2007.094961
122. Martin-Ventura JL, Nicolas V, Houard X, Blanco-Colio LM, Leclercq A, Egido J, et al. Biological significance of decreased HSP27 in human atherosclerosis. Arterioscler Thromb Vasc Biol (2006) 26(6):1337–43. doi:10.1161/01.ATV.0000220108.97208.67
123. Lamfers ML, Grimbergen JM, Aalders MC, Havenga MJ, de Vries MR, Huisman LG, et al. Gene transfer of the urokinase-type plasminogen activator receptor-targeted matrix metalloproteinase inhibitor TIMP-1.ATF suppresses neointima formation more efficiently than tissue inhibitor of metalloproteinase-1. Circ Res (2002) 91(10):945–52. doi:10.1161/01.RES.0000041418.51906.57
124. Sims JD, McCready J, Jay DG. Extracellular heat shock protein (Hsp)70 and Hsp90alpha assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS One (2011) 6(4):e18848. doi:10.1371/journal.pone.0018848
125. Song X, Wang X, Zhuo W, Shi H, Feng D, Sun Y, et al. The regulatory mechanism of extracellular Hsp90{alpha} on matrix metalloproteinase-2 processing and tumor angiogenesis. J Biol Chem (2010) 285(51):40039–49. doi:10.1074/jbc.M110.181941
126. Choi SH, Lee HJ, Jin YB, Jang J, Kang GY, Lee M, et al. MMP9 processing of HSPB1 regulates tumor progression. PLoS One (2014) 9(1):e85509. doi:10.1371/journal.pone.0085509
127. Osterloh A, Breloer M. Heat shock proteins: linking danger and pathogen recognition. Med Microbiol Immunol (2008) 197(1):1–8. doi:10.1007/s00430-007-0055-0
128. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell (2011) 145(3):341–55. doi:10.1016/j.cell.2011.04.005
129. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol (2006) 6(7):508–19. doi:10.1038/nri1882
130. Schwartz SM, deBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res (1995) 77(3):445–65. doi:10.1161/01.RES.77.3.445
131. Miller-Graziano CL, De A, Laudanski K, Herrmann T, Bandyopadhyay S. HSP27: an anti-inflammatory and immunomodulatory stress protein acting to dampen immune function. Novartis Found Symp (2008) 291:196–208; discussion 208–11, 221–4. doi:10.1002/9780470754030.ch15
132. Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res (2009) 50(Suppl):S382–7. doi:10.1194/jlr.R800032-JLR200
133. Campbell JH, Campbell GR. Cell biology of atherosclerosis. J Hypertens Suppl (1994) 12(10):S129–32.
134. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464(7293):1357–61. doi:10.1038/nature08938
135. Raizman JE, Chen YX, Seibert T, Hibbert B, Cuerrier CM, Salari S, et al. Heat shock protein-27 attenuates foam cell formation and atherogenesis by down-regulating scavenger receptor-A expression via NF-kappaB signaling. Biochim Biophys Acta (2013) 1831(12):1721–8. doi:10.1016/j.bbalip.2013.07.015
136. Laudanski K, De A, Miller-Graziano C. Exogenous heat shock protein 27 uniquely blocks differentiation of monocytes to dendritic cells. Eur J Immunol (2007) 37(10):2812–24. doi:10.1002/eji.200636993
137. Di Gregoli K, Johnson JL. Role of colony-stimulating factors in atherosclerosis. Curr Opin Lipidol (2012) 23(5):412–21. doi:10.1097/MOL.0b013e328357ca6e
138. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev (2014) 262(1):153–66. doi:10.1111/imr.12218
139. Nahrendorf M, Swirski FK. Innate immune cells in ischaemic heart disease: does myocardial infarction beget myocardial infarction? Eur Heart J (2016) 37(11):868–72. doi:10.1093/eurheartj/ehv453
140. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol (2016) 13(3):167–79. doi:10.1038/nrcardio.2015.169
141. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol (2015) 35(2):280–7. doi:10.1161/ATVBAHA.114.303568
142. Tselios K, Sarantopoulos A, Gkougkourelas I, Boura P. T regulatory cells: a promising new target in atherosclerosis. Crit Rev Immunol (2014) 34(5):389–97. doi:10.1615/CritRevImmunol.2014010802
143. Wick C. Tolerization against atherosclerosis using heat shock protein 60. Cell Stress Chaperones (2016) 21(2):201–211. doi:10.1007/s12192-015-0659-z
144. Conti P, Shaik-Dasthagirisaeb Y. Atherosclerosis: a chronic inflammatory disease mediated by mast cells. Cent Eur J Immunol (2015) 40(3):380–6. doi:10.5114/ceji.2015.54603
145. Roy A, Ganesh G, Sippola H, Bolin S, Sawesi O, Dagalv A, et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J Biol Chem (2014) 289(1):237–50. doi:10.1074/jbc.M112.435156
146. Beresford PJ, Jaju M, Friedman RS, Yoon MJ, Lieberman J. A role for heat shock protein 27 in CTL-mediated cell death. J Immunol (1998) 161(1):161–7.
147. Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science (1992) 258(5081):468–71. doi:10.1126/science.1411543
148. Cuerrier CM, Chen YX, Tremblay D, Rayner K, McNulty M, Zhao X, et al. Chronic over-expression of heat shock protein 27 attenuates atherogenesis and enhances plaque remodeling: a combined histological and mechanical assessment of aortic lesions. PLoS One (2013) 8(2):e55867. doi:10.1371/journal.pone.0055867
149. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem (2002) 277(51):49982–8. doi:10.1074/jbc.M209649200
150. Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature (1997) 386(6622):292–6. doi:10.1038/386292a0
151. Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med (2002) 8(11):1235–42. doi:10.1038/nm1102-1235
152. Makinen PI, Lappalainen JP, Heinonen SE, Leppanen P, Lahteenvuo MT, Aarnio JV, et al. Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc Res (2010) 88(3):530–8. doi:10.1093/cvr/cvq235
153. Venu VKP, Adijiang A, Shi C, Chen YX, O’Brien ER. HSP27 mediated atheroprotection requires GM-CSF induction: potential role in reverse cholesterol transport? (best of basic science abstract). Circulation (2015) 132(Suppl 3):A17586–17586.
154. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol (2007) 8(1):49–62. doi:10.1038/nrm2083
155. Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene (1999) 18(49):6853–66. doi:10.1038/sj.onc.1203239
156. Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene (1999) 18(49):6867–74. doi:10.1038/sj.onc.1203219
157. Dodd SL, Hain B, Senf SM, Judge AR. Hsp27 inhibits IKKbeta-induced NF-kappaB activity and skeletal muscle atrophy. FASEB J (2009) 23(10):3415–23. doi:10.1096/fj.08-124602
158. You W, Min X, Zhang X, Qian B, Pang S, Ding Z, et al. Cardiac-specific expression of heat shock protein 27 attenuated endotoxin-induced cardiac dysfunction and mortality in mice through a PI3K/Akt-dependent mechanism. Shock (2009) 32(1):108–17. doi:10.1097/SHK.0b013e318199165d
159. Chen Y, Ross BM, Currie RW. Heat shock treatment protects against angiotensin II-induced hypertension and inflammation in aorta. Cell Stress Chaperones (2004) 9(1):99–107. doi:10.1379/1466-1268(2004)009<0099:HSTPAA>2.0.CO;2
160. Chen Y, Arrigo AP, Currie RW. Heat shock treatment suppresses angiotensin II-induced activation of NF-kappaB pathway and heart inflammation: a role for IKK depletion by heat shock? Am J Physiol Heart Circ Physiol (2004) 287(3):H1104–14. doi:10.1152/ajpheart.00102.2004
161. Parcellier A, Schmitt E, Gurbuxani S, Seigneurin-Berny D, Pance A, Chantome A, et al. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol Cell Biol (2003) 23(16):5790–802. doi:10.1128/MCB.23.16.5790-5802.2003
162. Park KJ, Gaynor RB, Kwak YT. Heat shock protein 27 association with the I kappa B kinase complex regulates tumor necrosis factor alpha-induced NF-kappa B activation. J Biol Chem (2003) 278(37):35272–8. doi:10.1074/jbc.M305095200
163. Stepp DW, Ou J, Ackerman AW, Welak S, Klick D, Pritchard KA Jr. Native LDL and minimally oxidized LDL differentially regulate superoxide anion in vascular endothelium in situ. Am J Physiol Heart Circ Physiol (2002) 283(2):H750–9. doi:10.1152/ajpheart.00029.2002
164. Pritchard KA Jr, Groszek L, Smalley DM, Sessa WC, Wu M, Villalon P, et al. Native low-density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res (1995) 77(3):510–8. doi:10.1161/01.RES.77.3.510
165. Vergnani L, Hatrik S, Ricci F, Passaro A, Manzoli N, Zuliani G, et al. Effect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production: key role of l-arginine availability. Circulation (2000) 101(11):1261–6. doi:10.1161/01.CIR.101.11.1261
166. Arrigo AP, Virot S, Chaufour S, Firdaus W, Kretz-Remy C, Diaz-Latoud C. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid Redox Signal (2005) 7(3–4):414–22. doi:10.1089/ars.2005.7.414
167. Martin JL, Mestril R, Hilal-Dandan R, Brunton LL, Dillmann WH. Small heat shock proteins and protection against ischemic injury in cardiac myocytes. Circulation (1997) 96(12):4343–8. doi:10.1161/01.CIR.96.12.4343
168. Ghayour-Mobarhan M, Saber H, Ferns GA. The potential role of heat shock protein 27 in cardiovascular disease. Clin Chim Acta (2012) 413(1–2):15–24. doi:10.1016/j.cca.2011.04.005
169. Chen H, Wu XJ, Lu XY, Zhu L, Wang LP, Yang HT, et al. Phosphorylated heat shock protein 27 is involved in enhanced heart tolerance to ischemia in short-term type 1 diabetic rats. Acta Pharmacol Sin (2005) 26(7):806–12. doi:10.1111/j.1745-7254.2005.00113.x
170. Lee JS, Zhang MH, Yun EK, Geum D, Kim K, Kim TH, et al. Heat shock protein 27 interacts with vimentin and prevents insolubilization of vimentin subunits induced by cadmium. Exp Mol Med (2005) 37(5):427–35. doi:10.1038/emm.2005.53
171. Robinson AA, Dunn MJ, McCormack A, dos Remedios C, Rose ML. Protective effect of phosphorylated Hsp27 in coronary arteries through actin stabilization. J Mol Cell Cardiol (2010) 49(3):370–9. doi:10.1016/j.yjmcc.2010.06.004
172. Nahomi RB, Palmer A, Green KM, Fort PE, Nagaraj RH. Pro-inflammatory cytokines downregulate Hsp27 and cause apoptosis of human retinal capillary endothelial cells. Biochim Biophys Acta (2014) 1842(2):164–74. doi:10.1016/j.bbadis.2013.11.011
173. Wick G, Jakic B, Buszko M, Wick MC, Grundtman C. The role of heat shock proteins in atherosclerosis. Nat Rev Cardiol (2014) 11(9):516–29. doi:10.1038/nrcardio.2014.91
174. Conroy SE, Sasieni PD, Amin V, Wang DY, Smith P, Fentiman IS, et al. Antibodies to heat-shock protein 27 are associated with improved survival in patients with breast cancer. Br J Cancer (1998) 77(11):1875–9. doi:10.1038/bjc.1998.312
175. Ghayour-Mobarhan M, Sahebkar A, Parizadeh SM, Moohebati M, Tavallaie S, Rezakazemi-Bajestani SM, et al. Antibody titres to heat shock protein 27 are elevated in patients with acute coronary syndrome. Int J Exp Pathol (2008) 89(3):209–15. doi:10.1111/j.1365-2613.2008.00586.x
176. Pourghadamyari H, Moohebati M, Parizadeh SM, Falsoleiman H, Dehghani M, Fazlinezhad A, et al. Serum antibody titers against heat shock protein 27 are associated with the severity of coronary artery disease. Cell Stress Chaperones (2011) 16(3):309–16. doi:10.1007/s12192-010-0241-7
177. Sahebkar A, Pourghadamyari H, Moohebati M, Parizadeh SM, Falsoleiman H, Dehghani M, et al. A cross-sectional study of the association between heat shock protein 27 antibody titers, pro-oxidant-antioxidant balance and metabolic syndrome in patients with angiographically-defined coronary artery disease. Clin Biochem (2011) 44(17–18):1390–5. doi:10.1016/j.clinbiochem.2011.09.011
178. Chen YX, Rayner K, Simard T, Ramirez FD, O’Brien E. Abstract 494: heat shock protein 27: influence on endothelial cell homeostasis and correlation with coronary artery disease. Circulation (2006) 114(Suppl 18):II_75.
179. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136(2):215–33. doi:10.1016/j.cell.2009.01.002
180. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 116(2):281–97. doi:10.1016/S0092-8674(04)00045-5
181. Katta A, Thulluri S, Manne ND, Addagarla HS, Arvapalli R, Nalabotu SK, et al. Overload induced heat shock proteins (HSPs), MAPK and miRNA (miR-1 and miR133a) response in insulin-resistant skeletal muscle. Cell Physiol Biochem (2013) 31(2–3):219–29. doi:10.1159/000343363
182. Asea AA, Pedersen BK. Heat Shock Proteins and Whole Body Physiology. (Vol. 5). Berlin, Germany: Springer Science & Business Media (2009).
183. Laukkanen T, Khan H, Zaccardi F, Laukkanen JA. Association between sauna bathing and fatal cardiovascular and all-cause mortality events. JAMA Intern Med (2015) 175(4):542–8. doi:10.1001/jamainternmed.2014.8187
184. Srivastava PK, Menoret A, Basu S, Binder RJ, McQuade KL. Heat shock proteins come of age: primitive functions acquire new roles in an adaptive world. Immunity (1998) 8(6):657–65. doi:10.1016/S1074-7613(00)80570-1
Keywords: heat shock protein 27, inflammation, atherosclerosis, coronary artery disease, HSPB1, non-classical secretion, extracellular HSP27
Citation: Batulan Z, Pulakazhi Venu VK, Li Y, Koumbadinga G, Alvarez-Olmedo DG, Shi C and O’Brien ER (2016) Extracellular Release and Signaling by Heat Shock Protein 27: Role in Modifying Vascular Inflammation. Front. Immunol. 7:285. doi: 10.3389/fimmu.2016.00285
Received: 02 February 2016; Accepted: 14 July 2016;
Published: 26 July 2016
Edited by:
Stuart Keith Calderwood, Harvard Medical School, USAReviewed by:
Gabriele Multhoff, Technische Universität München, GermanyCopyright: © 2016 Batulan, Pulakazhi Venu, Li, Koumbadinga, Alvarez-Olmedo, Shi and O’Brien. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward R. O’Brien, ZXJtb2JyaWVAdWNhbGdhcnkuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.