
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 June 2016
Sec. T Cell Biology
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00222
This article is part of the Research Topic The Molecular Mechanisms of cyclic AMP in regulation of immunity and tolerance View all 8 articles
 Vanessa L. Wehbi1,2,3
Vanessa L. Wehbi1,2,3 Kjetil Taskén1,2,3,4,5*
Kjetil Taskén1,2,3,4,5*
The cyclic AMP/protein kinase A (cAMP/PKA) pathway is one of the most common and versatile signal pathways in eukaryotic cells. A-kinase anchoring proteins (AKAPs) target PKA to specific substrates and distinct subcellular compartments providing spatial and temporal specificity for mediation of biological effects channeled through the cAMP/PKA pathway. In the immune system, cAMP is a potent negative regulator of T cell receptor-mediated activation of effector T cells (Teff) acting through a proximal PKA/Csk/Lck pathway anchored via a scaffold consisting of the AKAP Ezrin holding PKA, the linker protein EBP50, and the anchoring protein phosphoprotein associated with glycosphingolipid-enriched microdomains holding Csk. As PKA activates Csk and Csk inhibits Lck, this pathway in response to cAMP shuts down proximal T cell activation. This immunomodulating pathway in Teff mediates clinically important responses to regulatory T cell (Treg) suppression and inflammatory mediators, such as prostaglandins (PGs), adrenergic stimuli, adenosine, and a number of other ligands. A major inducer of T cell cAMP levels is PG E2 (PGE2) acting through EP2 and EP4 prostanoid receptors. PGE2 plays a crucial role in the normal physiological control of immune homeostasis as well as in inflammation and cancer immune evasion. Peripherally induced Tregs express cyclooxygenase-2, secrete PGE2, and elicit the immunosuppressive cAMP pathway in Teff as one tumor immune evasion mechanism. Moreover, a cAMP increase can also be induced by indirect mechanisms, such as intercellular transfer between T cells. Indeed, Treg, known to have elevated levels of intracellular cAMP, may mediate their suppressive function by transferring cAMP to Teff through gap junctions, which we speculate could also be regulated by PKA/AKAP complexes. In this review, we present an updated overview on the influence of cAMP-mediated immunoregulatory mechanisms acting through localized cAMP signaling and the therapeutical increasing prospects of AKAPs disruptors in T-cell immune function.
Cyclic AMP (cAMP) is a second messenger, which relays signals from the outside to the inside of a cell and triggers downstream signaling cascades. Modulation of the intracellular cAMP concentration reflects changes in the cellular environment and creates changes in cellular function. In the T cell, cAMP is known as a potent negative regulator, which dampens T-cell immune function through the cAMP/protein kinase A (PKA) signaling pathway. Indeed, the cAMP/type I PKA/Csk/Lck [lymphocyte-specific protein tyrosine kinase (PTK)] signaling module has been defined as a dominant regulator driving the inhibition of T-cell function (1).
Specificity of cAMP signaling is achieved by compartmentalization through A-kinase anchoring proteins (AKAPs). Most cells express between 10 and 15 different AKAPs (2) and so far 7 AKAPs have been identified in T cells. AKAPs are characterized by their cellular localization mediated through a targeting domain and their binding partners, which define the spatiotemporal control of cAMP signaling, the interaction with other signaling pathways, and contribute to distinct cellular functions. In T cells ezrin functions as an AKAP and assembles a supramolecular signaling complex with PKA type I, EBP50 [ezrin–radixin–moesin (ERM)-binding phosphoprotein 50], phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), and Csk in the vicinity of the T cell receptor (TCR), which in turn modulates T cell immune responses (3).
The cAMP/PKA inhibitory signaling pathway controlling TCR signaling plays a key role in maintaining homeostasis in the T cell. However, any imbalance in TCR regulation can lead to T cell dysfunction and dramatic functional consequences. During diseases, such as cancer and chronic infections, T cells have high cAMP concentrations, which in turn cause excessive downregulation of TCR signaling and can favor disease development. Regulation of the cAMP/PKA pathway is crucial to protect against inappropriate regulation and immunological overshoot. Non-steroidal anti-inflammatory drugs (NSAIDs) are known to negatively regulate this pathway through their inhibitory action on the activity of cyclooxygenases (COX). NSAIDs, aspirin, and coxibs (selective COX-2 inhibitors) block prostaglandin E2 (PGE2) synthesis, which in turn downregulates the intracellular cAMP concentration in T cells. A number of studies have agreed on the beneficial use of COX inhibitors to enhance anti-tumor responses cancer (4–7). Despite their efficiency, the broad-spectrum activity of COX inhibitors can trigger unwanted effects, which may be avoided with new drugs that target the pathway at a different level. Because AKAPs scaffold supramolecular complexes acting as signal processing hubs that coordinate multiple signals in normal and aberrant conditions, protein–protein interaction disruptors that displace particular components of such complexes emerge as essential research tools and potentially targeted drugs complementary to the current therapeutic strategies.
Cyclic AMP is an intracellular second messenger (8) identified by the Nobel-Prize winning work of Earl Sutherland (9), able to trigger a plethora of signaling pathways leading to different biological outcomes. In effector T cells (Teff), PGs (10), adenosine (11), histamine (12), β-adrenergic agonists (13), neuropeptide hormones (14), and β-endorphin (15) induce cAMP, which acts as a potent negative regulator of TCR-mediated activation and proliferation (16–18). This contributes to regulation and maintenance of a healthy immune response. Any imbalance in regulatory mechanisms creates immune disorders and can lead to autoimmune diseases, chronic inflammation, and allergic responses.
Prostaglandin E2 is the most ubiquitous PG produced by the human body and plays a critical role in guiding and governing various aspects of the inflammatory response. The role of PGE2 in driving acute inflammation is well established. However, PGE2 also elicits powerful immunosuppressive properties that contribute to the resolution phase of acute inflammation, the tissue regeneration, and the return into homeostasis. These multifaceted properties of PGE2 are both cell-type- and context-specific. The production of PGs begins with the liberation of arachidonic acid from membrane phospholipids by phospholipase A2 in response to inflammatory stimuli. Arachidonic acid is converted to PGH2 by the COX enzymes COX-1 and COX-2, and then to PGE2 by cell-specific PG synthases. Whereas COX-1 is considered as a ubiquitous housekeeping enzyme constitutively expressed and responsible for maintaining basal PG levels important for tissue homeostasis, COX-2 is an inducible enzyme that produces PGs during inflammatory conditions (19, 20). In tumor cells, COX-2 is often overexpressed, which in turn upregulates PGE2 and contributes to the immune evasion and cancer immunotherapy resistance creating an environment rich in IL-10 and TGF-β, cytokines known to promote regulatory T cells (Tregs) differentiation and proliferation (21–29). Treg are known as a unique population of T cells that maintain peripheral immune tolerance and effectively inhibit autoreactive T cells (30–32) and Teff responses, such as cytokine production and proliferation (33). Treg produce and respond to PGE2, which acts as an autocrine factor and increases intracellular cAMP that in turn upregulates forkhead/winged helix transcription factor (FOXP3) expression. PGE2 enhances Treg induction and differentiation through FOXP3 upregulation (4, 34–38). Tregs have also been shown to have high endogenous cAMP levels, which can be explained by a FOXP3-dependent downregulation of phosphodiesterase 3 (PDE3 is known to decline cAMP levels) (39) and an adenylyl cyclase (AC) 9 upregulation (AC9 is known to synthetize cAMP) (40). Immunosuppressive activity can be mediated by intercellular transfer of cAMP from Treg to Teff via gap junctions (GJ) presumably formed by Cx43, which is the connexin in T cells (41–43). cAMP leakage into Teff have been found to exhibit suppressive activity by enhancing the expression and nuclear function of a potent transcriptional inhibitor, inducible cAMP early repressor (ICER) and modulating the levels of surface-expressed cytotoxic T lymphocyte antigen-4 (CTLA-4) (44). In this case, the suppressive mechanisms triggered by cAMP transfer are cell-contact dependent; a close proximity between donor and recipient cells is required for the transfer of the second messenger.
Prostaglandin E2 can also act in a paracrine manner through direct binding and activation of the E-prostanoid (EP) family of G protein-coupled receptors. PGE2 can activate four subtypes of EP receptor at the T-cell surface, called EP1–EP4. Upon activation, EP receptor couples to specific G protein and activates specific G protein-dependent signaling pathways. The active EP1 receptor coupled to Gq protein increases phosphatidylinositol hydrolysis and intracellular Ca2+ through activation of phospholipase-C. The active EP3 receptor coupled to Gi protein leads to the AC inhibition and intracellular cAMP decrease. Only active EP2 and EP4 coupled to Gs protein lead to cAMP production through AC activation. This in turn activates PKA that next can activate the transcription factor cAMP response element-binding protein (CREB). CREB is known for its role in cell proliferation, differentiation, and survival (45). CREB has been shown to induce transcription of immune-related genes that possess a CRE element, including interleukin-2 (IL-2), IL-6, IL-10, tumor necrosis factor alpha (TNF-α), and COX-2 (46, 47). CREB plays a role in T-cell function (48) and also drives the generation and maintenance of Treg in a TGF-β/FoxP3-dependent manner (45, 49–53). The paracrine effect of PGE2 is clearly EP receptor subtype dependent, but it seems also to depend on the level of EP receptor cell surface expression. Indeed, during mucosal inflammation IL-2 secretion has been found to be negatively correlated to the EP4 expression (54).
Most studies on T cells have focused on CD4+ cells and showed the roles of PGE2 in the modulation of proliferation, apoptosis, and cytokine production. Less is known about the effect on CD8+ T cells, but PGE2 can inhibit CD8+ T-cell proliferation (55) and decrease the production of interferon γ (IFN-γ) (56). The impact of PGE2 in immune cells can also be cell-type-specific. Indeed PGE2 suppresses the function of macrophages, neutrophils, T helper 1 cells (Th1) cells, and natural killers, whereas it promotes T helper 2 cell (Th2), Treg, and T helper 17 cell (Th17) responses (57, 58). PGE2 has no effect on or enhances the production of Th2 cytokines, such as IL-4, IL-5, and IL-10, but inhibits drastically the production of Th1 cytokines, such as IFN-γ, TNF-α, and IL-2 (59–63). The induction of the Th2 response by PGE2 was found to be mainly cAMP-dependent (64) and recently the transcription factor, cAMP response element modulator (CREM), was characterized as a negative regulator of Th2 responses and a key factor in allergic asthma (65). In addition, PGE2 has been shown to promote T-cell anergy (66), maintain memory T-cell survival (67), and inhibit γδ T-cell cytotoxicity through a cAMP/PKA-dependent mechanism (68).
Inhibitory mechanisms triggered by PGE2 in T cells started to be unraveled in the late 1980s [reviewed in Ref. (20, 69–71)]. PGE2 triggers anti-proliferative effects through interference with IL-2-mediated gene expression (72, 73) and inhibition of IL-2 receptor expression (74, 75). PGE2 inhibits IL-2 gene transcription by downregulating the activation of its promoter. Indeed cAMP inhibits nuclear transcription of the human interleukin-2 gene by targeting two transcription factors: the nuclear factor of activated T cells (NFAT) and the activator protein 1 (AP1) contained in IL-2 promoter in human T cells (76). Furthermore, recently Rodriguez and co-authors found that elevated intracellular cAMP through a PKA-dependent pathway can disrupt IL-2R complex formation, Jak3 catalytic activity, and the ability to phosphorylate Stat5, resulting in a severe reduction in IL-2R signaling and T lymphocyte proliferation (77).
Despite the well-established molecular mechanisms unraveling cAMP inhibitory effect on T-cell function, new studies continue to fulfill our understanding with additional and/or alternative mechanisms. A recent study has described a potential new role for exchange protein directly activated by cAMP (EPAC) in the T-cell suppressive process. Downstream effectors of cAMP can be PKA dependent (78), cAMP-regulated ion channels (79, 80), and EPAC dependent (81, 82). In T cells, effects mediated by cAMP seem to be most likely through PKA activation, since EPAC and cAMP-gated channels appear to be expressed at low level in T cells (82). However, the cAMP suppressive effects on Teff have been showed to require both PKA and EPAC-dependent pathways. The involvement of EPAC in Teff suppression has been proved by the ability to mimic the cAMP response with an EPAC-selective cAMP analog, combined with the insensitivity of the cAMP response to inhibitors of PKA. The authors suggested that EPAC may function as an alternative effector to promote cAMP-dependent but PKA-independent Teff suppression (83).
Cyclic AMP is mostly known as an immunosuppressant, however, the regulation of T-cell activation is not the result of a linear control driven by cAMP gradients but the sum of competitive mechanisms processing in parallel of the cAMP immunosuppressant effects. Besides the Gs/AC/cAMP pathway, active EP2 and EP4 receptors trigger multiple signaling pathways, which can counteract the immunosuppressant effect of cAMP. In addition, full T-cell activation generally requires accompanying signals as CD28 co-stimulation inducing several specific signaling pathways as well. Yao and co-authors have shown that cAMP-mediated suppression of TCR signaling can be overcome by simultaneous activation of PI3-kinase through activation of EP2/EP4 and/or CD28. PGE2 promotes Th1 differentiation via synergistic amplification of IL-12 signaling by cAMP and PI3-kinase (84). These findings underscore the complexity of the regulation of T-cell function, which is overall the result of positive and negative events on the TCR signaling pathway.
Activation of T cells is a key step in adaptive immunity and requires the coordination and organization of the components of the TCR complex with its surrounding signaling molecules leading to TCR signaling events. The molecular process begins when the TCR identifies a peptide presented by the major histocompatibility complex (MHC) expressed on the surface of antigen-presenting cells (APCs) (85) and that binds with high affinity to the TCR. The part of the receptor that recognizes the large variety of antigens is a highly polymorphic heterodimer of α and β chains, which is associated with polypeptides γ, δ, ϵ, and ζ. All CD3 chains contain immunoreceptor tyrosine-based activation motifs (ITAMs) (86, 87). Activation is accompanied by formation of the immunological synapse (88) where lipid rafts (membrane microdomains enriched in cholesterol and sphingolipids) and Src-family tyrosine kinase (SFK) Lck accumulate (89–91). Signal transduction requires that at least two ITAMs are phosphorylated by Lck (92–94). Phosphorylated ITAMs of CD3 chains serve as docking and activation sites for Syk family kinase members as the zeta-chain-associated protein kinase 70 (ZAP-70), which in turn is phosphorylated and activated by Lck (95). ZAP-70 activity is essential in conventional T cells (CD4+, CD8+ T cells) but apparently not in Tregs (96). Activated ZAP-70 phosphorylates another membrane raft component: the transmembrane adaptor protein “linker for activation of T cells” (LAT) (97, 98). LAT is phosphorylated on multiple tyrosine residues that next form docking sites for other adapter proteins, such as SLP-76, Grb-2, and Gads, and enzymes, such as PI3K and PLCγ1 (98–101), and is essential for downstream TCR signaling. Lack of LAT expression uncouples TCR-proximal tyrosine phosphorylation from these downstream signaling cascades (100, 102). Ultimately, TCR activation triggers induction of gene expression. The nuclear factors NFκB, NFAT, AP1, and CREB are activated (103), which in turn promotes transcription of important genes for immune activation (104). TCR activation also triggers secretion of cytokines, such as IL-2, IL-4, IL-6, and IL-12 (105), and controls cytokine receptor expression (106), leading to qualitatively different intracellular responses. Moreover, TCR activation and the cytokine milieu synergically influence cell fate determination. Indeed, weak signals through the TCR trigger CD4+ T cells to differentiate to Th2 cells (107), whereas strong signals lead to Th1 cells differentiation (105, 106). By guiding lineage commitment, the cytokine environment and TCR activation also tune the immune response. Th1 cells produce IFNγ, TNF-α, and IL-2, promote cell-mediated immune responses, and control intracellular pathogen infections. Th2 cells produce IL-4, IL-5, IL-9, IL-10, and IL-13, promote humoral immune responses, and mediate resistance to parasites, such as helminths (105, 108–110).
The aggregation of TCR microclusters upon activation forms the central supramolecular activation cluster (cSMAC) (111, 112), which is part of the immunological synapse formed between a T cell and the APC (113). Co-receptors, such as CD4, CD8, and CD28, and signaling molecules co-aggregate in the cSMACs; and, overall, this creates an environment that is conducive to precise T-cell activation. Appropriate spatiotemporal localization of proteins is a key factor determining signaling activity. To this end, lipid rafts play a critical role (114–116); they serve as signaling platforms that contain several key signaling components involved also in TCR signaling, such as SFKs, transmembrane adaptor proteins, phosphatidylinositol bis-phosphate (PIP2), and G-proteins, and aggregate to form the C-SMAC and IS. Spatiotemporal changes in this well-organized molecular machinery modulate TCR signaling pathways and, therefore, affect T-cell function.
Upon TCR activation, cAMP is rapidly produced in lipid rafts (117, 118) leading to the downregulation of TCR signaling, and then to the inhibition of T-cell proliferation and cytokine production. Several studies have unraveled molecular mechanisms involved in the inhibition of TCR signaling by cAMP and characterized the PTK, Csk, as a key player in TCR signaling downregulation (Figure 1) (1, 119–123).
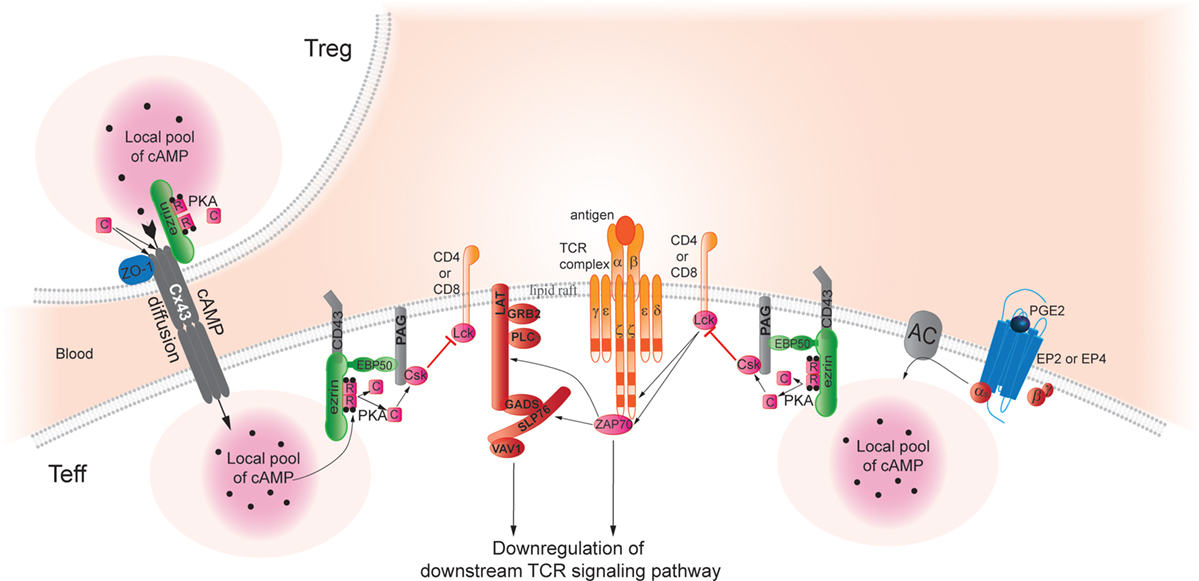
Figure 1. Cyclic AMP immunoregulatory pathways inhibit TCR signaling and T-cell activation. In Teff, pools of cAMP are generated after binding of PGE2 to its cognate receptors, which stimulates adenylyl cyclase (AC) activity and increases intracellular cAMP levels, thus, activating protein kinase A (PKA). Aided by an Ezrin/EBP50/PAG scaffold that holds both enzymes, PKA phosphorylates Csk, which in turn phosphorylates Lck to inhibit its activity. Lck normally acts to promote TCR signaling; thus, Lck inhibition through this PGE2-initiated pathway inhibits TCR signaling in effector T cells. Pools of cAMP can also be created by transfer from Treg to Teff through gap junctions (42). As a hypothetical model based on studies in trophoblasts (124), we speculate that this process may also require an AKAP bound to connexin 43 (Cx43) to facilitate PKA-mediated gap junction opening and cAMP transfer from Treg to Teff. Indeed, Pidoux and co-authors have found that ezrin binds to the C-terminal part of Cx43 and delivers PKA in the vicinity of gap junctions. Furthermore, the phosphorylation of Cx43 by PKA promotes opening of the gap junction and allows the passage of signal molecules. The authors suggested the PKA/ezrin/Cx43 macromolecular complex controlling the gap junction communication could be a general mechanism that regulates opening of Cx43 gap junctions in response to a cAMP increase also in other cell types. Thus, gap junctions may also deliver a local pool of cAMP that can dampen TCR signaling pathways by the same mechanisms described above and contributing to the Treg suppressive capacity of Teff.
Cyclic AMP serves as a second messenger within the cell and activates PKA, the dominant effector in T cells (82). The PKA is a heterotetrameric holoenzyme consisting of two regulatory (R) subunits that maintain two catalytic (C) subunits in an inactive state (125, 126). PKA exists in two classes, PKA type I and II that differ in the R subunit. Both R subunit and C subunit exist as multiple isoforms (RIα, RIβ, RIIα, RIIβ, Cα, Cβ, Cγ, and PrKX). The type I PKA is thought to be predominantly cytoplasmic and is most highly expressed in the immune system, whereas type II PKA associates with specific cellular structures and organelles and is abundant in the heart and the brain (127). When four molecules of cAMP bind its regulatory subunits, the PKA molecule releases the two catalytic subunits that have enzyme activity toward target proteins. Upon T-cell activation and formation of the immunological synapse, type I PKA is redistributed colocalizes with the TCR–CD3 complex (128). PKA type I phosphorylates the PTK, Csk, on serine 364 (1), which in turn initiates downregulation of the TCR signal. Indeed, Csk negatively regulates Lck by phosphorylation of a C-terminal inhibitory tyrosine residue, Y505 (129, 130) that contributes to stabilize Lck in an inactive conformation (131). Csk is recruited to membrane lipid rafts and is then activated (132) through its interaction with a transmembrane adaptor protein found in lipid rafts, PAG (133, 134), also known as Csk binding protein (Cbp). Upon TCR activation, PAG is dephosphorylated by a mechanism that appears to involve the activity of the phosphatase CD45 (135). Dissociation of Csk relieves Src kinases inhibition, enabling TCR downstream signaling pathways. Although Csk plays a critical role in the regulation of Lck activity, constant and abundant Lck fractions either phosphorylated on Y394, the active site, or double-phosphorylated on Y505 + Y394 reveals a more complex regulatory mechanism (136). Indeed, joint actions on phosphorylation state and spatial distribution of Lck are necessary for a balanced T-cell activity (137, 138).
The balance between the activities of two families of enzymes: ACs and cyclic nucleotide PDE regulate the intracellular cAMP level and its spatiotemporal distribution. PDEs are intimately coupled to limitation of cAMP gradients and termination of specific signals through local pools of cAMP and, therefore, multiple PDEs play important roles in modulating each cellular function [reviewed in Ref. (139, 140)].
Phosphodiesterases comprise a superfamily of enzymes classified into at least 11 families (PDE1–PDE11) and more than 50 isoforms that are distributed in different tissues at varying levels. PDEs have a highly conserved catalytic domain located near the C-terminus (>50% amino acid identity) flanked by regulatory domains in the N-terminus and the C-terminus. These family-specific regulatory domains include phosphorylation sites, membrane targeting domains, binding sites for small ligands, and dimerization domains [reviewed in Ref. (141, 142)]. Seven of the 11 families of PDEs have been reported to be present in T cells (143, 144). In most mammalian cells, PDE3 and PDE4 predominantly hydrolyze cAMP, and PDE4 is found as the major enzyme responsible for cAMP hydrolysis and dominant in inflammatory cells (145–147).
Phosphodiesterase 4 activity was detected in lipid rafts upon T-cell activation and especially after TCR and CD28 co-stimulation. Indeed, CD28 activation potentiates TCR signaling pathways and induces full TCR activation and clonal expansion (117, 148). Increase in PDE4 activity in response to CD28 co-ligation leads to negative regulation of the spatial and temporal cAMP gradient and thereby upregulates TCR signaling pathways. More specifically in human T cells PDE4A4, PDE4B2, and PDE4D1/2 are recruited to lipid rafts upon TCR and CD28 co-stimulation (117, 149). The scaffolding beta-arrestin protein has been reported to recruit PDE4 to the plasma membrane (150, 151), and the recruitment is mainly induced by CD28 stimulation (117). Moreover, the β-arrestin/PDE4 complex was shown to preexist prior to stimulation, indicating that the partners are recruited to the lipid rafts together (117). Stabilization of the β-arrestin/PDE4 complex in the lipid rafts is required for an efficient cAMP regulation by PDE4. Bjorgo and co-authors have found that recruitment of β-arrestin/PDE4 to the plasma membrane happens directly through protein kinase B (PKB) better known as Akt, with which β-arrestin and PDE4 form a complex (152, 153). Indeed, upon TCR/CD28 stimulation PI3K activity generates phosphatidylinositol-3,4,5-trisphosphate (PIP3) at the plasma membrane, which in turn directly recruits PH domain-containing PKB and, therefore, β-arrestin/PDE4 to the lipid raft (153).
Specificity of the cAMP/PKA signaling pathway is determined by generation of local gradients of cAMP controlled by PDEs and by spatially and temporally restricted activation of compartmentalized pools of PKA at different subcellular locations facilitated by AKAPs. Thus, AKAPs participate in organizing the functional complexity of cAMP signaling pathways.
A-kinase anchoring proteins bind to the regulatory subunit of PKA and ensure specificity and diversity in signal transduction by placing the enzyme close to relevant substrate (154–158). The functional importance involves the targeting of PKA to specific subcellular compartments, including the plasma membrane, nuclei, and mitochondria (159), and thereby provides spatial and temporal regulation of the PKA signaling events. Moreover, by interacting with additional signaling molecules, such as PDEs, protein kinase C (PKC), and phosphoproteins phosphatase 1 and 2B (PP1/2B) (160), AKAPs coordinate multiple signals transduction pathways and relay specific signals to downstream targets.
A-kinase anchoring proteins are a structurally diverse family of functionally related proteins that comprise more than 50 members [reviewed in Ref. (161, 162)]. All the anchoring proteins contain a PKA-anchoring domain, which binds the R subunit of PKA and a unique targeting domain directing the PKA–AKAP complex to subcellular structures, membranes, or organelles (Figure 2A). AKAPs are defined as PKA type I or type II specific or dual specific depending on whether they preferentially interact with PKA type I or PKA type II or interact with both. Treating cells with the anchoring disruptor peptide (Ht31) can disrupt PKA localization. The anchoring disruptor peptide binds to the R subunits of PKA, preventing their binding to AKAPs. Several studies have illustrated that delocalization of PKA blocks its ability to respond to cAMP level increases (163, 164).
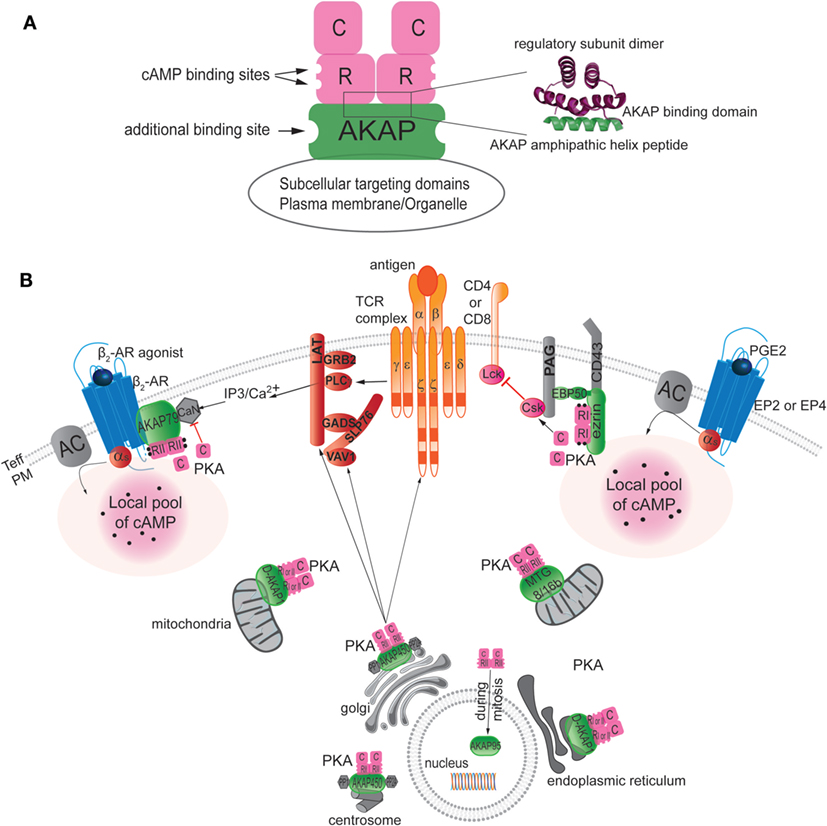
Figure 2. Localization of AKAPs in T cells. (A) Left: schematic diagram of an AKAP anchoring PKA through hydrophobic interaction between the amphipathic helical region of AKAP and the N-terminal dimerization region of the two R subunits of PKA. When cAMP binds to the R subunit, the C subunit of PKA is activated and released to phosphorylate nearby substrates. The AKAP signaling scaffold also typically has additional binding sites for other signaling proteins, such as kinases, phosphatases, phosphodiesterases, or potential substrates. Finally, the AKAP target the supramolecular signaling complex to the appropriate subcellular compartment via protein–protein or protein–lipid interactions. Right: ribbon representation of the NMR structure of the regulatory subunit (green) in complex with the AKAP amphipathic helix peptide (pink) [modified from (162)]. (B) AKAPs target PKA to specific compartments in T cells, including the plasma membrane (PM), mitochondria, endoplasmic reticulum, Golgi, nucleus, and centrosome. The same AKAP can be found in different compartments, as illustrated by the presence of D-AKAP-1 both in mitochondria and the endoplasmic reticulum and by finding AKAP450 in the Golgi and at the centrosome. AKAPs bind to specific partners and, hence, define specific supramolecular complex at discrete subcellular locales. For example, besides targeting PKA AKAP79 was shown to interact with beta 2-adrenergic receptor (β2AR) and calcineurin (CaN) at the plasma membrane and AKAP450 with PP1 and PP2A to the Golgi and the centrosome area. The role of each AKAP in T cells has not yet been reported. AKAP450 appears to be needed for early events as CD3, LAT, and Vav1 activation as well as late events as IL-2 production but the mechanism is still not determined. However, studies of the role of ezrin and AKAP79 have delineated their functions in downregulation of T-cell function by dampening signaling through the TCR pathway at the level of inhibition of Lck activity or by blocking IL-2 production through the inhibition of the CaN phosphatase activity, respectively.
A-kinase anchoring proteins have been identified in T cells and have been shown to contribute to the maintenance of T-cell homeostasis (3, 165). AKAPs are found in lipid rafts in T cells, as well as in dendritic cells, macrophages, and likely in platelets (3, 166–170). Among the 50 members of the AKAP family, seven different AKAPs have been detected in T cells: Ezrin, AKAP79, D-AKAP1, AKAP450, MTG8, MTG16b, AKAP95, and AKAP220, but their exact and individual functional roles in T cells have not been fully elucidated (Figure 2B).
Ezrin is a 78-kDa protein, which belongs to the ERM family of proteins that play structural and regulatory roles. Ezrin was originally identified as a component of sub-cortical structure underneath the cell membrane that contains an actin cytoskeleton (171, 172), and as a substrate of specific PTKs (173) [reviewed in Ref. (174)]. Ezrin and Moesin are expressed in lymphocytes (175, 176). They are cytoskeletal adaptor proteins that crosslink cell membrane and cytosolic proteins to the actin cytoskeleton and thereby govern membrane structure, its organization, and help to regulate diverse signaling routes. Ezrin has an N-terminal containing a four-point-one and ERM (FERM) domain, a central α-helical region spanning the A-kinase binding domain, AKB, linking ezrin to PKA (3, 177), and a C-terminal actin binding domain (178). The ERM family interacts with effectors of intracellular signaling either directly through the FERM domain or indirectly through adaptor molecules, such as EBP50 (179–181). In the cytoplasm, ezrin exists in a dormant form, which is unable to interact with its ligands; the binding sites for interaction partners are masked due to an intramolecular interaction between the FERM domain and the C-terminus. Phosphorylation by PKC or Rho kinase of the threonine residue, T567, at the C-terminal of ezrin, induces the conformational switch from the dormant to the active form, the intramolecular bond is released. In the active form, the N-terminal region binds to plasma membrane lipids and cytoplasmic tails of transmembrane proteins, while the C-terminal region binds to F-actin (174).
In T cells, ERM proteins control cell shape, cytokinesis, and cell adhesion (182–184) and participate in immune synapse formation (180, 185). In addition, the ERM family maintains lipid raft structures in T cells (179, 180) and contributes to control apoptosis signaling (186). Although generally described as functionally redundant, ezrin and moesin can display distinct and critical roles in the T-cell cortex during IS formation, thus promoting T-cell activation (187). Indeed, phosphorylated ezrin directly interacts and recruits ZAP-70 in the IS formation, whereas dephosphorylated moesin is removed, along with CD43, to prepare a region of the cell cortex for IS (188). The first evidence that ERM proteins play an important role in T-cell activation came precisely from studies on CD43, a large, glycosylated surface protein abundantly expressed on lymphocytes. CD43 seems acting in part as a negative regulator of T-cell activation by impeding the effective interactions of other surface receptors. Indeed, interaction with antigen-presenting cells leads to the removal of CD43 from the IS region (189, 190). ERM protein colocalizes with CD43 at the distal T-cell pole, and disruption of the interaction, either by overexpression of the FERM domain or by mutation of the relevant amino acids in CD43, leads to loss of CD43 movement and disruption of some aspects of T-cell activation.
Identified as an AKAP (177), the function of Ezrin was refined as the most important AKAP for PKA type I in T-cell lipid rafts (3). Indeed, the inhibitory effect of PKA on T-cell function is released by disruption of PKA and ezrin interaction by using specific PKA-anchoring disruptors [peptides Ht31, RI anchoring disruptor (RIAD)], which displace PKA type I from the lipid rafts (3, 191, 192). Furthermore, small interfering RNA (siRNA)-mediated knockdown of Ezrin abrogated cAMP regulation of IL-2 secretion (3), whereas reconstitution with siRNA-resistant wild-type Ezrin restored the cAMP regulation of IL-2 secretion (3, 192). Mapping studies of Ezrin reveal that the PKA RIα binding sequence is located in the α-helical region between the FERM domain and the C-terminus (3). Thus, Ezrin places PKA type I in the proximity of its TCR-proximal substrate Csk that is bound to PAG/Cbp. Moreover, ERM proteins through EBP50 have also been shown to interact with PAG/Cbp in lipid rafts (180). Together, the AKAP ezrin, EBP, and PAG/Cbp form a scaffold that holds and colocalizes PKA and Csk (3, 119, 193). Combinations of knockdown and reconstitution experiments with ezrin have demonstrated that cAMP/PKA regulation of Csk is heavily dependent on the supramolecular complex formation organized by ezrin (3, 192).
In the coming years, the role of ezrin in T cells regulation could be expanded. A new role for ezrin as an AKAP for the Connexin 43 has already been suggested by Pidoux and co-authors (124).
Connexins are a family of multiple-span membrane proteins, which construct GJ intercellular channels. Connexins have a short (~20 amino acid) cytoplasmic amino terminus and a highly variable (18 amino acids in Connexin 26, 156 amino acids in Connexin 43, and 275 amino acids in Connexin 57) cytoplasmic carboxyl terminus, which determines their overall size (194). Connexin43 (Cx43) has been described as a fundamental constituent of the immunological synapse (194), and as contributing in the regulation of proliferation of peripheral T cells (195). In Treg, Cx43 contributes not only to the formation of GJs with target cells (42), but has also been found to enhance the generation of Treg through the regulation of FoxP3+ expression (196). Besides intercellular communication, Cx43 through its C-terminal cytoplasmic domain interacts with cytoskeleton and signaling molecules, such as PKA and PKC (197). cAMP-enhanced GJ assembly has been reported to be PKA mediated (198). PKA activation increases intercellular communication, whereas PKC activation abrogates communication through GJs (199), suggesting regulatory mechanism balancing phosphorylation on GJ mediated by PKA and PKC. In human trophoblasts, Pidoux and co-authors proposed a model to explain the control and the regulation of communication through GJs. They have found that as an AKAP ezrin binds to the C-terminal part of Cx43 and delivers PKA in the vicinity of GJ (124). Indeed, upon local cAMP increase, after human chorionic gonadotropin (hCG) stimulation, PKA bound to ezrin is activated and phosphorylates Cx43. The phosphorylation of Cx43 by PKA promotes opening of the GJ and allows the passage of signal molecules. Ezrin promotes gap junctional communication by facilitating the spatiotemporal control of Cx43 phosphorylation by PKA, thereby controlling trophoblast cell fusion (124, 200).
The authors suggested that the PKA/ezrin/CX43 macromolecular complex control of GJ communication could be a general mechanism that regulates opening of Cx43 GJs in response to a cAMP increase also in others cell types (Figure 1). This study suggested an extended role of Ezrin as an AKAP in T cells, not only crucial to gather PKA type I, EBP50, PAG/Cbp, and Csk in the vicinity of TCR contributing to the control of receptor activity but also as a key regulator for the cAMP transfer from Treg to Teff, thereby contributing to Treg suppressive capacity.
A-kinase anchoring protein 5 was named AKAP79 in humans and AKAP150 in rodents. AKAP79 has been found in T lymphocytes (201) and more recently in dendritic cell lipid raft (167). The C-terminus of AKAP79 has been shown to interact with the PKA regulatory subunit II, PKC, and the protein phosphatase-2B/Calcineurin (CaN) (202, 203). NFAT proteins have crucial roles in the development and function of the immune system, and are regulated by the phosphatase activity of CaN. Indeed, CaN dephosphorylates the transcription factor NFAT, which facilitates its translocation into the nucleus and the IL-2 transcription (204). The association AKAP79/CaN with the T-cell plasma membrane has been shown to inhibit CaN phosphatase activity and, therefore, the NFAT activity (201, 203, 205).
A-kinase anchoring protein 79 has also been found to bind to the beta2-adrenergic receptor (β2AR) contributing to receptor phosphorylation and signaling (206). Riether and co-authors have found that upon β2AR stimulation on TCR-activated CD4+ T cells the cellular activity of the protein phosphatase CaN was drastically reduced along with a reduction in Th1-cytokine production and T-cell proliferation. Moreover, upon β2AR activation, the disruption of the interaction between PKA and AKAP79 by the inhibitor peptide St-Ht31 fully blocked CaN inhibition, demonstrating that PKA–AKAP79 interaction is essential for the β2AR-mediated CaN inhibition. These findings suggested that upon activation β2AR interacts with PKA and CaN through AKAP79 forming the supramolecular complex β2AR/AKAP79/PKA/CaN, which leads to inhibition of the CaN activity and, therefore, blocks IL-2 production and T-cell proliferation. These findings provide evidence for a link between the β2AR and TCR signaling pathways and describe a novel AKAP-dependent intracellular mechanism that can lead to the downregulation of T-cell function (207).
D-AKAP1 (also known as AKAP149, S-AKAP84, and AKAP121) is a member of the AKAP1 gene family (208). D-AKAP1 was reported as a dual-specific AKAP binding both RI and RII, and the N-terminus of PKA RI or RII is sufficient for its interaction with D-AKAP1 (209). D-AKAP1 is a differentially targeted AKAP, which can be localized to the mitochondrial membrane or to the endoplasmic reticulum (ER) depending on its NH2-terminal targeting motif (208). Experiments have found the presence of AKAP149 in T lymphocytes and more precisely in lipid rafts but so far no functional role was identified for AKAP149 in T-cell activation and regulation (3, 202). Lemay and co-authors have found an interaction between AKAP149 and HIV-1 reverse transcriptase in infected Jurkat T cells with a potential role in HIV-1 reverse transcription (210).
The scaffolding protein AKAP450 also known AKAP9, AKAP350, or CG-NAP (centrosome and Golgi localized protein kinase N-associated protein) is associated, as the name suggests, with the centrosome and the Golgi apparatus. AKAP450 has been found in T cells (211) anchoring several protein kinases (PKN and PKA RIIα) and phosphatases (PP1 and PP2A) (212, 213). Even if the mechanism is not fully understood, AKAP450 is reported to be required for T-cell activation by regulating the conformational activation of Lymphocyte function-associated antigen 1 and TCR/CD3 molecules at the immune synapse (214), which may have to do with its role in orchestrating cytoskeletal rearrangements. Indeed upon TCR activation, AKAP450 was needed for early events, such as CD3, LAT, and Vav1 activation, and late events, such as IL-2 production. AKAP450 was described as an important component of T-cell response to antigen stimulation (214).
Two members of the myeloid translocation gene family (MTG) family have been defined as AKAPs and are found in T cells: MTG8 and MTG16b. The proto-oncogene MTG8 was originally found as part of the leukemic fusion gene, AML1–MTG8 (215–217). MTG8 is expressed ubiquitously in human tissue but with varying levels of expression (high in brain, heart, and muscle and low in hematopoietic tissues) and cell-dependent localization (found in the nucleus or in the cytoplasm) (218). MTG8 was identified as a transcriptional suppressor by its tight association with the nuclear matrix (219). MTG16b, which is another MTG family member, was originally identified in patients with acute myeloid leukemia, but the normal physiological function of this protein has not been reported (220). MTG8 and MTG16b interact with the PKA RII subunit with some differences in location. Whereas MTG16b target PKA to the Golgi of T lymphocytes, MTG8 and PKARII were found in Golgi/centrosome area (202, 219). Their physiological functions as AKAPs in T cells have, however, not yet been reported.
A-kinase anchoring protein 95 was cloned and characterized in the rat (221) and human (222). AKAP95 specifically binds the RIIα subunit of PKA with high affinity and also has a DNA binding domain (221). The interaction between AKAP95 and PKARII is cell cycle-dependent and has only been detected during mitosis when the nuclear envelope is disassembled (222, 223) and the PKA/AKAP95 complex has a role in controlling chromosome condensation (224, 225).
The presence of AKAP95 has been reported in T cells (202) but no functional role has been identified for AKAP95 in regulating T-cell activation.
A 220-kDa AKAP220 was the first AKAP described able to coordinate the location of PKA and the type 1 protein phosphatase catalytic subunit (PP1c) (226, 227). AKAP220 has been found to interact with the PKA RII (2) and its presence was detected in Jurkat T cells but not in primary T cells (228).
The diversity of AKAPs in T cells and their functional role, when known, suggest critical new roles that could help to unravel the T-cell function. AKAPs, their interacting partners and appropriate targets shape the biological role of these scaffolds and supramolecular signaling complexes inside T cells; AKAPs are able to interact with PDEs, providing a route of cAMP degradation. The compartmentalization of such enzymes is crucial for the generation of intracellular cAMP gradients. The ability to form and shape intracellular cAMP pools depends on targeted PDE activity. In T cells, AKAP95, AKAP149, and MTG8/16b are in complex with PDE4A, AKAP450 with PDE4D3 (229), whereas only MTG8 is in complex with PDE7A. AKAP79 did not form a complex with either PDE4A or PDE7A (230). The specific interaction between PDE4A and selective AKAPs in T lymphocytes creates a regulatory-feedback signaling allowing localized PDE activity, compartmentalized cAMP production and PKA activity and, consequently, controlling T-cell activation with a specific pattern. This finely tuned regulatory process is not exempt from facing diverse defects, which can lead to abnormal conditions and, therefore, disease development (231, 232).
Several human tumors and infectious diseases are characterized by high levels of intracellular cAMP (233–236). Rising cAMP concentrations have been correlated with upregulation of COX-2 levels and PGE2 and modulation of immune responses (16, 29, 69, 236–238). For example, T cells from HIV-infected patients contain twice as much cAMP as those of healthy controls leading to downregulation of TCR signaling and immunosuppression through an aberrant activated cAMP/PKA signaling pathway (233, 234, 239, 240).
The tumor microenvironment may foster immune tolerance by attracting and/or inducing immunosuppressive networks to escape tumor-specific immunity in favor of disease progression (241). Several studies have defined Treg as a leading player in cancer progression through a PGE2/cAMP-dependent suppressive ability (4, 37, 38). Patients with several forms of cancer, including gastrointestinal, lung, and ovarian tumors, have been shown to display increasing numbers of circulating and tumor-associated Treg compared with healthy controls (242–245). Moreover, Treg depletion in animal models has been shown to enhance anti-tumor responses underscoring the role of Treg to the impaired anti-tumor immunity (246–248). In addition, the percentage of Treg has been associated not only with the disease progression but also with disease outcomes. Indeed, the percentage of Treg in peripheral blood was inversely correlated with disease prognosis in patients with gastrointestinal malignancies (244). Use of COX-2 inhibitors has been shown to reverse Treg suppressive effects (38). For example, in a murine lung cancer model, inhibition of COX-2 has been found to enhance anti-tumor immune responses (4, 5). COX-2 is overexpressed in 85% of human colorectal cancers (CRCs) and approximately 50% of colorectal adenomas leading to high PGE2 concentrations and chronic inflammation around the cancer (249). Regular use of COX inhibitors, including aspirin reduces the incidence of CRC by 30–45% (250–253). In CRC patients, the anti-tumor immune responses of Treg are reported to be COX-2/PGE2/cAMP dependent and can be reversed by COX-2 inhibitor, PKA inhibitor, or Treg depletion (6). COX-2 overexpression is also correlated with the development of CRC metastases (254). Indeed in CRC patients with recurrent disease, T cell phenotyping has revealed high frequencies of COX-2 and high plasma PGE2 levels after surgery. Brudvik and co-authors have established strong correlation between Treg level, PGE2-mediated suppressive anti-tumor activity and disease recurrence (69).
Prostaglandin E2 plays a crucial role in the neoplastic process by stimulating tumor cell proliferation, tissue invasion, promoting angiogenesis, and by suppressing tumor cell apoptosis (255–257). Through Gs-coupled EP receptor signaling pathways, PGE2 expands and recruits Treg in tumor environment, which in turn suppresses T-, B-, and NK-cell immune responses, and contributes to tumor immune tolerance (38, 119, 258, 259). This combined with EP receptor activation, which triggers the cAMP/PKA/Csk signaling pathway, leads to downregulated TCR signaling and then further decreased T-cell immune function (6, 259, 260). All together these findings highlight the significant impact of the cAMP/PKA/Csk pathway on PGE2 control of tumor immune responses.
Along the same line, hyper-activation of the cAMP/type I PKA pathway is involved in T-cell dysfunction in immunodeficiencies. HIV-1 infection is associated with increased levels of cAMP and enhanced activation of PKA (233, 234). The HIV-1 protein gp120 functionally activates Treg by binding to CD4 and inducing enhanced AC activity and elevated intracellular cAMP levels in Treg, thereby increasing their suppressive activity on Teff (42, 261, 262). Moreover, cytokine networks have been found to be under cAMP-mediated regulation in T cells from HIV-infected patients. These findings indicated that high intracellular cAMP concentrations contribute to T-cell anergy in HIV infection. Accordingly, drugs that decrease intracellular cAMP levels may restore T-cell proliferation and cytokine networks providing a stronger antiviral response and be beneficial in the treatment of AIDS (240, 263). A similar mechanism has been found to contribute to the T-cell dysfunction in a subset of patients with common variable immunodeficiency (CVID) (264). Low level of IL-10 secreted by T cells observed in CVID patients has been related to the cAMP/PKA type I signaling. This pathway could represent a novel target for therapeutic immunomodulation in CVID (265). Similarly, in the murine AIDS (MAIDS) model induced by the murine leukemia virus, hyper-activation of the cAMP/PKA pathway, related to high level of PGE2, was found to contribute to severe T-cell anergy, typical feature of the pathology (238, 266). All together these findings support the idea that the cAMP/PKA pathway in T cells is a target for treating immunodeficiency diseases, chronic infections, and cancer.
Control of the cAMP/PKA pathway could help to kill tumor or infected cells, restore, or build a specific environment for robust immune responses. To this end, different strategies are possible, either upstream of the cAMP/PKA cascade by using COX-2 inhibitor, EP receptor antagonist, or downstream, blocking the intracellular cAMP/PKA cascade or its anchoring or even combining these strategies. Alone or in combination with other clinical therapeutic strategies, COX-2 could be a target to improve the efficiency of cancer and HIV treatments. In vivo experiments in the MAIDS model have shown that treatment with COX-2 inhibitor reduces PGE2 levels, reverses T-cell anergy, and thereby restores T-cell immune function (238). Moreover, combination between COX-2 inhibitors and antiretroviral treatment of HIV-infected patients has contributed toward improving T-cell proliferation and persistent immune activation. The modulation of cAMP may represent a therapeutic strategy in HIV infection in addition to antiretroviral therapy (237, 267, 268). As mentioned earlier effectiveness of selective COX-2 inhibitors has also been supported by several studies in cancer treatment and often associated with reduction of mortality rate. However, COX-2 inhibitors have also been related to serious cardiovascular events, which has resulted in interruption of long-term trials for cancer prevention (269, 270). Modulation of targets downstream of COX-2 is expected to improve the drug efficacy, specificity, and safety. Indeed, COX-2 inhibitor activity, through reduction of PGE2 synthesis, is not exclusive to the cAMP/PKA pathway. Actually G protein-dependent and -independent EP signaling pathways as well as crosstalk between EP signaling and parallel signaling pathways are blocked by COX inhibitor treatment. Such broad action leads to unwanted effects and call to delineate appropriate targets in order to better define exclusive inhibitors. Current knowledge has already defined several proteins required in the cAMP/PKA signaling activation, all of which could become potential targets for inhibitors, each with presumably different biological consequences. A study in a mouse model developing multiple adenomas in the intestinal tract at an early age has illustrated these potential biological differences. Indeed, whereas the anti-tumorigenic effects were correlated to COX inhibitors, the anti-proliferative effects were linked to PKA antagonism (7). These findings have identified specific chemo-protective actions related to the nature of the inhibitor and more precisely to its target and its action in the PGE2 signaling pathway. In a previous study, a specific PKA type I antagonist, Rp-8-Br-cAMPS, has been found to increase T-cell proliferation and restore immune responses of T cells from HIV-infected patients. These findings suggested a novel strategy in treatment of HIV infection, which would combine treatment modalities counteracting PKA type I activity and antiretroviral therapy (233, 239). Given the importance of the cAMP/PKA pathway, compartmentalized cAMP signaling and PKA activity in immune responses regulation, the targeting of AKAPs complexes for new therapeutic intervention in cancer and chronic infection has become clearly apparent. All together, these findings underscore the importance to develop agents able to specifically disrupt AKAP type I complexes.
Generation of peptides that disrupt AKAP complexes is challenging especially for therapeutic purposes. One strategy to disrupt the interaction between AKAP and PKA is to selectively displace PKA subtype from the AKAP platform with peptides that mimic the amphipathic helices domain of AKAP. Such disruptors of the AKAP complex have to be cell permeable and require high specificity and high binding affinity for their target. Most AKAPs bind avidly to the RII isoform (271), whereas others, such as Ezrin, are RI-selective AKAPs (3). A third class of AKAPs, termed dual-specific AKAPs, can bind both the RI and RII isoforms, yet their preference for binding the RII isoform strongly predominates (209). The first AKAP disruptor peptide described, Ht31, is derived from the RII-binding domain of AKAP-Lbc (AKAP13) (272). Ht31 is a peptide, which forms an amphipathic helix mimicking that found in AKAPs (273). The helix binds to the regulatory subunits of PKA disrupting localization with both RI and RII from AKAPs (274). Disruption of the AKAP–PKA interaction with Ht31 was shown to induce cytokine production (increase of IL-2, IL-4, IL-5, and IFNγ secretion) and in synergy with Ag to enhance T-cell proliferation, suggesting that PKA is necessary for maintaining T cells in a resting state. Furthermore, Ht31 treated cells were insensitive to the inhibitory effects of cAMP on IL-2 production, indicating that anchored PKA activity is necessary for cAMP-mediated inhibition of T-cell activation (165). Moreover, use of Ht31 peptide as an inhibitor of the binding between ezrin and PKA type I has shown a release of the cAMP/PKA type I-mediated inhibition on T-cell proliferation (3).
Since then, studies have identified multiple high-affinity RII-selective disruptors. In 2003, the first high potent RII inhibitor peptide, AKAP-in silico (AKAP-IS), was designed by a bioinformatics approach combined with peptide array screening (275). This peptide was shown to have higher affinity for RII as compared to Ht31 peptide. The initial peptide had limited solubility in aqueous solution and was not cell permeable. The introduction of a peptide derived from the TAT protein of the HIV-1 greatly improved cell permeability (276). A later version called SuperAKAP-IS was developed with high affinity for RII and almost none for RI (277). Because of structure instability, disruptor peptides may lose their cell-penetration abilities, specificity for the target or binding affinity and become more susceptible to degradation. Stabilization of their bioactive structure, thus, rapidly became a priority for cell-based assays. The all-hydrocarbon staple has emerged as one solution combining two distinct conformational stabilization strategies [reviewed in Ref. (278)]. The stabilized conformation was associated with increase in target affinity, stability against proteolysis, and robust cell-penetration while remaining safe in in vivo models (279–281). Using this technique, two stapled AKAP disruptor (STAD) peptides were developed: STAD-2 and STAD-3 (282). The new class of AKAP disruptors, highly cell permeable, has been shown to block interactions between AKAPs and RII and was described as a promising tool to study compartmentalized RII-regulated PKA signaling in cells.
Designing peptides for RI-selective interaction has been more challenging than for RII-selective peptides. Indeed one RI-specific disruptor peptide, PV-38, was designed from D-AKAP2 (283). In parallel, a bioinformatics analysis combined with a peptide array screening had led to the development of a peptide that binds RIα with high affinity and specificity. This high-affinity binding peptide called RIAD has been found to specifically disrupt anchoring of PKA type I from intracellular locations and inhibited type I regulation of T-cell effector function and steroid biosynthesis (191). RIAD was proposed as a tool to define anchored PKA type I signaling events and has been used extensively like in a MAIDS model described later in this review (284). Wang and co-authors also designed a series of RI-Stapled Anchoring Disruptors (RI-STADs) where two peptides, RI-STAD-1 and RI-STAD-2, were reported to have improved cell permeability and to selectively disrupt the interactions between AKAPs and PKA-RI in biochemical and cell-based assays (285).
Despite technical advances improving the specificity for PKA subtype I or II, these disruptor peptides will non-specifically inhibit all AKAP interactions with either RI or RII isoforms. Taking into account that most of cell types express at least 10–15 different anchoring proteins (2), and so far seven identified in T cells, the specificity for PKA subtype I or II is obviously not enough to specifically displace interaction between PKA and one specific AKAP. Recently, a new approach based on a phage selection procedure was employed to engineer RII sequences (Rselect) able to selectively target particular AKAP. Biochemical and cell-based experiments validated the efficacy of Rselect mutants for AKAP2 and AKAP18 (286). Described as a new class of reagents, these genetically encoded AKAP-selective probes could help to design new compounds targeting specifically individual AKAP and to unravel the functions of different AKAP-targeted PKA. Another strategy would be to disrupt individual AKAP complexes by displacing interaction partners other than PKA, such as the substrate for PKA if that is bound to the AKAP. For example, interaction between AKAP18δ with phospholamban (PLN) was disrupted by using a short peptide derived from the PLN interaction site for AKAP18δ. This pharmacologic tool has allowed determining partners, conditions of formation, and biological consequences of the supramolecular complex formed in cardiac myocytes (287). Another possibility would be to disrupt the interaction between the AKAP targeting domain and the interaction partner providing its subcellular localization. As an example, a cell-permeant peptide of the Ezrin binding domain in EBP50 (EBP50pep) has been shown to displace Ezrin and reverse the cAMP/PKA-mediated inhibition of T-cell activation due to loss of PKA proximity to Csk (288). Along the same line, an inducible competing muscle-specific A-kinase anchoring protein (mAKAP) fragment (residues 585–1286) was used to displace the mAKAP from the perinuclear membrane highlighting the importance of its localization in the control of cardiomyocyte size (289). These strategies might offer higher selectivity.
Because peptides need to be administered parenterally, possess a short half-life hampered by limited stability in serum, and may generate immune responses, their potential therapeutic applications are limited. Non-peptidic agents, such as peptidomimetics and small molecules, could be one solution to counteract these drawbacks. Peptidomimetics are compounds whose essential elements mimic a natural peptide or protein in the three-dimensional space. Peptidomimetics, thus, provide a possible strategy for the modulation and regulation of AKAPs with the ability to interact with the biological target and, therefore, to displace other potential interactions without degradation. RIAD peptidomimetics were developed by adding unnatural amino acids at different positions, increasing the stability in serum while keeping their specificity to disrupt the AKAP/PKA-RI interaction (290). The RIAD peptidomimetic, RIAD-P3, was shown to limit HIV-1 viral replication and stabilize CD4 levels by disrupting AKAP/PKA-RI in human T cells and humanized mice (291). Thus, peptidomimetic research emerges as an indispensable tool of structure–activity relationships in drug discovery. Small molecules are also promising alternatives to disruptor peptides and offer several manufacturing and delivery advantages for drug discovery. Several examples have shown the ability of small molecule to disrupt protein–protein interactions (292). Small molecules interfering by orthosteric or allosteric binding have been identified (293, 294). In summary, the specificity and diversity of protein–protein interactions offer promising opportunities to develop highly selective inhibitors. However, their development requires detailed knowledge about the interaction between the two proteins (295). Displacing selected proteins from AKAP complexes could improve efficacy and specificity of anchoring disruptors with fewer side effects. This approach may lead to alternative strategies for the treatment of diseases associated with altered cAMP signaling. All together improved peptides, peptidomimetic and small molecules can help to characterize and understand molecular mechanism converging and emerging from AKAP platforms. These agents could help to define “druggable” target and alternative therapeutic strategies for the treatment of diseases associated with altered cAMP signaling.
Despite challenges to find and generate selective AKAP disruptor peptides, recent technical breakthroughs and findings from in vitro and in vivo studies strongly depict targeting protein–protein interaction as promising therapeutic strategies. Indeed, experiments using Ht31 peptides in in vitro experiments in T cells (3, 165) and a specific PKA type I antagonist, Rp-8-Br-cAMPS in T cells from HIV-infected patients as well as in a CRC mouse model (7, 233, 239) have underscored the major role of the AKAP, ezrin, in T-cell immune functions. Moreover, identification of an additional PKA-binding determinant, the RI specifier region (RISR), upstream of AKB in Ezrin has strengthened the role of Ezrin/PKA complex in T-cell signaling regulation. Indeed mutations in the RISR of Ezrin have been shown to perturb RI anchoring and alter the suppression of T-cell signaling through the cAMP/PKA type I/Csk pathway. The RISR was shown to act in synergy with the AKB to enhance anchoring of PKA type I (192). In an extended study, RIAD transgenic mice were generated by expressing a soluble ezrin fragment with the endogenous RISR and RIAD substituting the endogenous AKB domain under control of the lck distal promoter (284). Peripheral T cells from RIAD transgenic mice were resistant to cAMP-mediated inhibition and displayed enhanced T-cell signaling and responsiveness. Furthermore, these mice did not develop MAIDS when infected with the murine leukemia virus, as did wild-type littermates or mice infected with a mutated transgene that did not bind and displace PKA (284). These findings define the cAMP/type I PKA pathway in T cells as a putative target for therapeutic intervention through AKAP complex disruption in immunodeficiency diseases and cancer. Besides ezrin, D-AKAP1 is also involved in the progression of HIV infection. D-AKAP1 was shown to interact with the HIV-1 reverse transcriptase and to support viral replication during HIV infection (210). The full mechanism is still unknown but PKA and PDE4, key players in the cAMP signaling pathway and anchored by D-AKAP1 to mitochondria, can stimulate HIV-1 replication and infection (296–298). Hence, D-AKAP1 could affect reverse transcription through a PKA-dependent signaling pathway. Based on these results, D-AKAP1 could be another potential target in HIV therapy.
Cyclic AMP is a potent regulator of the immune response. Insights into molecular mechanisms causing and controlling the generation of cAMP and its propagation through the cAMP/PKA pathway are undoubtedly crucial to generate immunomodulatory agents. Several studies have assessed the efficiency of non-NSAIDs to regulate intracellular cAMP concentration and then further to control T-cell function. However, their efficiency could be impaired by lack of specificity. Indeed their early inhibitory action on PGE2 production blocks all EP receptor signaling pathways instead of specifically inhibiting the cAMP/PKA signaling pathway, which could create unwanted effects with biological consequences. Unraveling mechanisms surrounding PKA phosphorylation events and localization with AKAPs will hopefully support the development of more targeted therapies. Bolstered by both technological advances and learning from in vitro and in vivo experiments, AKAP disruptors emerge as essential tool, which selectively probe anchored PKA signaling and decode functions of AKAP/PKA interactions. AKAPs have crucial role in T-cell function and are involved in development and regulation of multiple chronic diseases, which make AKAPs potential targets for drug discovery. In a broader perspective, their ubiquitous nature and capacity to act as signaling hubs where multiple signals converge also offer other promising targeting perspectives.
VLW and KT wrote the article and reviewed and/or edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Our research is funded by the Research Council of Norway, the Norwegian Cancer Society, the Regional Health Authority for South Eastern Norway, the KG Jebsen Foundation, and Novo Nordic Foundation.
1. Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skalhegg BS, et al. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med (2001) 193:497–507. doi:10.1084/jem.193.4.497
2. Lester LB, Coghlan VM, Nauert B, Scott JD. Cloning and characterization of a novel A-kinase anchoring protein. AKAP 220, association with testicular peroxisomes. J Biol Chem (1996) 271:9460–5. doi:10.1074/jbc.271.16.9460
3. Ruppelt A, Mosenden R, Gronholm M, Aandahl EM, Tobin D, Carlson CR, et al. Inhibition of T cell activation by cyclic adenosine 5’-monophosphate requires lipid raft targeting of protein kinase A type I by the A-kinase anchoring protein ezrin. J Immunol (2007) 179:5159–68. doi:10.4049/jimmunol.179.8.5159
4. Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res (2005) 65:5211–20. doi:10.1158/0008-5472.CAN-05-0141
5. Sharma S, Zhu L, Yang SC, Zhang L, Lin J, Hillinger S, et al. Cyclooxygenase 2 inhibition promotes IFN-gamma-dependent enhancement of antitumor responses. J Immunol (2005) 175:813–9. doi:10.4049/jimmunol.175.2.813
6. Yaqub S, Henjum K, Mahic M, Jahnsen FL, Aandahl EM, Bjornbeth BA, et al. Regulatory T cells in colorectal cancer patients suppress anti-tumor immune activity in a COX-2 dependent manner. Cancer Immunol Immunother (2008) 57:813–21. doi:10.1007/s00262-007-0417-x
7. Brudvik KW, Paulsen JE, Aandahl EM, Roald B, Tasken K. Protein kinase A antagonist inhibits beta-catenin nuclear translocation, c-Myc and COX-2 expression and tumor promotion in Apc(Min/+) mice. Mol Cancer (2011) 10:149. doi:10.1186/1476-4598-10-149
8. Robison GA, Butcher RW, Sutherland EW. Cyclic AMP. Annu Rev Biochem (1968) 37:149–74. doi:10.1146/annurev.bi.37.070168.001053
9. Sutherland EW, Rall TW. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem (1958) 232:1077–91.
10. Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther (2004) 103:147–66. doi:10.1016/j.pharmthera.2004.06.003
11. Hershfield MS. New insights into adenosine-receptor-mediated immunosuppression and the role of adenosine in causing the immunodeficiency associated with adenosine deaminase deficiency. Eur J Immunol (2005) 35:25–30. doi:10.1002/eji.200425738
12. Jutel M, Watanabe T, Klunker S, Akdis M, Thomet OA, Malolepszy J, et al. Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature (2001) 413:420–5. doi:10.1038/35096564
13. Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev (2001) 53:487–525.
14. Delgado M, Abad C, Martinez C, Juarranz MG, Leceta J, Ganea D, et al. PACAP in immunity and inflammation. Ann N Y Acad Sci (2003) 992:141–57. doi:10.1111/j.1749-6632.2003.tb03145.x
15. Borner C, Warnick B, Smida M, Hartig R, Lindquist JA, Schraven B, et al. Mechanisms of opioid-mediated inhibition of human T cell receptor signaling. J Immunol (2009) 183:882–9. doi:10.4049/jimmunol.0802763
16. Kammer GM. The adenylate cyclase-cAMP-protein kinase A pathway and regulation of the immune response. Immunol Today (1988) 9:222–9. doi:10.1016/0167-5699(88)91220-0
17. Henney CS, Lichtenstein LM. The role of cyclic AMP in the cytolytic activity of lymphocytes. J Immunol (1971) 107:610–2.
18. Skalhegg BS, Landmark BF, Doskeland SO, Hansson V, Lea T, Jahnsen T. Cyclic AMP-dependent protein kinase type I mediates the inhibitory effects of 3’,5’-cyclic adenosine monophosphate on cell replication in human T lymphocytes. J Biol Chem (1992) 267:15707–14.
19. Kaur K, Harris SG, Padilla J, Graf BA, Phipps RP. Prostaglandin E2 as a modulator of lymphocyte mediated inflammatory and humoral responses. Adv Exp Med Biol (1999) 469:409–12. doi:10.1007/978-1-4615-4793-8_59
20. Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol (2002) 23:144–50. doi:10.1016/S1471-4906(01)02154-8
21. Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis (2009) 30:377–86. doi:10.1093/carcin/bgp014
22. Liu B, Qu L, Yan S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int (2015) 15:106. doi:10.1186/s12935-015-0260-7
23. Sharma S, Stolina M, Yang SC, Baratelli F, Lin JF, Atianzar K, et al. Tumor cyclooxygenase 2-dependent suppression of dendritic cell function. Clin Cancer Res (2003) 9:961–8.
24. Nzeako UC, Guicciardi ME, Yoon JH, Bronk SF, Gores GJ. COX-2 inhibits Fas-mediated apoptosis in cholangiocarcinoma cells. Hepatology (2002) 35:552–9. doi:10.1053/jhep.2002.31774
25. Tsujii M, DuBois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell (1995) 83:493–501. doi:10.1016/0092-8674(95)90127-2
26. Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell (1998) 93:705–16. doi:10.1016/S0092-8674(00)81433-6
27. Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res (2007) 67:8865–73. doi:10.1158/0008-5472.CAN-07-0767
28. Mougiakakos D, Johansson CC, Trocme E, All-Ericsson C, Economou MA, Larsson O, et al. Intratumoral forkhead box P3-positive regulatory T cells predict poor survival in cyclooxygenase-2-positive uveal melanoma. Cancer (2010) 116:2224–33. doi:10.1002/cncr.24999
29. Okano M, Sugata Y, Fujiwara T, Matsumoto R, Nishibori M, Shimizu K, et al. E prostanoid 2 (EP2)/EP4-mediated suppression of antigen-specific human T-cell responses by prostaglandin E2. Immunology (2006) 118:343–52. doi:10.1111/j.1365-2567.2006.02376.x
30. Powrie F, Correa-Oliveira R, Mauze S, Coffman RL. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J Exp Med (1994) 179:589–600. doi:10.1084/jem.179.2.589
31. Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell (2000) 101:455–8. doi:10.1016/S0092-8674(00)80856-9
32. Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol (2002) 2:389–400. doi:10.1038/nri821
33. Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med (1998) 188:287–96. doi:10.1084/jem.188.2.287
34. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
35. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
36. Bopp T, Dehzad N, Reuter S, Klein M, Ullrich N, Stassen M, et al. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J Immunol (2009) 182:4017–24. doi:10.4049/jimmunol.0803310
37. Baratelli F, Krysan K, Heuze-Vourc’h N, Zhu L, Escuadro B, Sharma S, et al. PGE2 confers survivin-dependent apoptosis resistance in human monocyte-derived dendritic cells. J Leukoc Biol (2005) 78:555–64. doi:10.1189/jlb.1004569
38. Mahic M, Yaqub S, Johansson CC, Tasken K, Aandahl EM. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol (2006) 177:246–54. doi:10.4049/jimmunol.177.1.246
39. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature (2007) 445:771–5. doi:10.1038/nature05543
40. Huang B, Zhao J, Lei Z, Shen S, Li D, Shen GX, et al. miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep (2009) 10:180–5. doi:10.1038/embor.2008.224
41. Bedner P, Niessen H, Odermatt B, Kretz M, Willecke K, Harz H. Selective permeability of different connexin channels to the second messenger cyclic AMP. J Biol Chem (2006) 281:6673–81. doi:10.1074/jbc.M511235200
42. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med (2007) 204:1303–10. doi:10.1084/jem.20062129
43. Neijssen J, Pang B, Neefjes J. Gap junction-mediated intercellular communication in the immune system. Prog Biophys Mol Biol (2007) 94:207–18. doi:10.1016/j.pbiomolbio.2007.03.008
44. Bodor J, Bopp T, Vaeth M, Klein M, Serfling E, Hunig T, et al. Cyclic AMP underpins suppression by regulatory T cells. Eur J Immunol (2012) 42:1375–84. doi:10.1002/eji.201141578
45. Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol (2010) 185:6413–9. doi:10.4049/jimmunol.1001829
46. Brenner S, Prosch S, Schenke-Layland K, Riese U, Gausmann U, Platzer C. cAMP-induced Interleukin-10 promoter activation depends on CCAAT/enhancer-binding protein expression and monocytic differentiation. J Biol Chem (2003) 278:5597–604. doi:10.1074/jbc.M207448200
47. Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol (2001) 2:599–609. doi:10.1038/35085068
48. Hughes-Fulford M, Sugano E, Schopper T, Li CF, Boonyaratanakornkit JB, Cogoli A. Early immune response and regulation of IL-2 receptor subunits. Cell Signal (2005) 17:1111–24. doi:10.1016/j.cellsig.2004.12.016
49. Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity (2010) 33:153–65. doi:10.1016/j.immuni.2010.08.004
50. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38:1654–63. doi:10.1002/eji.200838105
51. Polansky JK, Schreiber L, Thelemann C, Ludwig L, Kruger M, Baumgrass R, et al. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med (Berl) (2010) 88:1029–40. doi:10.1007/s00109-010-0642-1
52. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10:490–500. doi:10.1038/nri2785
53. Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med (2007) 204:1543–51. doi:10.1084/jem.20070109
54. Cosme R, Lublin D, Takafuji V, Lynch K, Roche JK. Prostanoids in human colonic mucosa: effects of inflammation on PGE(2) receptor expression. Hum Immunol (2000) 61:684–96. doi:10.1016/S0198-8859(00)00131-2
55. Hendricks A, Leibold W, Kaever V, Schuberth HJ. Prostaglandin E2 is variably induced by bacterial superantigens in bovine mononuclear cells and has a regulatory role for the T cell proliferative response. Immunobiology (2000) 201:493–505. doi:10.1016/S0171-2985(00)80069-8
56. Ganapathy V, Gurlo T, Jarstadmarken HO, von Grafenstein H. Regulation of TCR-induced IFN-gamma release from islet-reactive non-obese diabetic CD8(+) T cells by prostaglandin E(2) receptor signaling. Int Immunol (2000) 12:851–60. doi:10.1093/intimm/12.6.851
57. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol (2012) 188:21–8. doi:10.4049/jimmunol.1101029
58. Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med (2009) 206:535–48. doi:10.1084/jem.20082293
59. Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol (1991) 146:108–13.
60. Hilkens CM, Snijders A, Snijdewint FG, Wierenga EA, Kapsenberg ML. Modulation of T-cell cytokine secretion by accessory cell-derived products. Eur Respir J Suppl (1996) 22:90s–4s.
61. Snijdewint FG, Kalinski P, Wierenga EA, Bos JD, Kapsenberg ML. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. J Immunol (1993) 150:5321–9.
62. Demeure CE, Yang LP, Desjardins C, Raynauld P, Delespesse G. Prostaglandin E2 primes naive T cells for the production of anti-inflammatory cytokines. Eur J Immunol (1997) 27:3526–31. doi:10.1002/eji.1830271254
63. Wu CY, Wang K, McDyer JF, Seder RA. Prostaglandin E2 and dexamethasone inhibit IL-12 receptor expression and IL-12 responsiveness. J Immunol (1998) 161:2723–30.
64. Phipps RP, Stein SH, Roper RL. A new view of prostaglandin E regulation of the immune response. Immunol Today (1991) 12:349–52. doi:10.1016/0167-5699(91)90064-Z
65. Verjans E, Ohl K, Reiss LK, van Wijk F, Toncheva AA, Wiener A, et al. The cAMP response element modulator (CREM) regulates TH2 mediated inflammation. Oncotarget (2015) 6:38538–51. doi:10.18632/oncotarget.6041
66. Mannie MD, Prevost KD, Marinakis CA. Prostaglandin E2 promotes the induction of anergy during T helper cell recognition of myelin basic protein. Cell Immunol (1995) 160:132–8. doi:10.1016/0008-8749(95)80018-E
67. Pace E, Bruno TF, Berenger B, Mody CH, Melis M, Ferraro M, et al. Elevated expression of prostaglandin receptor and increased release of prostaglandin E2 maintain the survival of CD45RO+ T cells in the inflamed human pleural space. Immunology (2007) 121:427–36. doi:10.1111/j.1365-2567.2007.02593.x
68. Martinet L, Jean C, Dietrich G, Fournie JJ, Poupot R. PGE2 inhibits natural killer and gamma delta T cell cytotoxicity triggered by NKR and TCR through a cAMP-mediated PKA type I-dependent signaling. Biochem Pharmacol (2010) 80:838–45. doi:10.1016/j.bcp.2010.05.002
69. Brudvik KW, Tasken K. Modulation of T cell immune functions by the prostaglandin E(2) – cAMP pathway in chronic inflammatory states. Br J Pharmacol (2012) 166:411–9. doi:10.1111/j.1476-5381.2011.01800.x
70. Lone AM, Tasken K. Proinflammatory and immunoregulatory roles of eicosanoids in T cells. Front Immunol (2013) 4:130. doi:10.3389/fimmu.2013.00130
71. Sreeramkumar V, Fresno M, Cuesta N. Prostaglandin E2 and T cells: friends or foes? Immunol Cell Biol (2012) 90:579–86. doi:10.1038/icb.2011.75
72. Farrar WL, Evans SW, Rapp UR, Cleveland JL. Effects of anti-proliferative cyclic AMP on interleukin 2-stimulated gene expression. J Immunol (1987) 139:2075–80.
73. Mary D, Aussel C, Ferrua B, Fehlmann M. Regulation of interleukin 2 synthesis by cAMP in human T cells. J Immunol (1987) 139:1179–84.
74. Anastassiou ED, Paliogianni F, Balow JP, Yamada H, Boumpas DT. Prostaglandin E2 and other cyclic AMP-elevating agents modulate IL-2 and IL-2R alpha gene expression at multiple levels. J Immunol (1992) 148:2845–52.
75. Rincon M, Tugores A, Lopez-Rivas A, Silva A, Alonso M, De Landazuri MO, et al. Prostaglandin E2 and the increase of intracellular cAMP inhibit the expression of interleukin 2 receptors in human T cells. Eur J Immunol (1988) 18:1791–6. doi:10.1002/eji.1830181121
76. Paliogianni F, Boumpas DT. Prostaglandin E2 inhibits the nuclear transcription of the human interleukin 2, but not the Il-4, gene in human T cells by targeting transcription factors AP-1 and NF-AT. Cell Immunol (1996) 171:95–101. doi:10.1006/cimm.1996.0178
77. Rodriguez G, Ross JA, Nagy ZS, Kirken RA. Forskolin-inducible cAMP pathway negatively regulates T-cell proliferation by uncoupling the interleukin-2 receptor complex. J Biol Chem (2013) 288:7137–46. doi:10.1074/jbc.M112.408765
78. Walsh DA, Perkins JP, Krebs EG. An adenosine 3’,5’-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem (1968) 243:3763–5.
79. Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev (2002) 82:769–824. doi:10.1152/physrev.00008.2002
80. Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu Rev Cell Dev Biol (2003) 19:23–44. doi:10.1146/annurev.cellbio.19.110701.154854
81. de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature (1998) 396:474–7. doi:10.1038/24884
82. Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, et al. A family of cAMP-binding proteins that directly activate Rap1. Science (1998) 282:2275–9. doi:10.1126/science.282.5397.2275
83. Vang AG, Housley W, Dong H, Basole C, Ben-Sasson SZ, Kream BE, et al. Regulatory T-cells and cAMP suppress effector T-cells independently of PKA-CREM/ICER: a potential role for Epac. Biochem J (2013) 456:463–73. doi:10.1042/BJ20130064
84. Yao C, Hirata T, Soontrapa K, Ma X, Takemori H, Narumiya S. Prostaglandin E(2) promotes Th1 differentiation via synergistic amplification of IL-12 signalling by cAMP and PI3-kinase. Nat Commun (2013) 4:1685. doi:10.1038/ncomms3315
85. Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nat Rev Immunol (2003) 3:973–83. doi:10.1038/nri1245
86. Irving BA, Chan AC, Weiss A. Functional characterization of a signal transducing motif present in the T cell antigen receptor zeta chain. J Exp Med (1993) 177:1093–103. doi:10.1084/jem.177.4.1093
88. Cemerski S, Shaw A. Immune synapses in T-cell activation. Curr Opin Immunol (2006) 18:298–304. doi:10.1016/j.coi.2006.03.011
89. Burack WR, Lee KH, Holdorf AD, Dustin ML, Shaw AS. Cutting edge: quantitative imaging of raft accumulation in the immunological synapse. J Immunol (2002) 169:2837–41. doi:10.4049/jimmunol.169.6.2837
90. Tavano R, Gri G, Molon B, Marinari B, Rudd CE, Tuosto L, et al. CD28 and lipid rafts coordinate recruitment of Lck to the immunological synapse of human T lymphocytes. J Immunol (2004) 173:5392–7. doi:10.4049/jimmunol.173.9.5392
91. Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science (1999) 283:680–2. doi:10.1126/science.283.5402.680
92. Latour S, Veillette A. Proximal protein tyrosine kinases in immunoreceptor signaling. Curr Opin Immunol (2001) 13:299–306. doi:10.1016/S0952-7915(00)00219-3
93. Molina TJ, Kishihara K, Siderovski DP, van Ewijk W, Narendran A, Timms E, et al. Profound block in thymocyte development in mice lacking p56lck. Nature (1992) 357:161–4. doi:10.1038/357161a0
94. Straus DB, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell (1992) 70:585–93. doi:10.1016/0092-8674(92)90428-F
95. Chan AC, Iwashima M, Turck CW, Weiss A. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell (1992) 71:649–62. doi:10.1016/0092-8674(92)90598-7
96. Au-Yeung BB, Levin SE, Zhang C, Hsu LY, Cheng DA, Killeen N, et al. A genetically selective inhibitor demonstrates a function for the kinase Zap70 in regulatory T cells independent of its catalytic activity. Nat Immunol (2010) 11:1085–92. doi:10.1038/ni.1955
97. Weber JR, Orstavik S, Torgersen KM, Danbolt NC, Berg SF, Ryan JC, et al. Molecular cloning of the cDNA encoding pp36, a tyrosine-phosphorylated adaptor protein selectively expressed by T cells and natural killer cells. J Exp Med (1998) 187:1157–61. doi:10.1084/jem.187.7.1157
98. Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell (1998) 92:83–92. doi:10.1016/S0092-8674(00)80901-0
99. Finco TS, Kadlecek T, Zhang W, Samelson LE, Weiss A. LAT is required for TCR-mediated activation of PLCgamma1 and the Ras pathway. Immunity (1998) 9:617–26. doi:10.1016/S1074-7613(00)80659-7
100. Zhang W, Sommers CL, Burshtyn DN, Stebbins CC, DeJarnette JB, Trible RP, et al. Essential role of LAT in T cell development. Immunity (1999) 10:323–32. doi:10.1016/S1074-7613(00)80032-1
101. Zhang W, Trible RP, Zhu M, Liu SK, McGlade CJ, Samelson LE. Association of Grb2, Gads, and phospholipase C-gamma 1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell angigen receptor-mediated signaling. J Biol Chem (2000) 275:23355–61. doi:10.1074/jbc.M000404200
102. Torgersen KM, Vaage JT, Rolstad B, Tasken K. A soluble LAT deletion mutant inhibits T-cell activation: reduced recruitment of signalling molecules to glycolipid-enriched microdomains. Cell Signal (2001) 13:213–20. doi:10.1016/S0898-6568(01)00131-0
103. Kuo CT, Leiden JM. Transcriptional regulation of T lymphocyte development and function. Annu Rev Immunol (1999) 17:149–87. doi:10.1146/annurev.immunol.17.1.149
104. Zhang L, Nabel GJ. Positive and negative regulation of IL-2 gene expression: role of multiple regulatory sites. Cytokine (1994) 6:221–8. doi:10.1016/1043-4666(94)90016-7
105. Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev (2010) 238:247–62. doi:10.1111/j.1600-065X.2010.00951.x
106. van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization In Vivo. Immunity (2014) 41:63–74. doi:10.1016/j.immuni.2014.06.003
107. Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, et al. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity (2012) 37:660–73. doi:10.1016/j.immuni.2012.09.007
108. Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol (2006) 6:329–33. doi:10.1038/nri1807
109. van der Werf N, Redpath SA, Phythian-Adams AT, Azuma M, Allen JE, Maizels RM, et al. Th2 responses to helminth parasites can be therapeutically enhanced by, but are not dependent upon, GITR-GITR ligand costimulation in vivo. J Immunol (2011) 187:1411–20. doi:10.4049/jimmunol.1100834
110. Jankovic D, Feng CG. CD4(+) T cell differentiation in infection: amendments to the Th1/Th2 axiom. Front Immunol (2015) 6:198. doi:10.3389/fimmu.2015.00198
111. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity (2006) 25:117–27. doi:10.1016/j.immuni.2006.04.010
112. Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, et al. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol (2005) 6:1253–62. doi:10.1038/ni1272
113. Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, et al. The immunological synapse. Annu Rev Immunol (2001) 19:375–96. doi:10.1146/annurev.immunol.19.1.375
114. Jury EC, Isenberg DA, Mauri C, Ehrenstein MR. Atorvastatin restores Lck expression and lipid raft-associated signaling in T cells from patients with systemic lupus erythematosus. J Immunol (2006) 177:7416–22. doi:10.4049/jimmunol.177.10.7416
115. Kabouridis PS, Janzen J, Magee AL, Ley SC. Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur J Immunol (2000) 30:954–63. doi:10.1002/1521-4141(200003)30:3<954::AID-IMMU954>3.0.CO;2-Y
116. Xavier R, Brennan T, Li Q, McCormack C, Seed B. Membrane compartmentation is required for efficient T cell activation. Immunity (1998) 8:723–32. doi:10.1016/S1074-7613(00)80577-4
117. Abrahamsen H, Baillie G, Ngai J, Vang T, Nika K, Ruppelt A, et al. TCR- and CD28-mediated recruitment of phosphodiesterase 4 to lipid rafts potentiates TCR signaling. J Immunol (2004) 173:4847–58. doi:10.4049/jimmunol.173.8.4847
118. Ledbetter JA, Parsons M, Martin PJ, Hansen JA, Rabinovitch PS, June CH. Antibody binding to CD5 (Tp67) and Tp44 T cell surface molecules: effects on cyclic nucleotides, cytoplasmic free calcium, and cAMP-mediated suppression. J Immunol (1986) 137:3299–305.
119. Mosenden R, Tasken K. Cyclic AMP-mediated immune regulation – overview of mechanisms of action in T cells. Cell Signal (2011) 23:1009–16. doi:10.1016/j.cellsig.2010.11.018
120. Mustelin T, Tasken K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem J (2003) 371:15–27. doi:10.1042/bj20021637
121. Vang T, Abrahamsen H, Myklebust S, Enserink J, Prydz H, Mustelin T, et al. Knockdown of C-terminal Src kinase by siRNA-mediated RNA interference augments T cell receptor signaling in mature T cells. Eur J Immunol (2004) 34:2191–9. doi:10.1002/eji.200425036
122. Vang T, Abrahamsen H, Myklebust S, Horejsi V, Tasken K. Combined spatial and enzymatic regulation of Csk by cAMP and protein kinase a inhibits T cell receptor signaling. J Biol Chem (2003) 278:17597–600. doi:10.1074/jbc.C300077200
123. Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R, et al. LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol (2012) 8:437–46. doi:10.1038/nchembio.916
124. Pidoux G, Gerbaud P, Dompierre J, Lygren B, Solstad T, vain-Brion DE, et al. A PKA-ezrin-Cx43 signaling complex controls gap junction communication and thereby trophoblast cell fusion. J Cell Sci (2014) 127:4172–85. doi:10.1242/jcs.149609
125. Corbin JD, Soderling TR, Park CR. Regulation of adenosine 3’,5’-monophosphate-dependent protein kinase. I. Preliminary characterization of the adipose tissue enzyme in crude extracts. J Biol Chem (1973) 248:1813–21.
126. Potter RL, Stafford PH, Taylor S. Regulatory subunit of cyclic AMP-dependent protein kinase I from porcine skeletal muscle: purification and proteolysis. Arch Biochem Biophys (1978) 190:174–80. doi:10.1016/0003-9861(78)90265-5
127. Scott JD. Cyclic nucleotide-dependent protein kinases. Pharmacol Ther (1991) 50:123–45. doi:10.1016/0163-7258(91)90075-W
128. Skalhegg BS, Tasken K, Hansson V, Huitfeldt HS, Jahnsen T, Lea T. Location of cAMP-dependent protein kinase type I with the TCR-CD3 complex. Science (1994) 263:84–7. doi:10.1126/science.8272870
129. Chow LM, Fournel M, Davidson D, Veillette A. Negative regulation of T-cell receptor signalling by tyrosine protein kinase p50csk. Nature (1993) 365:156–60. doi:10.1038/365156a0
130. Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J Biol Chem (1991) 266:24249–52.
131. Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat Immunol (2014) 15:798–807. doi:10.1038/ni.2940
132. Takeuchi S, Takayama Y, Ogawa A, Tamura K, Okada M. Transmembrane phosphoprotein Cbp positively regulates the activity of the carboxyl-terminal Src kinase, Csk. J Biol Chem (2000) 275:29183–6. doi:10.1074/jbc.C000326200
133. Brdicka T, Pavlistova D, Leo A, Bruyns E, Korinek V, Angelisova P, et al. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J Exp Med (2000) 191:1591–604. doi:10.1084/jem.191.9.1591
134. Kawabuchi M, Satomi Y, Takao T, Shimonishi Y, Nada S, Nagai K, et al. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature (2000) 404:999–1003. doi:10.1038/35010121
135. Davidson D, Bakinowski M, Thomas ML, Horejsi V, Veillette A. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol Cell Biol (2003) 23:2017–28. doi:10.1128/MCB.23.6.2017-2028.2003
136. Nika K, Soldani C, Salek M, Paster W, Gray A, Etzensperger R, et al. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity (2010) 32:766–77. doi:10.1016/j.immuni.2010.05.011
137. Pfisterer K, Forster F, Paster W, Supper V, Ohradanova-Repic A, Eckerstorfer P, et al. The late endosomal transporter CD222 directs the spatial distribution and activity of Lck. J Immunol (2014) 193:2718–32. doi:10.4049/jimmunol.1303349
138. Rossy J, Pageon SV, Davis DM, Gaus K. Super-resolution microscopy of the immunological synapse. Curr Opin Immunol (2013) 25:307–12. doi:10.1016/j.coi.2013.04.002
139. BjØrgo E, Moltu K, Taskén K. Phosphodiesterases as targets for modulating T-cell responses. Handb Exp Pharmacol (2011) 204:345–63. doi:10.1007/978-3-642-17969-3_15
140. Bjorgo E, Tasken K. Role of cAMP phosphodiesterase 4 in regulation of T-cell function. Crit Rev Immunol (2006) 26:443–51. doi:10.1615/CritRevImmunol.v26.i5.40
141. Houslay MD, Milligan G. Tailoring cAMP-signalling responses through isoform multiplicity. Trends Biochem Sci (1997) 22:217–24. doi:10.1016/S0968-0004(97)01050-5
142. Mehats C, Andersen CB, Filopanti M, Jin SL, Conti M. Cyclic nucleotide phosphodiesterases and their role in endocrine cell signaling. Trends Endocrinol Metab (2002) 13:29–35. doi:10.1016/S1043-2760(01)00523-9
143. Essayan DM. Cyclic nucleotide phosphodiesterases. J Allergy Clin Immunol (2001) 108:671–80. doi:10.1067/mai.2001.119555
144. Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res (2007) 100:309–27. doi:10.1161/01.RES.0000256354.95791.f1
145. Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev (2011) 91:651–90. doi:10.1152/physrev.00030.2010
146. Salari P, Abdollahi M. Phosphodiesterase inhibitors in inflammatory bowel disease. Expert Opin Investig Drugs (2012) 21:261–4. doi:10.1517/13543784.2012.658915
147. Schafer P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol (2012) 83:1583–90. doi:10.1016/j.bcp.2012.01.001
148. Michel F, Attal-Bonnefoy G, Mangino G, Mise-Omata S, Acuto O. CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity (2001) 15:935–45. doi:10.1016/S1074-7613(01)00244-8
149. Arp J, Kirchhof MG, Baroja ML, Nazarian SH, Chau TA, Strathdee CA, et al. Regulation of T-cell activation by phosphodiesterase 4B2 requires its dynamic redistribution during immunological synapse formation. Mol Cell Biol (2003) 23:8042–57. doi:10.1128/MCB.23.22.8042-8057.2003
150. Bolger GB, McCahill A, Huston E, Cheung YF, McSorley T, Baillie GS, et al. The unique amino-terminal region of the PDE4D5 cAMP phosphodiesterase isoform confers preferential interaction with beta-arrestins. J Biol Chem (2003) 278:49230–8. doi:10.1074/jbc.M303772200
151. Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science (2002) 298:834–6. doi:10.1126/science.1074683
152. Bjorgo E, Solheim SA, Abrahamsen H, Baillie GS, Brown KM, Berge T, et al. Cross talk between phosphatidylinositol 3-kinase and cyclic AMP (cAMP)-protein kinase a signaling pathways at the level of a protein kinase B/beta-arrestin/cAMP phosphodiesterase 4 complex. Mol Cell Biol (2010) 30:1660–72. doi:10.1128/MCB.00696-09
153. Bjorgo E, Tasken K. Novel mechanism of signaling by CD28. Immunol Lett (2010) 129:1–6. doi:10.1016/j.imlet.2010.01.007
154. Michel JJ, Scott JD. AKAP mediated signal transduction. Annu Rev Pharmacol Toxicol (2002) 42:235–57. doi:10.1146/annurev.pharmtox.42.083101.135801
155. Colledge M, Scott JD. AKAPs: from structure to function. Trends Cell Biol (1999) 9:216–21. doi:10.1016/S0962-8924(99)01558-5
156. Dell’Acqua ML, Scott JD. Protein kinase A anchoring. J Biol Chem (1997) 272:12881–4. doi:10.1074/jbc.272.20.12881
157. Feliciello A, Gottesman ME, Avvedimento EV. The biological functions of A-kinase anchor proteins. J Mol Biol (2001) 308:99–114. doi:10.1006/jmbi.2001.4585
158. Rubin CS. A kinase anchor proteins and the intracellular targeting of signals carried by cyclic AMP. Biochim Biophys Acta (1994) 1224:467–79.
159. Wong L, Lieser SA, Miyashita O, Miller M, Tasken K, Onuchic JN, et al. Coupled motions in the SH2 and kinase domains of Csk control Src phosphorylation. J Mol Biol (2005) 351:131–43. doi:10.1016/j.jmb.2005.05.042
160. Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol (2004) 5:959–70. doi:10.1038/nrm1527
161. Tasken K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev (2004) 84:137–67. doi:10.1152/physrev.00021.2003
162. Pidoux G, Tasken K. Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J Mol Endocrinol (2010) 44:271–84. doi:10.1677/JME-10-0010
163. Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature (1994) 368:853–6. doi:10.1038/368853a0
164. Vijayaraghavan S, Goueli SA, Davey MP, Carr DW. Protein kinase A-anchoring inhibitor peptides arrest mammalian sperm motility. J Biol Chem (1997) 272:4747–52. doi:10.1074/jbc.272.8.4747
165. Williams RO. Cutting edge: a-kinase anchor proteins are involved in maintaining resting T cells in an inactive state. J Immunol (2002) 168:5392–6. doi:10.4049/jimmunol.168.11.5392
166. Delint-Ramirez I, Willoughby D, Hammond GR, Ayling LJ, Cooper DM. Palmitoylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium-sensitive adenylyl cyclase type 8. J Biol Chem (2011) 286:32962–75. doi:10.1074/jbc.M111.243899
167. Schillace RV, Miller CL, Carr DW. AKAPs in lipid rafts are required for optimal antigen presentation by dendritic cells. Immunol Cell Biol (2011) 89:650–8. doi:10.1038/icb.2010.148
168. Raslan Z, Magwenzi S, Aburima A, Tasken K, Naseem KM. Targeting of type I protein kinase A to lipid rafts is required for platelet inhibition by the 3’,5’-cyclic adenosine monophosphate-signaling pathway. J Thromb Haemost (2015) 13:1721–34. doi:10.1111/jth.13042
169. Torgersen KM, Aandahl EM, Tasken K. Molecular architecture of signal complexes regulating immune cell function. Handb Exp Pharmacol (2008) 186:327–63. doi:10.1007/978-3-540-72843-6_14
170. Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC, et al. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci Signal (2009) 2:ra28. doi:10.1126/scisignal.2000202
171. Bretscher A. Purification of an 80,000-dalton protein that is a component of the isolated microvillus cytoskeleton, and its localization in nonmuscle cells. J Cell Biol (1983) 97:425–32. doi:10.1083/jcb.97.2.425
172. Pakkanen R, Hedman K, Turunen O, Wahlstrom T, Vaheri A. Microvillus-specific Mr 75,000 plasma membrane protein of human choriocarcinoma cells. J Histochem Cytochem (1987) 35:809–16. doi:10.1177/35.8.3298422
173. Gould KL, Cooper JA, Bretscher A, Hunter T. The protein-tyrosine kinase substrate, p81, is homologous to a chicken microvillar core protein. J Cell Biol (1986) 102:660–9. doi:10.1083/jcb.102.2.660
174. Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol (2002) 3:586–99. doi:10.1038/nrm882
175. Shcherbina A, Bretscher A, Kenney DM, Remold-O’Donnell E. Moesin, the major ERM protein of lymphocytes and platelets, differs from ezrin in its insensitivity to calpain. FEBS Lett (1999) 443:31–6. doi:10.1016/S0014-5793(98)01674-3
176. Egerton M, Burgess WH, Chen D, Druker BJ, Bretscher A, Samelson LE. Identification of ezrin as an 81-kDa tyrosine-phosphorylated protein in T cells. J Immunol (1992) 149:1847–52.
177. Dransfield DT, Bradford AJ, Smith J, Martin M, Roy C, Mangeat PH, et al. Ezrin is a cyclic AMP-dependent protein kinase anchoring protein. EMBO J (1997) 16:35–43. doi:10.1093/emboj/16.1.35
178. Algrain M, Turunen O, Vaheri A, Louvard D, Arpin M. Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane-cytoskeletal linker. J Cell Biol (1993) 120:129–39. doi:10.1083/jcb.120.1.129
179. Brdickova N, Brdicka T, Andera L, Spicka J, Angelisova P, Milgram SL, et al. Interaction between two adapter proteins, PAG and EBP50: a possible link between membrane rafts and actin cytoskeleton. FEBS Lett (2001) 507:133–6. doi:10.1016/S0014-5793(01)02955-6
180. Itoh K, Sakakibara M, Yamasaki S, Takeuchi A, Arase H, Miyazaki M, et al. Cutting edge: negative regulation of immune synapse formation by anchoring lipid raft to cytoskeleton through Cbp-EBP50-ERM assembly. J Immunol (2002) 168:541–4. doi:10.4049/jimmunol.168.2.541
181. Reczek D, Bretscher A. The carboxyl-terminal region of EBP50 binds to a site in the amino-terminal domain of ezrin that is masked in the dormant molecule. J Biol Chem (1998) 273:18452–8. doi:10.1074/jbc.273.29.18452
182. Bretscher A. Regulation of cortical structure by the ezrin-radixin-moesin protein family. Curr Opin Cell Biol (1999) 11:109–16. doi:10.1016/S0955-0674(99)80013-1
183. Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M, et al. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol (2007) 178:1938–47. doi:10.4049/jimmunol.178.3.1938
184. Mangeat P, Roy C, Martin M. ERM proteins in cell adhesion and membrane dynamics: authors’ correction. Trends Cell Biol (1999) 9:289. doi:10.1016/S0962-8924(99)01607-4
185. Cullinan P, Sperling AI, Burkhardt JK. The distal pole complex: a novel membrane domain distal to the immunological synapse. Immunol Rev (2002) 189:111–22. doi:10.1034/j.1600-065X.2002.18910.x
186. Niggli V, Rossy J. Ezrin/radixin/moesin: versatile controllers of signaling molecules and of the cortical cytoskeleton. Int J Biochem Cell Biol (2008) 40:344–9. doi:10.1016/j.biocel.2007.02.012
187. Shaffer MH, Dupree RS, Zhu P, Saotome I, Schmidt RF, McClatchey AI, et al. Ezrin and moesin function together to promote T cell activation. J Immunol (2009) 182:1021–32. doi:10.4049/jimmunol.182.2.1021
188. Ilani T, Khanna C, Zhou M, Veenstra TD, Bretscher A. Immune synapse formation requires ZAP-70 recruitment by ezrin and CD43 removal by moesin. J Cell Biol (2007) 179:733–46. doi:10.1083/jcb.200707199
189. Delon J, Kaibuchi K, Germain RN. Exclusion of CD43 from the immunological synapse is mediated by phosphorylation-regulated relocation of the cytoskeletal adaptor moesin. Immunity (2001) 15:691–701. doi:10.1016/S1074-7613(01)00231-X
190. Sperling AI, Sedy JR, Manjunath N, Kupfer A, Ardman B, Burkhardt JK. TCR signaling induces selective exclusion of CD43 from the T cell-antigen-presenting cell contact site. J Immunol (1998) 161:6459–62.
191. Carlson CR, Lygren B, Berge T, Hoshi N, Wong W, Tasken K, et al. Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J Biol Chem (2006) 281:21535–45. doi:10.1074/jbc.M603223200
192. Jarnaess E, Ruppelt A, Stokka AJ, Lygren B, Scott JD, Tasken K. Dual specificity A-kinase anchoring proteins (AKAPs) contain an additional binding region that enhances targeting of protein kinase A type I. J Biol Chem (2008) 283:33708–18. doi:10.1074/jbc.M804807200
193. Cornez I, Tasken K. Spatiotemporal control of cyclic AMP immunomodulation through the PKA-Csk inhibitory pathway is achieved by anchoring to an Ezrin-EBP50-PAG scaffold in effector T cells. FEBS Lett (2010) 584:2681–8. doi:10.1016/j.febslet.2010.04.056
194. Oviedo-Orta E, Evans WH. Gap junctions and connexins: potential contributors to the immunological synapse. J Leukoc Biol (2002) 72:636–42.
195. Oviedo-Orta E, Perreau M, Evans WH, Potolicchio I. Control of the proliferation of activated CD4+ T cells by connexins. J Leukoc Biol (2010) 88:79–86. doi:10.1189/jlb.0909613
196. Kuczma M, Lee JR, Kraj P. Connexin 43 signaling enhances the generation of Foxp3+ regulatory T cells. J Immunol (2011) 187:248–57. doi:10.4049/jimmunol.1003785
197. Herve JC, Bourmeyster N, Sarrouilhe D, Duffy HS. Gap junctional complexes: from partners to functions. Prog Biophys Mol Biol (2007) 94:29–65. doi:10.1016/j.pbiomolbio.2007.03.010
198. Paulson AF, Lampe PD, Meyer RA, TenBroek E, Atkinson MM, Walseth TF, et al. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci (2000) 113(Pt 17):3037–49.
199. Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol (2004) 36:1171–86. doi:10.1016/S1357-2725(03)00264-4
200. Pidoux G, Tasken K. Anchored PKA as a gatekeeper for gap junctions. Commun Integr Biol (2015) 8:e1057361. doi:10.1080/19420889.2015.1057361
201. Kashishian A, Howard M, Loh C, Gallatin WM, Hoekstra MF, Lai Y. AKAP79 inhibits calcineurin through a site distinct from the immunophilin-binding region. J Biol Chem (1998) 273:27412–9. doi:10.1074/jbc.273.42.27412
202. Schillace RV, Andrews SF, Liberty GA, Davey MP, Carr DW. Identification and characterization of myeloid translocation gene 16b as a novel a kinase anchoring protein in T lymphocytes. J Immunol (2002) 168:1590–9. doi:10.4049/jimmunol.168.4.1590
203. Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, et al. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science (1995) 267:108–11. doi:10.1126/science.7528941
204. Northrop JP, Ho SN, Chen L, Thomas DJ, Timmerman LA, Nolan GP, et al. NF-AT components define a family of transcription factors targeted in T-cell activation. Nature (1994) 369:497–502. doi:10.1038/369497a0
205. Oliveria SF, Gomez LL, Dell’Acqua ML. Imaging kinase – AKAP79 – phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol (2003) 160:101–12. doi:10.1083/jcb.200209127
206. Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, et al. Assembly of an A kinase-anchoring protein-beta(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol (2000) 10:409–12. doi:10.1016/S0960-9822(00)00419-X
207. Riether C, Kavelaars A, Wirth T, Pacheco-Lopez G, Doenlen R, Willemen H, et al. Stimulation of beta(2)-adrenergic receptors inhibits calcineurin activity in CD4(+) T cells via PKA-AKAP interaction. Brain Behav Immun (2011) 25:59–66. doi:10.1016/j.bbi.2010.07.248
208. Huang LJ, Wang L, Ma Y, Durick K, Perkins G, Deerinck TJ, et al. NH2-Terminal targeting motifs direct dual specificity A-kinase-anchoring protein 1 (D-AKAP1) to either mitochondria or endoplasmic reticulum. J Cell Biol (1999) 145:951–9. doi:10.1083/jcb.145.5.951
209. Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem (1997) 272:8057–64. doi:10.1074/jbc.272.12.8057
210. Lemay J, Maidou-Peindara P, Cancio R, Ennifar E, Coadou G, Maga G, et al. AKAP149 binds to HIV-1 reverse transcriptase and is involved in the reverse transcription. J Mol Biol (2008) 383:783–96. doi:10.1016/j.jmb.2008.08.055
211. El Din El Homasany BS, Volkov Y, Takahashi M, Ono Y, Keryer G, Delouvee A, et al. The scaffolding protein CG-NAP/AKAP450 is a critical integrating component of the LFA-1-induced signaling complex in migratory T cells. J Immunol (2005) 175:7811–8. doi:10.4049/jimmunol.175.12.7811
212. Witczak O, Skalhegg BS, Keryer G, Bornens M, Tasken K, Jahnsen T, et al. Cloning and characterization of a cDNA encoding an A-kinase anchoring protein located in the centrosome, AKAP450. EMBO J (1999) 18:1858–68. doi:10.1093/emboj/18.7.1858
213. Takahashi M, Shibata H, Shimakawa M, Miyamoto M, Mukai H, Ono Y. Characterization of a novel giant scaffolding protein, CG-NAP, that anchors multiple signaling enzymes to centrosome and the Golgi apparatus. J Biol Chem (1999) 274:17267–74. doi:10.1074/jbc.274.24.17267
214. Robles-Valero J, Martin-Cofreces NB, Lamana A, Macdonald S, Volkov Y, Sanchez-Madrid F. Integrin and CD3/TCR activation are regulated by the scaffold protein AKAP450. Blood (2010) 115:4174–84. doi:10.1182/blood-2009-12-256222
215. Maeda M, Otsuka T, Kimura N, Kozu T, Fukuyama T, Uchida N, et al. Induction of MTG8-specific cytotoxic T-cell lines: MTG8 is probably a tumour antigen that is recognized by cytotoxic T cells in AML1-MTG8-fused gene-positive acute myelogenous leukaemia. Br J Haematol (2000) 111:570–9. doi:10.1111/j.1365-2141.2000.02400.x
216. Miyoshi H, Kozu T, Shimizu K, Enomoto K, Maseki N, Kaneko Y, et al. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J (1993) 12:2715–21.
217. Kozu T, Komori A, Sueoka E, Fujiki H, Kaneko Y, Matsui T, et al. Significance of MTG8 in leukemogenesis. Leukemia (1997) 11(Suppl 3):297–8.
218. Wolford JK, Prochazka M. Structure and expression of the human MTG8/ETO gene. Gene (1998) 212:103–9. doi:10.1016/S0378-1119(98)00141-3
219. Fukuyama T, Sueoka E, Sugio Y, Otsuka T, Niho Y, Akagi K, et al. MTG8 proto-oncoprotein interacts with the regulatory subunit of type II cyclic AMP-dependent protein kinase in lymphocytes. Oncogene (2001) 20:6225–32. doi:10.1038/sj.onc.1204794
220. Gamou T, Kitamura E, Hosoda F, Shimizu K, Shinohara K, Hayashi Y, et al. The partner gene of AML1 in t(16;21) myeloid malignancies is a novel member of the MTG8(ETO) family. Blood (1998) 91:4028–37.
221. Coghlan VM, Langeberg LK, Fernandez A, Lamb NJ, Scott JD. Cloning and characterization of AKAP 95, a nuclear protein that associates with the regulatory subunit of type II cAMP-dependent protein kinase. J Biol Chem (1994) 269:7658–65.
222. Eide T, Coghlan V, Orstavik S, Holsve C, Solberg R, Skalhegg BS, et al. Molecular cloning, chromosomal localization, and cell cycle-dependent subcellular distribution of the A-kinase anchoring protein, AKAP95. Exp Cell Res (1998) 238:305–16. doi:10.1006/excr.1997.3855
223. Landsverk HB, Carlson CR, Steen RL, Vossebein L, Herberg FW, Tasken K, et al. Regulation of anchoring of the RIIalpha regulatory subunit of PKA to AKAP95 by threonine phosphorylation of RIIalpha: implications for chromosome dynamics at mitosis. J Cell Sci (2001) 114:3255–64.
224. Collas P, Le Guellec K, Tasken K. The A-kinase-anchoring protein AKAP95 is a multivalent protein with a key role in chromatin condensation at mitosis. J Cell Biol (1999) 147:1167–80. doi:10.1083/jcb.147.6.1167
225. Steen RL, Cubizolles F, Le Guellec K, Collas P. A kinase-anchoring protein (AKAP)95 recruits human chromosome-associated protein (hCAP)-D2/Eg7 for chromosome condensation in mitotic extract. J Cell Biol (2000) 149:531–6. doi:10.1083/jcb.149.3.531
226. Schillace RV, Scott JD. Association of the type 1 protein phosphatase PP1 with the A-kinase anchoring protein AKAP220. Curr Biol (1999) 9:321–4. doi:10.1016/S0960-9822(99)80141-9
227. Schillace RV, Voltz JW, Sim AT, Shenolikar S, Scott JD. Multiple interactions within the AKAP220 signaling complex contribute to protein phosphatase 1 regulation. J Biol Chem (2001) 276:12128–34. doi:10.1074/jbc.M010398200
228. Schillace RV, Carr DW. The role of protein kinase A and A-kinase anchoring proteins in modulating T-cell activation: progress and future directions. Crit Rev Immunol (2006) 26:113–31. doi:10.1615/CritRevImmunol.v26.i2.20
229. Tasken KA, Collas P, Kemmner WA, Witczak O, Conti M, Tasken K. Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J Biol Chem (2001) 276:21999–2002. doi:10.1074/jbc.C000911200
230. Asirvatham AL, Galligan SG, Schillace RV, Davey MP, Vasta V, Beavo JA, et al. A-kinase anchoring proteins interact with phosphodiesterases in T lymphocyte cell lines. J Immunol (2004) 173:4806–14. doi:10.4049/jimmunol.173.8.4806
231. Dema A, Perets E, Schulz MS, Deak VA, Klussmann E. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell Signal (2015) 27:2474–87. doi:10.1016/j.cellsig.2015.09.008
232. Otero C, Penaloza JP, Rodas PI, Fernandez-Ramires R, Velasquez L, Jung JE. Temporal and spatial regulation of cAMP signaling in disease: role of cyclic nucleotide phosphodiesterases. Fundam Clin Pharmacol (2014) 28:593–607. doi:10.1111/fcp.12080
233. Aandahl EM, Aukrust P, Skalhegg BS, Muller F, Froland SS, Hansson V, et al. Protein kinase A type I antagonist restores immune responses of T cells from HIV-infected patients. FASEB J (1998) 12:855–62.
234. Hofmann B, Nishanian P, Nguyen T, Insixiengmay P, Fahey JL. Human immunodeficiency virus proteins induce the inhibitory cAMP/protein kinase A pathway in normal lymphocytes. Proc Natl Acad Sci U S A (1993) 90:6676–80. doi:10.1073/pnas.90.14.6676
235. Nokta M, Pollard R. Human immunodeficiency virus infection: association with altered intracellular levels of cAMP and cGMP in MT-4 cells. Virology (1991) 181:211–7. doi:10.1016/0042-6822(91)90486-U
236. Su Y, Huang X, Raskovalova T, Zacharia L, Lokshin A, Jackson E, et al. Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP-PKA signaling and immunosuppression. Cancer Immunol Immunother (2008) 57:1611–23. doi:10.1007/s00262-008-0494-5
237. Pettersen FO, Torheim EA, Dahm AE, Aaberge IS, Lind A, Holm M, et al. An exploratory trial of cyclooxygenase type 2 inhibitor in HIV-1 infection: downregulated immune activation and improved T cell-dependent vaccine responses. J Virol (2011) 85:6557–66. doi:10.1128/JVI.00073-11
238. Rahmouni S, Aandahl EM, Nayjib B, Zeddou M, Giannini S, Verlaet M, et al. Cyclo-oxygenase type 2-dependent prostaglandin E2 secretion is involved in retrovirus-induced T-cell dysfunction in mice. Biochem J (2004) 384:469–76. doi:10.1042/BJ20031859
239. Aandahl EM, Aukrust P, Muller F, Hansson V, Tasken K, Froland SS. Additive effects of IL-2 and protein kinase A type I antagonist on function of T cells from HIV-infected patients on HAART. AIDS (1999) 13:F109–14. doi:10.1097/00002030-199912030-00001
240. Hofmann B, Nishanian P, Nguyen T, Liu M, Fahey JL. Restoration of T-cell function in HIV infection by reduction of intracellular cAMP levels with adenosine analogues. AIDS (1993) 7:659–64. doi:10.1097/00002030-199305000-00008
241. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer (2005) 5:263–74. doi:10.1038/nrc1586
242. Clarke SL, Betts GJ, Plant A, Wright KL, El-Shanawany TM, Harrop R, et al. CD4+CD25+FOXP3+ regulatory T cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS One (2006) 1:e129. doi:10.1371/journal.pone.0000129
243. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10:942–9. doi:10.1038/nm1093
244. Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer (2003) 98:1089–99. doi:10.1002/cncr.11618
245. Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res (2003) 9:606–12.
246. Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev (2001) 182:18–32. doi:10.1034/j.1600-065X.2001.1820102.x
247. Tanaka H, Tanaka J, Kjaergaard J, Shu S. Depletion of CD4+ CD25+ regulatory cells augments the generation of specific immune T cells in tumor-draining lymph nodes. J Immunother (2002) 25:207–17. doi:10.1097/00002371-200205000-00003
248. Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol (2007) 178:2883–92. doi:10.4049/jimmunol.178.5.2883
249. Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology (1994) 107:1183–8.
250. Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer (2001) 1:11–21. doi:10.1038/35094017
251. Jacobs EJ, Thun MJ, Bain EB, Rodriguez C, Henley SJ, Calle EE. A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. J Natl Cancer Inst (2007) 99:608–15. doi:10.1093/jnci/djk132
252. Kraus S, Arber N. Cancer: do aspirin and other NSAIDs protect against colorectal cancer? Nat Rev Gastroenterol Hepatol (2011) 8:125–6. doi:10.1038/nrgastro.2010.217
253. Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, Morrow J, et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest (1997) 99:2254–9. doi:10.1172/JCI119400
254. Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A (1997) 94:3336–40. doi:10.1073/pnas.94.7.3336
255. Fujino H, Toyomura K, Chen XB, Regan JW, Murayama T. Prostaglandin E(2) regulates cellular migration via induction of vascular endothelial growth factor receptor-1 in HCA-7 human colon cancer cells. Biochem Pharmacol (2011) 81:379–87. doi:10.1016/j.bcp.2010.11.001
256. Sheng H, Shao J, Morrow JD, Beauchamp RD, DuBois RN. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res (1998) 58:362–6.
257. Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem (2001) 276:18075–81. doi:10.1074/jbc.M009689200
258. Kim R, Emi M, Tanabe K, Arihiro K. Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res (2006) 66:5527–36. doi:10.1158/0008-5472.CAN-05-4128
259. Yaqub S, Tasken K. Role for the cAMP-protein kinase A signaling pathway in suppression of antitumor immune responses by regulatory T cells. Crit Rev Oncog (2008) 14:57–77. doi:10.1615/CritRevOncog.v14.i1.40
260. Oberprieler NG, Lemeer S, Kalland ME, Torgersen KM, Heck AJ, Tasken K. High-resolution mapping of prostaglandin E2-dependent signaling networks identifies a constitutively active PKA signaling node in CD8+ CD45RO+ T cells. Blood (2010) 116:2253–65. doi:10.1182/blood-2010-01-266650
261. Nilsson J, Boasso A, Velilla PA, Zhang R, Vaccari M, Franchini G, et al. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood (2006) 108:3808–17. doi:10.1182/blood-2006-05-021576
262. Kinter A, McNally J, Riggin L, Jackson R, Roby G, Fauci AS. Suppression of HIV-specific T cell activity by lymph node CD25+ regulatory T cells from HIV-infected individuals. Proc Natl Acad Sci U S A (2007) 104:3390–5. doi:10.1073/pnas.0611423104
263. Johansson CC, Bryn T, Yndestad A, Eiken HG, Bjerkeli V, Froland SS, et al. Cytokine networks are pre-activated in T cells from HIV-infected patients on HAART and are under the control of cAMP. AIDS (2004) 18:171–9. doi:10.1097/00002030-200401230-00005
264. Aukrust P, Aandahl EM, Skalhegg BS, Nordoy I, Hansson V, Tasken K, et al. Increased activation of protein kinase A type I contributes to the T cell deficiency in common variable immunodeficiency. J Immunol (1999) 162:1178–85.
265. Holm AM, Aukrust P, Aandahl EM, Muller F, Tasken K, Froland SS. Impaired secretion of IL-10 by T cells from patients with common variable immunodeficiency – involvement of protein kinase A type I. J Immunol (2003) 170:5772–7. doi:10.4049/jimmunol.170.11.5772
266. Rahmouni S, Aandahl EM, Trebak M, Boniver J, Tasken K, Moutschen M. Increased cAMP levels and protein kinase (PKA) type I activation in CD4+ T cells and B cells contribute to retrovirus-induced immunodeficiency of mice (MAIDS): a useful in vivo model for drug testing. FASEB J (2001) 15:1466–8. doi:10.1096/fj.00-0813fje
267. Johansson CC, Bryn T, Aandahl EM, Areklett MA, Aukrust P, Tasken K, et al. Treatment with type-2 selective and non-selective cyclooxygenase inhibitors improves T-cell proliferation in HIV-infected patients on highly active antiretroviral therapy. AIDS (2004) 18:951–2. doi:10.1097/00002030-200404090-00015
268. Kvale D, Ormaasen V, Kran AM, Johansson CC, Aukrust P, Aandahl EM, et al. Immune modulatory effects of cyclooxygenase type 2 inhibitors in HIV patients on combination antiretroviral treatment. AIDS (2006) 20:813–20. doi:10.1097/01.aids.0000218544.54586.f1
269. Psaty BM, Potter JD. Risks and benefits of celecoxib to prevent recurrent adenomas. N Engl J Med (2006) 355:950–2. doi:10.1056/NEJMe068158
270. Patrignani P, Patrono C. Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochim Biophys Acta (2015) 1851:422–32. doi:10.1016/j.bbalip.2014.09.016
271. Welch EJ, Jones BW, Scott JD. Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol Interv (2010) 10:86–97. doi:10.1124/mi.10.2.6
272. Carr DW, Hausken ZE I, Fraser D, Stofko-Hahn RE, Scott JD. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem (1992) 267:13376–82.
273. Carr DW, Stofko-Hahn RE, Fraser ID, Bishop SM, Acott TS, Brennan RG, et al. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem (1991) 266:14188–92.
274. Herberg FW, Maleszka A, Eide T, Vossebein L, Tasken K. Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. J Mol Biol (2000) 298:329–39. doi:10.1006/jmbi.2000.3662
275. Alto NM, Soderling SH, Hoshi N, Langeberg LK, Fayos R, Jennings PA, et al. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc Natl Acad Sci U S A (2003) 100:4445–50. doi:10.1073/pnas.0330734100
276. Faruque OM, Le-Nguyen D, Lajoix AD, Vives E, Petit P, Bataille D, et al. Cell-permeable peptide-based disruption of endogenous PKA-AKAP complexes: a tool for studying the molecular roles of AKAP-mediated PKA subcellular anchoring. Am J Physiol Cell Physiol (2009) 296:C306–16. doi:10.1152/ajpcell.00216.2008
277. Gold MG, Lygren B, Dokurno P, Hoshi N, McConnachie G, Tasken K, et al. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell (2006) 24:383–95. doi:10.1016/j.molcel.2006.09.006
278. Henchey LK, Jochim AL, Arora PS. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr Opin Chem Biol (2008) 12:692–7. doi:10.1016/j.cbpa.2008.08.019
279. Walensky LD, Bird GH. Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem (2014) 57:6275–88. doi:10.1021/jm4011675
280. Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science (2004) 305:1466–70. doi:10.1126/science.1099191
281. Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol (2012) 503:3–33. doi:10.1016/B978-0-12-396962-0.00001-X
282. Wang Y, Ho TG, Bertinetti D, Neddermann M, Franz E, Mo GC, et al. Isoform-selective disruption of AKAP-localized PKA using hydrocarbon stapled peptides. ACS Chem Biol (2014) 9:635–42. doi:10.1021/cb500329z
283. Burns-Hamuro LL, Ma Y, Kammerer S, Reineke U, Self C, Cook C, et al. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci U S A (2003) 100:4072–7. doi:10.1073/pnas.2628038100
284. Mosenden R, Singh P, Cornez I, Heglind M, Ruppelt A, Moutschen M, et al. Mice with disrupted type I protein kinase A anchoring in T cells resist retrovirus-induced immunodeficiency. J Immunol (2011) 186:5119–30. doi:10.4049/jimmunol.1100003
285. Wang Y, Ho TG, Franz E, Hermann JS, Smith FD, Hehnly H, et al. PKA-type I selective constrained peptide disruptors of AKAP complexes. ACS Chem Biol (2015) 10:1502–10. doi:10.1021/acschembio.5b00009
286. Gold MG, Fowler DM, Means CK, Pawson CT, Stephany JJ, Langeberg LK, et al. Engineering A-kinase anchoring protein (AKAP)-selective regulatory subunits of protein kinase A (PKA) through structure-based phage selection. J Biol Chem (2013) 288:17111–21. doi:10.1074/jbc.M112.447326
287. Lygren B, Carlson CR, Santamaria K, Lissandron V, McSorley T, Litzenberg J, et al. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep (2007) 8:1061–7. doi:10.1038/sj.embor.7401081
288. Stokka AJ, Mosenden R, Ruppelt A, Lygren B, Tasken K. The adaptor protein EBP50 is important for localization of the protein kinase A-Ezrin complex in T-cells and the immunomodulating effect of cAMP. Biochem J (2010) 425:381–8. doi:10.1042/BJ20091136
289. Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, et al. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature (2005) 437:574–8. doi:10.1038/nature03966
290. Torheim EA, Jarnaess E, Lygren B, Tasken K. Design of proteolytically stable RI-anchoring disruptor peptidomimetics for in vivo studies of anchored type I protein kinase A-mediated signalling. Biochem J (2009) 424:69–78. doi:10.1042/BJ20090933
291. Singh M, Singh P, Vaira D, Torheim EA, Rahmouni S, Tasken K, et al. The RIAD peptidomimetic inhibits HIV-1 replication in humanized NSG mice. Eur J Clin Invest (2014) 44:146–52. doi:10.1111/eci.12200
292. Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov (2004) 3:301–17. doi:10.1038/nrd1343
293. Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature (2007) 450:1001–9. doi:10.1038/nature06526
294. Buchwald P. Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life (2010) 62:724–31. doi:10.1002/iub.383
295. Schachterle C, Christian F, Fernandes JM, Klussmann E. Screening for small molecule disruptors of AKAP-PKA interactions. Methods Mol Biol (2015) 1294:151–66. doi:10.1007/978-1-4939-2537-7_12
296. Rabbi MF, al-Harthi L, Saifuddin M, Roebuck KA. The cAMP-dependent protein kinase A and protein kinase C-beta pathways synergistically interact to activate HIV-1 transcription in latently infected cells of monocyte/macrophage lineage. Virology (1998) 245:257–69. doi:10.1006/viro.1998.9158
297. Skalhegg BS, Funderud A, Henanger HH, Hafte TT, Larsen AC, Kvissel AK, et al. Protein kinase A (PKA) – a potential target for therapeutic intervention of dysfunctional immune cells. Curr Drug Targets (2005) 6:655–64. doi:10.2174/1389450054863644
Keywords: cAMP, AKAP, protein–protein interaction, T cell, prostaglandin
Citation: Wehbi VL and Taskén K (2016) Molecular Mechanisms for cAMP-Mediated Immunoregulation in T cells – Role of Anchored Protein Kinase A Signaling Units. Front. Immunol. 7:222. doi: 10.3389/fimmu.2016.00222
Received: 15 March 2016; Accepted: 23 May 2016;
Published: 08 June 2016
Edited by:
Josef Bodor, Institute of Experimental Medicine, Academy of Sciences of the Czech Republic (IEM), Czech RepublicReviewed by:
Karsten Sauer, Pfizer World-Wide R&D, USACopyright: © 2016 Wehbi and Taskén. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kjetil Taskén, a2pldGlsLnRhc2tlbkBuY21tLnVpby5ubw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.