
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 April 2016
Sec. Alloimmunity and Transplantation
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00117
This article is part of the Research Topic Molecular Mechanisms of Immune Complex Pathophysiology View all 8 articles
 Barbora Knoppova1,2†
Barbora Knoppova1,2† Colin Reily3†
Colin Reily3† Nicolas Maillard4,5†Dana V. Rizk3
Nicolas Maillard4,5†Dana V. Rizk3 Zina Moldoveanu1
Zina Moldoveanu1 Jiri Mestecky1Milan Raska1,2Matthew B. Renfrow6Bruce A. Julian3
Jiri Mestecky1Milan Raska1,2Matthew B. Renfrow6Bruce A. Julian3 Jan Novak1*
Jan Novak1*
IgA nephropathy (IgAN) is the most common primary glomerulonephritis, frequently leading to end-stage renal disease, as there is no disease-specific therapy. IgAN is diagnosed from pathological assessment of a renal biopsy specimen based on predominant or codominant IgA-containing immunodeposits, usually with complement C3 co-deposits and with variable presence of IgG and/or IgM. The IgA in these renal deposits is galactose-deficient IgA1, with less than a full complement of galactose residues on the O-glycans in the hinge region of the heavy chains. Research from the past decade led to the definition of IgAN as an autoimmune disease with a multi-hit pathogenetic process with contributing genetic and environmental components. In this process, circulating galactose-deficient IgA1 (autoantigen) is bound by antiglycan IgG or IgA (autoantibodies) to form immune complexes. Some of these circulating complexes deposit in glomeruli, and thereby activate mesangial cells and induce renal injury through cellular proliferation and overproduction of extracellular matrix components and cytokines/chemokines. Glycosylation pathways associated with production of the autoantigen and the unique characteristics of the corresponding autoantibodies in patients with IgAN have been uncovered. Complement likely plays a significant role in the formation and the nephritogenic activities of these complexes. Complement activation is mediated through the alternative and lectin pathways and probably occurs systemically on IgA1-containing circulating immune complexes as well as locally in glomeruli. Incidence of IgAN varies greatly by geographical location; the disease is rare in central Africa but accounts for up to 40% of native-kidney biopsies in eastern Asia. Some of this variation may be explained by genetically determined influences on the pathogenesis of the disease. Genome-wide association studies to date have identified several loci associated with IgAN. Some of these loci are associated with the increased prevalence of IgAN, whereas others, such as deletion of complement factor H-related genes 1 and 3, are protective against the disease. Understanding the molecular mechanisms and genetic and biochemical factors involved in formation and activities of pathogenic IgA1-containing immune complexes will enable the development of future disease-specific therapies as well as identification of non-invasive disease-specific biomarkers.
IgA nephropathy (IgAN) is currently recognized as the most common primary glomerulonephritis in the world and is a frequent cause of end-stage renal disease. The diagnosis is established by immunofluorescence examination of cortical renal tissue that shows IgA as the dominant or codominant immunoglobulin in glomeruli (Figure 1A) (1, 2). Complement protein C3 is frequently present, often accompanied by IgG, IgM, or both. Confocal microscopy shows colocalization of these proteins, consistent with the presence of immune complexes. Light microscopy findings usually include mesangial hypercellularity and increased mesangial matrix (Figure 1B). Electron microscopy shows electron-dense deposits consistent with immune complexes in the mesangial and paramesangial areas (Figure 1C), occasionally with subepithelial or subendothelial deposits. In 2009, the Oxford classification of IgAN was published. This classification was put forth by an international group of nephrologists and renal pathologists to standardize pathologic findings and ascertain those that predict disease progression. Ultimately, four pathologic features were identified as being of prognostic value, independent of clinical data: mesangial hypercellularity, segmental glomerulosclerosis, endocapillary hypercellularity, and tubular atrophy/interstitial fibrosis. This classification allows the pathologist to give a score for each of these features that correlates to clinical outcome. Most cases used to develop the Oxford classification did not have significant crescents or necrosis, and therefore, neither of these findings was included in the assessment (3, 4).
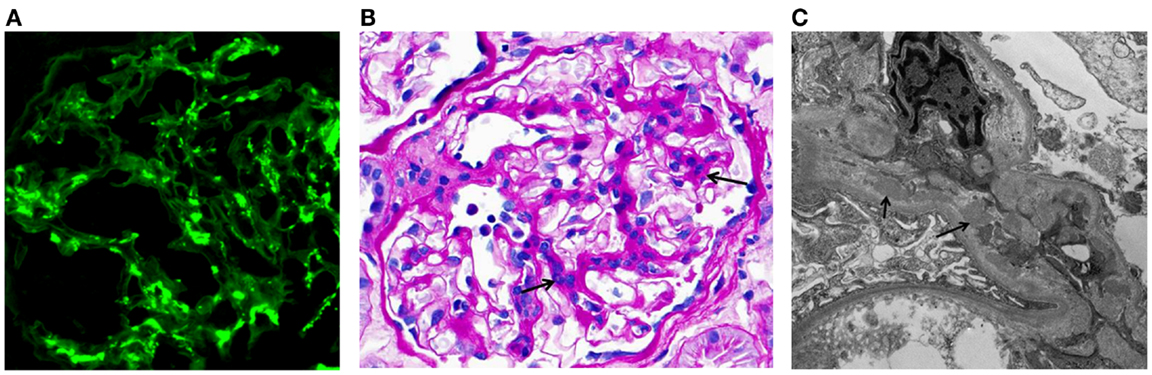
Figure 1. Examples of immunofluorescence-, light-, and electron-microscopy features of renal biopsy specimens from patients with IgAN. (A) Immunofluorescence staining for IgA in a kidney biopsy specimen from a patient with IgAN showing mesangial staining. (B) Periodic acid–Schiff staining of a kidney biopsy specimen from a patient with IgAN. Arrows indicate mesangial expansion and hypercellularity. (C) Electron micrograph of kidney biopsy specimen from a patient with IgAN. Arrows point to examples of electron-dense material representative of mesangial and paramesangial immune complex deposits. Images are courtesy of Dr. Huma Fatima (B,C) and Dr. Lea Novak (A), Department of Pathology, UAB.
IgA nephropathy may affect children as young as 4 years of age. The most common clinical presentation in children is visible hematuria accompanying a febrile illness, frequently an infection of the upper respiratory tract. Among adults, visible hematuria is much less common (extremely rare beyond age 40 years), and typical manifestations include microscopic hematuria, proteinuria, hypertension, and variable degrees of chronic kidney disease (5, 6). The gender distribution differs geographically, with a 2–3:1 male-to-female ratio in North America compared with a 1:1 ratio in Asia (6). About 5–8% of patients have a first- or second-degree relative with biopsy-proven IgAN or urinary abnormalities, suggesting that genetic factors influence the expression of disease. The prevalence of disease varies greatly between different regions of the world. East Asia has the highest rates, whereas the disease is rare in central Africa (7). A recent genome-wide association study (GWAS) found that the frequency of risk alleles in regional populations correlated with disease prevalence (8). The true prevalence of IgAN is impossible to establish because the diagnosis currently requires a kidney biopsy and criteria for undertaking the invasive procedure vary widely. Furthermore, IgAN is frequently subclinical, as evidenced by a study in Finland that found IgAN in 1.3% of autopsies of persons who had committed suicide or died violently (9) and a Japanese study in which biopsies of renal allografts at implantation showed IgAN in 1.6% of cases (10). Although clinical series published shortly after the discovery of IgAN in 1968 indicated generally a benign clinical course, later reports with longer observation have documented progression to end-stage renal disease in 14–39% of patients by 20 years after diagnosis (5). On the one hand, for those who undergo renal transplantation, the disease recurs in about 50% of allografts by 10 years after engraftment (11). On the other hand, the IgA immune deposits clear within several weeks from allografts with subclinical disease at the time of transplantation (12).
Henoch–Schönlein purpura (HSP) is the most common vasculitis in childhood with an incidence of 6–24 per 100,000 children per year (13, 14). Extrarenal involvement includes skin (palpable purpura), gastrointestinal tract (abdominal pain and bloody diarrhea), and musculoskeletal system (arthritis and arthralgia) (15). Renal disease affects a minority of HSP patients and typically manifests as hematuria and proteinuria about 6 weeks after the appearance of purpura. Histological features of HSP with nephritis are pathologically indistinguishable from those of IgAN, suggesting that the two entities share mechanisms of disease (16, 17). While most children with HSP with nephritis resolve their urinary abnormalities, some develop long-term kidney dysfunction and may progress to end-stage renal disease. HSP is relatively uncommon in adults, but the prognosis of HSP with nephritis is worse with increasing age. Patients with HSP with nephritis were excluded from the Oxford classification, so the prognostic value of their histopathologic findings has not been established (18).
IgA-dominant immune complex glomerulonephritis has also been described in patients with a variety of systemic diseases and is thought to be a secondary manifestation. The pathogenesis behind these associations has not been elucidated, but several theories have been proposed (19). In patients with cirrhosis due to alcohol abuse or chronic infection with hepatitis C virus, glomerular IgA is thought to result from decreased clearance of the immunoglobulin by hepatocytes (20, 21). Patients with inflammatory bowel disease or celiac disease may be exposed to increased loads and variety of antigens due to impaired integrity of the gastrointestinal mucosa, inciting increased synthesis of IgA as well as abnormalities of the IgA immune system (20). Finally, chronic infections, such as those caused by staphylococci, may increase production of pathogenic IgA (20).
Considerable evidence has suggested that mesangial immunodeposits in IgAN are derived from IgA-containing circulating immune complexes: (1) disease recurs in about 50% of IgAN patients after kidney transplantation (22–26); (2) immune deposits clear within weeks in kidney from a person with subclinical IgAN after transplantation into a patient with non-IgAN renal disease (12); (3) blood levels of IgA and IgA-containing immune complexes are elevated in many patients with IgAN (25, 27–32); and (4) circulating complexes and mesangial deposits share idiotypic determinants (33), although a disease-specific idiotype has not been identified (34). Thus, circulating immune complexes likely play a key role in IgAN, and kidneys are “innocent bystanders.”
The apparent key role of IgA-containing immune complexes in IgAN and HSP with nephritis has been supported by data from several other studies. Circulating immune complexes with IgA and C3 are elevated in approximately one half of patients with IgAN (28). Moreover, serum levels of IgA-containing immune complexes in patients with IgAN correlate to clinical and histological activity, such as magnitude of microscopic hematuria and percentage of glomeruli with florid crescents (27, 35). In IgAN, hematuria is typical and often includes episodes of macroscopic bleeding that coincide with mucosal infections, including those of the upper respiratory tract and digestive system. These and other observations, and the fact that IgA in immunodeposits is polymeric, have indicated potential involvement of mucosal system [for review, see Ref. (36)].
Circulating immune complexes containing IgA are present in serum of healthy individuals and patients with diseases other than IgAN. Although immune complexes in such subjects may form, for example, due to binding of IgA antibodies to food or microbial antigens, it was shown for patients with IgAN that the microbial and food antigens are not substantial components of IgA-containing glomerular immunodeposits (37).
Humans have two IgA subclasses, IgA1 and IgA2. IgA1 contains O-glycans attached to Ser or Thr, usually three to six, in the hinge region (HR) of the heavy chains (Figure 2). IgA1 HR has nine Ser and Thr amino-acid residues; those are missing in IgA2 HR and, thus, IgA2 does not have O-glycans (Figure 2). In normal human serum IgA1, HR glycoforms with four and five glycans are the most common [for review, see Ref. (38)]. Each heavy chain of IgA1 also contains two N-glycans, one in the CH2 domain (Asn263) and the second in the tailpiece portion (Asn459) (39, 40). Normal human circulatory IgA1 usually has core 1 O-glycans consisting of N-acetylgalactosamine (GalNAc) with β1,3-linked galactose. One or both saccharides can be sialylated, galactose with α2,3-linked and GalNAc with α2,6-linked sialic acid (Figure 3, right panel). The composition of the O-glycans on normal serum IgA1 is variable; prevailing forms include the GalNAc-galactose disaccharide and its mono- and di-sialylated forms (41–44). Normal serum IgA1 had been thought to contain little or no galactose-deficient O-glycans (44), but it is now considered that some terminal or sialylated GalNAc is likely present even in healthy individuals (45).
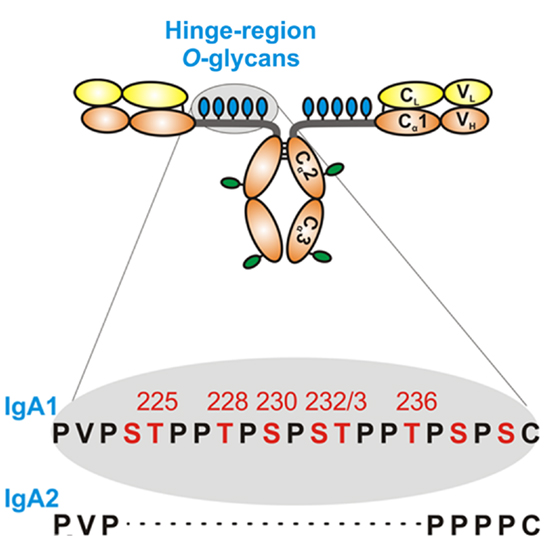
Figure 2. Hinge-region glycosylation of human IgA1 and comparison of amino-acid sequences of human IgA1 and IgA2. Human IgA1 has nine Ser (S) and Thr (T) amino-acid residues in the hinge-region segment (between constant regions C1 and C2 of the heavy chains). Usually, three to six clustered O-glycans are attached per hinge region. IgA2 hinge region is shorter compared to that of IgA1, does not have Ser and Thr residues and, thus, IgA2 does not have O-glycans. Moreover, each IgA1 heavy chain has two N-glycans, one in the C2 domain and the second in the tailpiece portion of the C3 domain.
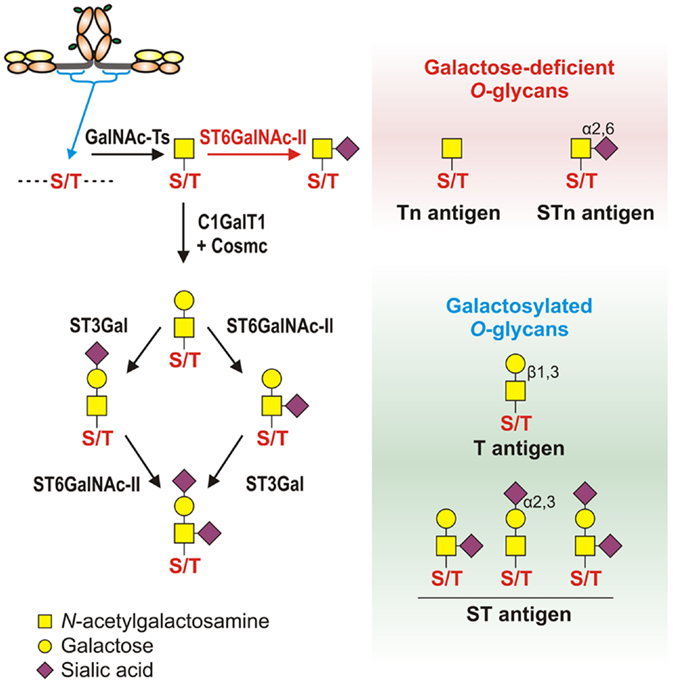
Figure 3. Pathways of O-glycosylation of IgA1 hinge region, including galactose-deficient and galactosylated O-glycans. Left panel: O-glycosylation of IgA1 hinge region occurs in the Golgi apparatus and begins with attachment of N-acetylgalactosamine (GalNAc) to Ser or Thr by an enzyme of UDP-GalNAc:polypeptide GalNAc-transferases family (GalNAc-Ts). In patients with IgAN, some terminal GalNAc residues may be prematurely sialylated by GalNAc α2,6-sialyltransferase (ST6GalNAc) (red arrow); this step prevents addition of galactose (the glycan thus remains galactose-deficient). In healthy individuals, GalNAc-α-Ser/Thr residue can be normally modified by addition of galactose, catalyzed by UDP-galactose:GalNAc-α-Ser/Thr β1,3-galactosyltransferase (C1GalT1); stability of C1GalT1 requires molecular chaperone Cosmc. Galβ1,3-GalNAc structures may be further modified by addition of sialic acid to galactose residues through the activity of Galβ1,3-GalNAc α2,3-sialyltransferase (ST3Gal) and/or to GalNAc residues through the activity of ST6GalNAc. Right panel: galactose-deficient O-glycans consist of terminal GalNAc, also known as Tn antigen, or GalNAc with α2,6-linked sialic acid, also known as STn antigen. Galactosylated O-glycans are disaccharides consisting of galactose and GalNAc (Galβ1,3-GalNAcα1-O-Ser/Thr, also known as T antigen) and may be modified by sialic acid (also known as ST antigen). T antigen does not carry sialic acid, but ST antigen has sialic acid attached to galactose and/or GalNAc.
Analysis of IgA in patients with IgAN revealed that abnormal O-glycosylation is a key step directing IgA1 immune complex formation and glomerular deposition (31, 46–52). The accumulated data suggest that circulating complexes in patients with IgAN contain galactose-deficient IgA1 (Gd-IgA1) (31, 49, 50, 53) and that the IgA in the mesangial deposits is exclusively of IgA1 subclass (54) and is enriched for Gd-IgA1 glycoforms (55, 56). Further insight about a relationship between Gd-IgA1 and nephritis has come from other observations: (1) Gd-IgA1 (57) and IgA–IgG circulating immune complexes (58) are in sera of patients with HSP with nephritis but not in sera of patients with HSP without nephritis and (2) patients with IgA1 myeloma have high circulating levels of IgA1, but only those with aberrantly glycosylated IgA1 develop immune complex glomerulonephritis (59, 60).
Human serum IgA, predominantly IgA1 with a small contribution of IgA2, is >90% in monomeric form and <10% in polymeric form, and a small fraction is bound in circulating immune complexes (61). Serum IgA1 is rapidly catabolized by hepatocytes (see below for more details) and thus has a short half-life (~5 days) (62). Hepatocytes express asialoglycoprotein receptor (ASGP-R) (63, 64) that binds IgA1 and other glycoproteins through terminal galactose or GalNAc residues (63–65). Gd-IgA1 remains in the circulation for a prolonged period of time (66). Galactose deficiency in itself should not hinder catabolism of IgA1 molecules because ASGP-R can recognize terminal GalNAc (65). However, if sialic acid is linked to GalNAc or IgA1 is bound by an antibody, then such IgA1 cannot be recognized by the receptor (53, 67). Serum Gd-IgA1 is bound primarily within immune complexes (31, 49). The large size of these complexes likely precludes entry into the space of Disse through relatively small endothelial fenestrae (68), hence preventing their hepatic clearance from the circulation (69–71). Immune complexes then deposit in the mesangium after passing through larger fenestrae in glomerular capillaries (36, 72–74). This postulate is consistent with observations that, in animals, large-molecular-mass immune complexes induce more severe glomerular lesions than do small complexes (75).
Abnormality of IgA1 O-glycans in patients with IgAN was first indicated by an observation of reduced reactivities of IgA1 with jacalin, a lectin-binding galactose-GalNAc disaccharide (46). Based on additional research, defective galactosylation of O-glycans of IgA1 molecules was proposed as an etiopathogenic factor in IgAN (47). Various lectin-binding assays were used to examine the presence of terminal galactose on N-glycans of purified serum IgG and IgA1 and O-glycans of IgA1 and C1 inhibitor (76). Serum IgA1 of patients with IgAN vs. controls had less galactose on GalNAc, whereas the glycosylation of C1 inhibitor did not exhibit this difference in glycosylation. Another study compared O-glycosylation of serum IgA1 and IgD, a second immunoglobulin with O-glycans, in patients with IgAN and healthy controls and found aberrant O-glycosylation only on IgA1 from patients with IgAN (77). Together, these data suggested that patients with IgAN had galactose-deficient O-glycans uniquely on circulatory IgA1 (78). These findings were confirmed using a panel of lectins, including that from Helix aspersa, specific for terminal GalNAc (31). Moreover, Gd-IgA1 was present in complexes with IgG, leading to speculation that formation of these complexes may reduce the rate of elimination of immune complex-bound IgA1 and lead to elevated serum levels of Gd-IgA1 (31, 79). These findings explained why serum levels of IgA1-containing immune complexes of patients with IgAN and HSP with nephritis are higher than those in healthy controls and, furthermore, why IgA1-containing immune complexes frequently contain also IgG (27, 31).
Additional assessments of IgA1 O-glycans used different analytical approaches, including lectins recognizing different O-glycans on intact IgA1 molecules, monosaccharide compositional analysis by gas–liquid chromatography (LC), mass spectrometric analysis of isolated O-glycosylated hinge-region glycopeptides, Edman sequencing, and separation and identification of free O-glycans released from IgA1 (31, 36, 41–44, 47, 70, 78, 80–96). Each technique presented advantages and disadvantages. For example, lectin ELISA allows high-throughput analyses in a quantitative manner (89, 90, 94) but does not provide information on sites of attachment and heterogeneity in the HR, whereas the more cumbersome methods of mass spectrometry will provide molecular-level details.
By the mid-1990s, mass spectrometry became the standard tool for analysis of IgA1 O-glycosylation, revealing variably O-glycosylated HR glycoforms. Two IgA1 HR glycopeptides containing four or five O-glycan chains were identified by MALDI-TOF mass spectrometry (97). Later analyses used normal serum IgA1 O-glycopeptides (98, 99), pooled serum of patients with IgAN (100), IgA1 isolated from pooled renal biopsies (56), and tonsillar IgA1 (101). Mass spectrometric analysis showed differences in HR O-glycopeptides of IgA1 from patients with IgAN vs. healthy controls (usually serum IgA1), consistent with less galactosylation in patients with IgAN (56, 100, 101). IgA-specific proteases that released IgA1 HR fragments of different lengths provided new tools for generating IgA1 HR O-glycopeptides for analysis (84). A method for direct localization of sites of O-glycan attachment in IgA1 myeloma protein was developed by the use of electron capture dissociation (ECD) tandem mass spectrometry (MS/MS) (102). For the first time, individual sites of O-glycan attachment were directly identified for individual IgA1 HR glycoforms. These data confirmed Thr225, Thr228, Ser230, Ser232, and Thr236 as sites of glycan attachment in a single IgA1 HR O-glycoform with five O-glycans and Thr225, Thr228, Ser230, and Ser232 as the sites of glycan attachment in two HR O-glycoforms with four O-glycans (102). The ability to localize all sites of glycosylation in a single IgA1 HR species expanded the possibilities of defining the heterogeneity and aberrant glycosylation of IgA1 from patients with IgAN.
Renfrow et al. pursued the O-glycan analysis of three distinct IgA1 myeloma proteins using reversed-phase LC separation of IgA1 O-glycopeptides and ECD fragmentation of a larger IgA1 HR tryptic fragment and the second fragment released by IgA-specific proteases (103), demonstrating the utility of high-resolution mass spectrometry. In 2010, they reported the complete localization of all sites of O-glycosylation in the six most abundant IgA1 O-glycoforms of an IgA1 myeloma protein (104). Three distinct IgA1 HR proteolytic fragments were analyzed, and the pattern of glycopeptides for each proteolytic fragment was assigned a relative distribution based on a label-free relative quantitative method developed for N-glycopeptides (105). Specific sites of galactose deficiency have been expressed as a percentage of the total distribution of all observed O-glycoforms. ECD and a newer ECD-type fragmentation method, electron transfer dissociation (ETD), were used to localize sites of O-glycan attachment with LC–MS/MS (103, 106). In 2012, a new type of heterogeneity was identified, representing IgA1 O-glycopeptide isomers, i.e., equally O-glycosylated IgA1 HRs with different sites of attachment (45), involving Ser230, Thr233, and Thr236 sites. With these 2010 and 2012 studies, the sites of attachment indicated a semi-ordered synthesis of the clustered IgA1 O-glycans and not a series of random attachments. Hopefully, these approaches will elucidate the structural basis of abnormal O-glycosylation of IgA1 in IgAN and provide clues as to whether specific isomers are associated with the clinical expression or course of the disease.
Molecules of monomeric IgA contain two α1 or two α2 chains, linked by inter α-chain disulfide bridges and two κ or two λ chains. A distinguishing feature of polymeric IgA, irrespective of its dimeric or tetrameric form, is the presence of a single molecule of joining (J) chain incorporated into polymeric IgA within IgA-producing cells (107). The role of J chain in the process of polymerization of IgA remains unresolved; polymeric IgA and IgM molecules devoid of J chain have been described [for review, see Ref. (108)]. Human α1 and α2 chains as well as α chains from other species comprise one variable- and three constant-region domains, each containing ~110 amino acids. Although comparable in its general structure to the γ chains of IgG, there are several important structural differences that are characteristic of α chains. These differences include the unique HR between Cα1 and Cα2 domains, the extension of the C terminus of the α over γ chains by 18 amino acids essential for the J chain binding and polymerization, and the glycan moieties characteristic of the α1 and α2 chains (107). All three constant-region domains of α1 and α2 chain have 90–98% primary structure homology; the difference is restricted to the HR and allotype-associated sequences in IgA2 molecules. There are 17 Cys residues that participate in the intradomain and interchain disulfide bridges. In polymeric IgA, the penultimate Cys residue of the α chain tailpiece is involved in the binding of J chain and formation of polymers. This small glycosylated peptide contains ~137 amino acids (107). The major structural difference between α1 and α2 heavy chains occurs in the HR that consists of 26 and 13 amino-acid residues in α1 and α2 chains, respectively. The additional 13 amino-acid residues in IgA1 HR consist of repeated sequences of Pro, Ser, and Thr residues. By its general structure, HR is reminiscent of mucin molecules.
The total circulating pools of IgA1 and IgA2 are 101 ± 26 and 14 ± 4 mg/kg body weight, respectively (109). Approximately 55% of total IgA is in the intravascular compartment; the remainder is in interstitial fluid. These data do not include IgA produced in mucosal tissues and selectively transported into the external secretions (secretory IgA). However, IgA from mucosal tissues contributes only small quantities to the circulatory pool (110).
IgA1 HR is a target of IgA-specific proteases produced by pathogenic bacteria, such as Haemophilus influenzae, Streptococcus pneumoniae, Neisseria meningitides, and Neisseria gonorrhoeae (111). Furthermore, the extended IgA1 HR confers greater flexibility of Fab “arms” (107) and facilitates interactions with antigens. The tailpiece of α chains and of μ chains of IgM contains a Cys residue to which J chain is attached. The presence of J chain in polymeric IgA and IgM is essential for the binding of polymeric immunoglobulin receptor (112). Not all polymeric IgA and IgM molecules contain J chain. For example, hexametric IgM produced in small quantities at the early phase of the immune response is devoid of J chain. Similarly, it appears that various human polymeric IgA myeloma proteins display variable J chain content. J chain is produced not only in plasma cells synthesizing polymeric IgA or IgM but also in IgG-, IgD-, or light-chain-producing multiple myeloma cells from mucosal tissues and bone marrow (107). The presence of J chain-containing polymeric IgA in circulating immune complexes and in mesangial deposits of IgAN patients suggests a mucosal origin of IgA1; however, the possibility that such polymeric IgA1 molecules are produced in the bone marrow of IgAN patients has been proposed (113). Further studies are needed to address this point.
Several investigators noted the effect of O-glycan heterogeneity on the propensity of some IgA1 glycoforms to aggregate under laboratory conditions using elevated temperatures (114). Moreover, non-galactosylated glycoforms of IgA1 exhibited binding with proteins of extracellular matrix (115). The authors of these studies suggested that IgA1 O-glycans played a protective role against aggregation and adhesion and that the underglycosylation of the IgA1 molecule may be involved in the non-immunologic glomerular accumulation of IgA1. It is not clear whether glomerular deposition of IgA1 that is not bound in complexes would lead to pathological consequences, i.e., mesangial proliferation and matrix expansion, and under what circumstances, and whether such a mechanism may play a role in the postulated heterogeneity of IgAN (116).
Immunohistochemical studies and results from short-term culture experiments of human tissues supported the above-described distribution of the form of IgA (polymeric or monomeric) and the isotype (IgA1 or IgA2) in several fluids that parallels the distribution of cells in various tissues and organs. Measurements of antigen-specific antibodies in individual external secretions mirrored the distribution of IgA1- or IgA2-producing cells in the corresponding mucosal tissues (107, 117). Furthermore, IgA-producing cells abundant in mucosal tissues secrete polymeric IgA that is efficiently transported through epithelial cells by a receptor-mediated mechanism into external secretions (112). Nevertheless, the contribution and location of polymeric IgA-producing cells to the circulating pool of IgA remain to be determined (107, 109).
The tissue origin of polymeric Gd-IgA1 bound in the circulating immune complexes and in mesangial immunodeposits of patients with IgAN is unclear. On the one hand, it is assumed that because of its polymeric character, Gd-IgA1 originates in mucosal tissues of the respiratory and/or gastrointestinal tracts. On the other hand, it is possible that the IgA-producing cells in the bone marrow may secrete, in addition to the dominant monomeric IgA1, also small quantities of polymeric IgA1 as a consequence of infection. The initial IgA responses to an infection or immunization, irrespective of the systemic or mucosal route of vaccination, are dominated by polymeric IgA in serum and secretions [for review, see Ref. (118–120)].
Studies of the association of naturally occurring or immunization-induced serum and secretory IgA antibodies to different types of antigens provided several highly relevant findings (107, 109). Antibodies specific for protein-, glycoprotein-, and virus-derived antigens (e.g., influenza and HIV) are dominantly of the IgA1 subclass; in contrast, antibodies against polysaccharides, lipopolysaccharides, and teichoic acid are associated with the IgA2 subclass. Notably, systemic or mucosal immunization with influenza virus vaccine induces a mainly polymeric IgA1 response in serum; polymeric IgA2-dominant responses are detected in individuals immunized with polysaccharide vaccines (107, 109). Thus, the type of the antigen substantially influences the IgA subclass-associated response. Some studies showed that patients with IgAN had reduced in IgA1 responses to challenges with some antigens (121, 122), whereas another study observed differential O-glycosylation of IgA1 antibodies against mucosal vs. systemic antigens (120). Moreover, some investigators have found secretory IgA1 (with secretory component) in renal deposits (123, 124) or polymeric IgA1 (125), suggesting that this IgA1 was generated during a mucosal immune response (126). It is not clear whether secretory IgA1, regardless of its O-glycosylation pattern, may be a major driver of the pathogenesis of IgAN.
The macroscopic hematuria associated with upper respiratory tract infections in patients with IgAN suggests that the synpharyngitic hematuria may reflect an inflammatory environment conducive to driving renal complications (127). IgA produced in the mucosal compartments is polymeric, the predominant form of Gd-IgA1 (128). Thus, circulatory Gd-IgA1 may originate from mucosal tissues, and local infections may accentuate Gd-IgA1 production. This concept is the subject of ongoing research that may elucidate mechanisms, which are responsible for increased levels of circulatory Gd-IgA1.
IgA1 production at mucosal tissues from resident IgA1-producing cells serves several functions; in this review, we will focus on mechanisms of aberrant IgA1 O-glycosylation in patients with IgAN. The Japanese Society of Nephrology now recommends tonsillectomy for treatment of IgAN, as tonsillectomy in combination with glucocorticoid pulse therapy improved renal outcomes in many patients with IgAN and macroscopic hematuria (129). However, a benefit of tonsillectomy on disease progression was not found in European cohorts (130), which could be due to genetic differences or early screening that is routinely done in Japan. Recent data suggest that B cells isolated from tonsils of patients with IgAN exhibit increased IL-4 and IFNγ production upon exposure to hemolytic streptococci and lipopolysaccharides when compared to tonsillar B cells from controls (131). Increased numbers of memory B cells were found in tonsils (5.7 vs. 1.8%) and peripheral blood (4.9 vs. 0.9%) of IgAN patients compared to controls; this finding correlated with proteinuria (r = 0.81) (132). Moreover, patients with IgAN after tonsillectomy had fewer peripheral blood memory B cells (4.9% regressed to 1.1%) (132). These studies highlight the role of inflammation and the importance of the mucosal-circulatory connection in patients with IgAN.
Other studies revealed increases in TLR9 and B-cell-activating factor (BAFF) mRNA expression in peripheral blood mononuclear cells as well as increased serum levels of BAFF protein (133). Mice overexpressing human BAFF develop a commensal microbiota-dependent IgA-associated nephropathy (134, 135). BAFF induces class-switch recombination in B cells and may drive the circulatory IgA1 levels in patients with IgAN (136). Moreover, more L-selectin was found in B and T cells derived from the circulation of patients with IgAN (137, 138). Together, these data suggest a proinflammatory state of B cells in patients with IgAN. This finding corroborates in vitro data, showing that certain cytokines can enhance production of Gd-IgA1 (139).
To study molecular mechanisms of production of Gd-IgA1, peripheral blood mononuclear cells and tonsillar B cells were isolated from IgAN patients and controls, and Epstein–Barr virus (EBV)-immortalized cells were generated. From these mixed cell lines, IgA1-producing cells were isolated through limiting dilution subcloning. Analysis of IgA1 secreted by these cell lines derived from blood of patients with IgAN showed enhanced production of Gd-IgA1 when compared to controls. The degree of galactose deficiency of IgA1 secreted by EBV-immortalized B cells corresponded to the serum Gd-IgA1 levels from the corresponding donors, indicating that glycosylation of IgA1 and Gd-IgA1 production had not been altered by EBV immortalization (140). These cell lines provide a new tool for studies of biosynthesis of Gd-IgA1 (93).
As noted above, patients with IgAN often exhibit macroscopic hematuria associated with mucosal infections. These infections may be associated with increased production of IgA and Gd-IgA1 (141). The exacerbation of kidney damage associated with acute infection/inflammation in patients with IgAN may be transient or permanent, and it indicates a connection with activated immune system (127). Increased levels of markers of inflammation, such as IL-6 and soluble vascular cell adhesion molecule-1 (sVCAM-1), have been found in the blood of patients with IgAN (142, 143). Some proinflammatory cytokines, such as IL-6 and leukemia inhibitory factor (LIF), increase production of Gd-IgA1 in B cells from patients but not controls (139). In IgA1-producing cells from patients with IgAN vs. healthy controls, IL-6 showed increased and prolonged activation of STAT3 (144). As STAT3 is the canonical transcription factor of IL-6 and other cytokines, changes in signaling and transcription driven by STAT3 may have an important role in Gd-IgA1 production (145).
Production of Gd-IgA1 in patients with IgAN has been linked to aberrant expression and activities of specific glycosylation enzymes in the Golgi apparatus for normal O-glycosylation (146, 147). Galactose deficiency of IgA1 O-glycans can be due to a reduced rate of galactosylation or premature sialylation that would prevent addition of galactose. Further dysregulation by IL-6 of the corresponding enzymes (see Mechanisms and Pathways Involved in Production of Aberrantly Glycosylated IgA1 for details) involved in these processes was observed (139), but the detailed mechanism that leads to these changes is unknown.
In addition to cytokines, it is also possible that other B-cell-stimulating factors may contribute to increased production of Gd-IgA1 (148). These factors, such as BAFF, may drive IgA class switching, B-cell differentiation and antibody production, and cellular proliferation (149). Such signaling ligands may share similar receptors (Figure 4). Several GWAS have implicated a locus encompassing the APRIL gene (TNFSF13) in IgAN, and serum levels of the expressed ligand were elevated in patients with IgAN (136, 150). Increased amounts of BAFF are also found in sera and tonsillar tissue of some patients with IgAN (151). Mice with BAFF overexpression exhibited a microbiota-dependent IgA-associated glomerulonephritis, further implicating B-cell activation in IgA glomerular deposition (135).
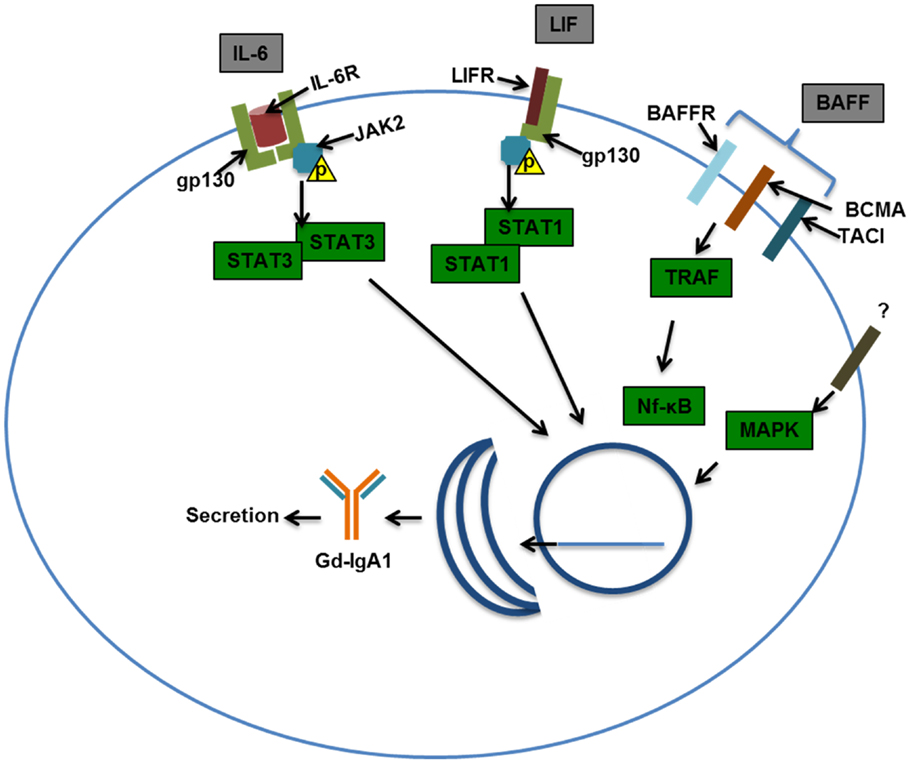
Figure 4. Examples of signaling pathways that may affect production of galactose-deficient IgA1. Interleukin-6 (IL-6) binds to the IL-6 receptor (IL-6R) and through co-receptor gp130 (glycoprotein 130) activates Janus kinase 2 (JAK2), which in turn phosphorylates signal transducer and activator transcription 3 (STAT3), leading to its dimerization and nuclear translocation. Leukemia inhibitory factor (LIF) binds gp130 and LIF receptor (LIFR) that also activates JAK2, leading to STAT1/3 activation and nuclear translocation. B-cell-activating factor (BAFF) can bind multiple receptors, BAFF receptor (BR), B-cell maturation antigen (BCMA), and transmembrane activator and calcium-modulating/cyclophilin ligand protein (TACI). Activation of TNF-receptor-associated factor (TRAF) by TACI, BAFFR, and BCMA leads to NF-kB activation and nuclear translocation. Mitogen-activated protein kinase (MAPK) activation has been shown to affect O-glycosylation and thus was included in this scheme. Nuclear translocation of the STATs and NF-kB may be important in driving production of Gd-IgA1 in IgA1-producing cells and/or maintaining viability/proliferation of these cells in patients with IgAN.
Production of Gd-IgA1 by IgA1-producing cells is enhanced in patients with IgAN, possibly through altered signaling of pathways involving STAT3. Contributing factors related to abnormal signaling could be genetic, as indicated by the increased serum levels of Gd-IgA1 in asymptomatic relatives of patients with IgAN (152). Environmental factors play a role as well, as there is a connection between infection/inflammation and disease activity. Future research is needed to define the interaction between environmental and genetic factors, and how it relates to signaling changes in IgA1-producing cells.
IgA from the circulation is primarily catabolized in the liver (107, 110, 112). IgA bound to ASGP-R expressed on hepatocytes in the presence of Ca2+ is internalized, and the IgA-containing vesicles fuse with lysosomes resulting in intracellular degradation (63, 64, 153). Experiments with human IgA1 and IgA2 myeloma proteins in their monomeric or polymeric forms (62) demonstrated that in monkeys, the liver has the highest uptake of IgA. Hepatocytes compared to non-parenchymal cells were more active in the catabolism of IgA (62). Only small quantities of IgA were catabolized in the kidneys, skin, and spleen. The importance of the ASGP-R in IgA catabolism was further confirmed using a human hepatoma cell line (64). Of note, autoantibodies specific for ASGP-R have been observed in patients with autoimmune hepatitis (154). The marked species-dependent differences in the structure, transport, metabolism, and catabolism of IgA of different molecular forms must be taken into consideration in animal models of IgAN as well as the fact that different molecular dimensions of monomeric and polymeric IgA and polymeric IgA-containing immune complexes affect the catabolism and distribution of free or complexed IgA.
Normal serum IgA1 O-glycans consist predominantly of galactose-β1-3GalNAc dissaccharide, also known as T antigen, and its mono- or di-sialylated forms [NeuAcα2-3-galactose-β1-3GalNAc and NeuAcα2-3-galactose-β1-3(NeuAcα2-6)GalNAc, commonly described as sialyl-T (ST) antigen] (Figure 3, left panel) (44, 45). O-glycosylation of IgA1 HR involves multiple glycosyltransferases that add one monosaccharide at a time in a stepwise manner to a growing O-glycan chain. O-glycosylation of IgA1 takes place in the Golgi apparatus (93). O-glycosylation is initiated by attachment of GalNAc to Ser/Thr residues by the activity of a UDP-GalNAc:polypeptide GalNAc-transferase family (ppGalNAc-Ts), consisting of 20 members in humans (155, 156). Dominant role during IgA1 HR O-glycosylation was attributed to the ubiquitous GalNAc-T2 (157). Further work indicates that GalNAc-T1 and GalNAc-T11 can also initiate O-glycosylation of IgA1 (158). Recently, we compared transcript levels of all known human GalNAc-Ts in IgA1-producing cells from IgAN patients and disease controls and identified significant differences for only GalNAc-T14 (159, 160). Preliminary data indicate that GalNAc-T14 could attach GalNAc to IgA1 HR and thus may contribute to the aberrant glycosylation of IgA1 (161). Interestingly, GalNAc-T14 is structurally the closest relative of GalNAc-T2 (162). Overexpression of GalNAc-T14 in IgA1-producing cells from IgAN patients could contribute to the increase in the overall number of O-glycans on IgA1 in IgAN patients (163).
After the initial addition of GalNAc to Ser/Thr residues, galactose is added by UDP-galactose:GalNAc-α-Ser/Thr β1,3-galactosyltransferase (C1GalT1) (164). A deficiency of C1GalT1 results in truncation of O-glycans (165). The biosynthesis of active C1GalT1 depends on molecular chaperone Cosmc (166, 167). Cosmc mutation(s) is associated with the expression of the terminal GalNAc and sialylated GalNAc (also called Tn and STn antigens, respectively) in various neoplastic lesions and Tn syndrome (167–169) but not in IgAN (170). Decreased levels of C1GalT1 transcript and protein activity were detected in subcloned Gd-IgA1-producing cells from IgAN patients (93). C1GalT1 deficiency is further accentuated after exposing the IgA1-producing cells to IL-6 (139). Together with the constitutionally increased activity of GalNAc-T14, IgA1-producing cells could, under local inflammatory conditions, insufficiently galactosylate GalNAc residues attached in the IgA1 HR.
Galactose-β1,3GalNAc structures are subsequently modified by attaching sialic acid from CMP-N-acetylneuraminic acid (CMP-NeuAc) to galactose residues by the activity of galactose-β1,3GalNAc α2,3-sialyltransferase (ST3Gal) and/or to the GalNAc residues by activity of an α2,6-sialyltransferase (ST6GalNAc) (171, 172). Neuraminidase-driven in vitro removal of sialic acid from IgA1 produced by EBV-immortalized cells from IgAN patients (93) and nasopharyngeal carcinoma (Dakiki cells) (146) enhanced reactivity with GalNAc-specific lectin (HAA). These studies suggested that some Tn O-glycans on IgA1 are capped with sialic acid (sialyl-Tn antigens) (93, 146, 173). The analysis of all known human ST6GalNAc transcripts (ST6GALNAC1–6) performed by real-time RT-PCR showed that ST6GalNAc-I, an enzyme described to be responsible for sialylation of Tn antigens, is not expressed in IgA1-producing cells; however, abundant transcription of ST6GALNAC2 was detected. Other ST6GALNAC genes were transcribed either in similar extent between Gd-IgA1- and normal IgA1-producing cells (ST6GALNAC3, ST6GALNAC4, and ST6GALNAC6) or were not detectable (ST6GALNAC5) (93, 146, 174). Recombinant human ST6GalNAc-II can sialylate terminal GalNAc of IgA1 in vitro (174). Involvement of ST6GalNAcII in sialylation of Tn antigens on IgA1 HR was confirmed by reduced HAA reactivity with IgA1 secreted from Gd-IgA1-producing cells lines, in which ST6GalNAc-II activity was suppressed by siRNA-driven ST6GALNAC2 knock-down (139). Subsequent in vitro experiments, in which α2,6-sialyltransferase and β1,3-galactosyltransferase enzymes were obtained as a Golgi extract from Gd-IgA1-producing cells, confirmed that sialylation of terminal GalNAc blocks effective galactosylation (139). Thus, premature sialylation, associated with increased transcriptional activity of ST6GALNAC2 in Gd-IgA1-producing cells, may contribute to Gd-IgA1 production in IgAN. Sialyltransferases are localized predominantly in trans-Golgi compartments, but the observation that galactose-deficient sialylated GalNAc-containing IgA1 is present throughout the Golgi (93) suggested a possible abnormal relocalization of sialyltransferases toward cis-Golgi. This abnormality may contribute to the galactose deficiency of IgA1 O-glycans. However, studies of subcellular localization of individual enzymes are needed to confirm this hypothesis.
In summary, Gd-IgA1-producing cells from IgAN patients have elevated expression GalNAc-T14 and ST6GalNAc-II, and decreased expression of C1GalT1 and Cosmc (93, 159). As macroscopic hematuria in IgAN patients often coincides with mucosal infections, inflammation may enhance galactose deficiency of IgA1. Indeed, IL-6 and, to a lesser extent, IL-4 accentuated galactose deficiency of IgA1 secreted by cell lines from IgAN patients (139). Stimulation of cells from IgAN patients with IL-6 increased α2,6-sialyltransferase activity and decreased activity of C1GalT1, whereas IL-4 only reduced the activity of C1GalT1 (139). These experiments indicate that IgA1-producing cells from IgAN patients accentuate production of Gd-IgA1 upon stimulation with IL-6. Aberrancies in JAK–STAT signaling pathways may be involved in these processes (144).
Comprehensive studies of the glycosylation abnormalities of IgA1 offered a potential phenotypic biomarker for IgAN, Gd-IgA1 (61, 69, 70, 88, 89, 175). A quantitative lectin-binding assay enabled assessment of the inheritance of Gd-IgA1 in familial and sporadic forms of IgAN (152). Elevated serum levels of Gd-IgA1 were found in most patients with IgAN, as well as many of their first-degree relatives, whereas levels in spouses were similar to those of healthy controls. Segregation analysis of Gd-IgA1 levels suggested inheritance of a major dominant gene with an additional polygenic component. The inheritance of Gd-IgA1 serum levels has been confirmed in patients with familial and sporadic IgAN (52, 176, 177), and in pediatric patients with IgAN and HSP with nephritis (178). Thus, aberrant IgA1 glycosylation is a common inherited defect that provides a unifying link in the pathogenesis of HSP with nephritis and IgAN in many populations worldwide (93, 179).
It is now well accepted that the circulation of patients with IgAN contains immune complexes consisting of Gd-IgA1 [for reviews, see Ref. (61, 180)]. Initial analyses showed that Gd-IgA1 was predominantly in large-molecular-mass fractions of serum and was associated with IgG, thus indicating a possibility that Gd-IgA1 was bound by IgG in an immune complex (31). A follow-up study confirmed that circulating immune complexes in patients with IgAN consist of polymeric Gd-IgA1 bound by IgG antibodies specific for GalNAc residues in the hinge-region O-glycans of IgA1 heavy chains (49).
Elevated serum levels of Gd-IgA1 are found not only in patients with IgAN but also in patients with HSP with nephritis (57, 88, 178). It is now proposed that the pathology of HSP with nephritis and IgAN is driven by glomerular deposition of large immune complexes from the circulation (6, 18). Importantly, patients with HSP without nephritis have only IgA–IgA immune complexes, whereas patients with HSP with nephritis have IgA–IgA and IgA–IgG immune complexes (58).
Autoantibodies forming complexes with Gd-IgA1 in the blood of IgAN patients are predominantly of the IgG isotype (31, 181). These autoantibodies recognize HR of IgA1 with terminal GalNAc residues (31, 182). This conclusion was based on several experiments. Binding of IgG autoantibodies from serum samples of IgAN patients was tested using ELISA with several antigens: enzymatically desialylated and degalactosylated IgA1 myeloma protein (dd-IgA1), Fab fragment of Gd-IgA1 containing part of the HR with O-glycans (Fab-IgA1), synthetic HR peptide linked to bovine albumin (HR-BSA), and a synthetic HR glycopeptide with three GalNAc residues linked to BSA (HR-GalNAc-BSA). Binding to dd-IgA1 and Fab-IgA1 was significantly higher for IgG from sera of patients with IgAN than that for IgG from sera of healthy controls. IgGs from IgAN patients recognized HR-GalNAc-BSA but not HR-BSA. These experiments thus confirmed that IgG autoantibodies from IgAN patients recognize terminal GalNAc on IgA1 HR (140, 182).
To better understand at a molecular level the nature of IgG autoantibodies specific for Gd-IgA1, panels of monoclonal IgG autoantibodies were cloned and characterized. EBV-immortalized IgG-secreting lymphocytes derived from peripheral blood of patients with IgAN and healthy controls were generated and, using limiting dilutions, single-cell clones producing IgG specific for Gd-IgA1 were isolated (182). Using single-cell RT-PCR, variable regions of heavy and light chains were amplified, cloned, and sequenced. Selected paired variable regions of heavy and light chains were also cloned and expressed as recombinant IgG, and binding to Gd-IgA1 was assessed. These experiments confirmed and extended previous observations that IgG autoantibodies bound to Gd-IgA1 and that such binding required terminal GalNAc. Moreover, sequence analysis of variable regions of heavy chains of IgG autoantibodies and comparison of the binding of the IgG to Gd-IgA1 pointed out some interesting features. For example, complementarity determining region 3 (CDR3) of variable region of heavy chain tended to be longer in IgG autoantibodies from patients with IgAN compared to that of IgG from healthy controls. Furthermore, a Ser residue was in the third position of CDR3 of autoantibodies in six of the seven studied patients with IgAN. In contrast, IgG from six healthy controls had Ala in that position (182). These observations thus implicated Ser residue in CDR3 in binding of Gd-IgA1. Recombinant IgG from a patient with IgAN was generated by site-directed mutagenesis to change the Ser residue in the third position of CDR3 of the heavy chains to Ala. This mutation reduced binding to Gd-IgA1. Conversely, introducing Ser residue in the third position of CDR3 of the heavy chains of IgG from a healthy control increased the binding of the IgG to Gd-IgA1 (182). Recent study has shown that Ser in CDR3 in the heavy chains of IgG autoantibodies originates from somatic mutations rather than from rare variants of VH genes (183).
It has been observed that levels of IgA1-containing immune complexes in patients with IgAN correlated with clinical and histological activity (27). It was later clarified that such complexes consist of Gd-IgA1 bound by antiglycan antibodies (49, 184, 185). To study biological activities of IgA1-containing immune complexes, a model of cultured primary human mesangial cells has been used (186). With this approach, it was shown that Gd-IgA1-containing immune complexes from patients with IgAN bound to the cells more efficiently than did uncomplexed IgA1 or immune complexes from healthy controls (53, 91). Moreover, large-molecular-mass complexes from sera of patients with IgAN stimulated cellular proliferation and production of cytokines (e.g., IL-6 and TGF-β) and components of extracellular matrix (50–52, 61, 91, 95, 187–192). The role of IgA1-containing immune complexes in these activities is confirmed by the fact that IgA1-depleted fractions are devoid of such stimulatory activities (50, 91, 95). Consistent with this finding, when sera of IgAN patients are supplemented with small quantities of polymeric Gd-IgA1, new IgA1-containing immune complexes are formed and, thus, the amount of stimulatory large-molecular-mass immune complexes increases (50, 91). These complexes contain IgG in addition to IgA1, just as do the native complexes (61, 95). In contrast, uncomplexed polymeric Gd-IgA1 or smaller immune complexes do not induce proliferation of cultured primary human mesangial cells.
Supplementation of Gd-IgA1 to serum from IgAN patients formed pathogenic immune complexes (50, 95), indicating an excess of antiglycan antibodies against Gd-IgA1. Following this approach, a new protocol for in vitro production of biologically active IgA1-containing immune complexes was developed. Cord blood serum, known to contain IgG but no other immunoglobulins, with high levels of antiglycan IgG was used to bind to Gd-IgA1 myeloma proteins to form immune complexes. Formation of biologically active immune complexes that stimulated cellular proliferation of cultured primary human mesangial cells required Gd-IgA1, antiglycan IgG antibody, and a heat-sensitive serum factor (193).
This model of formation of engineered immune complexes was later enhanced by using recombinant IgG specific for Gd-IgA1 from a patient with IgAN (182) with serum as the source of other factor(s) (193). Notably, these engineered immune complexes stimulate signaling in cultured primary human mesangial cells and increase cellular proliferation in a similar fashion as with native IgA1-containing complexes in sera of patients with IgAN (194, 195).
In IgAN, complement C3 frequently colocalizes with IgA in mesangial immunodeposits (2, 6, 196) and is also present in IgA1-containing circulating immune complexes of patients with IgAN (28). Moreover, a deletion of CFHR1 and CFHR3 genes encoding complement factor H-related factors 1 and 3, the factors involved in the regulation of factor H (197–199), protects against the occurrence of IgAN (8, 150, 179). These observations underscore the contribution of the complement alternative pathway (AP) to pathophysiology of the disease [recently reviewed Ref. (200)]. Using the abovementioned model of engineered immune complexes and targeted proteomic and immunologic analyses, complement C3 products associated with these Gd-IgA1–rIgG complexes were studied (201). Proteomic analysis revealed C3 α and β chain elements in the active large-molecular-mass Gd-IgA1–rIgG immune complexes and only low amounts of β chain in corresponding fractions in a negative control (serum only, not supplemented with Gd-IgA1 or rIgG). Amino-acid sequence by mass spectrometric analysis of specific bands from SDS-PAGE identified iC3b, C3c, and C3dg in the Gd-IgA1–rIgG immune complexes (201). Presence of these C3 fragments was confirmed by immunoblotting. Thus, biologically active Gd-IgA1–rIgG complexes activate complement C3 in vitro and associate with C3 degradation fragments. The observed C3 components (iC3b, C3c, and C3d) result from the combined action of factors I and H, suggesting a critical role of regulators in activation of the complement AP in IgAN (200). Thus, (1) addition of serum to Gd-IgA1 bound by anti-Gd-IgA1–IgG autoantibody results in dose-dependent formation of pathogenic immune complexes that activate cultured human mesangial cells and (2) stimulatory immune complexes contain activated C3 products. The relatively small size of these C3 fragments in the nephritogenic immune complexes (molecular mass ~800 kDa) and the association of these C3 fragments with Gd-IgA1–IgG immune complexes suggest direct binding of C3 and activation of the alternative complement pathway in this in vitro model of IgAN immune complexes (201).
The role of complement in the pathogenesis of IgAN has been suspected since 1980s, based on the commonly observed mesangial codeposition of C3 with IgA (2, 200, 202).
Complement can be activated through three pathways (Figure 5). The classical pathway (CP) is initiated by the recognition of some IgG subclasses (IgG1 and IgG3, and IgG2 to a lesser extent) and IgM by C1q. C1q then binds C1r and C1s and cleaves successively C2 and C4 to form C4b2a complex, a C3 convertase. The AP is activated continuously by spontaneous hydrolysis of C3, exposing an unstable thioester bond and changing C3 conformation to allow its interaction with complement factor B, forming C3(H2O)Bb, which cleaves C3 into C3a and C3b (Figure 6) (203). This process is tightly controlled by AP regulatory proteins, such as complement factors I and H, and DAF. Without these regulators, especially on an activating surface (such as a bacterial cell-wall glycan), an amplification loop develops in the presence of factor D and properdin, leading to the accumulation of C3bBb, the AP C3 convertase. The third pathway, the “lectin pathway,” is activated by some sugar moieties, such as mannose or glucosamine on the surface of bacterial cell walls, through interaction with mannan-binding lectin (MBL). The activation process is thereafter similar to the CP to generate C4b2a. Finally, C3 convertase cleaves C3 into C3b that is added to the complex to form C5 convertase. This complex cleaves C5 into C5a and C5b. The latter product binds C6, C7, C8, and C9 (C5b–9) to form the terminal complement complex that can insert into cell-membrane lipid bilayers. This final process can lead to cell lysis or, more commonly in nucleated cells, cellular stress (sublytic complement attack) (204, 205).
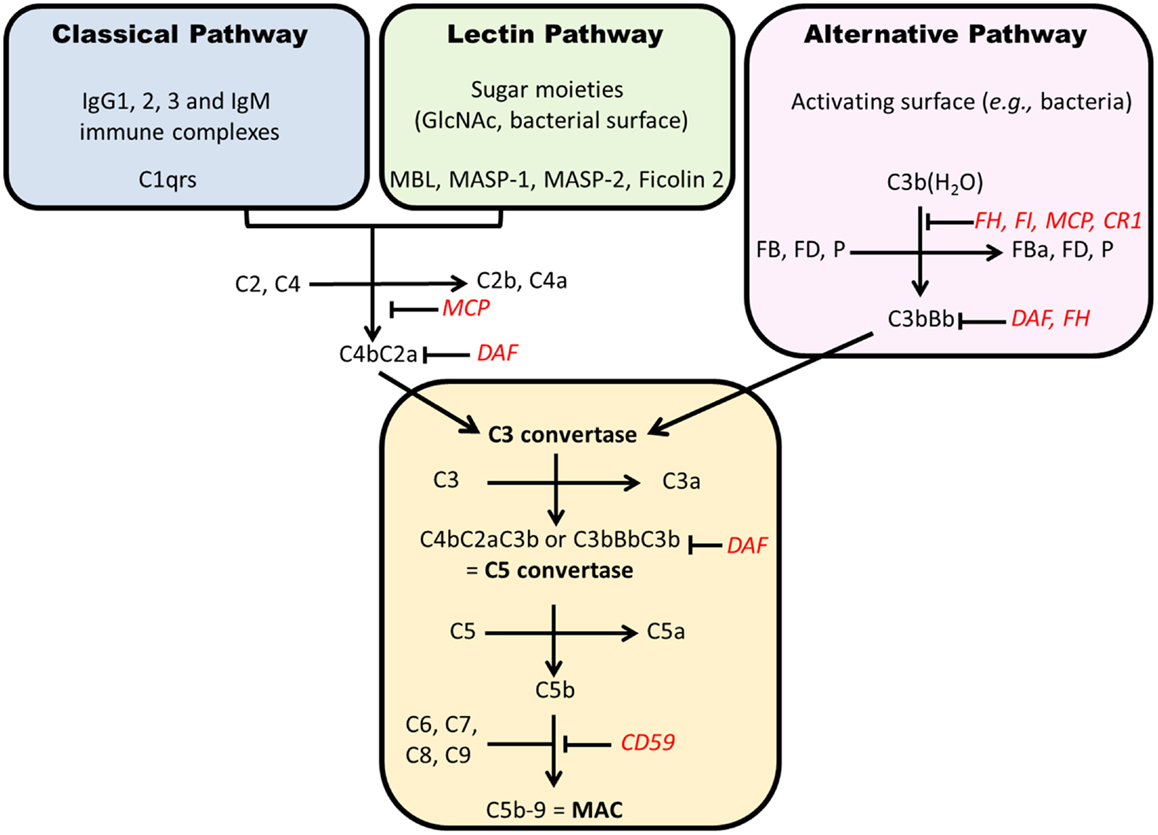
Figure 5. Complement activation pathways. Each pathway results in formation of a C3 convertase that, after addition of C3b, becomes a C5 convertase. The generation of C5b starts the formation of membrane attack complex (C5b–9). Regulatory factors are in red. CR1, complement receptor 1; FD, factor D; MAC, membrane attack complex; MCP, membrane cofactor protein; P, properdin; DAF, decay accelerating factor; MBL, mannan-binding lectin; MASP, MBL-associated serine proteases.
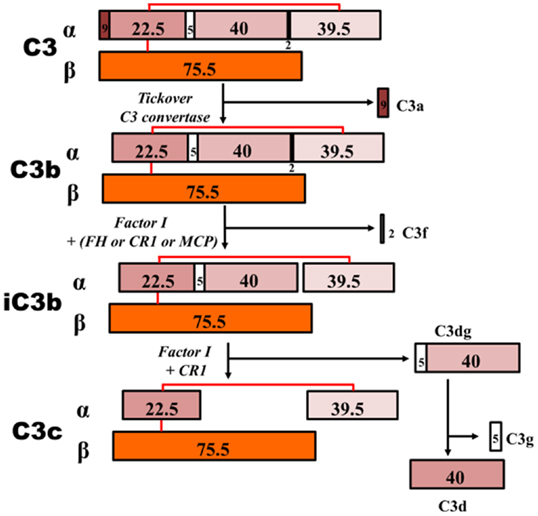
Figure 6. C3 proteolytic cascade. The hydrolysis of C3 leads to release of activation products C3a – an anaphylatoxin – and C3b. C3b binds activating surfaces, such as a bacterial cell wall, triggering the alternative pathway cascade. This activation is controlled by regulator molecules, such as FI, FH, and complement receptor 1 (CR1), that degrade C3b into products that cannot contribute to the formation of the C5 convertase (iC3b, C3c, C3dg, and C3d). Detection of these inactive breakdown products is considered evidence of activation of C3. The numbers, in kilodaltons, represent the molecular masses of the corresponding polypeptides. MCP, membrane cofactor protein.
Alternative pathway is considered an important player in the pathogenesis of IgAN. First, key AP components are codeposited with IgA in the glomerular mesangium. C3 is detected in the immunodeposits in kidney tissue in up to 90% of cases (206–208) as well as properdin (75–100%) and factor H (30–90%) (202, 209, 210). Plasma concentrations of C3 inactivation products (iC3b and C3d) are elevated, reflecting increased production of C3b (211–213). IgA can activate AP in vitro, especially while immobilized on a surface in a polymeric form (214, 215).
More recently, GWAS (150, 179) identified a single nucleotide polymorphism (SNP) at position 1q32 in factor H gene that was strongly protective against IgAN (odds ratio 0.74 for one allele and 0.55 for two alleles). This SNP was in total linkage disequilibrium with the large deletion of complement factor H-related genes 1 and 3 (CFHR1 and CFHR3), positioned downstream of factor H gene. The copy number association study confirmed the protective impact of this deletion on the risk to develop IgAN. Products of these genes are also AP regulatory proteins that can bind C3 in a similar way as with factor H (216). However, these proteins are less efficient than factor H to regulate AP, such that their absence could lead to a stronger factor H-mediated AP inhibition (198). A recent study has shown that CFHR1 and CFHR3 deletion was associated with higher serum levels of factor H and C3, lower serum C3a levels, and less C3 mesangial deposition in Chinese patients with IgAN (199).
The lectin pathway has been examined as a potential mediator for IgAN severity and/or progression of the disease (124, 217, 218). In vitro activation of this pathway by polymeric immobilized IgA certainly occurs (219). Several studies have confirmed the negative prognostic impact of the mesangial codeposition of lectin pathway elements, including MBL, MBL-associated serine proteases (MASP-1 and MASP-2), L-ficolin, C4d, and C4-binding protein (220, 221).
The CP is not considered to be a significant player in IgAN, as IgA cannot activate it and actually hinders its activation by IgG (215). C1q is usually missing in IgAN kidney biopsies (<10%, as trace) (207, 222), and the presence of C4 is more representative of lectin pathway activation (220).
The terminal complement complex is commonly codeposited with IgA (210, 223), and its urinary excretion is increased (224). Sublytic C5b–9 can induce mesangial stress, potentially leading to the elevated production of fibronectin, TGF-β, and IL-6 (205, 225). Podocytes can also be severely affected by C5b–9 that can cause cell injury (204, 226).
The elevated levels of plasma C3 breakdown products in IgAN patients suggest a soluble-phase activation of the AP. Similarly, a model of mixed IgA–IgG complexes supported this conclusion and indicated that C3 activation required IgG (227). Recently, proteomic analyses of patients’ circulating immune complexes, as well as engineered in vitro complexes (formed with polymeric Gd-IgA1, antiglycan IgG, and IgA/IgG-depleted normal serum), revealed cleavage products in high-molecular-mass fractions isolated by size-exclusion chromatography (201). Thus, IgAN immune complexes can act as a surface for AP activation, leading to cleavage of C3 into C3b and thereafter to factor I-dependent inactivated C3 products (iC3b, C3c, C3d, and C3dg).
C3 glomerulonephritis illustrates that AP activation leading to mesangial deposition of C3 products can induce a mesangioproliferative disorder by itself, without significant deposition of immunoglobulins (228). Mesangial cells are potent players in complement-driven glomerular inflammation. They produce factor H and, under inflammatory conditions (IL-1 and TNF-α), express C3 (229). Mesangial cells can express C3 after stimulation by Gd-IgA1-containing immune complexes (230). C3a, an anaphylatoxin produced by the cleavage of C3, can induce cultured human mesangial cells to switch to a secretory phenotype that leads to increased production of mesangial extracellular matrix elements (231).
Complement consumption and deposition in patients with kidney disease can be assessed with serum, urine, and kidney biopsy specimens. A decreased serum C3 level has been proposed as a disease activity biomarker in several studies from Asia. Serum IgA/C3 ratio has also been associated with IgAN severity (232, 233). A European pediatric study showed a positive correlation of IgA/C3 ratio with clinical- and Oxford classification-based kidney tissue injury (234). Whether plasma factor H level could be a reliable disease activity biomarker remains uncertain, as findings of other studies were inconsistent (235, 236).
Urinary excretion of complement elements has also been evaluated, mostly in Asia. Two studies showed greater excretion of factor H (237) and C5b–9 (224) compared to healthy controls, but without a disease-control group with proteinuria.
The deposition of complement elements in glomeruli could also be a valuable tool to predict IgAN progression. The intensity of mesangial C3 deposition was associated with worse clinical outcome (238, 239). Finally, activation of lectin pathway leading to C4d deposition in IgAN predicted worse outcomes in three retrospective studies (221, 240, 241).
Size and composition of immune complexes determine biological activities (50, 71, 95, 193). Based on the size, circulating IgA1-containing immune complexes in IgAN patients can be divided into two groups: immune complexes with high molecular mass (>800 kDa) and immune complexes with low molecular mass (≤800 kDa). Notably, the high-molecular-mass complexes activate cultured human mesangial cells, as indicated by cellular proliferation and overproduction of cytokines and components of extracellular matrix (50, 95). In contrast, the low-molecular-mass complexes exhibit an inhibitory effect (95). Circulating immune complexes with higher content of Gd-IgA1 have enhanced capacity to induce proliferation of mesangial cells, whereas complexes without Gd-IgA1 or Gd-IgA1 alone do not have proliferative effects (50). Stimulation of proliferation of mesangial cells by immune complexes containing Gd-IgA1 was confirmed by experiments with in vitro-formed immune complexes (193).
Stimulatory Gd-IgA1-containing complexes induce not only cellular proliferation but also production of laminin, a protein component of extracellular matrix (95). Similarly, production of laminin was increased by stimulation with TGF-β in a murine mesangial-cell model (242). Large-molecular-mass complexes bind to CD71 and activate mitogen-activated protein kinase/extracellular-signal-regulated kinase (MAPK/ERK) pathway (243). This cellular activation alters crosstalk between mesangial cells and podocytes through TNF-α and TGF-β. These cytokines are released from mesangial cells in elevated amounts and induce expression of nephrin, erzin, and podocin in podocytes (191, 192). Furthermore, elevated production of TGF-β could contribute to glomerular fibrosis by enhancing expression of profibrotic genes driving accumulation of extracellular matrix. TGF-β increases expression of profibrotic connective tissue growth factor (CTGF) via sphingosine 1-phosphate receptor 5 (S1P5) on cultured human mesangial cells (244, 245).
It is not known which receptor(s) on mesangial cells plays a key role in binding to Gd-IgA1-containing immune complexes and activation of human mesangial cells. Myeloid IgA Fc receptor (CD89) and ASGP-R are not expressed on human mesangial cells (53, 246–249). Additional details on Fc receptors, including those on mesangial cells, can be found in a recent review with an extensive list of references (250). Currently, it is thought that the main receptor is CD71, known as transferrin receptor. CD71 is highly expressed in glomeruli of IgAN patients, and its localization correlates to deposits of IgA (251, 252). Moreover, studies using mice expressing human IgA1 heavy chain and human CD89 indicated that complexes of IgA1–sCD89 could initiate an autoamplification process involving overexpression of transferrin receptor 1 (TFR1) and transglutaminase 2 (TGase2). Involvement of sCD89–IgA1 complexes and participation of TFR and TGase2 explain an alternative mechanism of mesangial-cell activation (253). Adding to the complexity, other receptor candidates from a family of integrins (integrin α1/β1 and integrin α2/β1) also bind IgA1 on mesangial cells (254).
Taken together, local inflammation, cellular proliferation, and increased production of extracellular matrix components by mesangial cells activated by IgA1-containing complexes considerably impact glomerular function, leading to hematuria and proteinuria. Without disease-specific therapy, many patients progress to end-stage renal disease and require renal replacement therapy.
Small-animal models of IgAN can be very helpful in studies of various aspects of disease pathogenesis or testing efficacy of new therapeutic approaches. However, development of such models for IgAN has been hindered because only humans and hominoid primates have IgA1 with its O-glycans, a pivotal component in the pathogenesis of human disease. For example, mice have only one subclass of IgA and it resembles human IgA2 (255). However, several different models have been developed that may elucidate various specific aspects of IgAN (256) (Table 1).

Table 1. Selected animal models of IgAN.
Spontaneous IgAN models include ddY mice, high-IgA (HIGA) mice, early-onset-grouped ddY mice (257–260), and marmosets’ wasting syndrome. The last model is associated with IgA antigliadin antibodies and IgA-containing circulating immune complexes that deposit in the mesangium (261). The ddY mouse is a model of spontaneous murine IgAN based on development of glomerulonephritis associated with mesangial deposition of IgA with co-deposits of IgG, IgM, and C3 (257). Based on the age of disease onset, ddY mice are categorized as early-onset, late-onset, and quiescent (i.e., no glomerulonephritis) phenotypes and are amenable to genomic analyses (259). GWAS identified four genetic susceptibility loci (D1Mit216, D1Mit16, D9Mit252, and D10Mit86) linked with the early-onset phenotype (259). The HIGA mouse strain was generated by interbreeding ddY mice with high serum IgA levels (258). However, serum IgA levels in HIGA mice were not associated with severity or incidence of disease (259). A more informative mouse model was developed by intercrossing early-onset ddY mice (260). These early-onset-grouped ddY mice develop proteinuria by 8 weeks of age and renal failure at 24 weeks of age. The grouped early-onset ddY mice show severe glomerular and tubulointerstitial lesions, characterized by mesangial proliferation, mesangial matrix expansion, and tubulointerstitial cellular infiltration. This model may provide useful insights into the pathogenesis of disease, to include identifying susceptibility genes, defining the role of IgA polymorphisms and IgA-containing immune complexes, and assessing the gender difference in progression of disease.
Knock-out and transgenic animal models include β1,4-galactosyltransferase-I-deficient mice, human BAFF-transgenic mice, and IgA1-CD89-transgenic mice (135, 253, 262, 263). The β1,4-galactosyltransferase-I-deficient mice show semi-lethality before weaning due to growth retardation and reduced inflammatory responses. The surviving β1,4-galactosyltransferase-I-deficient mice developed similarly as did control mice. However, starting from 10 weeks of age, the β1,4-galactosyltransferase-I-deficient mice developed an IgAN-like disease associated with high serum IgA levels with greater portions of polymeric IgA. Histological examination of kidneys showed IgA deposition, expanded mesangial matrix, and electron-dense deposits in the paramesangial regions.
The model of BAFF-transgenic mice showed high serum IgA levels with increased portions of polymeric IgA and IgA deposition in the glomeruli but only in mice with microbiota (not in mice without microbiota) (135). This finding emphasized the role of microbiota in driving IgA responses in species/individuals with a specific genetic background. Another transgenic model includes mice producing IgA consisting of heavy chains of human IgA1 with murine light chains. In mice with a transgene to produce the soluble fragment of human CD89, circulating IgA–CD89 complexes form (253, 263). These transgenic mice develop mesangial IgA deposits, glomerular and interstitial macrophage infiltration, mesangial matrix expansion, hematuria, and mild proteinuria. However, follow-up studies have raised questions whether CD89 is involved in a similar manner in human IgAN, as mice do not have a homolog of human CD89 (266, 267).
We have recently developed a passive mouse model of IgAN based on injection of SCID mice with preformed immune complexes consisting of human Gd-IgA1 bound by antiglycan antibodies (264, 265). These Gd-IgA1–IgG complexes deposit in the glomerular mesangium with murine C3 and induce mesangial proliferation, hematuria, and proteinuria. This model further supports the key roles of aberrant O-glycosylation of IgA1 and the corresponding autoantibodies specific for these IgA1 glycoforms in formation of glomerular immunodeposits in IgAN.
Clinical and laboratory research during recent years has led to a widely accepted definition of IgAN as an autoimmune disease with a complex multistep, also called multi-hit, pathogenetic process (Figure 7) (173). Specifically, circulatory Gd-IgA1 in patients with IgAN (Hit 1) is recognized by autoantibodies of IgG and/or IgA isotype (Hit 2). Subsequently, IgA1–IgG and IgA1–IgA1 immune complexes are formed (Hit 3) that contain additional proteins, including components of complement system (200, 201). Some of these immune complexes ultimately deposit in the glomerular mesangium to activate mesangial cells and induce renal injury (Hit 4) (38, 61, 173). An alternative hypothesis has been proposed to suggest that aberrantly glycosylated IgA1 accumulates in the mesangium as lanthanic deposits that are later bound by newly appearing autoantibodies, resulting in the in situ formation of immune complexes (268). The immune deposits stimulate the mesangial cells to proliferate and overproduce components of extracellular matrix, cytokines, and chemokines. Some of these cytokines may cause podocyte injury to induce proteinuria (191, 269). Complement likely plays a role in the formation and activities of these complexes in the circulation as well as in those that may be formed in situ [for review, see Role of Complement in IgA Nephropathy above, and Refs (230) and (200)]. Moreover, some of the hits in the pathogenesis may be modulated or controlled by various environmental and genetic factors [for review, see Ref. (270)].
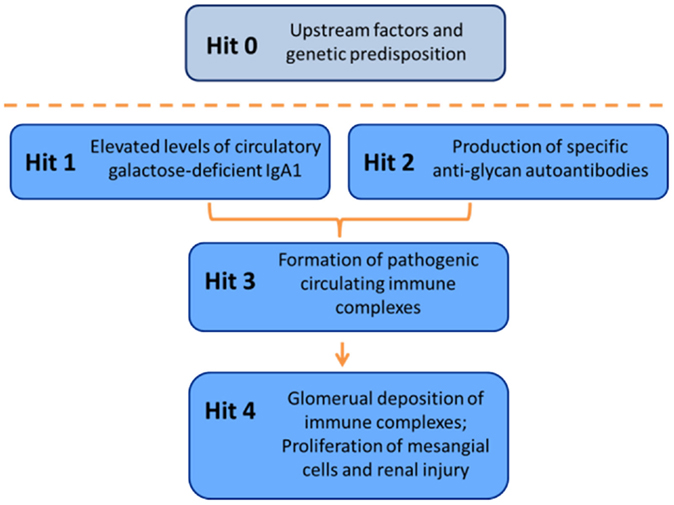
Figure 7. Multi-hit hypothesis for pathogenesis of IgAN. Several processes are involved in development of IgAN. Circulatory Gd-IgA1 (Hit 1) is recognized by Gd-IgA1-specific autoantibodies (Hit 2) that leads to formation of pathogenic Gd-IgA1-containing circulating immune complexes (Hit 3). Some of these complexes reach the renal glomeruli to bind to mesangial cells and activate them, thereby inducing renal injury (Hit 4).
Multiple other publications and findings lend credence to the multi-hit hypothesis on the pathogenesis of IgAN. For example, serum levels of Gd-IgA1 may predict disease progression (32), and serum levels of IgG and/or IgA autoantibodies specific for Gd-IgA1 correlate to disease severity and may also predict disease progression (182, 271). Moreover, serum levels of Gd-IgA1, IgG autoantibodies, and IgA1–IgG immune complexes predict disease recurrence in renal allografts (272).
Progress in the clinical and laboratory studies of IgAN has fueled a paradigm-shifting hypothesis on the autoimmune nature of the disease and identified some of the associated genetic factors (270). The multi-hit hypothesis not only describes the pathogenetic steps of IgAN but also serves as a “blueprint” for identifying targets of future disease-specific therapy and developing key biomarkers of the disease.
Clinical and laboratory studies in the last several years have identified several potential biomarkers for IgAN. It is hoped that some of these candidate markers can be developed into clinical assays to aid in the diagnosis, prognosis, patient stratification, monitoring of disease progression, and assessment of responses to treatment. Below, we briefly outline some of the candidate markers and also mention prospects for the development of disease-specific therapy.
Involvement of genetic factors in IgAN was first recognized through the discovery of familial forms of the disease (273). Specific loci and genes were later identified through linkage studies and GWAS [for review, see Ref. (274)]. Multiple susceptibility alleles have been identified by GWAS in cohorts from Europe, North America, and East Asia (179, 275–277). Disease susceptibility is affected by common variations in genes involved in antigen processing and presentation as well as in the mucosal defense system and alternative complement pathway. These findings further support an autoimmune nature of IgAN. GWAS data revealed that common genetic variants influence the risk of IgAN and suggest a multilocus adaptation process, possibly related to the variation in local pathogens across world populations (179). Moreover, serum levels of Gd-IgA1, the key autoantigen in IgAN, are genetically codetermined (152). Multiple risk and protective alleles among these disease-associated genes have been uncovered, and the cumulative number of risk alleles has been linked to the age of disease onset (179). However, additional genomic studies are needed to better define major genetic factors and their variants and to enable development of future individualized genetic/genomic approaches.
A better understanding of the causes of IgAN through combined clinical, biochemical, and molecular studies will identify candidates for developing disease-specific biochemical biomarkers. Candidate biomarkers include serum levels and/or specific characteristics of the autoantigen (Gd-IgA1), levels of autoantibodies specific for Gd-IgA1, and levels and/or specific characteristics of immune complexes consisting of IgG autoantibody bound to Gd-IgA1 (181, 272, 278) (Table 2). These biomarkers, whether used individually or in combination as panels, may have diagnostic and/or prognostic significance and would support future testing of disease-specific therapeutic approaches.
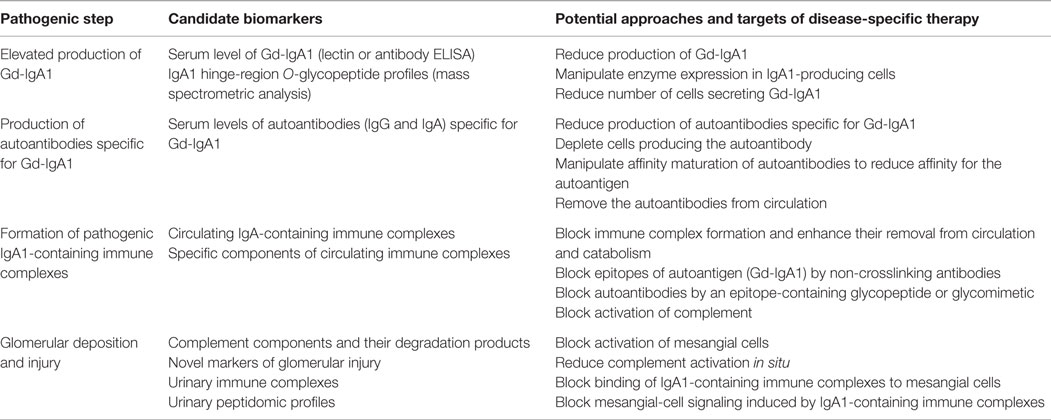
Table 2. Candidate biomarkers and disease-specific approaches for treatment of IgAN.
IgAN is diagnosed based on evaluation of a renal biopsy specimen. Laboratory screening for the possible presence of the disease include assessment of proteinuria and hematuria. These measurements are not disease-specific and, thus, there have been numerous efforts to identify urinary markers specific for IgAN (279–281). For example, urinary concentrations of several cytokines related to cellular proliferation were evaluated as potential markers of histopathologic glomerular and tubulointerstitial changes. Urinary IL-6 levels were elevated in patients with glomerulonephritis; however, the results did not define the type of primary glomerulonephritis (282). Nonetheless, urinary excretion of IL-6 predicted long-term renal outcome in patients with IgAN (283), and excretion of IL-6 and epidermal growth factor (EGF) has been correlated with degree of tubulointerstitial damage that itself predicts a poor long-term outcome (3, 4). Based on these results, the ratio of urinary IL-6/EGF was proposed as a prognostic marker for the progression of renal damage (284). In addition, urinary levels of monocyte chemotactic peptide-1 (MCP-1) and IL-8 correlated to tubulointerstitial damage (285, 286). However, cytokine/chemokine excretion again did not distinguish the specific types of glomerulonephritis. Other potential markers that have been evaluated include urinary α-1 antitrypsin in the α-1-globulin fraction (287) and urinary heparan sulfate (288), both of which were significantly higher in patients with IgAN. Urinary IgA concentrations are higher in patients with IgAN than in healthy individuals or in patients with other renal diseases and correlate with proteinuria (289). Immune complexes consisting of Gd-IgA1 and IgG were detected in the urine of patients with IgAN (290), but the prognostic value has not been defined. In contrast, excretion of the membrane attack complex was elevated in patients with membranous nephropathy (291) but not in patients with IgAN (292). Another study showed a correlation between glomerular filtration rate, urinary immunoglobulin excretion, and pathological grading of renal biopsies in patients with HSP with nephritis (293).
In addition to intact proteins, urine contains naturally occurring fragments (peptides) derived from serum and renal tubular or glomerular proteins (280, 294–301). Analysis of urinary peptides may offer an opportunity to develop a non-invasive and unbiased diagnostic tool without a priori assumptions as to the pathogenesis of disease (281, 300–305). Initial studies using urinary peptidomic techniques indicated the potential to differentiate patients with IgAN from patients with other glomerular diseases (302, 306).
Although many reports on urinary proteins and peptides demonstrated differential amounts of some proteins and protein complexes as well as peptides in the urine of patients with IgAN, none of the tests has been used in a routine clinical laboratory. It is hoped, however, that future studies will provide markers useful both for diagnosis and therapeutic monitoring of this disease (281, 296, 300, 301, 307).
As outlined in the previous sections, Gd-IgA1-containing immune complexes are considered to be a critical factor in the pathogenesis of IgAN. Theoretically, any intervention that would reduce production of Gd-IgA1 or autoantibodies specific for Gd-IgA1, block formation of the IgA1–IgG complexes, or otherwise reduce levels of pathogenic immune complexes would constitute effective disease-specific treatment. Similarly, any approach that would block activation of mesangial cells by the pathogenic IgA1–IgG complexes would be desirable. Examples of such approaches are listed in Table 2, and more details can be found in recent reviews (6, 180, 308). It is hoped that studies that discover the molecular defects of IgA1, the mechanisms of induction of the autoantibodies specific for Gd-IgA1, the composition and biological activities of the immune complexes, and the signaling pathways for activation of mesangial cells and glomerular injury will lead to disease-specific therapy.
Accumulated knowledge indicates that IgAN, the most common primary glomerulonephritis in the world, is an autoimmune disease driven by formation and glomerular deposition of IgA1-containing immune complexes. Currently, there is no disease-specific therapy, and many patients with IgAN progress to end-stage renal disease. The diagnosis of IgAN is established by determination of IgA as the dominant or codominant immunoglobulin in glomeruli. The IgA in glomerular deposits is exclusively of the IgA1 subclass and is enriched for glycoforms deficient in galactose on the hinge-region O-glycans. Multiple studies led to a hypothesis for a multi-hit pathogenetic process with contributing genetic and environmental components. In this process, circulatory Gd-IgA1 is recognized as an autoantigen by IgG or IgA autoantibodies, resulting in the formation of immune complexes. Some of these circulating complexes deposit in glomeruli, activate mesangial cells, and induce glomerular injury through cellular proliferation and overproduction of components of extracellular matrix and cytokines/chemokines. Glycosylation pathways associated with production of the autoantigen and the unique characteristics of the corresponding autoantibodies in patients with IgAN leading to the formation of pathogenic immune complexes have been uncovered, and genetic factors associated with IgAN have been identified. Complement plays a significant role in the formation and nephritogenic activities of these complexes; complement activation likely occurs systemically on IgA1-containing circulating immune complexes as well as locally in glomeruli. Multiple new models and approaches have been developed that will lead to a better understanding of the molecular mechanisms and factors involved in formation and activities of pathogenic IgA1-containing immune complexes. It is hoped that the ongoing and future studies will enable development of much needed disease-specific therapy (308).
JN and BAJ conceived the general outline, BK, CR, and NM further developed the outline and content. Each author contributed intellectually by writing assigned sections, editing and revising the drafts and proofreading.
The authors declare that the review was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by grants from the National Institutes of Health DK078244, DK082753, DK099228, DK105124, GM098539, DK079337, and DK106341, by a grant 15-33686A from Ministry of Health of the Czech Republic, and by a gift from the IGA Nephropathy Foundation of America.
1. Berger J, Hinglais N. [Intercapillary deposits of IgA-IgG]. J Urol Nephrol (Paris) (1968) 74:694–5.
2. Jennette JC. The immunohistology of IgA nephropathy. Am J Kidney Dis (1988) 12:348–52. doi:10.1016/S0272-6386(88)80022-2
3. Cattran DC, Coppo R, Cook HT, Feehally J, Roberts IS, Troyanov S, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int (2009) 76:534–45. doi:10.1038/ki.2009.243
4. Roberts IS, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int (2009) 76:546–56. doi:10.1038/ki.2009.168
5. Berthoux FC, Mohey H, Afiani A. Natural history of primary IgA nephropathy. Semin Nephrol (2008) 28:4–9. doi:10.1016/j.semnephrol.2007.10.001
7. McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol Dial Transplant (2011) 26:414–30. doi:10.1093/ndt/gfq665
8. Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet (2012) 8:e1002765. doi:10.1371/journal.pgen.1002765
9. Varis J, Rantala I, Pasternack A, Oksa H, Jantti M, Paunu ES, et al. Immunoglobulin and complement deposition in glomeruli of 756 subjects who had committed suicide or met with a violent death. J Clin Pathol (1993) 46:607–10. doi:10.1136/jcp.46.7.607
10. Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int (2003) 63:2286–94. doi:10.1046/j.1523-1755.63.6s.2.x
11. Ponticelli C, Glassock RJ. Posttransplant recurrence of primary glomerulonephritis. Clin J Am Soc Nephrol (2010) 5:2363–72. doi:10.2215/CJN.06720810
12. Silva FG, Chander P, Pirani CL, Hardy MA. Disappearance of glomerular mesangial IgA deposits after renal allograft transplantation. Transplantation (1982) 33:241–6.
15. Faille-Kuyber EH, Kater L, Kooiker CJ, Dorhout Mees EJ. IgA-deposits in cutaneous blood-vessel walls and mesangium in Henoch-Schönlein syndrome. Lancet (1973) 1:892–3. doi:10.1016/S0140-6736(73)91471-2
16. Davin JC, Ten Berge IJ, Weening JJ. What is the difference between IgA nephropathy and Henoch-Schönlein purpura nephritis? Kidney Int (2001) 59:823–34. doi:10.1046/j.1523-1755.2001.059003823.x
17. Davin JC. Henoch-Schönlein purpura nephritis: pathophysiology, treatment, and future strategy. Clin J Am Soc Nephrol (2011) 6:679–89. doi:10.2215/CJN.06710810
18. Pohl M. Henoch-Schönlein purpura nephritis. Pediatr Nephrol (2015) 30:245–52. doi:10.1007/s00467-014-2815-6
19. Mestecky J, Hammarström L. IgA-associated diseases. In: Kaetzel CS, editor. Mucosal Immune Defense: Immunoglobulin A. New York: Springer (2007). p. 321–44.
20. Pouria S, Barratt J. Secondary IgA nephropathy. Semin Nephrol (2008) 28:27–37. doi:10.1016/j.semnephrol.2007.10.004
21. Tissandié E, Morelle W, Berthelot L, Vrtovsnik F, Daugas E, Walker F, et al. Both IgA nephropathy and alcoholic cirrhosis feature abnormally glycosylated IgA1 and soluble CD89-IgA and IgG-IgA complexes: common mechanisms for distinct diseases. Kidney Int (2011) 80:1352–63. doi:10.1038/ki.2011.276
22. Berger J. Recurrence of IgA nephropathy in renal allografts. Am J Kidney Dis (1988) 12:371–2. doi:10.1016/S0272-6386(88)80027-1
23. Odum J, Peh CA, Clarkson AR, Bannister KM, Seymour AE, Gillis D, et al. Recurrent mesangial IgA nephritis following renal transplantation. Nephrol Dial Transplant (1994) 9:309–12.
24. Coppo R, Amore A, Cirina P, Messina M, Basolo B, Segoloni G, et al. Characteristics of IgA and macromolecular IgA in sera from IgA nephropathy transplanted patients with and without IgAN recurrence. Contrib Nephrol (1995) 111:85–92. doi:10.1159/000423881
25. Coppo R, Amore A, Cirina P, Messina M, Basolo B, Segoloni G, et al. IgA serology in recurrent and non-recurrent IgA nephropathy after renal transplantation. Nephrol Dial Transplant (1995) 10:2310–5.
26. Chandrakantan A, Ratanapanichkich P, Said M, Barker CV, Julian BA. Recurrent IgA nephropathy after renal transplantation despite immunosuppressive regimens with mycophenolate mofetil. Nephrol Dial Transplant (2005) 20:1214–21. doi:10.1093/ndt/gfh773
27. Coppo R, Basolo B, Martina G, Rollino C, De Marchi M, Giacchino F, et al. Circulating immune complexes containing IgA, IgG and IgM in patients with primary IgA nephropathy and with Henoch-Schönlein nephritis. Correlation with clinical and histologic signs of activity. Clin Nephrol (1982) 18:230–9.
28. Czerkinsky C, Koopman WJ, Jackson S, Collins JE, Crago SS, Schrohenloher RE, et al. Circulating immune complexes and immunoglobulin A rheumatoid factor in patients with mesangial immunoglobulin A nephropathies. J Clin Invest (1986) 77:1931–8. doi:10.1172/JCI112522
29. Schena FP, Pastore A, Ludovico N, Sinico RA, Benuzzi S, Montinaro V. Increased serum levels of IgA1-IgG immune complexes and anti-F(ab’)2 antibodies in patients with primary IgA nephropathy. Clin Exp Immunol (1989) 77:15–20.
30. Coppo R, Emancipator S. Pathogenesis of IgA nephropathy: established observations, new insights and perspectives in treatment. J Nephrol (1994) 7:5–15.
31. Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int (1997) 52:509–16. doi:10.1038/ki.1997.361
32. Zhao N, Hou P, Lv J, Moldoveanu Z, Li Y, Kiryluk K, et al. The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int (2012) 82:790–6. doi:10.1038/ki.2012.197
33. Gonzalez-Cabrero J, Egido J, Barat A, Gonzalez E. Detection and characterization of circulating and glomerular immune complexes in experimental IgA nephropathy. Immunology (1990) 70:296–302.
34. van den Wall Bake AWL, Bruijn JA, Accavitti MA, Crowley-Nowick PA, Schrohenloher RE, Julian BA, et al. Shared idiotypes in mesangial deposits in IgA nephropathy are not disease-specific. Kidney Int (1993) 44:65–74. doi:10.1038/ki.1993.214
35. Coppo R, Basolo B, Piccoli G, Mazzucco G, Bulzomi MR, Roccatello D, et al. IgA1 and IgA2 immune complexes in primary IgA nephropathy and Henoch-Schönlein nephritis. Clin Exp Immunol (1984) 57:583–90.
36. Novak J, Julian BA, Tomana M, Mestecky J. Progress in molecular and genetic studies of IgA nephropathy. J Clin Immunol (2001) 21:310–27. doi:10.1023/A:1012284402054
37. Russell MW, Mestecky J, Julian BA, Galla JH. IgA-associated renal diseases: antibodies to environmental antigens in sera and deposition of immunoglobulins and antigens in glomeruli. J Clin Immunol (1986) 6:74–86. doi:10.1007/BF00915367
38. Novak J, Julian BA, Mestecky J, Renfrow MB. Glycosylation of IgA1 and pathogenesis of IgA nephropathy. Semin Immunopathol (2012) 34:365–82. doi:10.1007/s00281-012-0306-z
39. Tanaka A, Iwase H, Hiki Y, Kokubo T, Ishii-Karakasa I, Toma K, et al. Evidence for a site-specific fucosylation of N-linked oligosaccharide of immunoglobulin A1 from normal human serum. Glycoconj J (1998) 15:995–1000. doi:10.1023/A:1006989910120
40. Gomes MM, Wall SB, Takahashi K, Novak J, Renfrow MB, Herr AB. Analysis of IgA1 N-glycosylation and its contribution to FcαRI binding. Biochemistry (2008) 47:11285–99. doi:10.1021/bi801185b
41. Tomana M, Niedermeier W, Mestecky J, Hammack WJ. The carbohydrate composition of human myeloma IgA. Immunochemistry (1972) 9:933–40. doi:10.1016/0019-2791(72)90166-8
42. Baenziger J, Kornfeld S. Structure of the carbohydrate units of IgA1 immunoglobulin II. Structure of the O-glycosidically linked oligosaccharide units. J Biol Chem (1974) 249:7270–81.
43. Field MC, Dwek RA, Edge CJ, Rademacher TW. O-linked oligosaccharides from human serum immunoglobulin A1. Biochem Soc Trans (1989) 17:1034–5. doi:10.1042/bst0171034
44. Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, et al. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcα receptor interactions. J Biol Chem (1998) 273:2260–72. doi:10.1074/jbc.273.4.2260
45. Takahashi K, Smith AD, Poulsen K, Kilian M, Julian BA, Mestecky J, et al. Identification of structural isomers in IgA1 hinge-region O-glycosylation using high-resolution mass spectrometry. J Proteome Res (2012) 11:692–702. doi:10.1021/pr200608q
46. Andre PM, Le Pogamp P, Chevet D. Impairment of jacalin binding to serum IgA in IgA nephropathy. J Clin Lab Anal (1990) 4:115–9. doi:10.1002/jcla.1860040208
47. Mestecky J, Tomana M, Crowley-Nowick PA, Moldoveanu Z, Julian BA, Jackson S. Defective galactosylation and clearance of IgA1 molecules as a possible etiopathogenic factor in IgA nephropathy. Contrib Nephrol (1993) 104:172–82. doi:10.1159/000422410
48. Allen AC. Abnormal glycosylation of IgA: is it related to the pathogenesis of IgA nephropathy? Nephrol Dial Transplant (1995) 10:1121–4.
49. Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest (1999) 104:73–81. doi:10.1172/JCI5535
50. Novak J, Tomana M, Matousovic K, Brown R, Hall S, Novak L, et al. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int (2005) 67:504–13. doi:10.1111/j.1523-1755.2005.67107.x
51. Leung JC, Tang SC, Chan LY, Chan WL, Lai KN. Synthesis of TNF-α by mesangial cells cultured with polymeric anionic IgA – role of MAPK and NF-κB. Nephrol Dial Transplant (2008) 23:72–81. doi:10.1093/ndt/gfm581
52. Tam KY, Leung JC, Chan LY, Lam MF, Tang SC, Lai KN. Macromolecular IgA1 taken from patients with familial IgA nephropathy or their asymptomatic relatives have higher reactivity to mesangial cells in vitro. Kidney Int (2009) 75:1330–9. doi:10.1038/ki.2009.71
53. Novak J, Vu HL, Novak L, Julian BA, Mestecky J, Tomana M. Interactions of human mesangial cells with IgA and IgA-containing immune complexes. Kidney Int (2002) 62:465–75. doi:10.1046/j.1523-1755.2002.00477.x
54. Conley ME, Cooper MD, Michael AF. Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. J Clin Invest (1980) 66:1432–6. doi:10.1172/JCI109998
55. Allen AC, Bailey EM, Brenchley PEC, Buck KS, Barratt J, Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: observations in three patients. Kidney Int (2001) 60:969–73. doi:10.1046/j.1523-1755.2001.060003969.x
56. Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A, Iwase H, et al. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int (2001) 59:1077–85. doi:10.1046/j.1523-1755.2001.0590031077.x
57. Allen AC, Willis FR, Beattie TJ, Feehally J. Abnormal IgA glycosylation in Henoch-Schönlein purpura restricted to patients with clinical nephritis. Nephrol Dial Transplant (1998) 13:930–4. doi:10.1093/ndt/13.4.930
58. Levinsky RJ, Barratt TM. IgA immune complexes in Henoch-Schönlein purpura. Lancet (1979) 2:1100–3. doi:10.1016/S0140-6736(79)92505-4
59. Zickerman AM, Allen AC, Talwar V, Olczak SA, Brownlee A, Holland M, et al. IgA myeloma presenting as Henoch-Schönlein purpura with nephritis. Am J Kidney Dis (2000) 36:E19. doi:10.1053/ajkd.2000.16221
60. van der Helm-van Mil AHM, Smith AC, Pouria S, Tarelli E, Brunskill NJ, Eikenboom HC. Immunoglobulin A multiple myeloma presenting with Henoch-Schönlein purpura associated with reduced sialylation of IgA1. Br J Haematol (2003) 122:915–7. doi:10.1046/j.1365-2141.2003.04539.x
61. Novak J, Mestecky J. IgA immune-complex. In: Lai KN, editor. Recent Advances in IgA Nephropathy. Hong Kong: Imperial College Press and the World Scientific Publisher (2009). p. 177–91.
62. Moldoveanu Z, Moro I, Radl J, Thorpe SR, Komiyama K, Mestecky J. Site of catabolism of autologous and heterologous IgA in non-human primates. Scand J Immunol (1990) 32:577–83. doi:10.1111/j.1365-3083.1990.tb03199.x
63. Stockert RJ, Kressner MS, Collins JC, Sternlieb I, Morell AG. IgA interaction with the asialoglycoprotein receptor. Proc Natl Acad Sci U S A (1982) 79:6229–31. doi:10.1073/pnas.79.20.6229
64. Tomana M, Kulhavy R, Mestecky J. Receptor-mediated binding and uptake of immunoglobulin A by human liver. Gastroenterology (1988) 94:887–92.
65. Baenziger JU, Fiete D. Galactose and N-acetylgalactosamine-specific endocytosis of glycopeptides by isolated rat hepatocytes. Cell (1980) 22:611–20. doi:10.1016/0092-8674(80)90371-2
66. Mestecky J, Hashim OH, Tomana M. Alterations in the IgA carbohydrate chains influence the cellular distribution of IgA1. Contrib Nephrol (1995) 111:66–72. doi:10.1159/000423879
67. Phillips JO, Komiyama K, Epps JM, Russell MW, Mestecky J. Role of hepatocytes in the uptake of IgA and IgA-containing immune complexes in mice. Mol Immunol (1988) 25:873–9. doi:10.1016/0161-5890(88)90124-1
68. Wisse E, Jacobs F, Topal B, Frederik P, De Geest B. The size of endothelial fenestrae in human liver sinusoids: implications for hepatocyte-directed gene transfer. Gene Ther (2008) 15:1193–9. doi:10.1038/gt.2008.60
69. Mestecky J, Tomana M, Moldoveanu Z, Julian BA, Suzuki H, Matousovic K, et al. The role of aberrant glycosylation of IgA1 molecules in the pathogenesis of IgA nephropathy. Kidney Blood Press Res (2008) 31:29–37. doi:10.1159/000112922
70. Novak J, Julian BA, Tomana M, Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol (2008) 28:78–87. doi:10.1016/j.semnephrol.2007.10.009
71. Mestecky J, Raska M, Julian BA, Gharavi AG, Renfrow MB, Moldoveanu Z, et al. IgA nephropathy: molecular mechanisms of the disease. Annu Rev Pathol (2013) 8:217–40. doi:10.1146/annurev-pathol-011110-130216
73. Coppo R, Amore A. Aberrant glycosylation in IgA nephropathy (IgAN). Kidney Int (2004) 65:1544–7. doi:10.1111/j.1523-1755.2004.05407.x
74. Julian BA, Novak J. IgA nephropathy: an update. Curr Opin Nephrol Hypertens (2004) 13:171–9. doi:10.1097/00041552-200403000-00005
75. Haakenstad AO, Mannik M. The biology of immune complexes. In: Talal N, editor. Autoimmunity. Genetic, Immunologic, Virologic, and Clinical Aspects. New York: Academic Press (1977). p. 277–360.
76. Allen AC, Harper SJ, Feehally J. Galactosylation of N- and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clin Exp Immunol (1995) 100:470–4. doi:10.1111/j.1365-2249.1995.tb03724.x
77. Smith AC, de Wolff JF, Molyneux K, Feehally J, Barratt J. O-glycosylation of serum IgD in IgA nephropathy. J Am Soc Nephrol (2006) 17:1192–9. doi:10.1681/ASN.2005101115
78. Allen AC, Bailey EM, Barratt J, Buck KS, Feehally J. Analysis of IgA1 O-glycans in IgA nephropathy by fluorophore-assisted carbohydrate electrophoresis. J Am Soc Nephrol (1999) 10:1763–71.
79. Tomana M, Novak J, Julian BA, Mestecky J. IgA1 glycosylation and the pathogenesis of IgA nephropathy. Am J Kidney Dis (2000) 35:555–6. doi:10.1016/S0272-6386(00)70215-0
80. Tomana M, Niedermeier W, Mestecky J, Skvaril F. The differences in carbohydrate composition between the subclasses of IgA immunoglobulins. Immunochemistry (1976) 13:325–8. doi:10.1016/0019-2791(76)90342-6
81. Hiki Y, Horii A, Iwase H, Tanaka A, Toda Y, Hotta K, et al. O-linked oligosaccharide on IgA1 hinge region in IgA nephropathy. Fundamental study for precise structure and possible role. Contrib Nephrol (1995) 111:73–84. doi:10.1159/000423880
82. Allen AC. Methodological approaches to the analysis of IgA1 O-glycosylation in IgA nephropathy. J Nephrol (1999) 12:76–84.
83. Hiki Y, Kokubo T, Iwase H, Masaki Y, Sano T, Tanaka A, et al. Underglycosylation of IgA1 hinge plays a certain role for its glomerular deposition in IgA nephropathy. J Am Soc Nephrol (1999) 10:760–9.
84. Novak J, Tomana M, Kilian M, Coward L, Kulhavy R, Barnes S, et al. Heterogeneity of O-glycosylation in the hinge region of human IgA1. Mol Immunol (2000) 37:1047–56. doi:10.1016/S0161-5890(01)00019-0
85. Leung JC, Tang SC, Chan DT, Lui SL, Lai KN. Increased sialylation of polymeric lambda-IgA1 in patients with IgA nephropathy. J Clin Lab Anal (2002) 16:11–9. doi:10.1002/jcla.2035
86. Tarelli E, Smith AC, Hendry BM, Challacombe SJ, Pouria S. Human serum IgA1 is substituted with up to six O-glycans as shown by matrix assisted laser desorption ionisation time-of-flight mass spectrometry. Carbohydr Res (2004) 339:2329–35. doi:10.1016/j.carres.2004.07.011
87. Takahashi K, Hiki Y, Odani H, Shimozato S, Iwase H, Sugiyama S, et al. Structural analyses of O-glycan sugar chains on IgA1 hinge region using SELDI-TOF MS with various lectins. Biochem Biophys Res Commun (2006) 350:580–7. doi:10.1016/j.bbrc.2006.09.075
88. Lau KK, Wyatt RJ, Moldoveanu Z, Tomana M, Julian BJ, Hogg RJ, et al. Serum levels of galactose-deficient IgA in children with IgA nephropathy and Henoch-Schönlein purpura. Pediatr Nephrol (2007) 22:2067–72. doi:10.1007/s00467-007-0623-y
89. Moldoveanu Z, Wyatt RJ, Lee J, Tomana M, Julian BA, Mestecky J, et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int (2007) 71:1148–54. doi:10.1038/sj.ki.5002185
90. Moore JS, Kulhavy R, Tomana M, Moldoveanu Z, Suzuki H, Brown R, et al. Reactivities of N-acetylgalactosamine-specific lectins with human IgA1 proteins. Mol Immunol (2007) 44:2598–604. doi:10.1016/j.molimm.2006.12.011
91. Novak J, Moldoveanu Z, Renfrow MB, Yanagihara T, Suzuki H, Raska M, et al. IgA nephropathy and Henoch-Schönlein purpura nephritis: aberrant glycosylation of IgA1, formation of IgA1-containing immune complexes, and activation of mesangial cells. Contrib Nephrol (2007) 157:134–8. doi:10.1159/000102455
92. Shimozato S, Hiki Y, Odani H, Takahashi K, Yamamoto K, Sugiyama S. Serum under-galactosylated IgA1 is increased in Japanese patients with IgA nephropathy. Nephrol Dial Transplant (2008) 23:1931–9. doi:10.1093/ndt/gfm913
93. Suzuki H, Moldoveanu Z, Hall S, Brown R, Vu HL, Novak L, et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest (2008) 118:629–39. doi:10.1172/JCI33189
94. Gomes MM, Suzuki H, Brooks MT, Tomana M, Moldoveanu Z, Mestecky J, et al. Recognition of galactose-deficient O-glycans in the hinge region of IgA1 by N-acetylgalactosamine-specific snail lectins: a comparative binding study. Biochemistry (2010) 49:5671–82. doi:10.1021/bi9019498
95. Novak J, Raskova Kafkova L, Suzuki H, Tomana M, Matousovic K, Brown R, et al. IgA1 immune complexes from pediatric patients with IgA nephropathy activate cultured mesangial cells. Nephrol Dial Transplant (2011) 26:3451–7. doi:10.1093/ndt/gfr448
96. Franc V, Rehulka P, Raus M, Stulik J, Novak J, Renfrow MB, et al. Elucidating heterogeneity of IgA1 hinge-region O-glycosylation by use of MALDI-TOF/TOF mass spectrometry: role of cysteine alkylation during sample processing. J Proteomics (2013) 92:299–312. doi:10.1016/j.jprot.2013.07.013
97. Iwase H, Tanaka A, Hiki Y, Kokubo T, Karakasa-Ishii I, Kobayashi Y, et al. Estimation of the number of O-linked oligosaccharides per heavy chain of human IgA1 by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOFMS) analysis of the hinge glycopeptide. J Biochem (1996) 120:393–7. doi:10.1093/oxfordjournals.jbchem.a021425
98. Iwase H, Tanaka A, Hiki Y, Kokubo T, Ishii-Karakasa I, Nishikido J, et al. Application of matrix-assisted laser desorption ionization time-of-flight mass spectrometry to the analysis of glycopeptide-containing multiple O-linked oligosaccharides. J Chromatogr B Biomed Sci Appl (1998) 709:145–9. doi:10.1016/S0378-4347(98)00050-4
99. Iwase H, Tanaka A, Hiki Y, Kokubo T, Sano T, Ishii-Karakasa I, et al. Aggregated human serum immunoglobulin A1 induced by neuraminidase treatment had a lower number of O-linked sugar chains on the hinge portion. J Chromatogr (1999) 724:1–7. doi:10.1016/S0378-4347(98)00552-0
100. Odani H, Hiki Y, Takahashi M, Nishimoto A, Yasuda Y, Iwase H, et al. Direct evidence for decreased sialylation and galactosylation of human serum IgA1 Fc O-glycosylated hinge peptides in IgA nephropathy by mass spectrometry. Biochem Biophys Res Commun (2000) 271:268–74. doi:10.1006/bbrc.2000.2613
101. Horie A, Hiki Y, Odani H, Yasuda Y, Takahashi M, Kato M, et al. IgA1 molecules produced by tonsillar lymphocytes are under-O-glycosylated in IgA nephropathy. Am J Kidney Dis (2003) 42:486–96. doi:10.1016/S0272-6386(03)00743-1
102. Renfrow MB, Cooper HJ, Tomana M, Kulhavy R, Hiki Y, Toma K, et al. Determination of aberrant O-glycosylation in the IgA1 hinge region by electron capture dissociation Fourier transform-ion cyclotron resonance mass spectrometry. J Biol Chem (2005) 280:19136–45. doi:10.1074/jbc.M411368200
103. Renfrow MB, MacKay CL, Chalmers MJ, Julian BA, Mestecky J, Kilian M, et al. Analysis of O-glycan heterogeneity in IgA1 myeloma proteins by Fourier transform ion cyclotron resonance mass spectrometry: implications for IgA nephropathy. Anal Bioanal Chem (2007) 389:1397–407. doi:10.1007/s00216-007-1500-z
104. Takahashi K, Wall SB, Suzuki H, Smith AD, Hall S, Poulsen K, et al. Clustered O-glycans of IgA1: defining macro- and micro-heterogeneity by use of electron capture/transfer dissociation. Mol Cell Proteomics (2010) 9:2545–57. doi:10.1074/mcp.M110.001834
105. Rebecchi KR, Wenke JL, Go EP, Desaire H. Label-free quantitation: a new glycoproteomics approach. J Am Soc Mass Spectrom (2009) 20:1048–59. doi:10.1016/j.jasms.2009.01.013
106. Wada Y, Dell A, Haslam SM, Tissot B, Canis K, Azadi P, et al. Comparison of methods for profiling O-glycosylation: human proteome organisation human disease glycomics/proteome initiative multi-institutional study of IgA1. Mol Cell Proteomics (2010) 9:719–27. doi:10.1074/mcp.M900450-MCP200
107. Woof JM, Mestecky J. Mucosal immunoglobulins. 4th ed. In: Mestecky J, Strober W, Russell MW, Kelsall BL, Cheroute H, Lambrecht BN, editors. Mucosal Immunology. Amsterdam: Elsevier/Academic Press (2015). p. 287–324.
108. Johansen FE, Braathen R, Brandtzaeg P. Role of J chain in secretory immunoglobulin formation. Scand J Immunol (2000) 52:240–8. doi:10.1046/j.1365-3083.2000.00790.x
110. Conley ME, Delacroix DL. Intravascular and mucosal immunoglobulin A: two separate but related systems of immune defense? Ann Intern Med (1987) 106:892–9. doi:10.7326/0003-4819-106-6-892
111. Kilian M, Russell MW. Microbial evasion of IgA functions. 4th ed. In: Mestecky J, Strober W, Russell MW, Kelsall BL, Cheroute H, Lambrecht BN, editors. Mucosal Immunology. Amsterdam: Elsevier/Academic Press (2015). p. 455–70.
112. Baker K, Blumberg RS, Kaetzel CS. Immunoglobulin transport and immunoglobulin receptors. 4th ed. In: Mestecky J, Strober W, Russell MW, Kelsall BL, Cheroute H, Lambrecht BN, editors. Mucosal Immunology. Amsterdam: Elsevier/Academic Press (2015). p. 349–408.
113. Harper SJ, Allen AC, Pringle JH, Feehally J. Increased dimeric IgA producing B cells in the bone marrow in IgA nephropathy determined by in situ hybridisation for J chain mRNA. J Clin Pathol (1996) 49:38–42. doi:10.1136/jcp.49.1.38
114. Hiki Y, Iwase H, Kokubo T, Horii A, Tanaka A, Nishikido J, et al. Association of asialo-galactosyl ß1-3N-acetylgalactosamine on the hinge with a conformational instability of Jacalin-reactive immunoglobulin A1 in immunoglobulin A nephropathy. J Am Soc Nephrol (1996) 7:955–60.
115. Kokubo T, Hiki Y, Iwase H, Tanaka A, Toma K, Hotta K, et al. Protective role of IgA1 glycans against IgA1 self-aggregation and adhesion to extracellular matrix proteins. J Am Soc Nephrol (1998) 9:2048–54.
116. Clarkson AR, Woodroffe AJ, Bannister KM, Lomax-Smith JD, Aarons I. The syndrome of IgA nephropathy. Clin Nephrol (1984) 21:7–14.
117. Pakkanen SH, Kantele JM, Moldoveanu Z, Hedges S, Hakkinen M, Mestecky J, et al. Expression of homing receptors on IgA1 and IgA2 plasmablasts in blood reflects differential distribution of IgA1 and IgA2 in various body fluids. Clin Vaccine Immunol (2010) 17:393–401. doi:10.1128/CVI.00475-09
118. Russell MW, Lue C, van den Wall Bake AW, Moldoveanu Z, Mestecky J. Molecular heterogeneity of human IgA antibodies during an immune response. Clin Exp Immunol (1992) 87:1–6. doi:10.1111/j.1365-2249.1992.tb06404.x
119. Russell MW, Kilian M, Mantis NJ, Corthésy B. Biological activities of IgA. 4th ed. In: Mestecky J, Strober W, Russell MW, Kelsall BL, Cheroute H, Lambrecht BN, editors. Mucosal Immunology. Amsterdam: Elsevier/Academic Press (2015). p. 429–54.
120. Smith AC, Molyneux K, Feehally J, Barratt J. O-glycosylation of serum IgA1 antibodies against mucosal and systemic antigens in IgA nephropathy. J Am Soc Nephrol (2006) 17:3520–8. doi:10.1681/ASN.2006060658
121. de Fijter JW, Eijgenraam JW, Braam CA, Holmgren J, Daha MR, van Es LA, et al. Deficient IgA1 immune response to nasal cholera toxin subunit B in primary IgA nephropathy. Kidney Int (1996) 50:952–61. doi:10.1038/ki.1996.396
122. Roodnat JI, de Fijter JW, van Kooten C, Daha MR, van Es LA. Decreased IgA1 response after primary oral immunization with live typhoid vaccine in primary IgA nephropathy. Nephrol Dial Transplant (1999) 14:353–9. doi:10.1093/ndt/14.2.353
123. Oortwijn BD, van der Boog PJ, Roos A, van der Geest RN, de Fijter JW, Daha MR, et al. A pathogenic role for secretory IgA in IgA nephropathy. Kidney Int (2006) 69:1131–8. doi:10.1038/sj.ki.5000074
124. Oortwijn BD, Rastaldi MP, Roos A, Mattinzoli D, Daha MR, van Kooten C. Demonstration of secretory IgA in kidneys of patients with IgA nephropathy. Nephrol Dial Transplant (2007) 22:3191–5. doi:10.1093/ndt/gfm346
125. Tomino Y, Sakai H, Miura M, Endoh M, Nomoto Y. Detection of polymeric IgA in glomeruli from patients with IgA nephropathy. Clin Exp Immunol (1982) 49:419–25.
126. Eijgenraam JW, Oortwijn BD, Kamerling SW, de Fijter JW, van den Wall Bake AW, Daha MR, et al. Secretory immunoglobulin A (IgA) responses in IgA nephropathy patients after mucosal immunization, as part of a polymeric IgA response. Clin Exp Immunol (2008) 152:227–32. doi:10.1111/j.1365-2249.2008.03616.x
127. Le W, Liang S, Chen H, Wang S, Zhang W, Wang X, et al. Long-term outcome of IgA nephropathy patients with recurrent macroscopic hematuria. Am J Nephrol (2014) 40:43–50. doi:10.1159/000364954
128. Feehally J, Allen AC. Structural features of IgA molecules which contribute to IgA nephropathy. J Nephrol (1999) 12:59–65.
129. Nagayama Y, Nishiwaki H, Hasegawa T, Komukai D, Kawashima E, Takayasu M, et al. Impact of the new risk stratification in the 2011 Japanese Society of Nephrology clinical guidelines for IgA nephropathy on incidence of early clinical remission with tonsillectomy plus steroid pulse therapy. Clin Exp Nephrol (2015) 19:646–52. doi:10.1007/s10157-014-1052-4
130. Feehally J, Coppo R, Troyanov S, Bellur SS, Cattran D, Cook T, et al. Tonsillectomy in a European cohort of 1,147 patients with IgA nephropathy. Nephron (2016) 132:15–24. doi:10.1159/000441852
131. Chen X, Liu H, Peng Y, He L, Zhang Y, Xie Y, et al. Expression and correlation analysis of IL-4, IFN-gamma and FcαRI in tonsillar mononuclear cells in patients with IgA nephropathy. Cell Immunol (2014) 289:70–5. doi:10.1016/j.cellimm.2014.03.004
132. Wu G, Peng YM, Liu FY, Xu D, Liu C. The role of memory B cell in tonsil and peripheral blood in the clinical progression of IgA nephropathy. Hum Immunol (2013) 74:708–12. doi:10.1016/j.humimm.2012.10.028
133. Li W, Peng X, Liu Y, Liu H, Liu F, He L, et al. TLR9 and BAFF: their expression in patients with IgA nephropathy. Mol Med Rep (2014) 10:1469–74. doi:10.3892/mmr.2014.2359
134. McCarthy DD, Chiu S, Gao Y, Summers-deLuca LE, Gommerman JL. BAFF induces a hyper-IgA syndrome in the intestinal lamina propria concomitant with IgA deposition in the kidney independent of LIGHT. Cell Immunol (2006) 241:85–94. doi:10.1016/j.cellimm.2006.08.002
135. McCarthy DD, Kujawa J, Wilson C, Papandile A, Poreci U, Porfilio EA, et al. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J Clin Invest (2011) 121:3991–4002. doi:10.1172/JCI45563
136. Xin G, Shi W, Xu LX, Su Y, Yan LJ, Li KS. Serum BAFF is elevated in patients with IgA nephropathy and associated with clinical and histopathological features. J Nephrol (2013) 26:683–90. doi:10.5301/jn.5000218
137. Kennel-de March A, Bene MC, Renoult E, Kessler M, Faure GC, Kolopp-Sarda MN. Enhanced expression of L-selectin on peripheral blood lymphocytes from patients with IgA nephropathy. Clin Exp Immunol (1999) 115:542–6. doi:10.1046/j.1365-2249.1999.00823.x
138. Takei T, Iida A, Nitta K, Tanaka T, Ohnishi Y, Yamada R, et al. Association between single-nucleotide polymorphisms in selectin genes and immunoglobulin A nephropathy. Am J Hum Genet (2002) 70:781–6. doi:10.1086/339077
139. Suzuki H, Raska M, Yamada K, Moldoveanu Z, Julian BA, Wyatt RJ, et al. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J Biol Chem (2014) 289:5330–9. doi:10.1074/jbc.M113.512277
140. Suzuki H, Moldoveanu Z, Hall S, Brown R, Julian BA, Wyatt RJ, et al. IgA nephropathy: characterization of IgG antibodies specific for galactose-deficient IgA1. Contrib Nephrol (2007) 157:129–33. doi:10.1159/0000102454
141. Yamaguchi K, Ozono Y, Harada T, Hara K. Changes in circulating immune complex and charge distribution with upper respiratory tract inflammation in IgA nephropathy. Nephron (1995) 69:384–90. doi:10.1159/000188507
142. Rostoker G, Rymer JC, Bagnard G, Petit-Phar M, Griuncelli M, Pilatte Y. Imbalances in serum proinflammatory cytokines and their soluble receptors: a putative role in the progression of idiopathic IgA nephropathy (IgAN) and Henoch-Schönlein purpura nephritis, and a potential target of immunoglobulin therapy? Clin Exp Immunol (1998) 114:468–76. doi:10.1046/j.1365-2249.1998.00745.x
143. Nelson CL, Karschimkus CS, Dragicevic G, Packham DK, Wilson AM, O’Neal D, et al. Systemic and vascular inflammation is elevated in early IgA and type 1 diabetic nephropathies and relates to vascular disease risk factors and renal function. Nephrol Dial Transplant (2005) 20:2420–6. doi:10.1093/ndt/gfi067
144. Yamada K, Reily C, Huang ZQ, Anderson JC, Raska M, Suzuki H, et al. Characterization of a signaling network that enhances production of galactose-deficient IgA1 in IgA1-secreting cells from patients with IgA Nephropathy. J Am Soc Nephrol (2015) 26:591A.
145. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J (2003) 374:1–20. doi:10.1042/bj20030407
146. Raska M, Moldoveanu Z, Suzuki H, Brown R, Kulhavy R, Andrasi J, et al. Identification and characterization of CMP-NeuAc:GalNAc-IgA1 α2,6-sialyltransferase in IgA1-producing cells. J Mol Biol (2007) 369:69–78. doi:10.1016/j.jmb.2007.03.002
147. Takahashi K, Raska M, Stuchlova Horynova M, Hall SD, Poulsen K, Kilian M, et al. Enzymatic sialylation of IgA1 O-glycans: implications for studies of IgA nephropathy. PLoS One (2014) 9:e99026. doi:10.1371/journal.pone.0099026
148. Reily C, Ueda H, Huang ZQ, Mestecky J, Julian BA, Willey CD, et al. Cellular signaling and production of galactose-deficient IgA1 in IgA nephropathy, an autoimmune disease. J Immunol Res (2014) 2014:197548. doi:10.1155/2014/197548
149. Stein JV, Lopez-Fraga M, Elustondo FA, Carvalho-Pinto CE, Rodriguez D, Gomez-Caro R, et al. APRIL modulates B and T cell immunity. J Clin Invest (2002) 109:1587–98. doi:10.1172/JCI0215034
150. Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet (2011) 43:321–7. doi:10.1038/ng.787
151. Meng HX, Ohe R, Li HN, Yang SR, Kabasawa T, Kato T, et al. Immunoglobulin and CD8(+) T-cell distribution in histologically distinctive tonsils of individuals with tonsillar focal infection. Acta Otolaryngol (2015) 135:264–70. doi:10.3109/00016489.2014.968802
152. Gharavi AG, Moldoveanu Z, Wyatt RJ, Barker CV, Woodford SY, Lifton RP, et al. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol (2008) 19:1008–14. doi:10.1681/ASN.2007091052
153. Ashwell G, Harford J. Carbohydrate-specific receptors of the liver. Annu Rev Biochem (1982) 51:531–54. doi:10.1146/annurev.bi.51.070182.002531
154. Roggenbuck D, Mytilinaiou MG, Lapin SV, Reinhold D, Conrad K. Asialoglycoprotein receptor (ASGPR): a peculiar target of liver-specific autoimmunity. Auto Immun Highlights (2012) 3:119–25. doi:10.1007/s13317-012-0041-4
155. Tabak LA. The role of mucin-type O-glycans in eukaryotic development. Semin Cell Dev Biol (2010) 21:616–21. doi:10.1016/j.semcdb.2010.02.001
156. Gerken TA, Jamison O, Perrine CL, Collette JC, Moinova H, Ravi L, et al. Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J Biol Chem (2011) 286:14493–507. doi:10.1074/jbc.M111.218701
157. Iwasaki H, Zhang Y, Tachibana K, Gotoh M, Kikuchi N, Kwon YD, et al. Initiation of O-glycan synthesis in IgA1 hinge region is determined by a single enzyme, UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 2. J Biol Chem (2003) 278:5613–21. doi:10.1074/jbc.M211097200
158. Wandall HH, Irazoqui F, Tarp MA, Bennett EP, Mandel U, Takeuchi H, et al. The lectin domains of polypeptide GalNAc-transferases exhibit carbohydrate-binding specificity for GalNAc: lectin binding to GalNAc-glycopeptide substrates is required for high density GalNAc-O-glycosylation. Glycobiology (2007) 17:374–87. doi:10.1093/glycob/cwl082
159. Raska M, Yamada K, Horynova M, Takahashi K, Suzuki H, Moldoveanu Z, et al. Role of GalNAc-transferases in the synthesis of aberrant IgA1 O-glycans in IgA nephropathy. J Am Soc Nephrol (2011) 22:625A.
160. Raska M, Yamada K, Stewart T, Stuchlova Horynova M, Huang Z, Suzuki H, et al. Role of N-acetylgalactosaminyl transferases in the synthesis of aberrant IgA1 O-glycans in IgA nephropathy. J Am Soc Nephrol (2012) 23:519A.
161. Novakova J, Stewart T, Yamada K, Suzuki H, Moldoveanu Z, Julian BA, et al. Overexpression of N-acetylgalactosaminyltransferase-14 contributes to galactose-deficient IgA1 production: relevance for IgA nephropathy. J Am Soc Nephrol (2013) 24:492A.
162. Wang H, Tachibana K, Zhang Y, Iwasaki H, Kameyama A, Cheng L, et al. Cloning and characterization of a novel UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase, pp-GalNAc-T14. Biochem Biophys Res Commun (2003) 300:738–44. doi:10.1016/S0006-291X(02)02908-X
163. Takahashi K, Suzuki H, Yamada K, Hall S, Moldoveanu Z, Poulsen K, et al. Molecular characterization of IgA1 secreted by IgA1-producing cell lines from patients with IgA nephropathy. J Am Soc Nephrol (2012) 23:853A.
164. Ju T, Brewer K, D’Souza A, Cummings RD, Canfield WM. Cloning and expression of human core 1 β1,3-galactosyltransferase. J Biol Chem (2002) 277:178–86. doi:10.1074/jbc.M109060200
165. Wang Y, Ju T, Ding X, Xia B, Wang W, Xia L, et al. Cosmc is an essential chaperone for correct protein O-glycosylation. Proc Natl Acad Sci U S A (2010) 107:9228–33. doi:10.1073/pnas.0914004107
166. Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 β3-galactosyltransferase. Proc Natl Acad Sci U S A (2002) 99:16613–8. doi:10.1073/pnas.262438199
167. Ju T, Cummings RD. Protein glycosylation: chaperone mutation in Tn syndrome. Nature (2005) 437:1252. doi:10.1038/4371252a
168. Ju T, Aryal RP, Stowell CJ, Cummings RD. Regulation of protein O-glycosylation by the endoplasmic reticulum-localized molecular chaperone Cosmc. J Cell Biol (2008) 182:531–42. doi:10.1083/jcb.200711151
169. Ju T, Lanneau GS, Gautam T, Wang Y, Xia B, Stowell SR, et al. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res (2008) 68:1636–46. doi:10.1158/0008-5472.CAN-07-2345
170. Malycha F, Eggermann T, Hristov M, Schena FP, Mertens PR, Zerres K, et al. No evidence for a role of cosmc-chaperone mutations in European IgA nephropathy patients. Nephrol Dial Transplant (2009) 24:321–4. doi:10.1093/ndt/gfn538
171. Dall’Olio F, Chiricolo M. Sialyltransferases in cancer. Glycoconj J (2001) 18:841–50. doi:10.1023/A:1022288022969
172. Harduin-Lepers A, Vallejo-Ruiz V, Krzewinski-Recchi MA, Samyn-Petit B, Julien S, Delannoy P. The human sialyltransferase family. Biochimie (2001) 83:727–37. doi:10.1016/S0300-9084(01)01301-3
173. Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol (2011) 22:1795–803. doi:10.1681/ASN.2011050464
174. Stuchlova Horynova M, Vrablikova A, Stewart TJ, Takahashi K, Czernekova L, Yamada K, et al. N-acetylgalactosaminide α2,6-sialyltransferase II is a candidate enzyme for sialylation of galactose-deficient IgA1, the key autoantigen in IgA nephropathy. Nephrol Dial Transplant (2015) 30:234–8. doi:10.1093/ndt/gfu308
175. Julian BA, Wyatt RJ, Matousovic K, Moldoveanu Z, Mestecky J, Novak J. IgA nephropathy: a clinical overview. Contrib Nephrol (2007) 157:19–26.
176. Lin X, Ding J, Zhu L, Shi S, Jiang L, Zhao M, et al. Aberrant galactosylation of IgA1 is involved in the genetic susceptibility of Chinese patients with IgA nephropathy. Nephrol Dial Transplant (2009) 24:3372–5. doi:10.1093/ndt/gfp294
177. Hastings MC, Moldoveanu Z, Julian BA, Novak J, Sanders JT, McGlothan KR, et al. Galactose-deficient IgA1 in African Americans with IgA nephropathy: serum levels and heritability. Clin J Am Soc Nephrol (2010) 5:2069–74. doi:10.2215/CJN.03270410
178. Kiryluk K, Moldoveanu Z, Sanders JT, Eison TM, Suzuki H, Julian BA, et al. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schönlein purpura nephritis. Kidney Int (2011) 80:79–87. doi:10.1038/ki.2011.16
179. Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet (2014) 46:1187–96. doi:10.1038/ng.3118
180. Novak J, Raska M, Mestecky J, Julian BA. IgA nephropathy and related diseases. 4th ed. In: Mestecky J, Strober W, Russell MW, Kelsall BL, Cheroute H, Lambrecht BN, editors. Mucosal Immunology. Amsterdam: Elsevier/Academic Press (2015). p. 2023–38.
181. Yanagawa H, Suzuki H, Suzuki Y, Kiryluk K, Gharavi AG, Matsuoka K, et al. A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PLoS One (2014) 9:e98081. doi:10.1371/journal.pone.0098081
182. Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest (2009) 119:1668–77. doi:10.1172/JCI38468
183. Huang ZQ, Raska M, Stewart T, Reily C, King RG, Crossman DK, et al. Somatic mutations modulate autoantibodies against galactose-deficient IgA1 in IgA nephropathy. J Am Soc Nephrol (2016). doi:10.1681/ASN.2014101044
184. Iwase H, Yokozeki Y, Hiki Y, Tanaka A, Kokubo T, Sano T, et al. Human serum immunoglobulin G3 subclass bound preferentially to asialo-, agalactoimmunoglobulin A1/sepharose. Biochem Biophys Res Commun (1999) 264:424–9. doi:10.1006/bbrc.1999.1369
185. Kokubo T, Hiki Y, Iwase H, Tanaka A, Nishikido J, Hotta K, et al. Exposed peptide core of IgA1 hinge region in IgA nephropathy. Nephrol Dial Transplant (1999) 14:81–5. doi:10.1093/ndt/14.1.81
186. Chen A, Chen WP, Sheu LF, Lin CY. Pathogenesis of IgA nephropathy: in vitro activation of human mesangial cells by IgA immune complex leads to cytokine secretion. J Pathol (1994) 173:119–26. doi:10.1002/path.1711730208
187. Gomez-Guerrero C, Alonso J, Lopez-Armada MJ, Ruiz-Ortega M, Gomez-Garre D, Alcazar R, et al. Potential factors governing extracellular matrix production by mesangial cells: their relevance for the pathogenesis of IgA nephropathy. Contrib Nephrol (1995) 111:45–54. doi:10.1159/000423876
188. Amore A, Cirina P, Conti G, Brusa P, Peruzzi L, Coppo R. Glycosylation of circulating IgA in patients with IgA nephropathy modulates proliferation and apoptosis of mesangial cells. J Am Soc Nephrol (2001) 12:1862–71.
189. Leung JC, Tsang AW, Chan LY, Tang SC, Lam MF, Lai KN. Size-dependent binding of IgA to HepG2, U937, and human mesangial cells. J Lab Clin Med (2002) 140:398–406. doi:10.1067/mlc.2002.129338
190. Leung JC, Tang SC, Chan LY, Tsang AW, Lan HY, Lai KN. Polymeric IgA increases the synthesis of macrophage migration inhibitory factor by human mesangial cells in IgA nephropathy. Nephrol Dial Transplant (2003) 18:36–45. doi:10.1093/ndt/18.1.36
191. Lai KN, Leung JC, Chan LY, Saleem MA, Mathieson PW, Lai FM, et al. Activation of podocytes by mesangial-derived TNF-α: glomerulo-podocytic communication in IgA nephropathy. Am J Physiol Renal Physiol (2008) 294:F945–55. doi:10.1152/ajprenal.00423.2007
192. Lai KN, Leung JC, Chan LY, Saleem MA, Mathieson PW, Tam KY, et al. Podocyte injury induced by mesangial-derived cytokines in IgA nephropathy. Nephrol Dial Transplant (2009) 24:62–72. doi:10.1093/ndt/gfn441
193. Yanagihara T, Brown R, Hall S, Moldoveanu Z, Goepfert A, Julian BA, et al. In vitro-formed immune complexes containing galactose-deficient IgA1 stimulate proliferation of mesangial cells. Results Immunol (2012) 2:166–72. doi:10.1016/j.rinim.2012.08.002
194. Huang ZQ, Anderson JC, Hall S, Rohrbach TD, Brown R, Julian BA, et al. Immune complexes from patients with IgA nephropathy containing galactose-deficient IgA1 and anti-glycan antibodies induce protein-kinase signaling and proliferation in cultured human mesangial cells. J Am Soc Nephrol (2011) 22:531A.
195. Huang ZQ, Anderson J, Rohrbach TD, Hall S, Brown R, Julian BA, et al. Characterization of signaling pathways in cultured human mesangial cells induced by IgA1-containing immune complexes from patients with IgA nephropathy. J Am Soc Nephrol (2012) 23:824A.
196. Muda AO, Feriozzi S, Rahimi S, Faraggiana T. Spatial arrangement of IgA and C3 as a prognostic indicator of IgA nephropathy. J Pathol (1995) 177:201–8. doi:10.1002/path.1711770214
197. Abrera-Abeleda MA, Nishimura C, Smith JL, Sethi S, McRae JL, Murphy BF, et al. Variations in the complement regulatory genes factor H (CFH) and factor H related 5 (CFHR5) are associated with membranoproliferative glomerulonephritis type II (dense deposit disease). J Med Genet (2006) 43:582–9. doi:10.1136/jmg.2005.038315
198. Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet (2010) 19:4694–704. doi:10.1093/hmg/ddq399
199. Zhu L, Zhai YL, Wang FM, Hou P, Lv JC, Xu DM, et al. Variants in complement factor H and complement factor H-related protein genes, CFHR3 and CFHR1, affect complement activation in IgA nephropathy. J Am Soc Nephrol (2015) 26:1195–204. doi:10.1681/ASN.2014010096
200. Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, et al. Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol (2015) 26:1503–12. doi:10.1681/ASN.2014101000
201. Maillard N, Boerma L, Hall S, Huang ZQ, Mrug M, Moldoveanu Z, et al. Proteomic analysis of engineered IgA1-IgG immune complexes reveals association with activated complement C3. J Am Soc Nephrol (2013) 24:490A.
202. Miyazaki R, Kuroda M, Akiyama T, Otani I, Tofuku Y, Takeda R. Glomerular deposition and serum levels of complement control proteins in patients with IgA nephropathy. Clin Nephrol (1984) 21:335–40.
203. Sahu A, Lambris JD. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol Rev (2001) 180:35–48. doi:10.1034/j.1600-065X.2001.1800103.x
204. Cybulsky AV, Takano T, Papillon J, McTavish AJ. Complement-induced phospholipase A2 activation in experimental membranous nephropathy. Kidney Int (2000) 57:1052–62. doi:10.1046/j.1523-1755.2000.00932.x
205. Qiu W, Zhou J, Zhu G, Zhao D, He F, Zhang J, et al. Sublytic C5b-9 triggers glomerular mesangial cell apoptosis via XAF1 gene activation mediated by p300-dependent IRF-1 acetylation. Cell Death Dis (2014) 5:e1176. doi:10.1038/cddis.2014.153
206. Evans DJ, Williams DG, Peters DK, Sissons JG, Boulton-Jones JM, Ogg CS, et al. Glomerular deposition of properdin in Henoch-Schönlein syndrome and idiopathic focal nephritis. Br Med J (1973) 3:326–8. doi:10.1136/bmj.3.5875.326
208. Wyatt RJ. The complement system in IgA nephropathy and Henoch-Schönlein purpura: functional and genetic aspects. Contrib Nephrol (1993) 104:82–91. doi:10.1159/000422400
209. Tomino Y, Sakai H, Nomoto Y, Endoh M, Arimori S, Fujita T. Deposition of C4-binding protein and ß 1H globulin in kidneys of patients with IgA nephropathy. Tokai J Exp Clin Med (1981) 6:217–22.
210. Rauterberg EW, Lieberknecht HM, Wingen AM, Ritz E. Complement membrane attack (MAC) in idiopathic IgA-glomerulonephritis. Kidney Int (1987) 31:820–9. doi:10.1038/ki.1987.72
211. Lagrue G, Branellec A, Intrator L, Moisy M, Sobel A. [Measurements of serum C3d in primitive chronic glomerular nephropathies (author’s transl)]. Nouv Presse Med (1979) 8:1153–6.
212. Solling J. Circulating immune complexes and complement breakdown product C3d in glomerulonephritis and kidney transplantation. Acta Pathol Microbiol Immunol Scand C (1984) 92:213–20.
213. Wyatt RJ, Kanayama Y, Julian BA, Negoro N, Sugimoto S, Hudson EC, et al. Complement activation in IgA nephropathy. Kidney Int (1987) 31:1019–23. doi:10.1038/ki.1987.101
214. Hiemstra PS, Gorter A, Stuurman ME, Van Es LA, Daha MR. Activation of the alternative pathway of complement by human serum IgA. Eur J Immunol (1987) 17:321–6. doi:10.1002/eji.1830170304
215. Russell MW, Mansa B. Complement-fixing properties of human IgA antibodies. Alternative pathway complement activation by plastic-bound, but not specific antigen-bound, IgA. Scand J Immunol (1989) 30:175–83. doi:10.1111/j.1365-3083.1989.tb01199.x
216. Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs). Mol Immunol (2013) 56:170–80. doi:10.1016/j.molimm.2013.06.001
217. Endo M, Ohi H, Ohsawa I, Fujita T, Matsushita M, Fujita T. Glomerular deposition of mannose-binding lectin (MBL) indicates a novel mechanism of complement activation in IgA nephropathy. Nephrol Dial Transplant (1998) 13:1984–90. doi:10.1093/ndt/13.8.1984
218. Faria B, Henriques C, Matos AC, Daha MR, Pestana M, Seelen M. Combined C4d and CD3 immunostaining predicts immunoglobulin (Ig)A nephropathy progression. Clin Exp Immunol (2015) 179:354–61. doi:10.1111/cei.12461
219. Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol (2001) 167:2861–8. doi:10.4049/jimmunol.167.5.2861
220. Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol (2006) 17:1724–34. doi:10.1681/ASN.2005090923
221. Espinosa M, Ortega R, Sanchez M, Segarra A, Salcedo MT, Gonzalez F, et al. Association of C4d deposition with clinical outcomes in IgA nephropathy. Clin J Am Soc Nephrol (2014) 9:897–904. doi:10.2215/CJN.09710913
222. D’Amico G, Imbasciati E, Barbiano Di Belgioioso G, Bertoli S, Fogazzi G, Ferrario F, et al. Idiopathic IgA mesangial nephropathy. Clinical and histological study of 374 patients. Medicine (Baltimore) (1985) 64:49–60. doi:10.1097/00005792-198501000-00004
223. Miyamoto H, Yoshioka K, Takemura T, Akano N, Maki S. Immunohistochemical study of the membrane attack complex of complement in IgA nephropathy. Virchows Arch A Pathol Anat Histopathol (1988) 413:77–86. doi:10.1007/BF00844284
224. Onda K, Ohsawa I, Ohi H, Tamano M, Mano S, Wakabayashi M, et al. Excretion of complement proteins and its activation marker C5b-9 in IgA nephropathy in relation to renal function. BMC Nephrol (2011) 12:64. doi:10.1186/1471-2369-12-64
225. Zhang J, Li Y, Shan K, Wang L, Qiu W, Lu Y, et al. Sublytic C5b-9 induces IL-6 and TGF-ß1 production by glomerular mesangial cells in rat Thy-1 nephritis through p300-mediated C/EBPß acetylation. FASEB J (2014) 28:1511–25. doi:10.1096/fj.13-242693
226. Nangaku M, Shankland SJ, Couser WG. Cellular response to injury in membranous nephropathy. J Am Soc Nephrol (2005) 16:1195–204. doi:10.1681/ASN.2004121098
227. Waldo FB, Cochran AM. Mixed IgA-IgG aggregates as a model of immune complexes in IgA nephropathy. J Immunol (1989) 142:3841–6.
228. Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al. C3 glomerulopathy: consensus report. Kidney Int (2013) 84:1079–89. doi:10.1038/ki.2013.377
229. van den Dobbelsteen ME, Verhasselt V, Kaashoek JG, Timmerman JJ, Schroeijers WE, Verweij CL, et al. Regulation of C3 and factor H synthesis of human glomerular mesangial cells by IL-1 and interferon-gamma. Clin Exp Immunol (1994) 95:173–80. doi:10.1111/j.1365-2249.1994.tb06033.x
230. Schmitt R, Stahl AL, Olin AI, Kristoffersson AC, Rebetz J, Novak J, et al. The combined role of galactose-deficient IgA1 and streptococcal IgA-binding M protein in inducing IL-6 and C3 secretion from human mesangial cells: implications for IgA nephropathy. J Immunol (2014) 193:317–26. doi:10.4049/jimmunol.1302249
231. Wan JX, Fukuda N, Endo M, Tahira Y, Yao EH, Matsuda H, et al. Complement 3 is involved in changing the phenotype of human glomerular mesangial cells. J Cell Physiol (2007) 213:495–501. doi:10.1002/jcp.21129
232. Komatsu H, Fujimoto S, Hara S, Sato Y, Yamada K, Eto T. Relationship between serum IgA/C3 ratio and progression of IgA nephropathy. Intern Med (2004) 43:1023–8. doi:10.2169/internalmedicine.43.1023
233. Zhang J, Wang C, Tang Y, Peng H, Ye ZC, Li CC, et al. Serum immunoglobulin A/C3 ratio predicts progression of immunoglobulin A nephropathy. Nephrology (2013) 18:125–31. doi:10.1111/nep.12010
234. Mizerska-Wasiak M, Maldyk J, Rybi-Szuminska A, Wasilewska A, Miklaszewska M, Pietrzyk J, et al. Relationship between serum IgA/C3 ratio and severity of histological lesions using the Oxford classification in children with IgA nephropathy. Pediatr Nephrol (2015) 30:1113–20. doi:10.1007/s00467-014-3024-z
235. Onda K, Ohi H, Tamano M, Ohsawa I, Wakabayashi M, Horikoshi S, et al. Hypercomplementemia in adult patients with IgA nephropathy. J Clin Lab Anal (2007) 21:77–84. doi:10.1002/jcla.20154
236. Edey M, Strain L, Ward R, Ahmed S, Thomas T, Goodship TH. Is complement factor H a susceptibility factor for IgA nephropathy? Mol Immunol (2009) 46:1405–8. doi:10.1016/j.molimm.2008.12.002
237. Zhang JJ, Jiang L, Liu G, Wang SX, Zou WZ, Zhang H, et al. Levels of urinary complement factor H in patients with IgA nephropathy are closely associated with disease activity. Scand J Immunol (2009) 69:457–64. doi:10.1111/j.1365-3083.2009.02234.x
238. Kim SJ, Koo HM, Lim BJ, Oh HJ, Yoo DE, Shin DH, et al. Decreased circulating C3 levels and mesangial C3 deposition predict renal outcome in patients with IgA nephropathy. PLoS One (2012) 7:e40495. doi:10.1371/journal.pone.0040495
239. Nasri H, Sajjadieh S, Mardani S, Momeni A, Merikhi A, Madihi Y, et al. Correlation of immunostaining findings with demographic data and variables of Oxford classification in IgA nephropathy. J Nephropathol (2013) 2:190–5. doi:10.12860/JNP.2013.30
240. Heinen S, Hartmann A, Lauer N, Wiehl U, Dahse HM, Schirmer S, et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood (2009) 114:2439–47. doi:10.1182/blood-2009-02-205641
241. Maeng YI, Kim MK, Park JB, Cho CH, Oh HK, Sung WJ, et al. Glomerular and tubular C4d depositions in IgA nephropathy: relations with histopathology and with albuminuria. Int J Clin Exp Pathol (2013) 6:904–10.
242. Ning L, Kurihara H, de Vega S, Ichikawa-Tomikawa N, Xu Z, Nonaka R, et al. Laminin α1 regulates age-related mesangial cell proliferation and mesangial matrix accumulation through the TGF-ß pathway. Am J Pathol (2014) 184:1683–94. doi:10.1016/j.ajpath.2014.02.006
243. Tamouza H, Chemouny JM, Raskova Kafkova L, Berthelot L, Flamant M, Demion M, et al. The IgA1 immune complex-mediated activation of the MAPK/ERK kinase pathway in mesangial cells is associated with glomerular damage in IgA nephropathy. Kidney Int (2012) 82:1284–96. doi:10.1038/ki.2012.192
244. Castro NE, Kato M, Park JT, Natarajan R. Transforming growth factor ß1 (TGF-ß1) enhances expression of profibrotic genes through a novel signaling cascade and microRNAs in renal mesangial cells. J Biol Chem (2014) 289:29001–13. doi:10.1074/jbc.M114.600783
245. Wunsche C, Koch A, Goldschmeding R, Schwalm S, Meyer Zu Heringdorf D, Huwiler A, et al. Transforming growth factor ß2 (TGF-ß2)-induced connective tissue growth factor (CTGF) expression requires sphingosine 1-phosphate receptor 5 (S1P5) in human mesangial cells. Biochim Biophys Acta (2015) 1851:519–26. doi:10.1016/j.bbalip.2015.01.003
246. Diven SC, Caflisch CR, Hammond DK, Weigel PH, Oka JA, Goldblum RM. IgA induced activation of human mesangial cells: Independent of FcαR1 (CD 89). Kidney Int (1998) 54:837–47. doi:10.1046/j.1523-1755.1998.00054.x
247. Westerhuis R, Van Zandbergen G, Verhagen NA, Klar-Mohamad N, Daha MR, van Kooten C. Human mesangial cells in culture and in kidney sections fail to express Fcα receptor (CD89). J Am Soc Nephrol (1999) 10:770–8.
248. Leung JC, Tsang AW, Chan DT, Lai KN. Absence of CD89, polymeric immunoglobulin receptor, and asialoglycoprotein receptor on human mesangial cells. J Am Soc Nephrol (2000) 11:241–9.
249. Barratt J, Greer MR, Pawluczyk IZ, Allen AC, Bailey EM, Buck KS, et al. Identification of a novel Fcα receptor expressed by human mesangial cells. Kidney Int (2000) 57:1936–48. doi:10.1046/j.1523-1755.2000.00043.x
250. van Egmond M, Bakema JE, Woof JM. Fc receptors in mucosal immunology. 4th ed. In: Mestecky J, Strober W, Russell MW, Kelsall BL, Cheroute H, Lambrecht BN, editors. Mucosal Immunology. Amsterdam: Elsevier/Academic Press (2015). p. 2023–38.
251. Haddad E, Moura IC, Arcos-Fajardo M, Macher MA, Baudouin V, Alberti C, et al. Enhanced expression of the CD71 mesangial IgA1 receptor in Berger disease and Henoch-Schönlein nephritis: association between CD71 expression and IgA deposits. J Am Soc Nephrol (2003) 14:327–37. doi:10.1097/01.ASN.0000046961.04917.83
252. Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, Benhamou M, Novak J, et al. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol (2004) 15:622–34. doi:10.1097/01.ASN.0000115401.07980.0C
253. Berthelot L, Papista C, Maciel TT, Biarnes-Pelicot M, Tissandie E, Wang PH, et al. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med (2012) 209:793–806. doi:10.1084/jem.20112005
254. Kaneko Y, Otsuka T, Tsuchida Y, Gejyo F, Narita I. Integrin α1/ß1 and α2/ß1 as a receptor for IgA1 in human glomerular mesangial cells in IgA nephropathy. Int Immunol (2012) 24:219–32. doi:10.1093/intimm/dxr125
255. Kaetzel C, Russell MW. Phylogeny and comparative physiology of mucosal immunoglobulins. 4th ed. In: Mestecky J, Strober W, Russell MW, Kelsall BL, Cheroute H, Lambrecht BN, editors. Mucosal Immunology. Amsterdam: Elsevier/Academic Press (2015). p. 325–48.
256. Suzuki H, Suzuki Y, Novak J, Tomino Y. Development of animal models of human IgA nephropathy. Drug Discov Today Dis Models (2014) 11:5–11. doi:10.1016/j.ddmod.2014.07.002
257. Imai H, Nakamoto Y, Asakura K, Miki K, Yasuda T, Miura AB. Spontaneous glomerular IgA deposition in ddY mice: an animal model of IgA nephritis. Kidney Int (1985) 27:756–61. doi:10.1038/ki.1985.76
258. Muso E, Yoshida H, Takeuchi E, Yashiro M, Matsushima H, Oyama A, et al. Enhanced production of glomerular extracellular matrix in a new mouse strain of high serum IgA ddY mice. Kidney Int (1996) 50:1946–57. doi:10.1038/ki.1996.517
259. Suzuki H, Suzuki Y, Yamanaka T, Hirose S, Nishimura H, Toei J, et al. Genome-wide scan in a novel IgA nephropathy model identifies a susceptibility locus on murine chromosome 10, in a region syntenic to human IGAN1 on chromosome 6q22-23. J Am Soc Nephrol (2005) 16:1289–99. doi:10.1681/ASN.2004030219
260. Okazaki K, Suzuki Y, Otsuji M, Suzuki H, Kihara M, Kajiyama T, et al. Development of a model of early-onset IgA nephropathy. J Am Soc Nephrol (2012) 23:1364–74. doi:10.1681/ASN.2011121160
261. Schroeder C, Osman AA, Roggenbuck D, Mothes T. IgA-gliadin antibodies, IgA-containing circulating immune complexes, and IgA glomerular deposits in wasting marmoset syndrome. Nephrol Dial Transplant (1999) 14:1875–80. doi:10.1093/ndt/14.8.1875
262. Nishie T, Miyaishi O, Azuma H, Kameyama A, Naruse C, Hashimoto N, et al. Development of immunoglobulin A nephropathy-like disease in ß-1,4-galactosyltransferase-I-deficient mice. Am J Pathol (2007) 170:447–56. doi:10.2353/ajpath.2007.060559
263. Launay P, Grossetete B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, et al. Fcα receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease): evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J Exp Med (2000) 191:1999–2009. doi:10.1084/jem.191.11.1999
264. Moldoveanu Z, Suzuki H, Satake K, Suzuki Y, Novak L, Huang ZQ, et al. IgA nephropathy: a murine model that displays typical IgAN pathology after passive administration of immune complexes. J Am Soc Nephrol (2012) 23:519A.
265. Novak L, Moldoveanu Z, Huang ZQ, Winstead CJ, Hall S, Brown R, et al. Assessment of glomerular changes in a passive mouse model of IgA nephropathy. J Am Soc Nephrol (2013) 24:570A.
266. Boyd JK, Barratt J. Immune complex formation in IgA nephropathy: CD89 a ‘saint’ or a ‘sinner’? Kidney Int (2010) 78:1211–3. doi:10.1038/ki.2010.365
267. Vuong MT, Hahn-Zoric M, Lundberg S, Gunnarsson I, van Kooten C, Wramner L, et al. Association of soluble CD89 levels with disease progression but not susceptibility in IgA nephropathy. Kidney Int (2010) 78:1281–7. doi:10.1038/ki.2010.314
268. Glassock RJ. The pathogenesis of IgA nephropathy. Curr Opin Nephrol Hypertens (2011) 20:153–60. doi:10.1097/MNH.0b013e3283436f5c
269. Lai KN. Pathogenesis of IgA nephropathy. Nat Rev Nephrol (2012) 8:275–83. doi:10.1038/nrneph.2012.58
270. Kiryluk K, Novak J. The genetics and immunobiology of IgA nephropathy. J Clin Invest (2014) 124:2325–32. doi:10.1172/JCI74475
271. Berthoux F, Suzuki H, Thibaudin L, Yanagawa H, Maillard N, Mariat C, et al. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol (2012) 23:1579–87. doi:10.1681/ASN.2012010053
272. Berthelot L, Robert T, Vuiblet V, Tabary T, Braconnier A, Drame M, et al. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int (2015) 88:815–22. doi:10.1038/ki.2015.158
273. Julian BA, Quiggins PA, Thompson JS, Woodford SY, Gleason K, Wyatt RJ. Familial IgA nephropathy. Evidence of an inherited mechanism of disease. N Engl J Med (1985) 312:202–8. doi:10.1056/NEJM198501243120403
274. Kiryluk K, Novak J, Gharavi AG. Pathogenesis of immunoglobulin A nephropathy: recent insight from genetic studies. Annu Rev Med (2013) 64:339–56. doi:10.1146/annurev-med-041811-142014
275. Feehally J, Farrall M, Boland A, Gale DP, Gut I, Heath S, et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol (2010) 21:1791–7. doi:10.1681/ASN.2010010076
276. Yu XQ, Li M, Zhang H, Low HQ, Wei X, Wang JQ, et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet (2012) 44:178–82. doi:10.1038/ng.1047
277. Li M, Foo JN, Wang JQ, Low HQ, Tang XQ, Toh KY, et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat Commun (2015) 6:7270. doi:10.1038/ncomms8270
278. Hastings MC, Moldoveanu Z, Suzuki H, Berthoux F, Julian BA, Sanders JT, et al. Biomarkers in IgA nephropathy: relationship to pathogenetic hits. Expert Opin Med Diagn (2013) 7:615–27. doi:10.1517/17530059.2013.856878
279. Hewitt SM, Dear J, Star RA. Discovery of protein biomarkers for renal diseases. J Am Soc Nephrol (2004) 15:1677–89. doi:10.1097/01.ASN.0000129114.92265.32
280. Fliser D, Novak J, Thongboonkerd V, Argiles A, Jankowski V, Girolami MA, et al. Advances in urinary proteome analysis and biomarker discovery. J Am Soc Nephrol (2007) 18:1057–71. doi:10.1681/ASN.2006090956
281. Julian BA, Suzuki H, Suzuki Y, Tomino Y, Spasovski G, Novak J. Sources of urinary proteins and their analysis by urinary proteomics for the detection of biomarkers of disease. Proteomics Clin Appl (2009) 3:1029–43. doi:10.1002/prca.200800243
282. Hrvacevic R, Topalov D, Stojanovic R, Lilic D, Dimitrijevic J, Maksic D, et al. [Serum and urinary interleukin-6 levels in patients with primary glomerulonephritis]. Srp Arh Celok Lek (1996) 124(Suppl 1):40–2.
283. Harada K, Akai Y, Kurumatani N, Iwano M, Saito Y. Prognostic value of urinary interleukin 6 in patients with IgA nephropathy: an 8-year follow-up study. Nephron (2002) 92:824–6. doi:10.1159/000065465
284. Ranieri E, Gesualdo L, Petrarulo F, Schena FP. Urinary IL-6/EGF ratio: a useful prognostic marker for the progression of renal damage in IgA nephropathy. Kidney Int (1996) 50:1990–2001. doi:10.1038/ki.1996.521
285. Grandaliano G, Gesualdo L, Ranieri E, Monno R, Montinaro V, Marra F, et al. Monocyte chemotactic peptide-1 expression in acute and chronic human nephritides: a pathogenetic role in interstitial monocytes recruitment. J Am Soc Nephrol (1996) 7:906–13.
286. Huang F, Horikoshi S, Kurusu A, Shibata T, Suzuki S, Funabiki K, et al. Urinary levels of interleukin-8 (IL-8) and disease activity in patients with IgA nephropathy. J Clin Lab Anal (2001) 15:30–4. doi:10.1002/1098-2825(2001)15:1<30::AID-JCLA6>3.0.CO;2-X
287. Machii R, Sakatume M, Kubota R, Kobayashi S, Gejyo F, Shiba K. Examination of the molecular diversity of α1 antitrypsin in urine: deficit of an α1 globulin fraction on cellulose acetate membrane electrophoresis. J Clin Lab Anal (2005) 19:16–21. doi:10.1002/jcla.20049
288. Mitsuhashi H, Tsukada Y, Ono K, Yano S, Naruse T. Urine glycosaminoglycans and heparan sulfate excretions in adult patients with glomerular diseases. Clin Nephrol (1993) 39:231–8.
289. Galla JH, Spotswood MF, Harrison LA, Mestecky J. Urinary IgA in IgA nephropathy and Henoch-Schönlein purpura. J Clin Immunol (1985) 5:298–306. doi:10.1007/BF00918248
290. Matousovic K, Novak J, Yanagihara T, Tomana M, Moldoveanu Z, Kulhavy R, et al. IgA-containing immune complexes in the urine of IgA nephropathy patients. Nephrol Dial Transplant (2006) 21:2478–84. doi:10.1093/ndt/gfl240
291. Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S. Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol (2000) 11:700–7.
292. Kusunoki Y. [Terminal complement complex (TTC) levels in plasma and urine from glomerular diseases: enzyme-linked immunosorbent assay (ELISA) using monoclonal antibody against neoantigens of TCC]. Hokkaido Igaku Zasshi (1990) 65:74–85.
293. Halling SF, Soderberg MP, Berg UB. Henoch Schönlein nephritis: clinical findings related to renal function and morphology. Pediatr Nephrol (2005) 20:46–51. doi:10.1007/s00467-004-1650-6
294. Good DM, Thongboonkerd V, Novak J, Bascands JL, Schanstra JP, Coon JJ, et al. Body fluid proteomics for biomarker discovery: lessons from the past hold the key to success in the future. J Proteome Res (2007) 6:4549–55. doi:10.1021/pr070529w
295. Coon JJ, Zurbig P, Dakna M, Dominiczak AF, Decramer S, Fliser D, et al. CE-MS analysis of the human urinary proteome for biomarker discovery and disease diagnostics. Proteomics Clin Appl (2008) 2:964. doi:10.1002/prca.200800024
296. Decramer S, Gonzalez de Peredo A, Breuil B, Mischak H, Monsarrat B, Bascands JL, et al. Urine in clinical proteomics. Mol Cell Proteomics (2008) 7:1850–62. doi:10.1074/mcp.R800001-MCP200
297. Julian BA, Suzuki H, Spasovski G, Suzuki Y, Tomino Y, Novak J. Application of proteomic analysis to renal disease in the clinic. Proteomics Clin Appl (2009) 3:1023–8. doi:10.1002/prca.200800244
298. Mischak H, Coon JJ, Novak J, Weissinger EM, Schanstra JP, Dominiczak AF. Capillary electrophoresis-mass spectrometry as a powerful tool in biomarker discovery and clinical diagnosis: an update of recent developments. Mass Spectrom Rev (2009) 28:703–24. doi:10.1002/mas.20205
299. Good DM, Zurbig P, Argiles A, Bauer HW, Behrens G, Coon JJ, et al. Naturally occurring human urinary peptides for use in diagnosis of chronic kidney disease. Mol Cell Proteomics (2010) 9:2424–37. doi:10.1074/mcp.M110.001917
300. Mischak H, Allmaier G, Apweiler R, Attwood T, Baumann M, Benigni A, et al. Recommendations for biomarker identification and qualification in clinical proteomics. Sci Transl Med (2010) 2:46s42. doi:10.1126/scitranslmed.3001249
301. Mischak H, Ioannidis JP, Argiles A, Attwood TK, Bongcam-Rudloff E, Broenstrup M, et al. Implementation of proteomic biomarkers: making it work. Eur J Clin Invest (2012) 42:1027–36. doi:10.1111/j.1365-2362.2012.02674.x
302. Haubitz M, Wittke S, Weissinger EM, Walden M, Rupprecht HD, Floege J, et al. Urine protein patterns can serve as diagnostic tools in patients with IgA nephropathy. Kidney Int (2005) 67:2313–20. doi:10.1111/j.1523-1755.2005.00335.x
303. Rossing K, Mischak H, Parving HH, Christensen PK, Walden M, Hillmann M, et al. Impact of diabetic nephropathy and angiotensin II receptor blockade on urinary polypeptide patterns. Kidney Int (2005) 68:193–205. doi:10.1111/j.1523-1755.2005.00394.x
304. Decramer S, Wittke S, Mischak H, Zurbig P, Walden M, Bouissou F, et al. Predicting the clinical outcome of congenital unilateral ureteropelvic junction obstruction in newborn by urinary proteome analysis. Nat Med (2006) 12:398–400. doi:10.1038/nm1384
305. Theodorescu D, Wittke S, Ross MM, Walden M, Conaway M, Just I, et al. Discovery and validation of new protein biomarkers for urothelial cancer: a prospective analysis. Lancet Oncol (2006) 7:230–40. doi:10.1016/S1470-2045(06)70584-8
306. Julian BA, Wittke S, Novak J, Good DM, Coon JJ, Kellmann M, et al. Electrophoretic methods for analysis of urinary polypeptides in IgA-associated renal diseases. Electrophoresis (2007) 28:4469–83. doi:10.1002/elps.200700237
307. Mischak H, Kolch W, Aivaliotis M, Bouyssie D, Court M, Dihazi H, et al. Comprehensive human urine standards for comparability and standardization in clinical proteome analysis. Proteomics Clin Appl (2010) 4:464–78. doi:10.1002/prca.200900189
Keywords: IgA, nephropathy, immune complexes, autoantibodies, complement C3
Citation: Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, Raska M, Renfrow MB, Julian BA and Novak J (2016) The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front. Immunol. 7:117. doi: 10.3389/fimmu.2016.00117
Received: 30 January 2016; Accepted: 15 March 2016;
Published: 12 April 2016
Edited by:
Anil Chauhan, Saint Louis University, USAReviewed by:
Cees Van Kooten, Leiden University Medical Center, NetherlandsCopyright: © 2016 Knoppova, Reily, Maillard, Rizk, Moldoveanu, Mestecky, Raska, Renfrow, Julian and Novak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Novak, amFubm92YWtAdWFiLmVkdQ==
†Barbora Knoppova, Colin Reily, and Nicolas Maillard have contributed equally.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.