 Guo-Min Deng
Guo-Min Deng Vasileios C. Kyttaris3
Vasileios C. Kyttaris3 George C. Tsokos
George C. Tsokos
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 07 March 2016
Sec. Alloimmunity and Transplantation
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00078
This article is part of the Research Topic Molecular Mechanisms of Immune Complex Pathophysiology View all 8 articles
Spleen tyrosine kinase (Syk) is a member of the Src family of non-receptor tyrosine kinases, which associates directly with surface receptors, including B-cell receptor and Fcγ receptor, and is involved in a variety of signal transduction pathways. Rheumatoid arthritis (RA) and systemic lupus erythematosus are autoimmune diseases in which autoantibodies, immune complexes, and autoreactive T cells account for the expression of tissue inflammation and damage. Syk inhibitors efficiently suppress RA in patients albeit in the expression of unwanted side effects, including gastrointestinal effects, hypertension, and neutropenia. Syk inhibitors also inhibit clinical manifestations in lupus-prone mice. Here, we review the evidence that supports the use of Syk inhibitors to treat rheumatic and other autoimmune diseases.
Spleen tyrosine kinase (Syk) is a cytoplasmic protein-tyrosine kinase and a member of the Src family of non-receptor tyrosine kinases (1). The Syk protein contains a pair of Src homology 2 (SH2) domains at the N-terminus that are joined to each other by linker A and are separated by a longer linker B from the catalytic domain (2, 3). Syk is activated when the tandem SH2 domains are engaged or when tyrosines participating in the linker–kinase sandwich become phosphorylated. SH2 domains are structural motifs that bind phosphotyrosine to enhance protein–protein interactions (4, 5). These high affinity Syk-binding sites are known as immunoreceptor tyrosine-based activation motifs or ITAMs, which are located in many important receptors (6). Syk physically docks to the doubly phosphorylated ITAM via its tandem SH2 domains in a head-to-tail orientation. Conformational changes disrupt the “linker–kinase sandwich” and activate the enzyme (7).
Spleen tyrosine kinase catalyzes the phosphorylation of proteins on tyrosines located at sites (8). Signals are further transmitted from the Syk-receptor complex through the phosphorylation of adapter proteins, such as BLNK/SLP-65, SLP-76, and LAT (3, 9). These phosphorylated proteins serve as scaffolds to which effectors dock with SH2 or other related phosphotyrosine-binding motifs. Effectors include members of the Tec-family of tyrosine kinases, lipid kinases, phospholipases, and guanine nucleotide exchange factors that further propagate the signal allowing for the activation of multiple pathways, including PI3K/Akt, Ras/ERK, PLCγ/NFAT, Vav-1/Rac, and IKK/NFκB (2, 3).
Spleen tyrosine kinase is widely expressed in the hematopoietic system and is involved in a variety of signal transduction pathways, including receptor signaling in mast cells, monocytes, osteoclasts, and T, B cells (10–16) (Figure 1). In this review, we discuss the role of Syk in Fcγ receptor (FcγR) signaling and the effect of Syk inhibitor in treatment of autoimmune diseases.
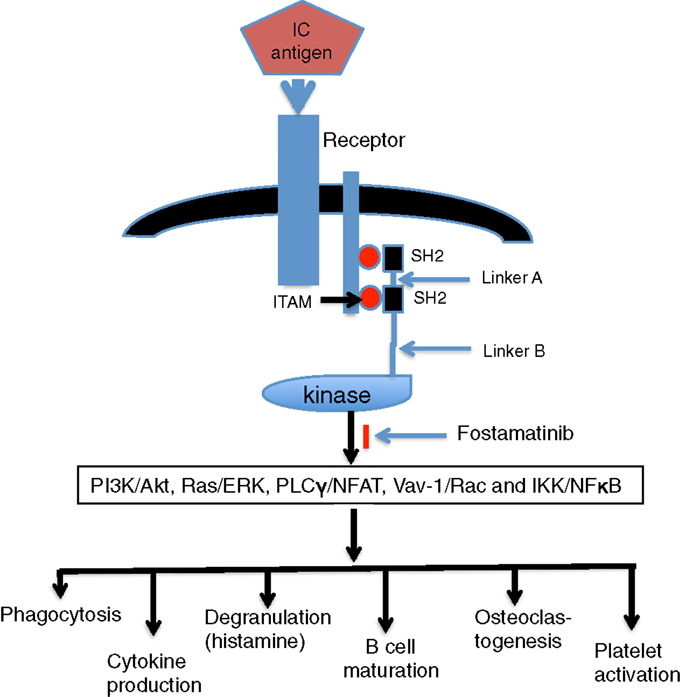
Figure 1. Engagement of Syk and downstream effects. Following aggregation of FcR by immune complex (IC), the phosphorylation of ITAM tyrosine leads to the recruitment of Syk to the receptor in an interaction mediated by its tandem pair of SH2 domains. Active Syk initiates signaling pathways of PI3K/Akt, Ras/ERK, PLCγ/NFAT, Vav-1/Rac, and IKK/NFκB and then generates downstream effects, such as phagocytosis, cytokine production, degranulation, B-cell maturation, osteoclastogenesis, and platelet activation.
IgG is recognized by FcγR, and IgG–antigen (Ags) complexes bind to FcγR on immune cells to mediate inflammatory immune responses. There are three kinds of FcγR: FcγRI, FcγRIIA, and FcγRIIIA. IgG-binding FcγR induces activation of Syk through ITAMs defined by these receptors (17). Receptor engagement enhances the phagocytosis of IgG-opsonized particles and the production of cytokines, nitric oxide, and reactive oxygen species, which promote the killing of microbes and cause tissue inflammatory damage. Syk-deficient macrophages cannot phagocytose IgG-coated particles, and Syk-deficient neutrophils fail to undergo an oxidative burst in response to the engagement of FcγRs (18, 19). In neutrophils, integrins signal through an association with either FcγR or DAP12, another ITAM-containing accessory protein, and Syk is required for adhesion-dependent activation (20).
Type II and Type III hypersensitivity reactions are mediated by IgG that interacts with bound and soluble Ags, respectively, and are responsible for the inflammation that accompanies many autoimmune diseases.
B and T cells have been shown to exert an important role in the pathogenesis of autoimmune diseases (21). The T cell receptor (TCR) is associated with the CD3 complex, which includes a dimer of ζ chains each of which contains three ITAMs (15). TCR engagement triggers the phosphorylation of ζ chain ITAM tyrosines that leads to the binding of Zap-70. B cells are responsible for production of IgG and are activated through the B-cell receptor (BCR). BCR consists of a membrane spanning immunoglobulin in association with two signaling adaptors: CD79a (Ig-α) and CD79b (Ig-β), each of which contains a single ITAM (2, 3). Syk-deficient mice lack mature B cells (22). Disruption of the Syk gene in DT40 B cells blocks essentially all BCR-stimulated signaling pathways (23).
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by high levels of autoantibodies and multiorgan tissue damage. The TCR–CD3 complex in SLE T cells is rewired in that the levels of CD3ζ is decreased, and its place is taken by FcγR, which recruits Syk and not Zap-70 as its signaling partner (24). Much of the altered gene expression that characterizes SLE T cells (e.g., increased expression of IL-21, CD44, PP2A, and OAS2) can be induced by the overexpression of Syk in normal T cells (25). High level of autoantibodies in serum and IgG deposition in tissues typify SLE. Circulating immune complexes (ICs) and primarily those formed in situ are important in the expression of the inflammatory response (20).
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by joint inflammation and bone destruction (26). T cells (especially Th1 and Th17 cells) are important in the pathogenesis of RA (27, 28). Recently, follicular helper T (Tfh) cells, whose primary task is to drive the formation of B cell responses, have been recognized as critical regulators of autoimmunity (29, 30). Levels of pSyk in peripheral blood B cells are preferentially higher in patients with RA compared to healthy subjects. Patients with significantly higher pSyk levels are strongly positive for anti-citrullinated protein antibodies (31). Mice deficient in FcγR or FcγRIII fail to develop collagen-induced arthritis (32), and genetic deficiency of Syk protects mice from autoantibody-induced arthritis (33). The depletion of Syk from neutrophils alone is effective in blocking joint inflammation in autoantibody-induced arthritis (34), and direct injection of naked Syk siRNA into joints inhibits the development of arthritis (35).
Systemic sclerosis (SSc) is a chronic autoimmune disease with a high morbidity and mortality. Skin and organ fibrosis are key manifestations of SSc, and pathogenesis remains unclear (36). Syk inhibitor fostamatinib was demonstrated to limit tissue damage and fibrosis in a scleroderma mouse model (37). It indicates that the Syk pathway appears as a potential molecular target for therapeutic intervention in SSc.
Thrombocytopenic purpura (ITP) and heparin-induced thrombocytopenia (HIT) are autoimmune diseases in which autoantibodies against Ags on platelets result in platelet activation and the opsonization and phagocytosis of both platelets and megakaryocytes by macrophages. Syk inhibitors block IC-mediated platelet activation through FcγRIIA in a mouse model of HIT (38). Fostamatinib (a Syk inhibitor) blocks platelet loss induced by an antibody (Ab) against integrin αIIβ in a mouse model of ITP (39). A Phase II clinical trial in patients demonstrated that fostamatinib can restore platelet counts in approximately 50% of patients with ITP (39).
A highly specific Syk inhibitor, known as R406, has been shown to block Fc receptor signaling (40, 41). R788 (renamed fostamatinib) is a small molecule, water-soluble prodrug of the biologically active R406 and a potent inhibitor of Syk (42). The small molecule, R406 as well as R788, has been shown to inhibit the development of experimental arthritis (43, 44). In a randomized clinical Phase II trial, fostamatinib when added to background treatment with methotrexate at a stable dose was effective in the treatment of patients with RA (45, 46). Side effects included diarrhea, neutropenia, alanine transferase elevation, and increased blood pressure. Most side effects were associated with the higher doses of fostamatinib. Thus, although fostamatinib is a useful DMARD, its clinical use has been precluded by the recorded unexpected side effects.
Increased expression of Syk in SLE T cells affect the expression of a number of enzymes, cytokines, and receptors, which are important in disease pathogenesis, suggesting Syk may become therapeutic target in SLE patients (25). In addition, IgG is involved in the skin and kidney injury in SLE patients (21, 47), and intradermal injection of lupus serum IgG induces skin inflammation (47). The expression of Syk is increased in the skin lesion of lupus MRL/lpr mice (48), and the Syk inhibitor R788 completely abrogates skin inflammation induced by lupus serum (Deng, unpublished data). The Syk inhibitor R788 can prevent skin injury and also suppress established skin injury in lupus MRL/lpr mice. Interestingly, discontinuation of treatment results in extended suppression of skin disease for at least 8 weeks (48). Finally, a Syk inhibitor has also been demonstrated to prevent and improve, if administered after the beginning of the disease, of kidney damage in lupus-prone mice (48, 49).
Spleen tyrosine kinase is expressed in rheumatoid synovium, with activated phosphorylated Syk being differentially expressed between RA and OA synovium (41). Syk activation plays an essential role in TNF-α-induced cytokine production in fibroblast-like synoviocytes and RANKL-induced osteoclastogenesis (3, 41). Expression of Syk is abnormally increased in T cells of SLE patients (24) and skin lesion in lupus-prone mice (48). Expression of Syk is associated with disease progression in lupus-prone mice (48), thus expression of phosphorylated Syk may be worked as a parameter of pathology of RA and SLE.
In the trials of RA patients, side effects of Syk inhibitor fostamatinib (R788) were observed. These side effects include diarrhea, nausea, hypertension, dizziness, headaches, neutropenia, upper respiratory tract infections, and increased serum alanine transaminase (ALT) levels (45, 46, 50). Diarrhea and neutropenia are the two most common adverse events in the overall safety population. These side effects were dose dependent and were often reported with the 150 mg bd dose of fostamatinib. Diarrhea occurred in 6 (13%), 5 (11%), 8 (16%), and 21 (45%) of the patients in the placebo and R788 50, 100, and 150 mg groups, respectively (45, 50). The number of neutrophil returned to normal in all patients within 3–7 days after interruption or reduction of the fostamatinib dose (46). Neutropenia caused by Syk may be by Syk-impairing bone marrow neutrophil release, and concurrent MTX use may also play a role (51). Hypertension was a potential side effect of concern. The increase in blood pressure was observed at month 1 in the fostamatinib groups (45, 50). Increases in blood pressure were more pronounced in patients with existing hypertension at screening or baseline. All cases responded to conventional antihypertensive medication or reduction in fostamatinib dose. It has been postulated that an off target effect on vascular endothelial growth factor receptor 2 (VEGFR) may be responsible for hypertension (52).
Follicular dendritic cells (FDCs) are unique immune cells that contribute to the regulation of humoral immune responses. FDCs are located in the B-cell follicles of secondary lymphoid tissues, where they trap and retain Ags in the form of highly immunogenic ICs consisting of Ag plus specific Ab and/or complement proteins through Fc and C receptor (53, 54). FDC–FcγRIIB exerts an essential role in mediating IC periodicity, Ag-presentation, inducing germinal center (GC) reaction, and generating specific Ab responses. Binding of ICs to FDC–FcγRIIB induces FDC activation that leads to significant upregulation of FDC–ICAM-1, FDC–VCAM-1, and FDC–FcγRIIB itself (54). IC-bearing FDCs and autoreactive GCs frequently exist in autoimmune diseases (55, 56). Interference with FDC-reticula attenuates autoreactive GC formation, reduces pathogenic auto-Ab titers and memory B cells, and ameliorates arthritis (56–58). It has been recently demonstrated that FDC follicular units develop in RA synovium (56, 59). The high levels of FcγRIIB in FDCs protects the immunogenicity of FDC–ICs by minimizing serious inhibition of B-cell activation upon BCR/FcγRIIB crosslinking (54, 60). Actually, the expression of FcγRIIB is significantly reduced on RA memory B cells and plasmablasts, and these alterations on FcγRIIB are associated with high levels of anti-citrullinated vimentin auto-Abs (61). It is not clear whether Syk inhibitor fostamatinib blocks FDC activation and signal transduction.
Based on the evidence, Syk exerts an important role in the IgG/FcγR signaling pathway and in the aberrant signaling of SLE T cells. There is ample evidence from the study of human samples preclinical experiments that signaling involving Syk contributes to the pathogenesis of autoimmune diseases. Syk inhibitors efficiently suppress RA in patients albeit in the expression of unwanted side effects and raise platelet counts in patients with immune thrombocytopenia. In lupus-prone mice, systemic administration of Syk inhibitors results in the prevention or treatment of skin and kidney injury. It is hoped that more specific inhibitors of Syk devoid of side effects should prove of great clinical value.
G-MD organized and wrote manuscript, GT organized and edited the manuscript, and VK organized the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by fund from Nanjing Medical University (KY101RC071203).
1. Kurosaki T, Takata M, Yamanashi Y, Inazu T, Taniguchi T, Yamamoto T, et al. Syk activation by the Src-family tyrosine kinase in the B cell receptor signaling. J Exp Med (1994) 179(5):1725–9. doi: 10.1084/jem.179.5.1725
2. Geahlen RL. Syk and pTyr’d: signaling through the B cell antigen receptor. Biochim Biophys Acta (2009) 1793:1115–27. doi:10.1016/j.bbamcr.2009.03.004
3. Mócsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol (2010) 10:387–402. doi:10.1038/nri2765
4. Liu BA, Jablonowski K, Raina M, Arcé M, Pawson T, Nash PD. The human and mouse complement of SH2 domain proteins establishing the boundaries of phosphotyrosine signaling. Mol Cell (2006) 22:851–68. doi:10.1016/j.molcel.2006.06.001
5. Fütterer K, Wong J, Grucza RA, Chan AC, Waksman G. Structural basis for Syk tyrosine kinase ubiquity in signal transduction pathways revealed by the crystal structure of its regulatory SH2 domains bound to a dually phosphorylated ITAM peptide. J Mol Biol (1998) 281:523–37. doi:10.1006/jmbi.1998.1964
6. Abram CL, Lowell CA. The expanding role for ITAM-based signaling pathways in immune cells. Sci STKE (2007) 377:re2. doi:10.1126/stke.3772007re2
7. Grädler U, Schwarz D, Dresing V, Musil D, Bomke J, Frech M, et al. Structural and biophysical characterization of the Syk activation switch. J Mol Biol (2013) 425:309–33. doi:10.1016/j.jmb.2012.11.007
8. Xue L, Geahlen RL, Tao WA. Identification of direct kinase substrates based on protein kinase assay linked phosphoproteomics. Mol Cell Proteomics (2013) 12:2969–80. doi:10.1074/mcp.O113.027722
9. Yablonski D, Weiss A. Mechanisms of signaling by the hematopoietic-specific adaptor proteins, SLP-76 and LAT and their B cell counterpart, BLNK/SLP-65. Adv Immunol (2001) 79:93–128. doi:10.1016/S0065-2776(01)79003-7
10. LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol (2007) 8(6):630–8. doi:10.1038/ni1460
11. Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature (1995) 378(6554):303–6. doi:10.1038/378303a0
12. Choi OH, Kim JH, Kinet JP. Calcium mobilization via sphingosine kinase in signalling by the Fc epsilon RI antigen receptor. Nature (1996) 380(6575):634–6. doi:10.1038/380634a0
13. Darby C, Geahlen RL, Schreiber AD. Stimulation of macrophage Fc gamma RIIIA activates the receptor-associated protein tyrosine kinase Syk and induces phosphorylation of multiple proteins including p95Vav and p62/GAP-associated protein. J Immunol (1994) 152(11):5429–37.
14. Reeve JL, Zou W, Liu Y, Maltzman JS, Ross FP, Teitelbaum SL. SLP-76 couples Syk to the osteoclast cytoskeleton. J Immunol (2009) 183(3):1804–12. doi:10.4049/jimmunol.0804206
15. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol (2009) 27:591–619. doi:10.1146/annurev.immunol.021908.132706
16. Kurosaki T, Hikida M. Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev (2009) 228(1):132–48. doi:10.1111/j.1600-065X.2008.00748.x
17. Sanchez-Mejorada G, Rosales C. Signal transduction by immunoglobulin Fc receptors. J Leukoc Biol (1998) 63:521–33.
18. Van Ziffle JA, Lowell CA. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood (2009) 114:4871–82. doi:10.1182/blood-2009-05-220806
19. Kiefer F, Brumell J, Al-Alawi N, Latour S, Cheng A, Veillette A, et al. The Syk protein tyrosine kinase is essential for Fc receptor signaling in macrophages and neutrophils. Mol Cell Biol (1998) 18:4209–20. doi:10.1128/MCB.18.7.4209
20. Mócsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol (2006) 7:1326–33. doi:10.1038/ni1407
21. Tsokos GC. Systemic lupus erythematosus. N Engl J Med (2011) 365(22):2110–21. doi:10.1056/NEJMra1100359
22. Turner M, Mee PJ, Costello PS, Williams O, Price AA, Duddy LP, et al. Perinatal lethality and a block in the development of B cells in mice lacking the tyrosine kinase p72syk. Nature (1995) 378:298–302. doi:10.1038/378298a0
23. Takata M, Sabe H, Hata A, Inazu T, Homma Y, Nukada T, et al. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J (1994) 13:1341–9.
24. Moulton VR, Tsokos GC. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis Res Ther (2011) 13:207. doi:10.1186/ar3251
25. Grammatikos AP, Ghosh D, Devlin A, Kyttaris VC, Tsokos GC. Spleen tyrosine kinase (Syk) regulates systemic lupus erythematosus (SLE) T cell signaling. PLoS One (2013) 8:e74550. doi:10.1371/journal.pone.0074550
26. Deane KD, El-Gabalawy H. Pathogenesis and prevention of rheumatic disease: focus on preclinical RA and SLE. Nat Rev Rheumatol (2014) 10(4):212–28. doi:10.1038/nrrheum.2014.6
27. Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, et al. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis (2008) 67:1299–304. doi:10.1136/ard.2007.080341
28. Sato K. Th17 cells and rheumatoid arthritis – from the standpoint of osteoclast differentiation. Allergol Int (2008) 57:109–14. doi:10.2332/allergolint.R-07-158
29. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
30. Wang J, Shan Y, Jiang Z, Feng J, Li C, Ma L, et al. High frequencies of activated B cells and follicular helper T cells are correlated with disease activity in patients with new onset rheumatoid arthritis. Clin Exp Immunol (2013) 174:212–20. doi:10.1111/cei.12162
31. Iwata S, Nakayamada S, Fukuyo S, Kubo S, Yunoue N, Wang SP, et al. Activation of Syk in peripheral blood B cells in patients with rheumatoid arthritis: a potential target for abatacept therapy. Arthritis Rheumatol (2015) 67(1):63–73. doi:10.1002/art.38895
32. Díaz de Ståhl T, Andrén M, Martinsson P, Verbeek JS, Kleinau S. Expression of FcgammaRIII is required for development of collagen-induced arthritis. Eur J Immunol (2002) 32:2915–22. doi:10.1002/1521-4141(2002010)32:10<2915::AID-IMMU2915>3.0.CO;2-4
33. Jakus Z, Simon E, Balázs B, Mócsai A. Genetic deficiency of Syk protects mice from autoantibody-induced arthritis. Arthritis Rheum (2010) 62:1899–910. doi:10.1002/art.27438
34. Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM, Lowell CA. Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol (2011) 187:4319–30. doi:10.4049/jimmunol.1100341
35. Huang ZY, Kim MK, Kim-Han TH, Indik ZK, Schreiber AD. Effect of locally administered Syk siRNA on allergen-induced arthritis and asthma. Mol Immunol (2013) 53:52–9. doi:10.1016/j.molimm.2012.06.011
36. Boren JC, Radstake TR, Rossato M. The role of genetics and epigenetics in the pathogenesis of systemic sclerosis. Nat Rev Rheumatol (2014) 10:671–81. doi:10.1038/nrrheum.2014.128
37. Pamuk ON, Can G, Awaz S, Karaca T, Pamuk GE, Demirtas S, et al. Spleen tyrosine kinase (Syk) inhibitor fostamatinib limits tissue damage and fibrosis in a bleomycin-induced scleroderma mouse model. Clin Exp Rheumatol (2015) 33:S15–22.
38. Reilly MP, Sinha U, André P, Taylor SM, Pak Y, Deguzman FR, et al. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood (2011) 117:2241–6. doi:10.1182/blood-2010-03-274969
39. Podolanczuk A, Lazarus AH, Crow AR, Grossbard E, Bussel JB. Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood (2009) 113:3154–60. doi:10.1182/blood-2008-07-166439
40. Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther (2006) 319(3):998–1008. doi:10.1124/jpet.106.109058
41. Cha HS, Boyle DL, Inoue T, Schoot R, Tak PP, Pine P, et al. A novel spleen tyrosine kinase inhibitor blocks c-Jun N-terminal kinase-mediated gene expression in synoviocytes. J Pharmacol Exp Ther (2006) 317(2):571–8. doi:10.1124/jpet.105.097436
42. Sheridan C. Small molecule challenges dominance of TNF-alpha inhibitors. Nat Biotechnol (2008) 26(2):143–4. doi:10.1038/nbt0208-143
43. Pine PR, Chang B, Schoettler N, Banquerigo ML, Wang S, Lau A, et al. Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clin Immunol (2007) 124:244–57. doi:10.1016/j.clim.2007.03.543
44. Coffey G, DeGuzman F, Inagaki M, Pak Y, Delaney SM, Ives D, et al. Specific inhibition of spleen tyrosine kinase suppresses leukocyte immune function and inflammation in animal models of rheumatoid arthritis. J Pharmacol Exp Ther (2012) 340:350–9. doi:10.1124/jpet.111.188441
45. Weinblatt ME, Kavanaugh A, Burgos-Vargas R, Dikranian AH, Medrano-Ramirez G, Morales-Torres JL, et al. Treatment of rheumatoid arthritis with a Syk inhibitor: a twelve week, randomized, placebo-controlled trial. Arthritis Rheum (2008) 58:3309–18. doi:10.1002/art.23992
46. Genovese MC, Kavanaugh A, Weinblatt ME, Peterfy C, DiCarlo J, White ML, et al. An oral Syk kinase inhibitor in the treatment of rheumatoid arthritis: a three month randomized, placebo controlled phase II study in patients with active rheumatoid arthritis that did not respond to biologic agents. Arthritis Rheum (2011) 63:337–45. doi:10.1002/art.30114
47. Deng GM, Liu L, Kyttaris VC, Tsokos GC. Lupus serum IgG induces skin inflammation through the TNFR1 signaling pathway. J Immunol (2010) 184(12):7154–61. doi:10.4049/jimmunol.0902514
48. Deng GM, Liu L, Bahjat FR, Pine PR, Tsokos GC. Suppression of skin and kidney disease by inhibition of spleen tyrosine kinase in lupus-prone mice. Arthritis Rheum (2010) 62:2086–92. doi:10.1002/art.27452
49. Bahjat FR, Pine PR, Reitsma A, Cassafer G, Baluom M, Grillo S, et al. An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus. Arthritis Rheum (2008) 58:1433–44. doi:10.1002/art.23428
50. Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med (2010) 363:1303–12. doi:10.1056/NEJMoa1000500
51. Okamoto H, Kobayashi A. Spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med (2011) 364:83–4. doi:10.1056/NEJMc1012187
52. Kazerounian S, Duquette M, Reyes MA, Lawler JT, Song K, Perruzzi C, et al. Priming of the vascular endothelial growth factor signaling pathway by thrombospondin-1, CD36, and spleen tyrosine kinase. Blood (2011) 117:4658–66. doi:10.1182/blood-2010-09-305284
53. Tew JG, Wu J, Fakher M, Szakal AK, Qin D. Follicular dendritic cells: beyond the necessity of T-cell help. Trends Immunol (2001) 22:361–7. doi:10.1016/S1471-4906(01)01942-1
54. EI Shikh ME, El Sayed R, Szakal AK, Tew JG. Follicular dendritic cell (FDC)-FcgammaRIIB engagement via immune complexes induces the activated FDC phenotype associated with secondary follicle development. Eur J Immunol (2006) 36:2715–24. doi:10.1002/eji.200636122
55. Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol (2006) 6:205–17. doi:10.1038/nri1786
56. Manzo A, Bombardieri M, Humby F, Pitzalis C. Secondary and ectopic lymphoid tissue responses in rheumatoid arthritis: from inflammation to autoimmunity and tissue damage/remodeling. Immunol Rev (2010) 233:267–85. doi:10.1111/j.0105-2896.2009.00861.x
57. Anolik JH, Ravikumar R, Barnard J, Owen T, Almudevar A, Milner EC, et al. Cutting edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol (2008) 180:688–92. doi:10.4049/jimmunol.180.2.688
58. Victoratos P, Kollias G. Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity (2009) 30:130–42. doi:10.1016/j.immuni.2008.10.019
59. Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med (2009) 6:e1. doi:10.1371/journal.pmed.0060001
60. EI Shikh ME, El Sayed RM, Sukumar S, Szakal AK, Tew JG. Activation of B cells by antigens on follicular dendritic cells. Trends Immunol (2010) 31:205–11. doi:10.1016/j.it.2010.03.002
Keywords: Syk, autoimmune disease, Syk inhibitor, IgG, Fcgamma receptors
Citation: Deng G-M, Kyttaris VC and Tsokos GC (2016) Targeting Syk in Autoimmune Rheumatic Diseases. Front. Immunol. 7:78. doi: 10.3389/fimmu.2016.00078
Received: 23 November 2015; Accepted: 16 February 2016;
Published: 07 March 2016
Edited by:
Anil Chauhan, Saint Louis University, USAReviewed by:
Mohey Eldin El Shikh, Queen Mary University of London, UKCopyright: © 2016 Deng, Kyttaris and Tsokos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guo-Min Deng, Z21kZW5nQG5qbXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.