 Mário Alberto C. Silva-Neto
Mário Alberto C. Silva-Neto Angela H. Lopes
Angela H. Lopes Georgia C. Atella
Georgia C. Atella- 1Programa de Biologia Molecular e Biotecnologia, CCS, Instituto de Bioquímica Médica Leopoldo de Meis, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 2Centro de Ciências da Saúde, Instituto de Microbiologia Prof. Paulo de Góes, Universidade Federal do Rio de Janeiro, Cidade Universitária – Ilha do Fundão, Rio de Janeiro, Brazil
Chagas disease is a severe illness, which can lead to death if the patients are not promptly treated. The disease is caused by the protozoan parasite Trypanosoma cruzi, which is mostly transmitted by a triatomine insect vector. There are 8–10 million people infected with T. cruzi in the world, but the transmission of such disease by bugs occurs only in the Americas, especially Latin America. Chronically infected patients will develop cardiac diseases (30%) and up digestive, neurological, or mixed disorders (10%). Lysophosphatidylcholine (LPC) is the major phospholipid component of oxidized low-density lipoproteins associated with atherosclerosis-related tissue damage. Insect-derived LPC powerfully attracts inflammatory cells to the site of the insect bite, enhances parasite invasion, and inhibits the production of nitric oxide by T. cruzi-stimulated macrophages. The recognition of the ubiquitous presence of LPC on the vector saliva, its production by the parasite itself and its presence both on patient plasma and its role on diverse host × parasite interaction systems lead us to compare its distribution in nature with the title of the famous Beatles song “Here, There and Everywhere” recorded exactly 50 years ago in 1966. Here, we review the major findings pointing out the role of such molecule as an immunosignaling modulator of Chagas disease transmission. Also, we believe that future investigation of the connection of this ubiquity and the immune role of such molecule may lead in the future to novel methods to control parasite transmission, infection, and pathogenesis.
Introduction
Lysophosphatidylcholine (LPC) is a major regulator of several biological processes. It is produced by the hydrolysis of the fatty acid at sn-2 position of phosphatidylcholine (PC) catalyzed by phospholipase A2 (PLA2) or alternatively by its transfer from PC to cholesterol by the action of another enzyme a cholesterol acetyltransferase (1–3). Such molecule was originally believed to play a role exclusively on membrane structure but their involvement on the pathogenesis of several human diseases is increasingly clear in the last few years. Its recognition as a causative factor has occurred by mainly by two different set of data. The first line of evidence toward a functional and signaling role of LPC is due to the close link between plasma lipid profile and pro-oxidant reactions common in inflammatory diseases, especially in atherosclerosis. The second route of evidences lies on the effects of LPC on cells involved in innate immunity. We will discuss the growing of such concepts and how they overlap and support the role of such lysophospholipid in parasitic diseases as follows.
Lysophosphatidylcholine and the bioactive lipids lysophosphatidic acid (LPA), sphingosine-1-phosphate (S1P), and sphingosylphosphorylcholine (SPC) modulate a wide number of biological processes on mammalian cells. LPC has been classically involved in various physiological events and is already known as a central molecule in various pathological conditions but is especially present during the deposition and infiltration of inflammatory cells and deposition of atheroma, as discussed above (4–6). Research aimed at LPC increased substantially from the fact that these molecules are involved in atherosclerosis (7). The idea that several phospholipases secreted by circulating leukocytes may participate in this pathology was soon proposed. Thus, the current model suggests that diabetes and hypercholesterolemia contribute to generate a large number of low-density lipoprotein (LDL) particles in plasma, which can undergo oxidation of unsaturated fatty acids, generating an oxidized particle (ox-LDL). Since on average, 50% of LDL fatty acids are arachidonic acid and linoleic acid, the chances of an oxidative event like this are huge. OxLDL is a potential cause of increased expression of inflammatory markers. TNF-α, MCP-1, and MCSF expression are increased by oxLDL and attract and differentiate monocytes to the lesion site. Accordingly, LPC is a powerful chemoattractant molecule to macrophages. It is also generated by cells during apoptosis, as mentioned above. oxLDL particles are recognized by PLA2 secreted by different cells in plasma, including the types IIa, V, and X. In conclusion, plasma LPC is derived from the hydrolysis of PC mainly present in LDL and cell membranes by several different subfamilies of PLA2s usually following and oxidative event on its fatty acids.
Trypanosoma cruzi infects the vertebrate host through skin bites produced by bugs during their feed or by interaction with conjunctival mucosa. This interaction sometimes produces the Romaña signal or chagoma whose histology is defined by a large number of mononuclear cells (8). It is likely that T. cruzi stimulates skin cells to produce mediators that trigger a local inflammatory response. Chagas disease displays an acute phase, followed by a chronic phase where the parasites are physically linked to inflammation sites in the heart and esophagus (9–15). The disease is considered today as multifactorial once pathogen and host are continuously interacting throughout the whole patient life (16). Following the first 2–3 weeks of the vector bite, T. cruzi infection is manifested by a high load of parasites in the blood and tissues. Acute infection is characterized by a huge activation of the immune system. Such activation includes the production of high levels of cytokines, a large activation of T and B cells, lymphadenopathy, and splenomegaly. Also, it displays a visible inflammation due to the infection in tissue niches. The acute phase leads to the development of acquired immunity. Such mechanism ensures the effective control of parasitemia. The chronic phase takes place throughout the patient’s life and is associated with low levels of parasite in the host. The beginning of chronic infection in Chagas disease is asymptomatic in most patients. However, clinical manifestations will range from no symptoms to the involvement of the cardiovascular symptoms and/or gastrointestinal complications as the disease progresses (17, 18). The innate immune system appears to be essential for at least two important aspects of Chagas disease: control of parasite replication in host tissue and the progress of the inflammatory reaction. The inflammatory reaction itself may be a major cause of tissue damage and dysfunction of certain organs in the patient (18). Experiments in T. cruzi infection models have shown that a potent Th-1 CD4 and CD8 immune response controls parasitemia during the initial stage of the disease. Such immune response is characterized by the production of IFN-γ, TNF-α, and IL-12, as well as the production of nitric oxide (NO). Additionally, cells of innate immunity, such as “natural killer” (NK) cells, dendritic cells, and macrophages, are also key factors in the initial control parasite replication (11, 17, 19).
In recent years, research on Chagas disease has focused on the investigation of the role of pathogen-associated molecular patterns (PAMPs) protozoa, which are the targets of the innate immune response receptors. Also the problem of identification of relevant receptors for the innate immunity to the parasite during the course of the disease in the host has been addressed by several laboratories. This strategy ultimately aimed at developing therapeutic interventions through the use of derivatives of PAMPs present in the parasites. Glycosyl-phosphatidylinositol (GPI) is the name given to the first glycoconjugate identified in Trypanosoma brucei. This molecule was identified with the function of anchoring proteins on the cell surface (20–23). T. cruzi express on their surface glycoproteins anchored by GPIs (20–23). GPI anchors from trypomastigotes include mucin-like glycoproteins (mucin-GPI) and are key molecules responsible for stimulating the host’s immune system (24–27). These mucin-GPIs activate macrophages and lead to the synthesis of proinflammatory cytokines, chemokines, and NO (28). The innate immune response to T. cruzi infection largely results from the activation of signaling pathways triggered by Toll-like receptors (TLRs).
Toll-like receptors recognize conserved structural motifs present on different pathogens. Following their activation, TLRs trigger intracellular signaling cascades that build up the host immune response (18, 29). There are 10 TLRs described in human and 12 in mice (18, 29). GPI-induced stimulus occurs during the initial phase of infection, where macrophages respond to trypomastigotes in a TLR-dependent mechanism. At this point in time the production of IL-12, TNF-α and the activation of CD4 and CD8 responses through IFN-γ will take place (30). TNF-α and IFN-γ-activated macrophages seems to have an important role in the control of parasite growth. Anchors free of proteins or GPI or glicoinositol phospholipids (GIPLs) are also able to stimulate the host immune system. GIPLs are similar to GPIs but contain ceramide in their lipid portion (24, 25). TLRs 2, 4, and 9 are the main TLRs involved in the innate immune response to T. cruzi (24, 31–36). TLR2 was found to be activated by mucin-GPI (24, 31–36). The heterodimer composed of TLR2 and TRL6 receptor is activated by GPI mucin and its co-receptor CD14 (34). T. cruzi GIPL activates the inflammatory response via TLR4. It induces neutrophil recruitment to the peritoneal cavity of mice and this effect is partly dependent on IL-1β production (32, 36). The T. cruzi genomic DNA also plays an important role in the vertebrate host proinflammatory response. Finally, TLR 9 is activated by CpG motifs in non-methylated DNA (34, 35). In the innate immune response mediated by TLRs, T. cruzi can also stimulate a TLR-independent response leading to the production of IFN-β and IFN-γ. This is due a rise on intracellular calcium concentration that activates calcineurin and calmodulin-dependent proteins (37–39).
Here: LPC-Mediated Handling of Innate Immunity at the Site of Parasite Infection
Our group demonstrated, for the first time, the presence of phospholipids and lysophospholipids in saliva and stool hematophagous organism Rhodnius prolixus, a triatomine vector of Chagas disease (40, 41). Such finding engaged salivary LPC into the host plasma environment and uncovered its role as both an anti-hemostatic and immunomodulatory molecule. The main lipids present in the saliva of R. prolixus are PC and LPC (40, 41). Salivary LPC is an additional anti-hemostatic molecule that is part of the pharmacological arsenal injected into the bite site to allow the insect feeds. It inhibits platelet aggregation and increases the production of NO in endothelial cells. Thus, the LPC was originally described as a molecule with antiplatelet and vasodilatory activity, and few years later, its effect as an immunomodulator of T. cruzi infection has been demonstrated (40, 41) (Figure 1).
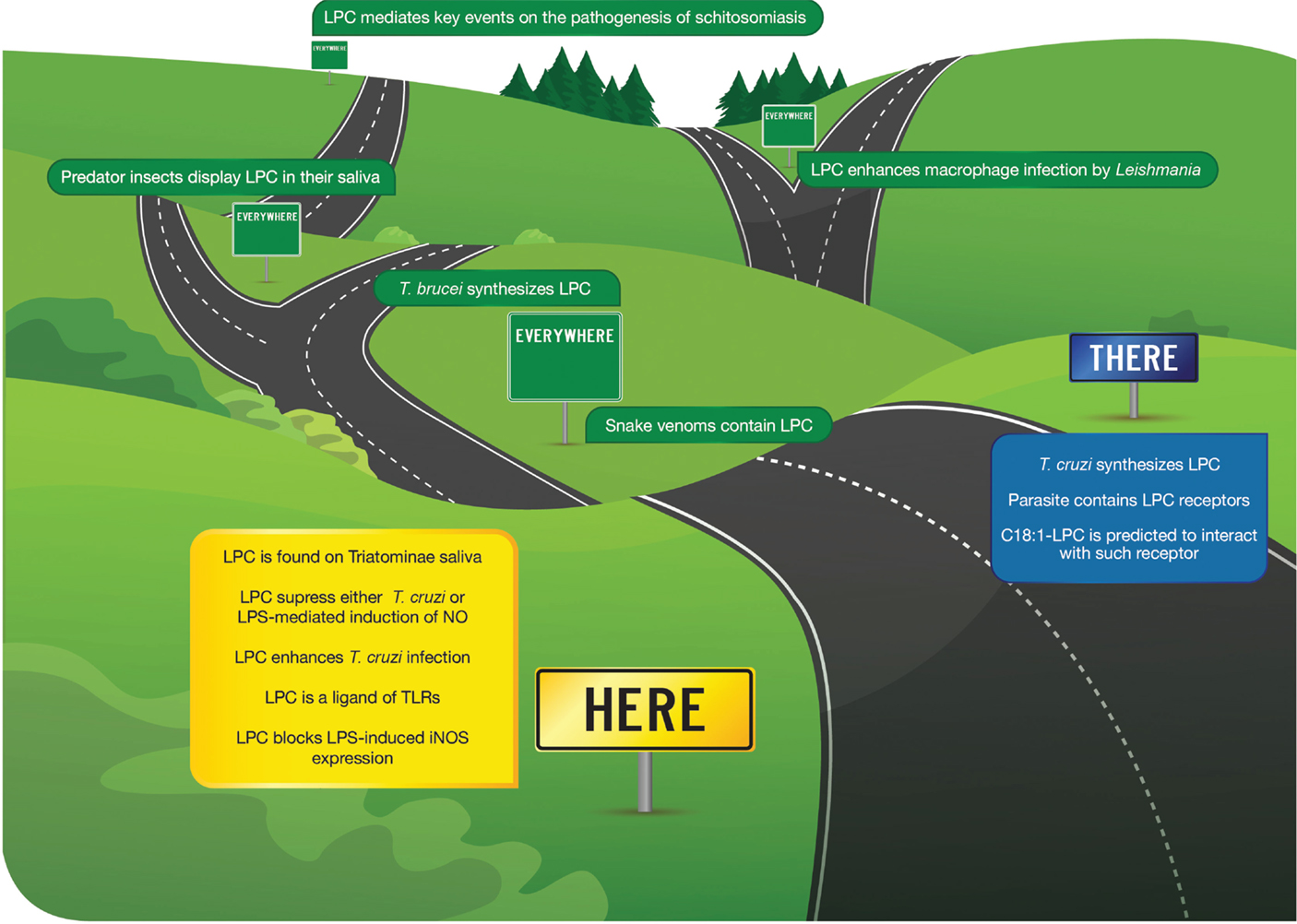
Figure 1. Here, there, and everywhere. This cartoon depicts the major findings in recent years regarding the role of LPC on infectious diseases. Here, originally found as a vector, salivary phospholipid able to modulate Chagas disease through the suppression of NO production. There, LPC was recently described as the product of parasite synthesis. Everywhere, several reports have involved LPC on different aspects of parasitic diseases but also a relevant mediator molecule used to subvert the prey by hunting insects. Finally, its role as a major component of snake venom is also shown.
The role of LPC as a modulator of T. cruzi infection is caused by three main mechanisms summarized as follows:
1. LPC is a vector-derived molecule. Following attraction of inflammatory cells to the insect bite site, it enhances the chance of infection by the latter arrival of T. cruzi (40, 41).
2. LPC produces a surge on intracellular calcium concentrations in macrophages. This will also enhance parasite invasion. It also modulates the phosphorylation of key signaling molecules such as mitogen-activated kinases (MAPKs) (41, 42).
3. LPC inhibits the production of NO by T. cruzi-stimulated macrophages, lipopolysaccharide (LPS), or LPS in the presence of IFN-γ. Thus, it handles the immune system of the vertebrate host (41, 42).
Such findings implied that LPC is a multifactorial signaling molecule, with signaling, anti-hemostatic, and immune effects on the context of Chagas disease transmission (43–46). The establishment of infection by T. cruzi depends on the initial invasion of these cells by the parasite (47–49). This leads to the assumption that the salivary LPC can facilitate parasitic infection, favoring not only insect feeding but also the preparation of macrophage susceptibility for the arrival of the parasite, minutes or hours after the initial bite. Salivary LPC injection into the host’s skin followed by inoculation of the parasite leaded to an increase of threefold to sixfold on blood parasitemia (41). LPC effect on parasitemia relies on the activation of macrophage chemotaxis and suppression of NO synthesis. We also show an increase in parasite loads within macrophages induced by either 500-fold diluted saliva or LPC. Such results were the first demonstration of a factor able to potentiate the transmission of Chagas disease and the first implication of a lysophospholipid as a modulator of an infectious disease (41, 42).
Activation of TLRs on host cells and the production of TNF-α, IL-12, and NO, required the adapter protein MyD88 (20–24, 30). TLRs 2, 4, and 9 have been implicated in the control of T. cruzi infection (20–24, 30). The involvement of TLR2 in the interaction between the parasite and macrophages was also demonstrated (24, 30). TLR2 expression is essential for the induction of IL-12, TNF-α, and NO. This receptor is activated by the parasite molecules, such as GPI anchors, that are isolated from the surface trypomastigotes of T. cruzi. Curiously, IL-12 synthesis in macrophages exposed to T. cruzi is not affected by saliva (41). Interestingly, in bone marrow derived macrophages derived from TLR2-deficient mice IL-12 production is largely suppressed by LPC (41). These data indicate that in some types of cells this cytokine production can be affected by LPC through a TLR2-independent mechanism.
Furthermore, T. cruzi GIPLs are TLR4 agonists with proinflammatory effects (32, 36). We have shown that NO production induced by the parasite or LPS, another ligand of TLR4 or in peritoneal murine macrophages or bone marrow-derived macrophages is blocked in both cases, the LPC even in the presence of IFN-γ vitro (41). The LPC’s ability to reverse the induction of NO production in every case, almost regardless of the type of ligand suggests that this bioactive lipid should act on a single pathway. Usually, LPC-sensitive receptors exhibit remarkable overlap in their specificity to the ligands they interact with and vice versa. Different receptors have been proposed for LPC. This includes G2A a G protein-coupled receptor, and GPR4, another important candidate (50). It is noteworthy the low reproducibility of LPC studies where radioactive lipids were used. Together with the capacity of G2A to bind to fatty acids and protons, such findings brought a high complexity to this field (51). Thus, G2A is still recognized as the most known receptor to convey signals mediated by LPC presence in the chemical environment of cells. The redistribution of G2A receptor itself and the exposure of TLR4 on cell surface are highly influenced by LPC metabolism (51, 52). In this latter case, intracellular LPC amount is under control due to the activity of LPC-acyltransferase (LPCAT). This enzyme uses LPC to produce phospholipids. The incubation of monocytes with LPS activates the enzyme and increases the transport of TLR4 to cell-membrane rafts (52). The LPCAT inhibitor, thiophenyl 5-hydroxyethyl 5′3 pyridine (HETP), increases the ratio LPC/PC while it reverses the effect of LPS. Thus, T. cruzi may promote a redistribution of G2A and TLR4 on cell surface during infection. This should be investigated in the future by both proteomic and immunological approaches.
In apoptotic cells, LPC is generated by a PLA2- calcium-independent activated caspase-3. LPC then attracts phagocytes to cells and represents a sign of recognition of ongoing apoptosis (50, 53, 54). LPC-induced chemotaxis in Chagas’ disease is intriguing once the uptake of apoptotic cells by macrophages infected with T. cruzi parasite stimulates their growth (55). Furthermore, the infective stages of T. cruzi are capable of generating lipid messengers, including LPC, that modulate the signaling of the host cell (56). LPC activates some protein kinase C isoforms (57). It is probably that different PKC isoforms are activated in different cell types. Also, when combined with different types of TLRs and adapters, the LPC-mediated signaling repertoire may produce a specific and yet to be studied particular mechanism of immunosuppression (Figure 1).
In order to evaluate some of these questions, we have addressed the role of LPC on TLR-mediated signaling pathway using HEK 293A cells. Such cells were transfected with TLRs constructs and stimulated with LPCs displaying different fatty acid chain lengths and saturation levels (42). All tested LPCs activated both TLR4 and TLR2-1 signaling pathways. Such results were confirmed through the evaluation of NF-κB activation and IL-8 production. Similar results were obtained when using peritoneal murine macrophages. These cells responded to LPC stimulation by displaying NF-κB translocation. Curiously, when incubated in the presence of LPS, LPC counteracted several features of TLR4 signaling. In this case, NF-κB translocation, NO synthesis, and the expression of inducible nitric oxide synthase (iNOS) were blocked. Such phenotypes occur concomitantly with a hierarchical activation of the MAPKs p38 and JNK, but not ERK, in murine macrophages. Also, LPC blocked LPS-induced ERK activation in peritoneal macrophages but not in TLR-transfected cells. Such results indicated that LPC behaves as a proinflammatory molecule in the absence of traditional TLR ligands. However, when LPS and LPC are present at the same time, a partial silencing of the canonical TLR4 pathway takes place. Such silencing involves the downregulation of ERK pathway and does not affect p32 and JNK. Under these conditions, LPC assumes an anti-inflammatory phenotype through yet unknown ligands on cell surface (42).
There: LPC is Synthesized by Trypanosomatid Parasites
Phosphatidylcholine and LPC are synthesized by T. cruzi (58, 59), T. brucei (60), Leishmania spp. (61, 62), as well as by the malaria parasite, Plasmodium falciparum (63). Intriguingly, more than 50% of the total lipids secreted by T. cruzi were identified as PC and LPC (58). Recently, we have purified and structurally characterized a C18:1-LPC in T. cruzi, which present a platelet-activating factor (PAF)-like activity, as it aggregates rabbit platelets (59). Comparable to PAF, platelet aggregation was completely blocked by the PAF receptor antagonist, WEB 2086. Considering that increased platelet aggregation related to myocarditis is observed in Chagas disease, it is possible that C18:1-LPC is an important lipid mediator in the progression of this disease. Nevertheless, what functions endogenous T. cruzi LPC have in the infection is yet to be unveiled. Such findings enhance our view that during the complete cycle of trypanosomatids transmission LPC will be able to handle surrounding cells, therefore enhancing parasite survival (Figure 1).
Everywhere: Role of LPC in Other Parasitic Infections
In recent years, our group and others have explored the role of lysophospholipids, and especially LPC on the transmission and pathogenesis of different parasitic diseases. Also, it was demonstrated that LPC plays a role on prey × predator. Such models lead us to the view that LPC is a ubiquitous modulator of host–parasite interaction (Figure 1).
Leishmaniasis
A key step on Leishmania infection is the invasion of macrophages, which usually occurs through an endocytic process. Such process leads to the formation of the parasitophorous vacuole (PV) that will harbor the parasite. PV was isolated from Leishmania-infected macrophages, and the analysis of its phospholipid composition was conducted (64). The levels of LPC increased as compared with non-infected cells. Such results were obtained by allowing either the promastigote or amastigote forms of the parasite to interact with macrophages previously labeled with radioactive phosphate. Thus, these findings indicate that following invasion there is a remodeling of host cell phospholipids, which enhances the production of LPC. The results obtained in the T. cruzi model (41, 42), where LPC was pointed out as a negative modulator of NO production suggest that Leishmania-induced remodeling of PV may handle the microbicide production and protects the parasite. If this mechanism is essential to allow parasite escape from PV should be investigated in the future. It is important to consider that the use of miltefosine, the first orally active anti-leishmanial drug when directly used to treat Leishmania donovani promastigotes, induced at higher doses the increase on LPC production on total parasite membrane extract, probably due to the activation of PLA2s (65). Curiously, such effect is confined to some specific membranar structure of the parasite since it is lost when the plasma membrane is evaluated separately. The involvement on LPC on Leishmania-infected was recently busted by the finding that such molecule in BALB/c mouse-derived DC infection (66). LPC enhanced by 92% the infection index was correlated to a delay on cell maturation as evaluated by the expression of CD86+/CD11c+. Treatment of such cells with LPS displayed the opposite effects. Also, the authors found that LPC reversed the immune balance between IL-10, TNF-α, and IL-6 once it induced a huge increase on the first cytokine while not affecting of slightly reducing the proinflammatory ones (66). Finally, they observed that LPC is also able to selectively increase arginase1 expression by 395%. This enzyme is commonly induced by Th2 cytokine response set and enhances parasite proliferation. Such effect is paralleled by a suppression of iNOS activation, which keeps the levels of the NO quite low in Leishmania-infected cells. Indoleamine 2,3-dioxygenase (IDO) catalyzes tryptophan catabolism through kynurenine pathway. The depletion of tryptophan blocks parasite proliferation. Leishmania and LPC induced IDO activation, which usually leads to the accumulation of kynurenine metabolites blocking Th1 response. Also, the increase on arginase expression and downregulation of iNOS production depletes invaded cells of GSH, which induced acute oxidative stress. LPC has recently been described as a major inducer of macrophage polarization toward the M2 phenotype (67–69). In monocytes obtained from patients with Post Kala-Azar Dermal Leishmanisis (68), it was noticed a strong downregulation of TLR2/4 receptors and an increased expression of PPAR-γ and arginase-1. Also, IL-4, IL-10, and IL-13 were significantly raised as compared to healthy individuals. Finally, it increased classical M2 markers CD206, arginase-1, and PPARG in monocytes. Such findings strongly demonstrate that M2 polarization of macrophages is a hallmark of Leishmania infection in vivo and suggest that a bridge to the LPC-induced effects is the next step toward the development of drugs that modulate disease chronicity. These drugs should be anti-leishmanial molecules that ensure the repolarization of M2 macrophages toward the M1 proinflammatory and protective phenotype (68).
Human African Trypanosomiasis
A global metabolic profiling of plasma of patients carrying T. brucei rhodesiense [human African trypanosomiasis (HAT)] was performed and differences in the lipid, amino acid and metabolite profiles were identified. The most striking difference between the HAT patients and the control group was that six significantly altered amounts of LPC species were lower in patients than in controls (69). The interpretation of these data suggests that even though T. brucei rhodesiense is known to synthesize LPC de novo, the parasite probably scavenges several lipids from the host, as needed. Bearing in mind the results from our group on the key roles LPC play in T. cruzi infection, it is not surprising that LPC most likely engage crucial functions in other diseases caused by trypanosomatids.
Schistosomiasis
The first connection between LPC and the biology of schistosomiasis was noticed by the modifications on lipid plasma composition in infected patients (70). Curiously, in the plasma of infected hamsters, the levels of LPC are similar in both control and infected patients but major differences were found in the composition of the fatty acid acyl chains (71). LPC fatty acid composition showed a decrease on 18:2 and 20:5, but less marked changes in 20:4 (71). Usually, human red blood cells (RBCs) are lysed by schistosomula of Schistosoma mansoni. LPC-derived from the parasite is used to lyse RBCs (72). The source of this LPC is probably the parasite itself once schistosomula synthesize LCP and release it in the culture medium as evaluated with experiments using [3H]-phospholipids (72). The dynamics of such process was further through the use of precursors to label phospholipid and neutral lipid with the use of acyl chains ([3H]palmitic acid and [3H]oleic acid) or phospholipid polar head groups ([3H]choline and [14C]ethanolamine) (73). Schistosomal-derived lipids and LPC were shown to stimulate macrophages in vitro and induced TLR2-dependent NF-κB activation and cytokine production (74). Administration of LPC induced eosinophil recruitment and cytokine production, in a mechanism largely dependent on TLR2. Thus, parasite-derived LPC mediated key events on the pathogenesis and lethality of schistosomiasis.
Everywhere
The use of LPC on host × parasite interaction seems to be a ubiquous strategy to allow the handling of intracellular signaling pathways. Indeed, LPC may be a handful molecule also used by predators while hunting their daily meal. Snakes and bugs contain lysophospholipids in their saliva (75), Belostoma anurum saliva. This predator insect injects its saliva into the prey, which becomes immobilized (76). LPC isolated from saliva is neuroparalytic in vitro and in vivo (76), and it is highly likely that induce membrane depolarization and calcium entry into sensory nerves (76, 77). Indeed, LPC represents 25% of the total phospholipids present in the saliva of such insect. Since Belostoma employs extra-oral digestion LPC also helps in the digestion of preys larger than this insect such as amphibians and small fishes. Thus, it was shown that LPC have paralytic activity in zebrafish, which was the first evidence that lysophospholipids might play an important role in prey immobilization (76). Lysophospholipids and specially LPC paralyze the neuromuscular junction and are also present on snake venoms (78, 79). LPC can also modulate the activity of NK cells (79). Indeed, snake presynaptic phospholipase A2 neurotoxins are commonly present on snake venoms. Mass spectromic analysis of the lipids resulting from SPAN action demonstrated several forms of LPC including myristoyl-LPC and fatty acids (77). Altogether the above finding suggest that LPC use both as a immune and neuromodulator is an ancient biochemical and pharmacological use for such molecule, which probably arose from the eventual leak of modified phospholipids from membranes of interacting organisms whether in the mammalian × protozoan model or in the insect × protozoan model (80, 81).
Conclusion
In the future, drugs that target LPC of the signaling systems conveying LPC-mediated signals may largely affect the transmission and pathogenesis of several of the above mentioned diseases. The development of such drugs has been hampered by the yet unknown true LPC receptor. A functional analysis of novel LPC receptor candidates and the test of hypothesis regarding the parasite receptor recently described will provide a wider view of the potential of such molecule to trigger investigations in this area. It is quite interesting that the potential possibility of LPC modulates the several steps of TLR-mediated signaling or the interaction of this cascade with downstream effectors. Due to its major role as a suppressor of NO production in the presence of LPS, it is likely that LPC mechanism of action provides a novel system to understand the functionality of still unknown transcriptional factors as well as the hierarchical mechanism of MAPK regulation. It is quite challenging to imagine that the previously known structural phospholipids are yet a potential source of yet not completed understood signaling modules. The seminal discover of lysophospholipid role in human atherosclerosis combined with its potential selection as an immunosupressor, pharmacological, and neurological modulator by bugs, parasites, worms, and snakes leads to an increase interest on how far the ubiquity of such molecules has been shaped during evolution. The detailed investigation of such models at the molecular level in the future will certainly provide in the near future several novel targets for blocking the transmission and pathogenesis of such diseases.
Author Contributions
MS-N, AL, and GA discussed the data and wrote the paper. MS-N and GA are both the corresponding authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kabarowski JH, Xu Y, Witte ON. Lysophosphatidylcholine as a ligand for immunoregulation. Biochem Pharmacol (2002) 64:161–7. doi: 10.1016/S0006-2952(02)01179-6
2. Xu H, Valenzuela N, Fai S, Figeys D, Bennett SA. Targeted lipidomics – advances in profiling lysophosphocholine and platelet-activating factor second messengers. FEBS J (2013) 280:5652–67. doi:10.1111/febs.12423
3. Drzazga A, Sowińska A, Koziołkiewicz M. Lysophosphatidylcholine and lysophosphatidylinosiol – novel promissing signaling molecules and their possible therapeutic activity. Acta Pol Pharm (2014) 71:887–99.
4. Wang J, Zhang Y, Wang H, Han H, Nattel S, Yang B, et al. Potential mechanisms for the enhancement of HERG K+ channel function by phospholipid metabolites. Br J Pharmacol (2004) 141:586–99. doi:10.1038/sj.bjp.0705646
5. Wang L, Radu CG, Yang LV, Bentolila LA, Riedinger M, Witte ON. Lysophosphatidylcholine-induced surface redistribution regulates signaling of the murine G protein-coupled receptor G2A. Mol Biol Cell (2005) 16:2234–47. doi:10.1091/mbc.E04-12-1044
6. Drobnik W, Liebisch G, Audebert FX, Frohlich D, Gluck T, Vogel P, et al. Plasma ceramide and lysophosphatidylcholine inversely correlate with mortality in sepsis patients. J Lipid Res (2003) 44:754–61. doi:10.1194/jlr.M200401-JLR200
7. Kugiyama K, Kerns SA, Morrisett JD, Roberts R, Henry PD. Impairment of endothelium-dependent arterial relaxation by lysolecithin in modified low-density lipoproteins. Nature (1990) 344:160–2. doi:10.1038/344160a0
8. Dias JC. Cecilio Romana, Romana’s sign and Chagas’ disease. Rev Soc Bras Med Trop (1997) 30:407–13.
9. Andrade ZA. Pathogenesis of Chagas’ disease. Res Immunol (1991) 142:126–9. doi:10.1016/0923-2494(91)90021-A
10. Tarleton RL. Chagas disease: a role for autoimmunity? Trends Parasitol (2003) 19:447–51. doi:10.1016/j.pt.2003.08.008
11. Golgher D, Gazzinelli RT. Innate and acquired immunity in the pathogenesis of Chagas disease. Autoimmunity (2004) 37:399–409. doi:10.1080/08916930410001713115
12. Andrade LO, Andrews NW. The Trypanosoma cruzi-host-cell interplay: location, invasion, retention. Nat Rev Microbiol (2005) 3:819–23. doi:10.1038/nrmicro1249
13. Jones EM, Colley DG, Tostes S, Lopes ER, Vnencak-Jones CL, McCurley TL. Amplification of a Trypanosoma cruzi DNA sequence from inflammatory lesions in human chagasic cardiomyopathy. Am J Trop Med Hyg (1993) 48:348–57.
14. Vago AR, Macedo AM, Oliveira RP, Andrade LO, Chiari E, Galvão LM, et al. Kinetoplast DNA signatures of Trypanosoma cruzi strains obtained directly from infected tissues. Am J Pathol (1996) 149:2153–9.
15. Benvenuti LA, Roggério A, Freitas HF, Mansur AJ, Fiorelli A, Higuchi ML. Chronic American trypanosomiasis: parasite persistence in endomyocardial biopsies is associated with high-grade myocarditis. Ann Trop Med Parasitol (2008) 102:481–7. doi:10.1179/136485908X311740
16. Dutra WO, Gollob KJ. Current concepts in immunoregulation and pathology of human Chagas disease. Curr Opin Infect Dis (2008) 21:287–92. doi:10.1097/QCO.0b013e3282f88b80
17. Rassi A Jr, Rassi A, Marin-Neto JA. Chagas disease. Lancet (2010) 375:1388–402. doi:10.1016/S0140-6736(10)60061-X
18. Junqueira C, Caetano B, Bartholomeu DC, Melo MB, Ropert C, Rodrigues MM, et al. The endless race between Trypanosoma cruzi and host immunity: lessons for and beyond Chagas disease. Expert Rev Mol Med (2010) 12:e29. doi:10.1017/S1462399410001560
19. Brener Z, Gazzinelli RT. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas’ disease. Int Arch Allergy Immunol (1997) 114:103–10. doi:10.1159/000237653
20. Ferguson MA, Low MG, Cross GA. Glycosyl-sn-1,2-dimyristylphosphatidylinositol is covalently linked to Trypanosoma brucei variant surface glycoprotein. J Biol Chem (1985) 260:14547–55.
21. Ferguson MA. The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. J Cell Sci (1999) 112:2799–809.
22. Almeida IC, Camargo MM, Procópio DO, Silva LS, Mehlert A, Travassos LR, et al. Highly purified glycosylphosphatidylinositols from Trypanosoma cruzi are potent proinflammatory agents. EMBO J (2000) 19:1476–85. doi:10.1093/emboj/19.7.1476
23. Almeida IC, Gazzinelli RT. Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J Leukoc Biol (2001) 70:467–77.
24. Campos MA, Almeida IC, Takeuchi O, Akira S, Valente EP, Procópio DO, et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol (2001) 167:416–23. doi:10.4049/jimmunol.167.1.416
25. Coelho PS, Klein A, Talvani A, Coutinho SF, Takeuchi O, Akira S, et al. Glycosylphosphatidylinositol-anchored mucin-like glycoproteins isolated from Trypanosoma cruzi trypomastigotes induce in vivo leukocyte recruitment dependent on MCP-1 production by IFN-gamma-primed-macrophages. J Leukoc Biol (2002) 71:837–44.
26. Aliberti JC, Machado FS, Souto JT, Campanelli AP, Teixeira MM, Gazzinelli RT, et al. beta-Chemokines enhance parasite uptake and promote nitric oxide-dependent microbiostatic activity in murine inflammatory macrophages infected with Trypanosoma cruzi. Infect Immun (1999) 67:4819–26.
27. Shoda LK, Kegerreis KA, Suarez CE, Roditi I, Corral RS, Bertot GM, et al. DNA from protozoan parasites Babesia bovis, Trypanosoma cruzi, and T. brucei is mitogenic for B lymphocytes and stimulates macrophage expression of interleukin-12, tumor necrosis factor alpha, and nitric oxide. Infect Immun (2001) 69:2162–71. doi:10.1128/IAI.69.4.2162-2171.2001
28. Gutierrez FR, Mineo TW, Pavanelli WR, Guedes PM, Silva JS. The effects of nitric oxide on the immune system during Trypanosoma cruzi infection. Mem Inst Oswaldo Cruz (2009) 1:236–45. doi:10.1590/S0074-02762009000900030
29. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
30. Campos MA, Closel M, Valente EP, Cardoso JE, Akira S, Alvarez-Leite JI, et al. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J Immunol (2004) 172:1711–8. doi:10.4049/jimmunol.172.3.1711
31. Martin D, Tarleton R. Generation, specificity, and function of CD8+ T cells in Trypanosoma cruzi infection. Immunol Rev (2004) 201:304–17. doi:10.1111/j.0105-2896.2004.00183.x
32. Oliveira AC, Peixoto JR, de Arruda LB, Campos MA, Gazzinelli RT, Golenbock DT, et al. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J Immunol (2004) 173:5688–96. doi:10.4049/jimmunol.173.9.5688
33. Ropert C, Gazzinelli RT. Regulatory role of Toll-like receptor 2 during infection with Trypanosoma cruzi. J Endotoxin Res (2004) 10:425–30. doi:10.1177/09680519040100060801
34. Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol (2006) 6:895–906. doi:10.1038/nri1978
35. Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol (2006) 177:3515–9. doi:10.4049/jimmunol.177.6.3515
36. Medeiros MM, Peixoto JR, Oliveira AC, Cardilo-Reis L, Koatz VL, Van Kaer L, et al. Toll-like receptor 4 (TLR4)-dependent proinflammatory and immunomodulatory properties of the glycoinositolphospholipid (GIPL) from Trypanosoma cruzi. J Leukoc Biol (2007) 82:488–96. doi:10.1189/jlb.0706478
37. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. Toll-like receptor recognizes bacterial DNA. Nature (2000) 408:740–5. doi:10.1038/35047123
38. Chessler AD, Ferreira LR, Chang TH, Fitzgerald KA, Burleigh BA. A novel IFN regulatory factor 3-dependent pathway activated by trypanosomes triggers IFN-beta in macrophages and fibroblasts. J Immunol (2008) 181:7917–24. doi:10.4049/jimmunol.181.11.7917
39. Chessler AD, Unnikrishnan M, Bei AK, Daily JP, Burleigh BA. Trypanosoma cruzi triggers an early type I IFN response in vivo at the site of intradermal infection. J Immunol (2009) 182:2288–96. doi:10.4049/jimmunol.0800621
40. Golodne DM, Monteiro RQ, Graca-Souza AV, Silva-Neto MA, Atella GC. Lysophosphatidylcholine acts as an anti-hemostatic molecule in the saliva of the blood-sucking bug Rhodnius prolixus. J Biol Chem (2003) 278:27766–71. doi:10.1074/jbc.M212421200
41. Mesquita RD, Carneiro AB, Bafica A, Gazos-Lopes F, Takiya CM, Souto-Padron T, et al. Trypanosoma cruzi infection is enhanced by vector saliva through immunosuppressant mechanisms mediated by lysophosphatidylcholine. Infect Immun (2008) 76:5543–52. doi:10.1128/IAI.00683-08
42. Carneiro AB, Iaciura BM, Nohara LL, Lopes CD, Veas EM, Mariano VS, et al. Lysophosphatidylcholine triggers TLR2- and TLR4-mediated signaling pathways but counteracts LPS-induced NO synthesis in peritoneal macrophages by inhibiting NF-κB translocation and MAPK/ERK phosphorylation. PLoS One (2013) 8:e76233. doi:10.1371/journal.pone.0076233
43. Murugesan G, Sandhya Rani MR, Gerber CE, Mukhopadhyay C, Ransohoff RM, Chisolm GM, et al. Lysophosphatidylcholine regulates human microvascular endothelial cell expression of chemokines. J Mol Cell Cardiol (2003) 35:1375–84. doi:10.1016/j.yjmcc.2003.08.004
44. Radu CG, Yang LV, Riedinger M, Au M, Witte ON. T cell chemotaxis to lysophosphatidylcholine through the G2A receptor. Proc Natl Acad Sci U S A (2004) 101:245–50. doi:10.1073/pnas.2536801100
45. Yang LV, Radu CG, Wang L, Riedinger M, Witte ON. Gi-independent macrophage chemotaxis to lysophosphatidylcholine via the immunoregulatory GPCR G2A. Blood (2005) 105:1127–34. doi:10.1182/blood-2004-05-1916
46. Silva-Neto MA, Carneiro AB, Silva-Cardoso L, Atella GC. Lysophosphatidylcholine: a novel modulator of Trypanosoma cruzi transmission. J Parasitol Res (2012) 2012:625838. doi:10.1155/2012/625838
47. Connelly MC, Kierszenbaum F. Modulation of macrophage interaction with Trypanosoma cruzi by phospholipase A2-sensitive components of the parasite membrane. Biochem Biophys Res Commun (1984) 121:931–9. doi:10.1016/0006-291X(84)90766-6
48. Kollien AH, Schaub GA. The development of Trypanosoma cruzi in triatominae. Parasitol Today (2000) 16:381–7. doi:10.1016/S0169-4758(00)01724-5
49. Denkers EY, Butcher BA. Sabotage and exploitation in macrophages parasitized by intracellular protozoans. Trends Parasitol (2005) 21:35–41. doi:10.1016/j.pt.2004.10.004
50. Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, et al. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem (2008) 283:5296–305. doi:10.1074/jbc.M706586200
51. Kabarowski JH. G2A and LPC: regulatory functions in immunity. Prostaglandins Other Lipid Mediat (2009) 89:73–81. doi:10.1016/j.prostaglandins.2009.04.007
52. Jackson SK, Abate W, Parton J, Jones S, Harwood JL. Lysophospholipid metabolism facilitates Toll-like receptor 4 membrane translocation to regulate the inflammatory response. J Leukoc Biol (2008) 84:86–92. doi:10.1189/jlb.0907601
53. Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med (2002) 196:655–65. doi:10.1084/jem.20020542
54. Lauber K, Bohn E, Kröber SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell (2003) 113:717–30. doi:10.1016/S0092-8674(03)00422-7
55. Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature (2000) 403:199–203. doi:10.1038/35003208
56. Belaunzarán ML, Wainszelbaum MJ, Lammel EM, Gimenez G, Aloise MM, Florin-Christensen J, et al. Phospholipase A1 from Trypanosoma cruzi infective stages generates lipid messengers that activate host cell protein kinase c. Parasitology (2007) 134:491–502. doi:10.1017/S0031182006001740
57. Oishi K, Raynor RL, Charp PA, Kuo JF. Regulation of protein kinase C by lysophospholipids. Potential role in signal transduction. J Biol Chem (1988) 263:6865–71.
58. Agusti R, Couto AS, Alves MJ, Colli W, Lederkremer RM. Lipids shed into the culture medium by trypomastigotes of Trypanosoma cruzi. Mem Inst Oswaldo Cruz (2000) 95:97–102. doi:10.1590/S0074-02762000000100016
59. Gazos-Lopes F, Oliveira MM, Hoelz LV, Vieira DP, Marques AF, Nakayasu ES, et al. Structural and functional analysis of a platelet-activating lysophosphatidylcholine of Trypanosoma cruzi. PLoS Negl Trop Dis (2014) 8:e3077. doi:10.1371/journal.pntd.0003077
60. Werbovetz KA, Englund PT. Lipid metabolism in Trypanosoma brucei: utilization of myristate and myristoyllysophosphatidylcholine for myristoylation of glycosyl phosphatidylinositols. Biochem J (1996) 318:575–81. doi:10.1042/bj3180575
61. Ramos RG, Libong D, Rakotomanga M, Gaudin K, Loiseau PM, Chaminade P. Comparison between charged aerosol detection and light scattering detection for the analysis of Leishmania membrane phospholipids. J Chromatogr A (2008) 1209:88–94. doi:10.1016/j.chroma.2008.07.080
62. Zheng L, T’Kind R, Decuypere S, von Freyend SJ, Coombs GH, Watson DG. Profiling of lipids in Leishmania donovani using hydrophilic interaction chromatography in combination with Fourier transform mass spectrometry. Rapid Commun Mass Spectrom (2010) 24:2074–82. doi:10.1002/rcm.4618
63. Asahi H. Plasmodium falciparum: chemically defined medium for continuous intraerythrocytic growth using lipids and recombinant albumin. Exp Parasitol (2009) 121:22–8. doi:10.1016/j.exppara.2008.09.009
64. Henriques C, Atella GC, Bonilha VL, de Souza W. Biochemical analysis of proteins and lipids found in parasitophorous vacuoles containing Leishmania amazonensis. Parasitol Res (2003) 89:123–33. doi:10.1007/s00436-002-0728-y
65. Rakotomanga M, Blanc S, Gaudin K, Chaminade P, Loiseau PM. Miltefosine affects lipid metabolism in Leishmania donovani promastigotes. Antimicrob Agents Chemother (2007) 51:1425–30. doi:10.1128/AAC.01123-06
66. Tounsi N, Meghari S, Moser M, Djerdjouri B. Lysophosphatidylcholine exacerbates Leishmania major-dendritic cell infection through interleukin-10 and a burst in arginase1 and indoleamine 2,3-dioxygenase activities. Int Immunopharmacol (2015) 25:1–9. doi:10.1016/j.intimp.2015.01.006
67. Ishihara K, Kuroda A, Sugihara K, Kanai S, Nabe T, Akiba S. Regulation of macrophage differentiation and polarization by group IVC phospholipase A2. Biochem Biophys Res Commun (2011) 416:325–30. doi:10.1016/j.bbrc.2011.11.034
68. Mukhopadhyay D, Mukherjee S, Roy S, Dalton JE, Kundu S, Sarkar A, et al. M2 polarization of monocytes-macrophages is a hallmark of Indian post kala-azar dermal leishmaniasis. PLoS Negl Trop Dis (2015) 9:e0004145. doi:10.1371/journal.pntd.0004145
69. Lamour SD, Gomez-Romero M, Vorkas PA, Alibu VP, Saric J, Holmes E, et al. Discovery of infection associated metabolic markers in human African trypanosomiasis. PLoS Negl Trop Dis (2015) 9:e0004200. doi:10.1371/journal.pntd.0004200
70. Gillett MP, Besterman EM. Plasma and erythrocyte lysolecithin Schistosomiasis mansioni. Atherosclerosis (1977) 26:389–90. doi:10.1016/0021-9150(77)90094-6
71. Allan D, Evans WH. Changes in unsaturated fatty acids in serum lipids of hamsters infected with schistosomes (S. mansoni). Biochim Biophys Acta (1987) 919:199–202. doi:10.1016/0005-2760(87)90208-6
72. Golan DE, Brown CS, Cianci CM, Furlong ST, Caulfield JP. Schistosomula of Schistosoma mansoni use lysophosphatidylcholine to lyse adherent human red blood cells and immobilize red cell membrane components. J Cell Biol (1986) 103:819–28. doi:10.1083/jcb.103.3.819
73. Furlong ST, Caulfield JP. Schistosoma mansoni: synthesis and release of phospholipids, lysophospholipids, and neutral lipids by schistosomula. Exp Parasitol (1989) 69:65–77. doi:10.1016/0014-4894(89)90172-0
74. Magalhães K, Almeida PE, Atella G, Maya-Monteiro CM, Castro-Faria-Neto H, Pelajo-Machado M, et al. Schistosomal-derived lysophosphatidylcholine are involved in eosinophil activation and recruitment through toll-like receptor-2-dependent mechanisms. J Infect Dis (2010) 202:1369–79. doi:10.1086/656477
75. Caccin P, Magnabosco A, Tedesco E, Silva-Cardoso L, Atella GC, Montecucco C. Lysophospholipids are evolutionary ancient venom components. Toxicon (2010) 56:1525–7. doi:10.1016/j.toxicon.2010.06.025
76. Silva-Cardoso L, Caccin P, Magnabosco A, Patrón M, Targino M, Fuly A, et al. Paralytic activity of lysophosphatidylcholine from saliva of the waterbug Belostoma anurum. J Exp Biol (2010) 213:3305–10. doi:10.1242/jeb.041954
77. Rigoni M, Caccin P, Gschmeissner S, Koster G, Postle AD, Rossetto O, et al. Equivalent effects of snake PLA2 neurotoxins and lysophospholipid-fatty acid mixtures. Science (2005) 310:1678–80. doi:10.1126/science.1120640
78. Caccin P, Rigoni M, Bisceglie A, Rossetto O, Montecucco C. Reversible skeletal neuromuscular paralysis induced by different lysophospholipids. FEBS Lett (2006) 580:6317–21. doi:10.1016/j.febslet.2006.10.039
79. Fuly AL, Machado AL, Castro P, Abrahão A, Redner P, Lopes UG, et al. Lysophosphatidylcholine produced by the phospholipase A2 isolated from Lachesis muta snake venom modulates natural killer activity as a protein kinase C effector. Toxicon (2007) 50:400–10. doi:10.1016/j.toxicon.2007.04.008
80. Qin X, Qiu C, Zhao L. Lysophosphatidylcholine perpetuates macrophage polarization toward classically activated phenotype in inflammation. Cell Immunol (2014) 289:185–90. doi:10.1016/j.cellimm.2014.04.010
Keywords: lysophosphatidylcholine, Chagas disease, nitric oxide, MAPK, Rhodnius prolixus, macrophage
Citation: Silva-Neto MAC, Lopes AH and Atella GC (2016) Here, There, and Everywhere: The Ubiquitous Distribution of the Immunosignaling Molecule Lysophosphatidylcholine and Its Role on Chagas Disease. Front. Immunol. 7:62. doi: 10.3389/fimmu.2016.00062
Received: 04 December 2015; Accepted: 08 February 2016;
Published: 19 February 2016
Edited by:
Ute Frevert, New York University School of Medicine, USAReviewed by:
Daniel Pinero, Universidad Nacional Autónoma de México, MéxicoAna Rosa Pérez, CONICET, Argentina
Copyright: © 2016 Silva-Neto, Lopes and Atella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mário Alberto C. Silva-Neto, maneto@bioqmed.ufrj.br;
Georgia C. Atella, atella@bioqmed.ufrj.br