
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 February 2016
Sec. Immunological Tolerance and Regulation
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00036
This article is part of the Research Topic The Early Components of Complement and Systemic Autoimmune Disease View all 7 articles
 Katherine E. Lintner1
Katherine E. Lintner1 Yee Ling Wu1
Yee Ling Wu1 Yan Yang1Charles H. Spencer1Georges Hauptmann2Lee A. Hebert3
Yan Yang1Charles H. Spencer1Georges Hauptmann2Lee A. Hebert3 John P. Atkinson4
John P. Atkinson4 C. Yung Yu1*
C. Yung Yu1*
The complement system consists of effector proteins, regulators, and receptors that participate in host defense against pathogens. Activation of the complement system, via the classical pathway (CP), has long been recognized in immune complex-mediated tissue injury, most notably systemic lupus erythematosus (SLE). Paradoxically, a complete deficiency of an early component of the CP, as evidenced by homozygous genetic deficiencies reported in human, are strongly associated with the risk of developing SLE or a lupus-like disease. Similarly, isotype deficiency attributable to a gene copy-number (GCN) variation and/or the presence of autoantibodies directed against a CP component or a regulatory protein that result in an acquired deficiency are relatively common in SLE patients. Applying accurate assay methodologies with rigorous data validations, low GCNs of total C4, and heterozygous and homozygous deficiencies of C4A have been shown as medium to large effect size risk factors, while high copy numbers of total C4 or C4A as prevalent protective factors, of European and East-Asian SLE. Here, we summarize the current knowledge related to genetic deficiency and insufficiency, and acquired protein deficiencies for C1q, C1r, C1s, C4A/C4B, and C2 in disease pathogenesis and prognosis of SLE, and, briefly, for other systemic autoimmune diseases. As the complement system is increasingly found to be associated with autoimmune diseases and immune-mediated diseases, it has become an attractive therapeutic target. We highlight the recent developments and offer a balanced perspective concerning future investigations and therapeutic applications with a focus on early components of the CP in human systemic autoimmune diseases.
The complement system is a humoral recognition and effector system that facilitates in the elimination of invading pathogens. The activation pathways of the complement system converge at C3 and progress to the formation of the membrane attack complexes (MAC or C5b–C9) on a target membrane. The cascades of complement system are mediated and adjusted according to the type of initiator and the microenvironment in which complement activation is occurring. Proteins of the complement system cooperate and coordinate to differentiate among invading microbes, immune complexes, apoptotic cells, cellular debris, and physiologic host cells (1–4).
Three distinct pathways trigger cascades of activation through proteolysis of zymogens or precursors present in the circulation (Figure 1). The classical pathway (CP) is predominantly triggered by IgM or IgG immune complexes. The formation of antigen–antibody complexes exposes binding sites for C1q on Fc-regions of immunoglobulins, triggering the assembly and activation of the multi-molecular C1 complex, C1q–C1r2–C1s2 (pathway 3, Figure 1). Conformational changes in the C1 complex are induced upon binding of C1q to antibody, leading to activation of the serine protease subunits C1r and then C1s. As a result, C1s next activates C4 and C2, leading to the formation of the CP C3 convertase (abbreviated C4b2a). Following the early components activation, later components of the complement cascade form the MAC, which perturbs membranes, including the creation of pores across the target membrane, inducing cell lysis, loss of cytoplasm, and osmotic shock.
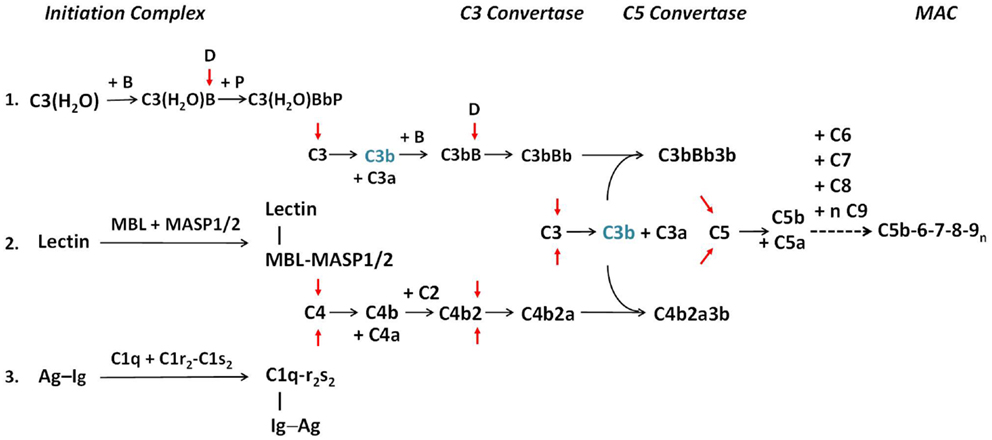
Figure 1. The activation pathways of the complement system. The three activation pathways of the complement system are shown according to evolution and physiologic sequences. Pathway 1 is known as the alternative pathway. It is activated through a tick-over mechanism because of continuous hydrolysis of the thioester bond in C3, which enables the formation with factor B to form a weak C3 convertase. Pathway 2 is known as the MBL or lectin pathway. It is initiated through the binding of mannan-binding lectin (MBL) or ficolin to arrays of simple sugar molecules in glycosylated antigens on microbes. This is a pattern recognition mechanism characteristic of the innate immune system. Pathway 3 is known as the classical pathway and is initiated through the binding of specific antibodies IgM or IgG to antigens. It is an effector arm of the humoral adaptive immune system. Each activation pathway engages the formation of a multi-molecular initiation complex, followed by the assembly of a C3 convertase and a C5 convertase for activation of C3 and C5, respectively and culminates in the formation of membrane attack complexes (MAC) on the target membrane. All three pathways can be amplified through a positive feedback mechanism, as C3b (in blue) generated by any C3 convertase can feed to the alternative pathway through association with factor B to form new C3 convertase after activation by factor D (pathway 1). Anaphylatoxins C3a and C5a are produced during the activation process. For brevity, by-products generated during the activation of C2 and factor B are not shown. Red arrows show activation of component proteins through cleavage by serine proteases. A dotted horizontal arrow denotes multiple steps are involved in the formation of the membrane attack complex. Early components of the classical pathway C1q, C1r, C1s, and C4 are engaged in the differentiation of immunity and autoimmunity, as genetic or acquired deficiency in any of these components are linked to pathogenesis of SLE. Complement C2 is also involved in the protection against autoimmunity but its effect size is smaller [modified from Ref. (2)].
In the mid-1950s, Pillemer and colleagues of Case Western Reserve University observed that complement activation could occur in the absence of a specific antibody (5). The existence of such an “alternative” pathway (AP) of activation was challenged but was confirmed more than two decades later (6). Specific protein factors involved in this AP are named factors, such as factor B, factor D, factor H (FH), and factor P (properdin). This pathway is initiated by a “tick-over” mechanism, in which a small proportion of complement C3 in the circulation is continuously hydrolyzed at slow rate (~1–2%/h) by water to form C3(H2O). C3(H2O) binds to factor B, which is activated by factor D, to form C3(H2O)Bb. C3(H2O)Bb accordingly acts as a relatively labile C3 convertase, constantly initiating C3 cleavage. Properdin stabilizes the short-lived C3 convertase. Under the appropriate circumstances, a C5 convertase (C3bBbP) is formed, and the cascade progresses to MAC formation on a foreign cell surface, similar to that of the CP (pathway 1, Figure 1). The binding of P to C3bBb on a microbial (or protected) surface will stabilize and protect the convertase from inactivation by regulatory proteins, thereby enhancing the convertase activity. The AP actually represents an ancient mechanism of innate immune host defense. The tick-over mechanism of complement activation enables a continuous surveillance for the host, executing the first line of defense against foreign invaders. With the development of a circulatory system, a system of host defense that both worked in seconds and was pathogen-destructing became mandatory.
A third pathway of complement activation involves the specific pattern recognition of biomolecules. One strategy for organisms to achieve species-specific diversity is by modification of biomolecules such as glycolipids and glycoproteins with different complexities of sugars. Typically, carbohydrate moieties on glycoproteins among vertebrates consist of complex sugars with secondary modifications (biantennary type) and ending with sialic acids. By contrast, the carbohydrate moieties in prokaryotes generally consist of simpler polymers of saccharides such as mannose. Pattern recognition of biomolecules is a universal theme of innate immunity. This pathway of complement activation is initiated by the binding of pattern recognition molecules including mannan-binding lectin (MBL) or ficolins to a bacterial membrane that express arrays of simple carbohydrates such as mannose and N-acetylglucosamine (7, 8). Such binding triggers the assembly of MBL/MASP2 and ficolin/MASP2 or MBL/MASP1 and ficolin/MASP1 complexes (pathway 2, Figure 1). MASP2 and MASP1 are both serine proteases. MASP2, associated with MBL or ficolin, activates both C4 and C2. As a result, a C3 convertase identical to that generated by the CP is formed.
Thus, all three complement activation pathways pass through the focal point on the activation of C3 to C3a and C3b, and then C5a and C5b, leading to the assembly of sublytic or lytic complexes on target membrane. It is noteworthy that all three activation pathways can be amplified by the positive feedback mechanism of the AP. In addition to cell lysis, effects of complement activation include opsonization to enhance phagocytosis of target cells, clearance of apoptotic bodies, solubilization and removal of immune complexes, stimulation of cytokine production, and anaphylatoxin-mediated effects. To summarize, the complement system has been designed in evolution primarily to activate on the membranes of bacteria and certain viruses. Opsonization via C3b and cellular activation via the anaphylatoxins C3a and C5a are its two primary functions.
Because the activated complement components C4b, C3b, and C5b67 can attach to any nearby cell surfaces including host cells, regulatory mechanisms have evolved to contain complement activation to damaged self and foreign targets. Inherently, all activated complement proteins spontaneously undergo intrinsic decay or inactivation when not stabilized by other pathway components or factors. In addition, several regulatory proteins in plasma or on the cell membrane can dissociate (decay) multi-molecular (activated) complexes and also proteolytically degrade the anchor proteins such as C4b and C3b. Upon initiation of classical or lectin pathways, C1-inhibitor (C1-INH) is a serine protease inhibitor that mimics the substrates for C1s in the C1 complex, and MASP2 or MASP1 in the MBL or ficolin complex. C1-INH forms a complex with activated C1r and C1s leading to the dissociation of the enzymatic C1r/C1s subunit from the recognition C1q subunit and preventing further activation of C4 and C2.
Importantly, there are also key strategies of regulation that act upon the assembly and stability of the C3 convertases. Fluid phase proteins, C4b-binding protein (C4bp) and FH and membrane proteins complement receptor 1 (CR1) and decay accelerating factor (DAF) all dissociate the recognition and enzymatic subunits of C3 convertases. Moreover, C4bp, CR1, and membrane cofactor protein (MCP) serve as cofactor proteins for factor I-mediated degradation of C4b, while FH, CR1, and MCP each serves as a cofactor for the factor I-mediated proteolysis of C3b. Notably, C4bp and FH recognize exposed host glycoproteins with glycosaminoglycans and sialic acids. The presence of such regulatory molecules on self-surfaces, but absence from most foreign particle surfaces, allows the regulators to prevent activation on host tissues while restricting complement activity to designated, foreign targets. Dysfunctional or uncontrolled complement activation can lead to destruction of body cells, overt release of inflammatory mediators, and tissue damage, as evidenced by clinical complications experienced by systemic lupus erythematosus (SLE) patients.
Systemic lupus erythematosus patients commonly present with evidence of complement consumption leading to low serum levels of C4 and C3 (9, 10). Initially, up to one-half of SLE patients will have low C4 and C3. In most established patients, serum C4 levels are biomarkers for lupus disease activity; low levels correlate with a flare, while normal levels correspond with remission (11). Longitudinal studies of serum C4 protein levels in SLE patients revealed different expression profiles characterized by three distinct groups (Figure 2) (12). The first group exhibited persistently low C4 levels throughout the course of the study, and many of these patients had a low copy number of C4 genes. The second group featured periodic fluctuations of C4 that paralleled disease activity while the third group had normal C4 levels most of the time. The typical pattern in active SLE patients is that both C4 and C3 are low simultaneously. However, exceptions occur. C3 levels are usually three- to sixfold higher than C4 levels; therefore, consumption of complement by immune complexes could reduce C4 below normal but leave C3 in the normal range. With a positive response to treatment, both C4 and C3 levels will rise. As noted, up to one-half of SLE patients will present with serum C4 and C3 in a normal range, which obviously does not rule out a lupus diagnosis. The CH50 and AP50 measure the lysis of red blood cells by the respective pathway and thus are functional tests. Furthermore, in vivo complement activation can also be assayed by testing for complexes or split products formed during activation (3).
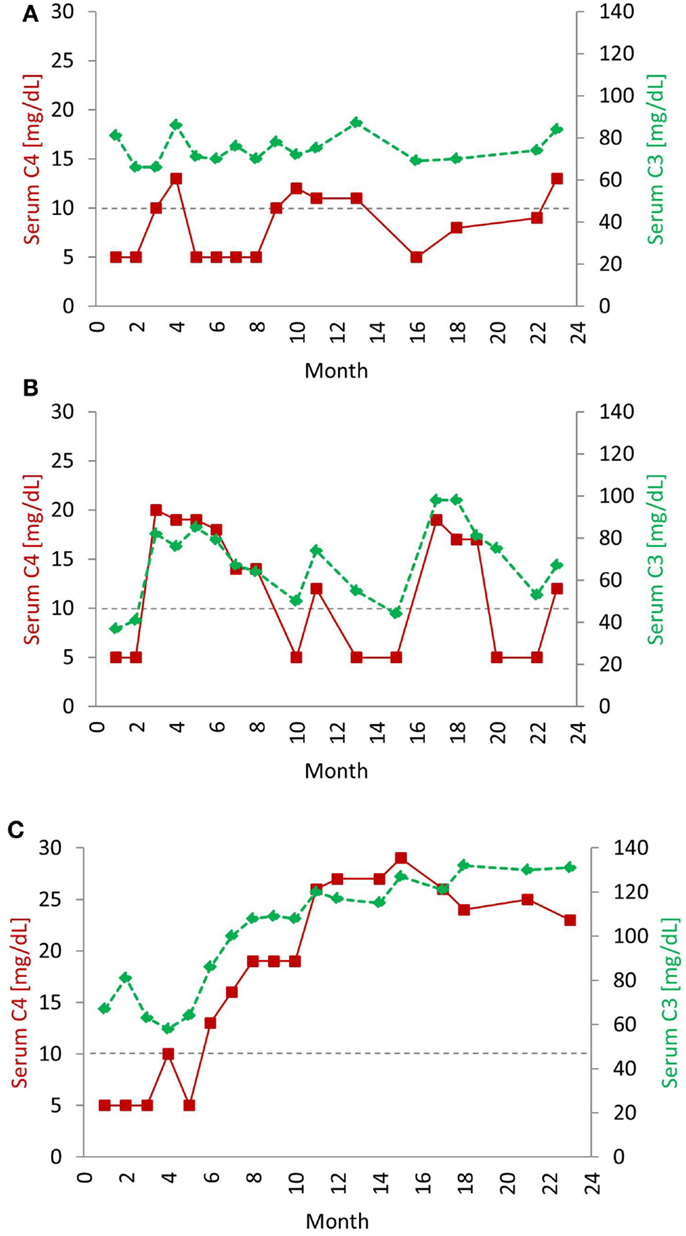
Figure 2. Typical serial serum protein profiles of complement C4 and C3 in human SLE patients. Serum C4 (red, solid line) and C3 (green, dashed line) protein levels tend to go up and down together in most SLE patients. The horizontal dotted line indicates the low level of serum C4 (<10 mg/dL), below which usually requires clinical attention. The profiles shown are taken from three individual patients over a 24-month period and represent three common profiles typically observed in SLE patients. In the first profile (A), levels of C4 and C3 were chronically low. In some patients, even if C3 levels rose to normal range, C4 levels remained low. Patients with this profile are often characterized by low copy-number of C4 genes. (B) The second profile had frequent and parallel fluctuations of serum C3 and C4. Patients with this profile had active disease, and low C3 and low C4 roughly correlated with disease activity. In the third profile (C), C4 and C3 protein levels stayed in the normal range most of the time, except at the time of diagnosis and during a disease relapse. Patients with this profile had relatively inactive disease. Patients with the second and third profiles have normal gene copy-number of total C4 but may have a heterozygous deficiency of C4A [modified from Reference (12)].
Copy-number variation (CNV) of C4 can affect serum C4 protein concentrations. In an American Caucasian populations, about 60% of individuals have four copies of the C4 gene, 28.5% have three (or less), and 12.5% have five (or more). In lupus, the number of patients with three or less C4 genes may increase to 42.2% (13). If an individual has low copy of C4 genes, the baseline C4 antigenic level may be 12–18 (~6 to 8 mg/dL per copy of a C4 gene). In this situation, it does not take much activation to lower the C4 out of the normal range. Additionally, a subject’s body mass index (BMI) is positively associated with serum C4 or C3 protein concentrations (14, 15). All things considered, the care of each patient must be individualized. Repeated, longitudinal serum measurements of C3 and C4 are usually clinically utilized.
In the past decade, cell-bound levels of processed complement activation products (CBCAPS), especially erythrocyte-bound C4 (E-C4d), has been proposed to assist in the diagnosis and clinical monitoring of SLE (16, 17). E-C4d is a stable, proteolytic end fragment of C4, which is covalently bound to the surface of erythrocytes following activation of the CP or MBL pathway. On cell surfaces, the activated C4b is processed to the cell-bound C4d with the release of soluble C4c through factor-I mediated proteolysis in the presence of a cofactor (i.e., CR1 or MCP on the plasma membrane or C4bp from plasma). In 2004, Manzi et al. found that erythrocytes from SLE patients had markedly higher levels of E-C4d when compared to healthy controls or patients with other diseases (16). Additional studies explored the utility of CBCAPS on T and B lymphocytes, platelets, and reticulocytes in SLE (18–21). The levels of C4d bound to the membrane of these cells were significantly higher in SLE than healthy controls or patients with other diseases. In a study of 304 SLE patients, 285 patients with other rheumatic diseases and 205 healthy controls, Putterman and colleagues reported that CBCAPS on erythrocytes or B cells had higher sensitivity than standard complement levels (serum C3 and C4) and anti-dsDNA measurements when distinguishing between SLE and non-SLE, suggesting that CBCAPS could be more specific and sensitive biomarkers for diagnosis and prognosis of SLE (17). However, given the relative simplicity and low cost of serum C4 and C3 measurements, it remains to be seen how assessment of cell-bound C4d and C3d will contribute to the clinical care of patients with SLE.
Historically, cell-bound complement activation proteins, particularly those of C4d, have offered clues to several important discoveries. Between 1960 and 1990, it was found that blood group antigens, Chido (Ch) and Rodgers (Rg), were polymorphic variants of complement C4 (22–24). Alloantibodies generated against Ch/Rg antigens after blood transfusion in certain recipients were mostly directed against polymorphic amino acids present in the C4d region of C4B and C4A proteins, respectively. The mapping of the Ch/Rg variants to the HLA contributed to the understanding of MHC genetics. The polymorphisms of C4A and C4B protein allotypes are readily demonstrated by immunofixation of EDTA-plasma resolved by high voltage, agarose gel electrophoresis based on gross differences of electric charge of protein molecules resulting in variations in electrophoretic mobilities (25–27). Using an assay that involved an overlay of “sensitized” sheep red blood cells on the described C4-allotyping gels, it was found that C4B proteins are functionally 4–10 times more active on the hemolysis of sheep red blood cells (26, 28, 29). While most C4A are associated with Rg antigens and C4B with Ch antigens, reverse associations such as C4A1 with Ch and C4B5 with Rg were demonstrated (23, 30). It was the cloning and sequencing of those functional and serological variants that enabled the identification of specific isotype residues at positions 1120–1125, PCPVLD for C4A and LSPVIH for C4B, encoded by exon 26 of the C4 gene (31, 32). The major epitopes for Rg and Ch blood groups were mapped to positions 1207–1210, VDLL for Rg and ADLR for Ch, encoded by exon 28 of C4 genes (32). Detailed characterization of C4A and C4B genetic polymorphisms unraveled a surprising phenomenon: CNVs and gene size dichotomy among human subjects with specific distribution patterns among different racial populations (13–15, 33, 34).
In parallel to the discovery of common CNV for C4A and C4B genes that contribute to quantitative and qualitative phenotypic diversities of innate immunity and associations with autoimmune diseases, another far-reaching observation related to application to cell-bound C4d occurred in the field of organ transplantation. The deposition of C4d on the endothelium of capillaries for renal, heart, and lung grafts is recognized to be a diagnostic biomarker or “immunohistochemical imprint” of alloantibody-mediated complement activation that leads to graft rejection. In transplant recipients, preexisting or de novo donor specific alloantibodies binding to graft cells may activate complement and cause graft injury. In acute and chronic rejections of renal grafts, peritubular capillary (PTC) deposition of C4d occurred in about 30% of biopsy specimens and detection of diffuse PTC-C4d often associates with poor renal graft outcome. Reviews of C4d-deposition on graft capillaries as a result of humoral alloreactivity can be found in Ref. (35–37).
Deficiencies or genetic polymorphisms of early complement components are strongly associated with increased risk of developing SLE or a lupus-like disease (Figure 3). Complement deficiencies are hypothesized to be associated with increased susceptibility of SLE for several reasons. Functionally, the role of complement includes the identification, opsonization, and proper disposal of apoptotic cells and immune complexes formed regularly between antibodies and foreign or self-antigens (1, 38). An inability to efficiently clear apoptotic cells could render them a source of autoantigens and thereby drive autoantibody production. Impaired clearance of immune complexes and “self” debris provides a logical explanation for complement deficiency in the induction of SLE (39). While there are multiple complement pathways to assist the host with clearance of these types of materials that accumulate continuously in healthy subjects, the CP is essential through at least C4, and to a lesser degree C2, to properly handle and dispose of immune complexes and apoptotic debris.
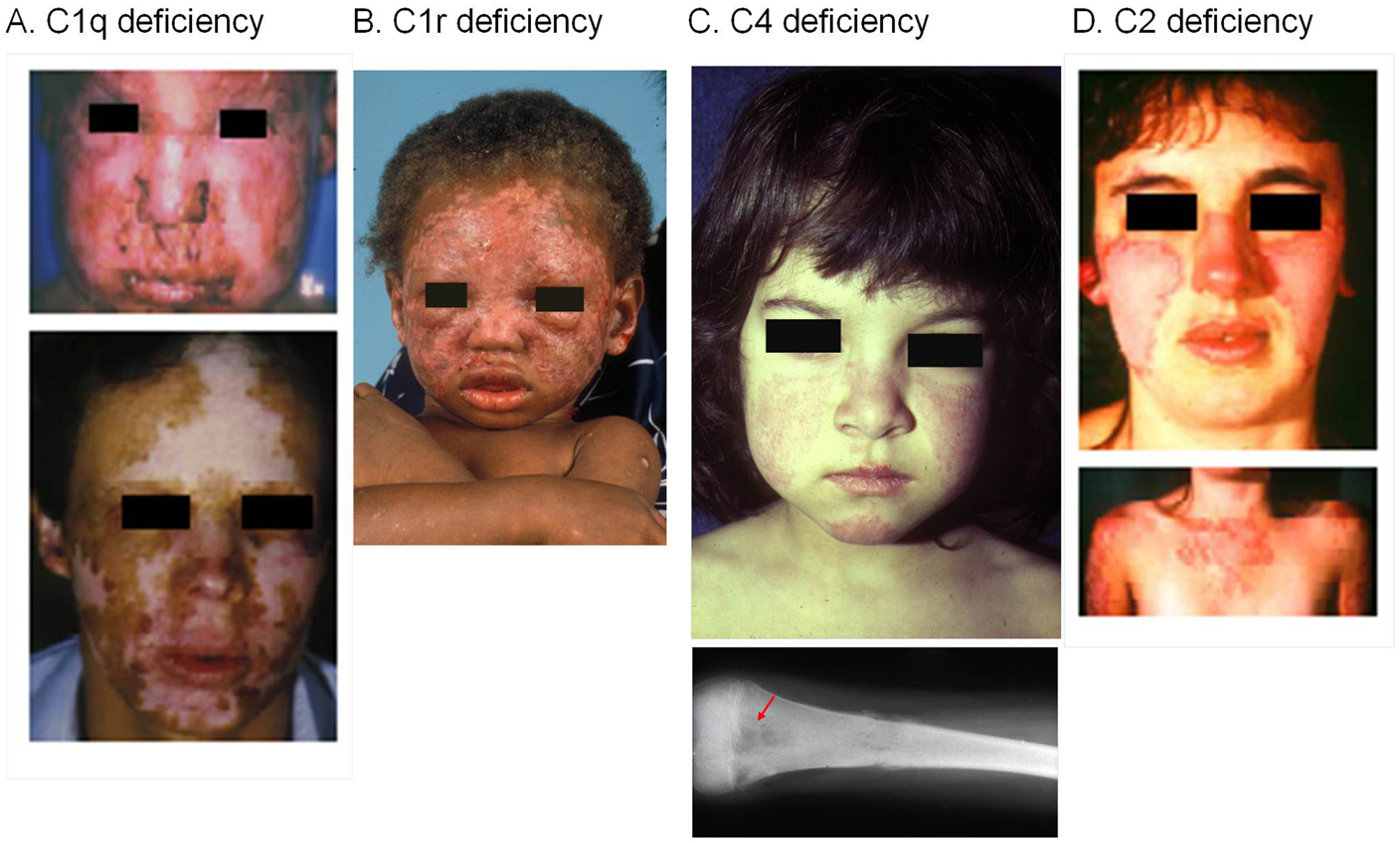
Figure 3. SLE patients with a homozygous deficiency of early components for the classical pathway of complement activation. Severe cutaneous lesions are common clinical presentations in SLE patients with a complete complement deficiency. (A) A homozygous C1q-deficient male child with cutaneous infection (upper panel) and with discoid lupus erythematosus and scarring lesions on face when he was 22 years old (lower panel). (B) A male child with discoid lupus at 16-month old with homozygous C1r-deficiency. This patient experienced generalized seizures, developed a scissoring gait with toe walking, spasticity and weakness of the legs. At 18 years old, he was diagnosed with class IV lupus nephritis and progressed to end-stage renal disease. (C) A complete C4-deficient girl at 3 years old with butterfly rash and cheilitis (upper panel), and osteomyelitis of the femur at 10 years old (lower panel). This patient died at age 12 because of pulmonary infection and cardiovascular failure. (D) A homozygous C2-deficient young woman with acute cutaneous lupus erythematosus. The upper panel shows the butterfly rash, and the lower panel shows photosensitive lesions on sun-exposed areas [adopted from Ref. (40–43)].
Another hypothesis that attempts to explain the association of complement deficiency with SLE suggests that the complement system is involved in immune tolerance (44). In other words, early components of the CP are engaged in the “cross-talk” to the adaptive immune system to achieve tolerance against self-antigens, or in the discrimination of “self” versus “non-self.” A complement deficiency that results in a breach of self-tolerance provides a reasonable explanation for association with SLE (45, 46). Normally, complement receptor 1 (CR1/CD35) and complement receptor 2 (CR2/CD21) on follicular dendritic cells of peripheral lymphoid tissues (such as the spleen and lymph nodes) bind to and deliver self-antigens coated with complement fragments to the autoreactive B-cells, which are anergized or kept away from germinal center reactions. Previously, it was demonstrated in mouse models that complement was necessary for the elimination of self-reactive lymphocytes during the maturation of the immune system (46). Specifically, Prodeus et al. showed that deficiency of either CD21/CD35 or of C4 in a well-defined mouse model of peripheral tolerance resulted in high titers of anti-nuclear antibodies (ANAs) and a severe lupus-like disease.
These two concepts of debris clearance and regulation of self-tolerance are not exclusive and likely overlap, or together are responsible for the development of SLE in humans deficient in early components of the CP.
In the following sections, we will describe genetic complement deficiency states associated with SLE for each component of the early CP (C1q, C1r, C1s, C4, and C2).
Three different genes (C1qA, C1qB, C1qC) closely linked on the short arm of chromosome 1 encode for the C1q protein, which is composed of 18 polypeptide chains. Inter-chain disulfide bonds are formed via the cysteine residue in the N-terminal region of each chain. Following the N-terminal region is a collagen-like region (CLR) of ~81 residues. One A chain and one B chain form a heterodimer during biosynthesis, and two C chains form a homodimer, both through disulfide linkages via conserved cysteine residues. Two A–B heterodimers associate with one C–C homodimer to form a hexameric structure, of the composition ABC–CBA. Three of these hexamers, with a total of 18 polypeptide chains together, form the tulip-like structure of C1q with a collagenous tail and six globular regions, each with globular heads ghA, ghB, and ghC.
The CP of activation is initiated by the C1 complex, of which C1q is the first subcomponent. When C1q in the C1 complex binds to IgM or IgG present in an immune complex, a binding site of C1r/C1s is exposed, allowing further activation of the complement pathway (47–49). C1q is an important opsonin to promote phagocytosis of apoptotic cells or debris, which can be archived directly without complement activation through binding at the collagenous region of C1q to calreticulin (CRT) in apoptotic cell blebs and to CD91 on phagocytes; or indirectly with activation of the CP as C1q binds to CRP ligated to phosphorylcholine/phosphatidylserine or to SAP ligated to fragmented chromatin from apoptotic cells, generating processed products C4b, C3b, iC3b, C3dg, and C3d that are ligands for CR3 and CR4 on myeloid cells to initiate phagocytosis.
The number of reported cases of homozygous deficiency of C1q has increased to 74 (3, 50–52). Among the reported C1q-deficient subjects, the median age of (any) disease onset was 6 years. The clinical presentations among C1q-deficient patients varied considerably, but the two common observations were: (a) SLE or lupus-like disease in 88% and (b) recurrent bacterial infections in 41% (40, 52). Some patients (17%) died at a young age secondary to septicemia. Among the C1q-deficient patients with SLE or lupus-like disease, cutaneous disorders, especially photosensitivity, were prominent with a frequency of 84%. Glomerulonephritis and neurologic disease affected about 30 and 19% of patients, respectively. Oral ulceration occurred in 22% and arthritis/arthralgia in 16%. Immunologically, most C1q-deficient patients had normal serum levels of complement C4 and C3, high frequency of ANAs (particularly anti-Ro/SSA) but a low frequency of anti-dsDNA.
Several different causative mutations have been identified in patients with complete C1q deficiency. A variety of mutations (including non-sense, frameshift indels, and splice site) result in the absence of biosynthesis of one of the three C1q (A, B, or C) chains. Other mechanisms have been identified by which C1q protein is defective in secretion, structure, or function. In Table 1, the molecular basis of the genetic mutations leading to complete absence or complete functional deficiency of C1q protein for which genetic information is reported, as well as the accompanying clinical manifestations.
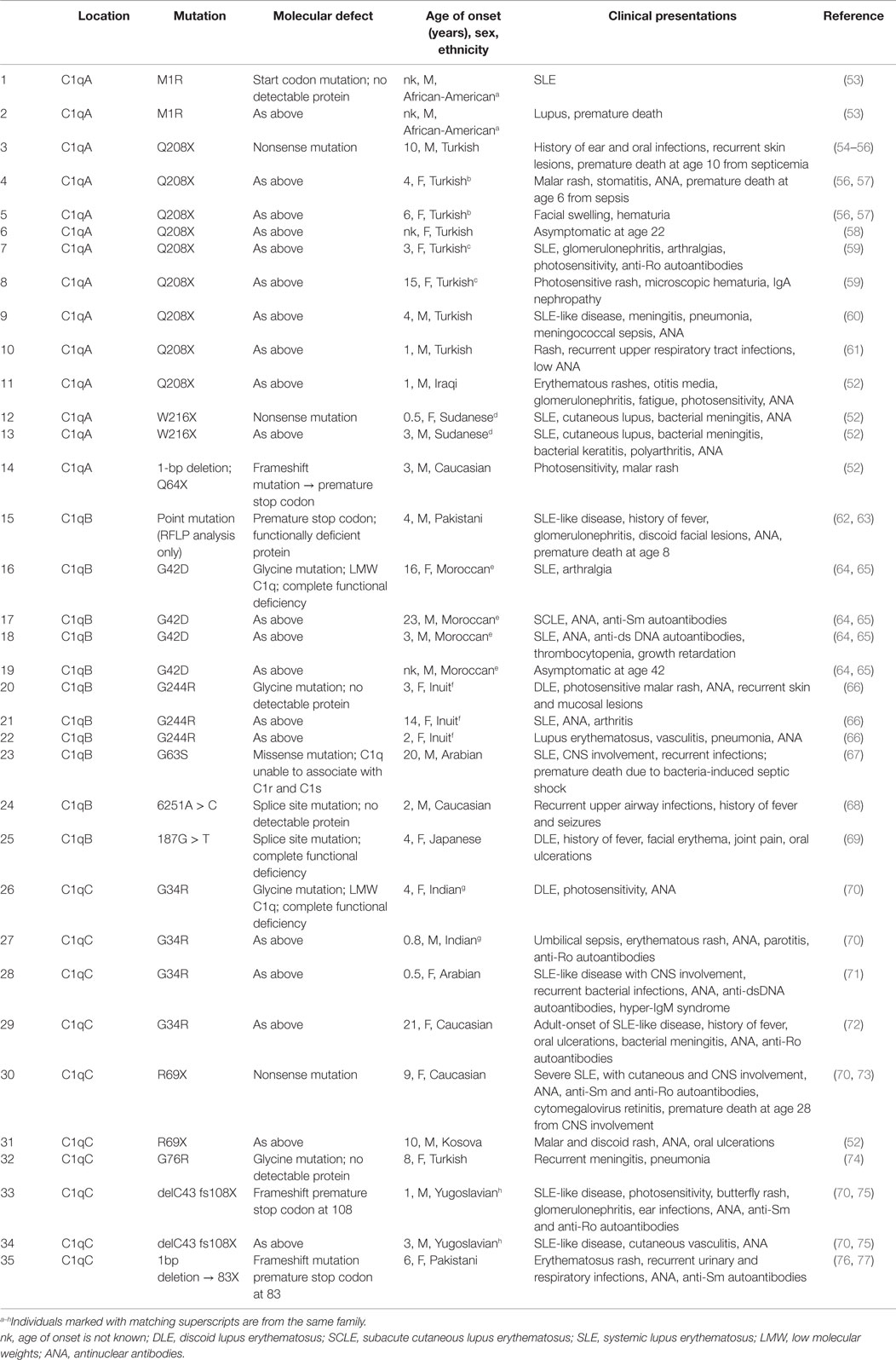
Table 1. Complete deficiency of A, B, or C chain genes of C1q.
Most of the causative mutations associated with homozygous C1q genetic deficiency are the result of a consanguineous marriage. The affected patients are likely close descendants of ancestors carrying the specific deleterious mutation. Surprisingly, screening of large SLE cohorts from those countries with reported cases of C1q deficiency to determine the prevalence of C1q deleterious mutations have yielded negative results (59, 78), suggesting that the mutations are “private” and rare but with very large effect size, as documented in many complex diseases (79, 80).
The genes for human C1s and C1r are located on the short arm of chromosome 12 (81). According to bioinformatics studies and previous publications (82), C1r and C1s are configured in a tail-to-tail orientation with their 3′ ends separated by ~9 kb. The DNA sequence for the genomic region harboring human C1s and C1r coding sequences is still incomplete in the Reference Genome (Annotation release 106, January 2015) and consists of gaps.
C1r and C1s are paralogous proteins that share 38% identity and 55% similarity. Each mature protein is a proenzyme consisting of six distinct modules: two CUB domains separated by an EGF domain with a binding site for Ca2+, followed by two complement controlling protein repeats CCP1 and CCP2, a linker segment and then a chymotrypsin-like serine protease domain SP at the carboxyl-terminus region. In circulation, C1r and C1s are proenzymes that exist as a tetrameric structure, C1s–C1r–C1r–C1s, which assembles in the presence of Ca2+ with C1q to form the multi-molecular C1 complex. Upon activation of C1q (e.g., through binding of its globular heads to the Fc-regions of IgG or IgM in an immune complex), the tetramer interacts with the hinge region of C1q to form the activated C1 complex. Autoactivation of the two C1r by proteolytic cleavages between Arg-463 and Ile-464 is followed by activation of C1s by proteolysis between Arg438 and Ile-439, which release the enzymatic activity of the nascent C1 complex. The C1s in this C1 complex activates C4 and then C2 which together form the CP C3 convertase, C4b2a.
Deficiencies in subcomponents C1r and C1s were among the earliest reports linking complement deficiency with human glomerulonephritis or a lupus-like disease (83–85). A total of 20 cases of C1r and/or C1s deficiencies have been reported, which include 12 cases of C1r deficiency from eight families and eight cases of C1s deficiency from five families. Among the C1r-deficient patients, there was a consistent reduction in the serum protein levels of C1s to 30% of its normal level, but highly elevated serum protein levels of C4, C2, and C1-inhibitor (200–400% of their corresponding normal ranges). C3 was also elevated by ~50%, but C1q levels were normal. A similar phenomenon was observable among C1s-deficient patients; C1s-deficient patients had greatly reduced serum levels of C1r, markedly elevated levels of C4, C2, C1-inhibitor, and C3, and normal levels of C1q.
Among the C1r/C1s deficient subjects, all but three had recurrent bacterial, viral, or fungal infections (85%), and many patients died at young age because of a severe infection. Thirteen subjects (65%) developed SLE or a lupus-like disease. The prevalence of ANA among these patients was about 60%. Mortality at young age from infections likely explains the slightly lower frequency of lupus disease association compared to C1q deficiency. Most C1r/C1s deficient patients had severe cutaneous lesions (Figure 3). Eight patients (40%) had renal disease due to lupus nephritis. Such presentations underscore the inter-dependence of C1r and C1s in sustaining a stable tetrameric structure that would otherwise be susceptible to a high turnover rate. A deficiency of C1r or C1s prevents the formation of the C1 complex and diminishes the need for engagement of C1-inhibitor and other regulators of complement activation. When C1 is not functional, the CP is not activated, and consumption of C4, C2, and C3 is greatly reduced, resulting in high levels of these proteins in the circulation (41). This and other results strongly indicate a chronic turnover of component proteins for the CP (86).
The molecular defects leading to C1r or C1s deficiency have been determined in one case of C1r deficiency and seven cases of C1s deficiency (Table 2). Relative to C1r deficiency, the defect was a homozygous C to T substitution in exon 10 resulting in the R380X non-sense mutation in the second CCP domain, resulting in no detectable protein in the serum (41). The proband developed SLE at 3 months of age and presented with reduced levels of C1s (similar to other C1r-deficient patients), but highly elevated protein levels of C4, C2, and C1 inhibitor.
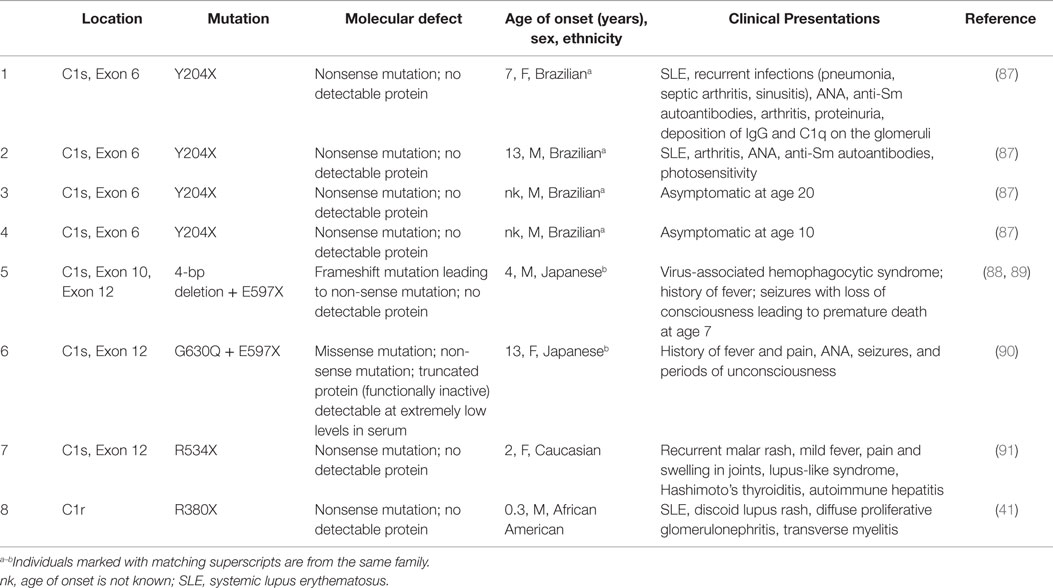
Table 2. Molecular defects and clinical presentation of complete deficiency for C1s or C1r.
For C1s-deficiency, several deleterious mutations have been identified. A C→G mutation in exon 6 (Y204X) resulted in a premature stop codon and abrogated any protein production (87). This particular non-sense mutation was homozygous in four siblings, and all showed no detectable C1s protein, but only two developed SLE at the ages of 7 and 13 years. The other two siblings, at ages 10 and 20, did not have clinical symptoms of SLE. Significantly reduced levels of C1r and elevated serum C4 were detected in all four siblings. In another case report, a 4-bp deletion (TTTG) in exon 10 that led to a frameshift and a non-sense mutation in exon 12 (E597X) was detected in a single patient (88, 89). This patient developed unique symptoms including virus-associated hemophagocytic syndrome and died after a long period of a comatose state. A mutation documented in another patient from the same family was a heterozygous G → T mutation in exon 12 leading to E597X and on the other allele, a novel missense mutation G630Q (90). This patient displayed symptoms that were similar to the other, related patient, including fever of unknown origin and short-term disturbances of consciousness. A second C1s genetic variant is a non-sense C → T mutation in exon 12 (R534X) (91). This patient had undetectable serum C1s, normal C1r and C1q, and absence of CH50 activity. The patient was 2 years of age and presented with several autoimmune diseases, including a lupus-like syndrome, Hashimoto’s thyroiditis, and autoimmune hepatitis. Each of these clinical observations and the similarities in autoimmune presentation with C1r/C1s deficiencies highlight the role of a dysfunction in the CP of complement leading to systemic autoimmune disease.
The complement C4 gene is located in the HLA class III region on the short arm of chromosome 6, telomeric of the C2 gene (92). Remarkably, there are extensive inter-individual gene CNVs for complement C4. Two to eight copies of C4 genes can be present in a diploid genome (93, 94) (Figure 4). Such common CNVs are an uncommon phenomenon in mammalian genetics, as they deviate from the conventional “one-to-one” concept for a gene and a polypeptide/gene product. Here, one to four copies of nearly identical genes co-exist on a single chromosome, thereby creating a gene dosage effect for a quantitative phenotype. Segmental duplications for C4 always include the RP (STK19) gene upstream of C4, and the downstream genes CYP21 and TNX (94). Each C4 gene either encodes for an acidic C4A or a basic C4B protein, with only four amino acid changes (PCPVLD 1120–1125 for the C4A isotype and LSPVIH for the C4B isotype), but this results in substantial differences in chemical reactivity for peptide and carbohydrate antigens (28, 29, 31, 95). C4A favors binding to amino groups (i.e., immune complexes) while C4B favors binding to hydroxyl or carbohydrate-rich groups. Approximately 40 protein variants for complement C4 have been documented (25). Technically, it is noteworthy to mention that the differential binding to hydroxyl- or amide-group containing substrates/immune complexes, or hemolytic activities between activated C4A and C4B can be readily demonstrated using purified component proteins or proteins resolved by gel electrophoresis, but not from sera when many regulatory proteins are present (15, 96).
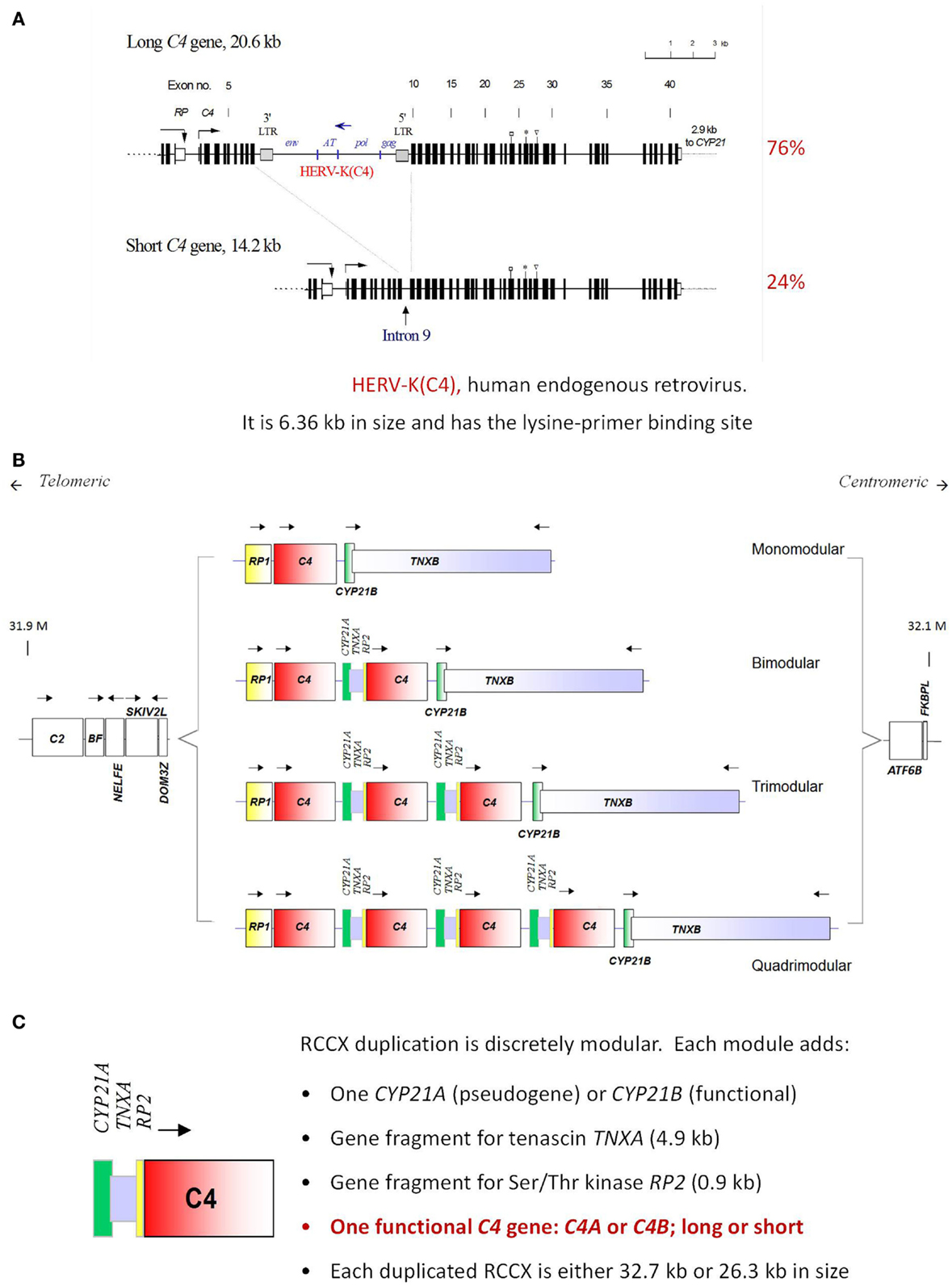
Figure 4. Gene size dichotomy and gene copy-number variation of complement C4. A human C4 gene consists of 41 exons coding for a precursor protein of 1744 amino acids including a signal peptide of 19 amino acids. (A) There are two forms of C4 genes. The long gene is 20.6 kb, and the short gene is 14.2 kb. In a long C4 gene, an endogenous retrovirus HERV-K (C4), which is 6.4 kb in size, integrated into its ninth intron. Among healthy subjects of European ancestry, 76% of C4 genes belong to the long form and 24% belong to the short form. (B) Among European subjects, one to four copies of C4 genes are present in the central region of the major histocompatibility complex (MHC) located on chromosome 6p21.3. Thus, there is a continuous variation in copy number of C4 genes from two to eight copies among different human subjects. (C) The duplication of a C4 gene occurs in a modular fashion, with a 0.9 kb fragment of RP (STK19) upstream of complement C4, a full steroid cytochrome P450 21-hydroxylase (CYP21) and a 4.0 kb fragment of the tenascin (TNX) at the downstream region of C4 (known as a RCCX module). The duplication of CYP21 gene can be a pseudogene (CYP21A or CYP21A1P) or an intact functional gene (CYP21B or CYP21A2). Each C4 gene in the RCCX module may either code for an acidic C4A or a basic C4B. Each C4 gene may be either long or short [adopted from Ref. (97–99)].
The primary site for C4 biosynthesis is in the liver. However, multiple tissues also synthesize C4, presumably for local consumption, particularly after stimulation by interferon-gamma (100). A thioester bond is present but hidden in native C4. A proteolytic cleavage by activated C1s removes a 74 amino acid C4a peptide and leads to a remarkable change of conformation in C4 (101). Consequently, the protected thioester bond becomes exposed to the exterior. In activated C4B, one of the four isotypic residues (Histidine-1125) serves as a catalyst and facilitates a rapid nucleophilic attack, resulting in formation of a covalent ester linkage between C4B and the target surface (102, 103). In activated C4A, Histidine-1125 is not present, and such a catalytic reaction does not occur. Instead, C4A reacts effectively with an amino group on an immune complex or a protein molecule to form a covalent amide bond. Such a difference in chemical reactivity appears to diversify the functional roles of C4A and C4B in the clearance of immune complexes and the propagation of activation pathways, respectively.
A complete or homozygous genetic deficiency of both complement C4A and C4B has been reported in 28 individuals (42, 104–107). The subjects came from 19 families with different racial backgrounds were characterized by 16 different HLA haplotypes. The female to male ratio was 1:1. SLE or lupus-like disease was diagnosed in 22 (78.6%) of the C4-deficient subjects, and four others had renal disease including glomerulonephritis. Early disease onset, severe photosensitive skin rash, the presence of autoantibodies against ribonuclear protein Ro/SSA, and high titers of ANA were common clinical features of the subjects. Many of the C4-deficient patients also had severe proliferative glomerulonephritis. We have summarized the molecular basis of complete C4 deficiency determined in 15 cases (Table 3).
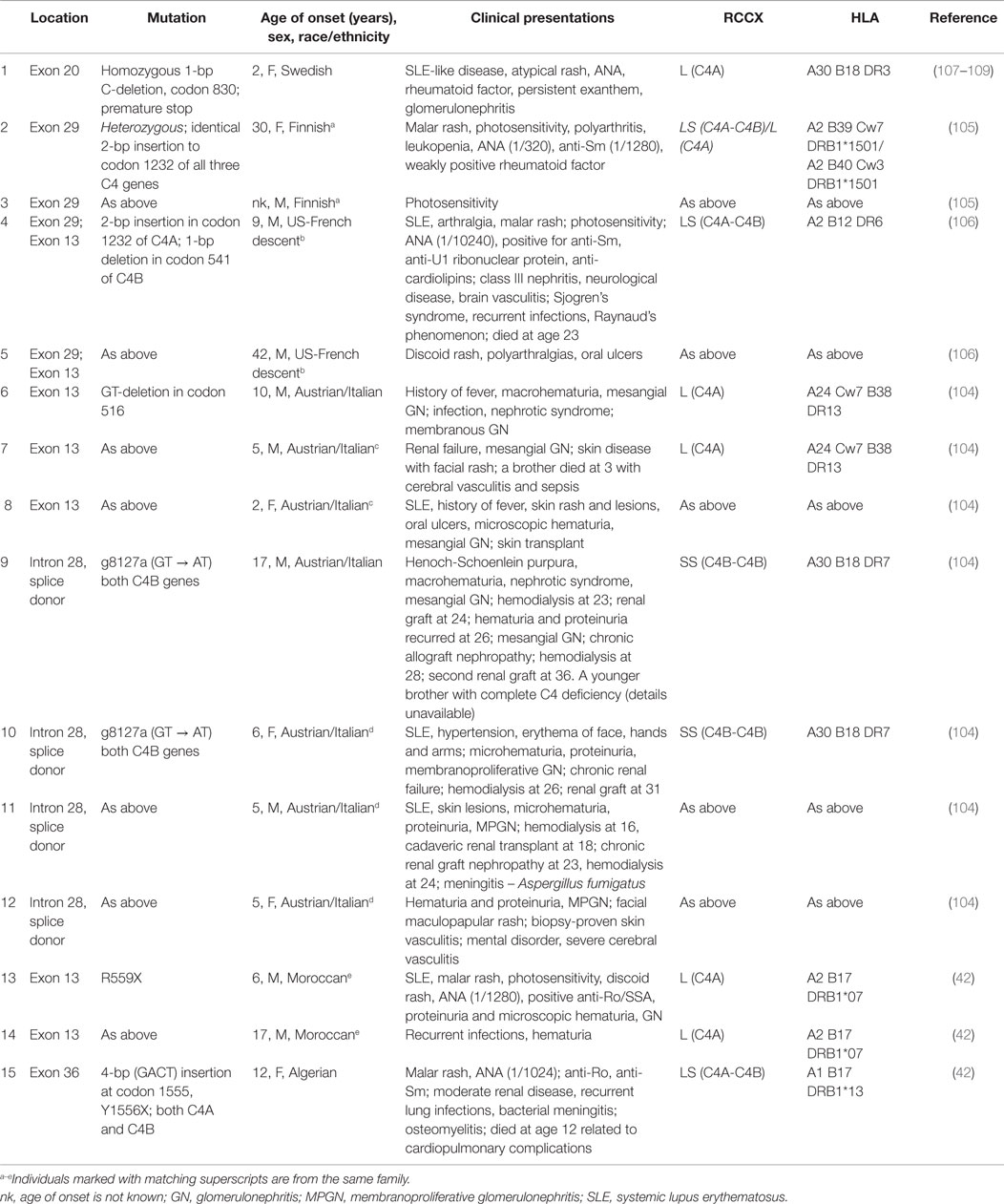
Table 3. Molecular defects and clinical presentations of complete deficiency of complement C4A and C4B.
While a complete deficiency of C4 is rare, an isotype deficiency of either C4A or C4B is much more commonly observed and has been implicated in several autoimmune diseases (13, 110–113). To investigate the C4 genetic diversities in SLE, a study population consisting of 216 female SLE patients, 17 male SLE patients, 362 first degree relatives, and 389 unrelated healthy female controls, and 128 male controls was investigated rigorously (13). In the study group of European Americans, total gene copy-number (GCN) of C4 ranged from 2 to 6 copies, GCN of C4A ranged from 0 to 5 copies, and GCN of C4B ranged from 0 to 4 copies. In comparison to healthy controls, SLE patients had significant reductions of GCN of total C4 (Figure 5). Among the SLE patients, 9.3% had only two copies of C4 genes, compared to 1.5% in healthy controls. The effect size of SLE disease risk (or odds ratio) for subjects who had only two copies of C4 genes was 6.51. Of the two C4 isotypes C4A and C4B, there were no significant differences detected among GCN of C4B between SLE and controls. However, significant decreases of GCN of C4A were noted in SLE patients. Among SLE patients, 6.5% had a homozygous deficiency (i.e., 0 copy) of C4A and 26.4% had a heterozygous deficiency (i.e., 1 copy), compared to 1.3 and 18.2%, respectively, in healthy controls. The odds ratio for SLE for a subject with C4A homozygous deficiency of C4A was 5.27. In other words, a total C4 GCN = 2 or a C4A GCN = 0 (C4A deficiency) are large effect size genetic risk factors for human SLE. Moreover, family based association tests revealed that monomodular RCCX haplotypes with a single short C4B gene and C4A deficiency were more frequently transmitted to the SLE patients than normal heritance pattern (p = 0.005). To date, no other common genetic variant has been identified to be so strongly associated with SLE. Remarkably, 32.9% of SLE subjects carried the risk factor of low GCN of C4A. As with most common genetic variants associated with autoimmune disease, the risk factor is also present in the general population with considerable frequency (19.5% in this study).
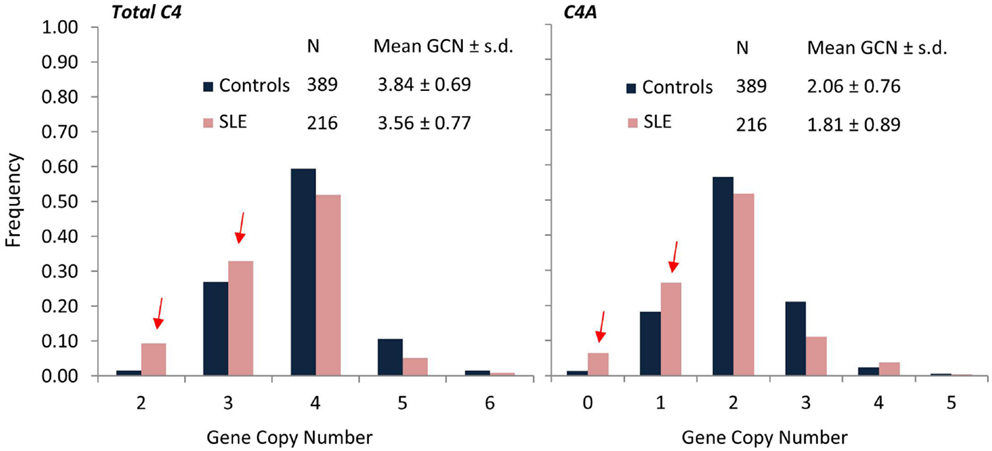
Figure 5. Comparisons of frequencies for total C4, C4A, and C4B gene copy-number groups in SLE (red) and controls (blue). SLE patients (N = 216) of European ancestry showed significantly higher frequencies for lower copy-numbers of total C4 (GCN = 2 or 3) and C4A (GCN = 0 or 1) compared to healthy, race-matched controls (N = 389). Mean GCN for Total C4 in SLE (3.56 ± 0.77) was significantly lower than in controls (3.84 ± 0.69; p = 5.3 × 10−6, t-test). Similarly, mean GCN for C4A in SLE (1.81 ± 0.89) was significantly lower than in controls (2.06 ± 0.76; p = 2.0 × 10−4, t-test) [modified from Ref. (13)].
From another way of statistical analysis, C4 GCN variations are continuous variations, and therefore, the mean of GCNs for total C4, C4A, C4B long genes and short genes can each be compared by Student’s t-test between patients and controls. The mean GCN (±SD) for total C4 between female controls and female patients were 3.81 ± 0.75 and 3.56 ± 0.77, respectively (p = 0.0001). The mean C4A GCN (±SD) was 2.05 ± 0.79 in controls and 1.81 ± 0.89 in SLE (p = 0.0005). The mean long C4 was 2.91 ± 1.03 in controls and 2.66 ± 1.14 in SLE (p = 0.005). In other words, when compared with controls, SLE showed reductions of 0.25 copies of total C4, 0.24 copies of C4A, or 0.25 copies of long C4. For C4B or short C4 genes, no significant differences were observed between the European American SLE and race-matched controls.
As the first original publication to document the highly prevalent, multi-allelic gene CNVs of complement C4 in SLE, this work (13) went through independent and rigorous data generation and validation processes to confirm the CNV calls. Those processes included (a) pulsed-field gel electrophoresis to resolve PmeI-digested genomic DNA fragments and Southern blot analysis for long-range mapping to elucidate the physical size of RCCX modules in haplotypes; (b) TaqI restriction digests and genomic Southern blot analyses (restriction fragment length polymorphisms) to resolve the relative dosages of RP1-C4L (7.0 kb), RP1-C4S (6.4 kb), RP2-C4L (6.0 kb), RP2-C4S (5.4 kb), plus relative dosages of CYP21B to CYP21A, and relative dosages of TNXB to TNXA; (c) PshAI-PvuII digests of genomic DNA and Southern blot analysis to segregate C4A and C4B and yield their relative dosages; (d) immunofixation of EDTA-plasma for polymorphic variants of C4A and C4B proteins resolved by high voltage agarose gel electrophoresis, based on differences in electric charges of C4 allotypes; and (e) corroboration of C4 genotypes and phenotypes of study subjects from data of family members.
Subsequently, TaqMan-based quantitative PCR amplicons for total C4, C4A, C4B long genes and short genes were developed and applied for replication studies, particularly when quantities of genomic DNA for patients and controls are limiting. In this later case, internal data validation is achieved when GCNs of total C4 = GCNs of C4A + C4B = GCNs of C4 long + C4 short. Such qPCR strategy is sensitive and highly robust when the quality of genomic DNA is excellent, which can be reflected by internal data validation of the independent amplicons. Our experience suggested that genomic DNA samples at low concentrations (≤15 ng/μl) are relatively unstable in storage, particularly if they had gone through rounds of freeze-thaws, and tend to yield inconsistent data in analyses of multi-allelic CNVs. This makes individual internal data validation crucial for data accuracy.
Association of lower GCN of total C4 and C4A-deficiency as a risk factor of SLE have also been observed in three independent East-Asian studies (114–116). C4A deficiency in subjects of European ancestry is primarily attributed to the presence of a single short C4B gene (mono-S) in the HLA that is predominantly in linkage disequilibrium with HLA A*01, B*08, and DRB1*0301 that is dubbed ancestral haplotype 8.1 (AH8.1) (117). Intriguingly, such AH8.1 haplotype is basically absent among East-Asian subjects (Figure 6), supporting the evidence that C4A-deficiency association with SLE is not due to linkage disequilibrium with certain HLA haplotypes (e.g., DRB1*0301). Different mechanisms leading to C4A deficiency in East Asians include (a) monomodular RCCX with a single long C4B gene, (b) bimodular RCCX haplotypes encoding C4B1-C4B96 or C4B1-C4B1 are prevalent in Asian SLE (116). Certainly other genetic or environmental risk factors, combined with the C4A deficiency, contributed to SLE development in genetically predisposed patients. Still, it is of interest to examine if restoring C4A in such (C4A-deficient) patients would result in positive therapeutic outcomes.
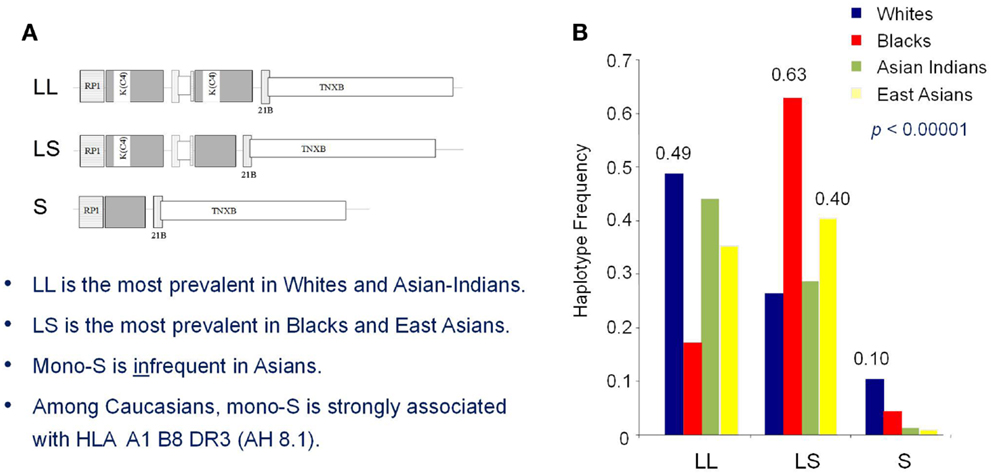
Figure 6. Race-specific distribution patterns of RCCX modules in human populations. The size dichotomy of C4 genes and copy-number variation of RCCX modules on an MHC haplotype together create a repertoire of length variants among different human subjects, which also exist with race-specific distribution patterns. (A) The most prevalent haplotypes of RCCX in Whites and Asian-Indians are the bimodular long-long (LL) and bimodular long-short (LS) in Blacks and East-Asians. (B) Notably, monomodular-short (mono-S or S) haplotypes with a single short C4B gene and C4A deficiency is relatively common in White and Black subjects but almost absent in Asians [modified from Ref. (118)].
On a recent case/control study of British SLE (cases, N = 501; controls, N = 719), total C4 GCNs were determined by a paralog ratio test, which employed a set of primers (16-mer) that hybridized to and PCR-amplified the duplicated regions of complement C4 in the MHC plus a unique and non-variable region in chromosome 19 as reference control (for two copies). C4A and C4B were deduced from the ratios of C4A and C4B that is determined by NlaIV restriction digest of a different PCR product spanning the C4-isotypic site (158 bp for C4B and 91 bp for C4A after NlaIV digest) and were resolved by capillary electrophoresis (119). While extensive GCN variations and associated polymorphisms were observable for complement C4, the difference on the mean copy number of total C4 between British SLE and controls was only marginal (3.79 ± 0.98 in SLE; 3.89 ± 0.98 in controls; p = 0.046). On the other hand, the mean copy numbers of C4A (1.82 ± 0.93 in SLE, 2.08 ± 0.93 in controls; p < 0.001) and C4B (1.96 ± 0.93 in SLE and 1.81 ± 0.72 in controls; p < 0.001) were both significantly reduced in SLE. However, in multiple logistic regression models, deficiencies of C4A or C4B both became insignificant in the presence of DRB1*03. This led the investigators to conclude that “…partial complement C4 deficiency states are not independent risk factors for SLE in UK….” It is notable that there were significant differences in the architecture of GCN group distributions between the British (119) and the US study populations (13).
Going through the previously mentioned Boteva study on C4-CNV determination, we note two issues that probably led to data misinterpretation. The first was a lack of an internal data validation for GCN calls. The second was an additional procedure to overcome the artifacts introduced by heteroduplex formation between multiple alleles during PCR, which would be resistant to restriction digest (NlaIV). Resistance to NlaIV digestions would therefore skew relative dosage of C4A and C4B and lead to misinterpretation of C4 copy-number calls (97, 120). As to dissecting the relative roles of C4-CNVs or C4A-deficiency and HLA-DRB1 alleles in SLE, the issues on the lack of internal validation for the reliability of C4-CNV calls cannot be ignored.
The unprecedented variations of GCNs with high frequencies of homozygous or heterozygous deficiency of C4A or C4B, and continuous variations in copy-numbers from one to four copies of C4 genes on each copy of chromosome 6 (or haplotype) among healthy subjects and SLE patients inevitably pose great challenges both technically and conceptually: the former for accurate data acquisition and the latter for accurate data interpretation. Without deliberate design and rigorous and independent validation strategies, unfortunately, many studies were inherently tainted with misinterpretations or partially correct or inappropriate conclusions. On determining the roles of common and multiallelic CNVs in health and disease such as those for complement C4A and C4B, immunoglobulin Fcγ receptors FCGR3A and FCBR3B, and neutrophil alpha-defensins DEFA3 and DEFA1, it is critically important to have meticulous experimental design and methods to acquire accurate and consistent data. Realization of the DNA sequence basis that causes a phenotype or functional diversity is essential for specific experimental method. As mentioned above, the heteroduplexes issue during the PCR process needs to be considered and resolved. Moreover, genetic studies of a complex disease usually require hundreds to thousands of genomic DNA samples from cases and controls, which are generally obtained from multiple sources. For determination of common and continuous CNVs, the high quality of genomic DNA samples is essential. Heterogeneous quality of DNA samples has a high tendency to yield inconsistent data. Under those conditions, independent replication and rigorous internal validation methods of samples from every subject becomes a necessity.
The complement C2 gene is located in the HLA class III region on the short arm of chromosome 6. Serum C2 is a precursor protein that is cleaved by activated C1 into two fragments: C2b and C2a. C2a is a serine protease and forms the C3 convertase along with C4b (denoted C4b2a) (38, 121). C2 also functions as a critical component in the lectin pathway. MBL or ficolins in complex with MASP-1 bind to relevant carbohydrate molecules and activate MASP-2, which then cleaves C2 and C4, forming a C3 convertase identical to that formed in the CP (122). Overall, C2 functions as a key component in the classical and lectin pathway, thereby providing defense against microbial infection and assisting in removal of immune complexes.
Among individuals of European descent, C2 deficiency occurs with an estimated prevalence of 1/20,000, which probably accounts for <1% of SLE patients. There are two types of C2 deficiency (123, 124). Type 1 C2 deficiency is caused by non-sense mutations leading to the absence of protein biosynthesis. The predominant form of such type 1 deficiency was a 28-bp deletion that removed 9 bp from the 3′ end of exon 6 and 19 bp from the 5′ end of intron 6 in the C2 gene, leading to a skipping of exon 6 in the C2 mRNA and generation of premature stop codon (123). Such 28-bp deletion is present in the HLA haplotype with A10 (A25) and B18 in the class I region, BF-S, C2Q0, C4A4, and C4B2 in the class III region, and DRB1*15 (DR2) in the class II region. The second form of Type 1 deficiency is present in HLA A3, B35, DR4, BF-F, C2Q0, C4A3, and C4A2 (125). The cause is a 2-bp deletion in exon 2 of C2 gene that leads to a non-sense mutation.
About 10% of C2 deficiency is secondary to the Type II deficiency in which the C2 protein is synthesized but not secreted. The molecular defects identified as missense mutations are C111Y, S189F, and G444R (126, 127). It is not clear how these mutations block the secretion of C2 protein.
Unlike a deficiency of proteins for the C1 complex or C4 described earlier, the penetrance of C2 deficiency on SLE is about 10%. Similar to other risk factors for SLE, there is a female predominance for patients with C2 deficiency. C2-deficient SLE patients tend to have early childhood onset but a milder disease process with prominent photosensitive dermatologic manifestations, speckled ANAs (the autoantibody specificity is common for the Ro/SSA antigen), and a family history of SLE. Anti-DNA antibody tests are usually negative, and severe kidney disease is rare.
Autoantibodies have been reported that bind with high affinity to complement proteins, particularly in SLE patients (128). Most of these antibodies are not directed against native proteins, but instead directed against neoepitopes. Such epitopes becomes exposed in active or inactivated proteins or upon multi-molecular complex assembly in the activation process or following proteolytic cleavage. The binding of these autoantibodies to complement proteins could lead to a state of an acquired deficiency and contribute to disease pathogenesis similar to the way genetic deficiencies do so. We will describe a series of autoantibodies that have been detected in systemic autoimmune disease against early components and regulators of the CP.
Approximately 30% of SLE patients synthesize autoantibodies against C1q. Their presence correlates with anti-dsDNA, nephritis, and low C4 and C3 in about 75% of such patients (129–131). A relevant question is whether the anti-C1q amplify the complement activation by immune complexes. The development of anti-C1q antibodies may arise in response to activation of the CP. Following CP activation, C1q remains attached to immune complexes and therefore is located at the site of inflammation. Proteases at the inflammatory site may degrade IgG and C1q (autoantigen), generating multiple proteolytic fragments of IgG and C1q. This may be an explanation for C1q antibodies and anti-IgG and anti-IgM (rheumatoid factors) that develop in SLE patients.
A large study was recently reported in which one objective was to assess the specificity of anti-C1q antibodies and their associations with SLE manifestations and diagnostic tests (132). The authors confirmed the association of anti-C1q antibodies, low complement (C4 and C3), and anti-dsDNA antibodies. Further, this combination had the highest serological association with renal disease. Anti-C1q antibodies were detected in 28% of all SLE patients, but were observed more frequently in patients with renal disease in ~68%. Anti-C1q autoantibodies were observed in 5–10% of patients with related systemic rheumatic diseases. It was suggested that the presence of anti-C1q antibodies contributes to a nephritis flare (130).
The presence of autoantibodies against other components of the C1 complex is less well established. A report of 15 SLE patients demonstrated that seven of them had autoantibodies to C1s (133). The binding of these antibodies to C1s was shown to enhance its enzymatic activity for C4, providing possible additional explanation for low serum C4.
An IgG autoantibody that inactivates C1-inhibitor (anti-C1-Inh) was initially described in a patient with the acquired angioedema (AAE) syndrome that mimics hereditary angioedema (134). Anti-C1-Inh autoantibodies have also been described in SLE, especially those exhibiting symptoms of angioedema (135, 136). Anti-C1-Inh antibodies bind and inactivate C1-Inh so that it is no longer available to participate in the regulation of C1. As a result, the CP is excessively activated, leading to development of angioedema as well as possibly more severe renal diseases. A more recent study of 202 SLE patients and 134 healthy controls detected anti-C1-Inh autoantibodies in 17% of SLE patients and 4% of controls (137). In SLE patients, the anti-C1-Inh levels correlated with the duration and activity of SLE but did not correlate with SLE laboratory parameters, including serum levels of C3 and C4. Conversely, 1–2% of hereditary angioedema patients develop SLE, probably related to the chronically very low C4 and C2.
C3 and C4 nephritic factors are IgG autoantibodies that bind to and stabilize the AP C3 convertase and the CP C3 convertase, respectively. By binding the C3 convertases, C3 and C4 nephritic factors prolong the half-life by preventing the regulation of C3 convertases. This results in uncontrolled complement activation and increased consumption and depletion of serum C3. C3 and C4 nephritic factors are associated with membranoproliferative glomerulonephritis, acquired partial lipodystrophy, and post-infectious acute glomerulonephritis (138–140). Both of these autoantibodies have been detected in SLE patients, and suggested to be associated with renal disease, but their prevalence and role in pathogenesis of systemic autoimmune disease is not well documented (139–141).
Because the complement system is increasingly found to be associated with autoimmune diseases, it is an attractive therapeutic target. However, as discussed earlier, complement deficiencies are overwhelmingly associated with increased susceptibility to autoimmune disease, most notably SLE. Therefore, at first glance, the idea that treatment with complement inhibitors would provide a general relief in such diseases seems illogical. Processing of immune complexes is facilitated by deposition of complement fragments, the ensuing binding of complement-decorated immune complexes to CR1 on erythrocytes, and transfer to resident monocytes or macrophages for destruction and/or antigen presentation. For that reason, blocking any step up to and including C3 could inhibit immune complex processing and possibly exacerbate disease in autoimmune patients. Moreover, inhibiting the complement system would further enhance susceptibility to infections, on top of the increased frequency of infections already observed in patients under immunosuppressive therapy.
On the other hand, complement activation is quite evident and partly responsible for tissue damage seen in autoimmune patients with established disease. In lupus, key mediators of tissue damage include C4b/C3b, C5a/C3a, and the MAC, all of which modulate membrane integrity or trigger inflammation in a setting where autoantibodies are already present in large amounts. Therapeutic goals should be aimed at inhibiting this cellular injury and preventing production of the proinflammatory peptide fragments. In atypical hemolytic uremic syndrome (aHUS), endothelial damage is mediated by over-activation of the AP. Inhibition of C5 cleavage has proven to be an effective therapy (142). Anti-C5 therapy has been effective in treatment of antigen-induced arthritis or experimental autoimmune uveoretinitis in animal models (143, 144). Other studies in animal models of arthritis that showed success in reducing inflammation and preventing disease progression include treatment with CR1 (145), antagonists of C3a receptor (146), and the complement regulator CD59 (147). Soluble CR1 prevented dysregulation of C3 convertase in sera from dense deposit disease patients (148). The same study reported that short-term use of soluble CR1 in a pediatric patient with end-stage renal failure normalized the activity of the terminal complement pathway. Inhibition of complement activation specifically in SLE with renal diseases deserves to be assessed.
Another treatment approach is to replenish complement proteins in patients with a complete complement deficiency. As discussed earlier, complete genetic deficiencies of complement are quite rare, but isotype deficiency and functional or acquired deficiencies are quite common. An obvious approach seems to be supplement or replacement of the missing component, systematically or locally. Currently, however, purified or recombinant complement proteins are not available for treatment purposes. One report indicated successful treatment of a C2-deficient patient with SLE using whole plasma preparations (149). Moreover, in patients with established disease and low C4 or C3, an increase in C4 and C3 is associated with a favorable response to treatment.
Fresh frozen plasma is able to restore C1q activity in C1q-deficient patients temporarily, but such activity drops off rapidly within 2 weeks. Thus, weekly infusions of plasma become necessary, which is burdensome, and confers its own risk of infections and thrombotic complications (69). Unlike most other component proteins of complement in blood, the primary site of biosynthesis for C1q is not in the liver, but rather in myeloid cells including macrophages, monocytes, and dendritic cells, which originate in the bone marrow. The effect of restoring the C1q protein to reconstitute complement function has been tested in mouse models. Bone marrow transplantation (BMT) of hematopoietic stem cells from wild-type animals has been shown to be effective in treating C1q deficient animals (150, 151).
Recently, BMT for hematopoietic stem cell therapy (HSCT) has been performed in a single case of a Pakistani C1q-deficient patient (152). In this particular case, involving a consanguineous family, the father and five of his six sons had C1q-deficiency. The father died of chronic glomerulonephritis at age 38, and one of his sons died at age 17 months from Streptococcus pneumonia meningitis. Another son was incapacitated by a CNS vasculopathy at 17 years of age. The index patient is the third son who survived S. pneumonia-induced meningitis at age 3 but developed an acute CNS vasculopathy at age 10. At age 16, his disease was brought under control through treatment with intravenous cyclophosphamide (pulse therapy) and B-cell depletion with rituximab. However, he had a persistent lupus rash and increased levels of anti-Ro (SSA-60) in addition to anticardiolipin antibodies. In view of his poor prognosis, HSCT was performed using bone marrow from his HLA-matched healthy brother, with graft-versus-host disease prophylaxis. Restoration of hematopoiesis, myelopoiesis, platelet production, and complement function with normal levels of C1q and CH50 were observed 2–4 weeks after transplantation. Such a result gives hope to SLE-patients with a C1q deficiency and a severe clinical course, although further studies are desirable.
Complement activation products and genetic polymorphisms have been observed and shown to contribute to inflammation and tissue damage of autoimmune diseases besides SLE. We will briefly summarize findings of complement activation and involvement in a few other autoimmune diseases.
Anti-phospholipid syndrome (APS) is characterized by arterial or venous thrombosis and recurrent pregnancy loss. The complement profile in general parallels that observed in SLE with CP activation being predominantly involved (153–156). Hypocomplementemia has been associated with fetal loss, pre-term delivery, and low-birth weight (157). Serum complement levels were significantly lower in patients with primary APS compared to patients with APS associated with a connective tissue disease (155). The patients with primary APS also had higher levels of complement activation fragments C3a and C4a demonstrating that the hypocomplementemia is, as expected, due to complement consumption (154, 155).
Of the inflammatory myopathies, dermatomyositis (DM) is the one most reported to possibly be associated with a complement-mediated pathogenesis (158). Complement-mediated destruction of perivascular endothelium and perifascicular ischemia of muscle fibers in biopsies from DM patients have been demonstrated by multiple investigators (159–164). Circulating immune complexes, IgG and IgM, complement C3, and late components of complement activation C5b–C9 MAC were detected in DM muscle and skin biopsies. A single case of an individual with a complete C2 genetic deficiency was reported to have DM (165). The observation of complement components and complement fragments indicates the possible involvement of complement pathways in tissue damage. Previous studies revealed that HLA class II gene DRB1 allele *0301 (also known as DR3) is the major immunogenetic risk factor for juvenile DM (JDM) (166–170). However, also present in the HLA region is the locus for complement C4. Specifically, DR3 in European subjects is in strong linkage disequilibrium with a particular C4 haplotype: a single C4B gene but the absence of a C4A gene (118, 171). A recent study found that C4A deficiency was independent of DR3 in association with JDM in a study of 95 patients and 500 race-matched controls of European ancestry (113). In addition, the authors demonstrated through multiple logistic regression analyses that the concurrence of C4A deficiency and DR3 together contributed toward the highest risk of JDM with an odds ratio of 3.2.
Several autoantibodies that play a major role in the autoimmune attack of synovia have been identified in rheumatoid arthritis (RA). There are three major types of RA-associated autoantibodies (i) anti-cyclic citrullinated peptide/protein autoantibodies (anti-CCP), (ii) autoantibodies against the Fc-fragments of IgG (rheumatoid factor, RF), and (iii) anti-type II collagen or glucose-6-phosphoisomerase autoantibodies (anti-G6PI). All of these autoantibodies bind their respective self-antigen, leading to increased levels of ICs and activation of the CP observed in RA patients (172–174). CP activation in RA patients has also been reported to be mediated by C-reactive protein (CRP) or fibromodulin (175, 176). Complexes formed between CRP and activated complement components were increased in the majority of RA patients and were further increased in patients with active disease versus patients with inactive disease. Agents/components that mediate and trigger inflammation in RA are likely derived from cartilage since disease activity subsides after joint replacement. Sjoberg et al. showed that the cartilage component fibromodulin can activate complement specifically by binding C1q, thereby activating the C1 complex and the remainder of the complement cascade (176). The evidence of complement activation in synovial fluid and the identification of several complement-triggering agents in RA support complement-mediated disease pathogenesis.
Depressed levels of complement proteins and elevated levels of complement cleavage products and late components MAC C5b–C9 were noted in tissue specific to RA pathogenesis, such as synovial fluid (177–180). Furthermore, the subsequent generation of anaphylatoxins C3a and C5a in synovial fluid creates an inflammatory state that attracts and activates neutrophils and other myeloid cells to the site of complement activation in the inflamed joints. The harmful effects of complement activation in RA are remarkable and evident. A recent study of C4 genetics in RA patients revealed that a deficiency of C4B genes was significantly more frequent in RA patients compared to non-RA patients or healthy controls (110). In other words, 40% of RA patients had a C4B deficiency compared to only 21.6% of controls, resulting in an odds ratio of 2.99 for RA subjects. Replication cohorts of RA patients are needed to validate and strengthen the possible association of C4B deficiency with RA, which could be secondary to the prevalence of HLA-DR4.
As with many autoimmune diseases, there are a number of autoantibodies that have been reported in primary Sjogren’s syndrome (SS). Therefore, complement activation via the CP may be involved in the development and/or pathogenesis of primary SS. A frequent clinical observation in primary SS is hypocomplementemia, particularly low serum levels of C3 and C4. Some studies reported that in the most severe cases of primary SS, the complement inhibitor C4b-binding protein was decreased in parallel with C3 and C4 levels, suggesting that dysregulation of complement may mediate disease pathogenesis (181). It is generally hypothesized that low C3 and C4 levels are a result of consumption by CP activation mediated by ICs in primary SS patients. Recent studies have showed that lymphoproliferative disease, mortality rates, and other severe disease manifestations were significantly higher in patients with low levels of C3 or C4 (182–184). Therefore, complement levels may be more a marker or predictor of disease activity in established SS patients.
A complete genetic deficiency in any one of the early components engaged in the CP of complement activation, C1q, C1r, C1s, or C4 almost always leads to SLE in humans, irrespective of race or sex. This phenomenon underscores the necessity for all of these early complement components, probably acting in concert, to achieve immune tolerance or prevent autoimmunity. While the incidence of a homozygous deficiency for one of these early acting complement components is extremely rare, partial genetic deficiency due to gene CNV of complement C4A (and total C4) and acquired deficiency such as secondary to anti-C1q autoantibodies are common. In combination, inherited insufficiency and acquired deficiency for early components of the CP may exist in over half of SLE patients with European ancestry. On the other hand, high copy numbers of total C4 and C4A are prevalent among healthy subjects and protective against SLE. Although less conclusively demonstrated, less explored topics include the single nucleotide polymorphisms (SNPs) in those complement genes that may modulate functional protein activities or gene expression levels, the presence of other complement autoantibodies or nephritic factors, dysfunctional regulatory proteins, or toxic side effects of drugs that may also contribute to inappropriate complement activation.
Genetic insufficiency and acquired deficiency of complement have medium to large effect size on disease susceptibility in European SLE. The frequency and effect size of specific genetic variants for a complex disease tend to be race specific. Considering the high prevalence of renal disease in African, Asian, and Hispanic SLE patients, it is of interest to further extend investigations on the roles of complement risk factors in these racial/ethnic groups.
Fluctuations in either hemolytic complement activity or levels of serum C3 and C4 in SLE patients were noticed over half of a century ago. More recently, high levels of processed C4d activation product attached to multiple cell types of hematopoietic origin are present in most SLE patients. Comprehensive data on genetic and acquired risk factors, plus cross-sectional and longitudinal profiling of autoantibodies and serum levels of native and activated complement proteins, and cell-bound levels of processed activation products hold promise as more sensitive biomarkers for SLE. Additionally, similar investigations should be extended to other autoimmune or inflammatory diseases including Sjögren’s syndrome, DM, RA, and antiphospholipid syndrome.
KL, YW, YY, CS, GH, LH, JA, and CYY all contributed to the acquisition of data, drafting and revision of the content, and final approval of the version to be published.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by National Institutes of Arthritis, Musculoskeletal and Skin Diseases grant 5R01 AR054459 (CYY), National Institutes of Digestive, Diabetes and Kidney Disease grant 5P01 DK55546 (LAH), CureJM Foundation (CYY), and National Institutes of Health/National Heart, Lung, and Blood Institute grant U54 HL112303, and National Institutes of Health/National Institutes of Allergy and Infectious Diseases grant 5 R01 AI041592 (JPA).
1. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi:10.1038/ni.1923
2. Yu C, Driest K, Wu Y, Lintner K, Patwardhan A, Spencer CH, et al. Complement in rheumatic diseases. In: Diamond B, Davidson A, editors. Encyclopedia of Medical Immunology: Autoimmune Diseases. New York, NY: Springer (2014). p. 286–302.
3. Atkinson J, Yu C. The complement system in systemic lupus erythematosus. In: Tsokos GC, editor. Systemic Lupus Erythematosus. Chapter 12, New York, NY: Elsevier (2016). p. 81–112. doi:10.1016/B978-0-12-801917-7.00012-7
4. Sturfelt G, Truedsson L. Complement in the immunopathogenesis of rheumatic disease. Nat Rev Rheumatol (2012) 8(8):458–68. doi:10.1038/nrrheum.2012.75
5. Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC. The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science (1954) 120(3112):279–85. doi:10.1126/science.120.3112.279
6. Fearon DT, Austen KF, Ruddy S. Formation of a hemolytically active cellular intermediate by the interaction between properdin factors B and D and the activated third component of complement. J Exp Med (1973) 138(6):1305–13. doi:10.1084/jem.138.6.1305
7. Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, et al. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J Immunol (2012) 189(8):3957–69. doi:10.4049/jimmunol.1201736
8. Jensenius JC. The mannan-binding lectin (MBL) pathway of complement activation: biochemistry, biology and clinical implications. Adv Exp Med Biol (2005) 564:21–2. doi:10.1007/0-387-25515-X_6
9. Elliott JA Jr, Mathieson DR. Complement in disseminated (systemic) lupus erythematosus. AMA Arch Derm Syphilol (1953) 68(2):119–28. doi:10.1001/archderm.1953.01540080003001
10. Lewis EJ, Carpenter CB, Schur PH. Serum complement component levels in human glomerulonephritis. Ann Intern Med (1971) 75(4):555–60. doi:10.7326/0003-4819-75-4-555
11. Birmingham DJ, Irshaid F, Nagaraja HN, Zou X, Tsao BP, Wu H, et al. The complex nature of serum C3 and C4 as biomarkers of lupus renal flare. Lupus (2010) 19(11):1272–80. doi:10.1177/0961203310371154
12. Wu YL, Higgins GC, Rennebohm RM, Chung EK, Yang Y, Zhou B, et al. Three distinct profiles of serum complement C4 proteins in pediatric systemic lupus erythematosus (SLE) patients: tight associations of complement C4 and C3 protein levels in SLE but not in healthy subjects. Adv Exp Med Biol (2006) 586:227–47. doi:10.1007/0-387-34134-X_16
13. Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet (2007) 80(6):1037–54. doi:10.1086/518257
14. Saxena K, Kitzmiller KJ, Wu YL, Zhou B, Esack N, Hiremath L, et al. Great genotypic and phenotypic diversities associated with copy-number variations of complement C4 and RP-C4-CYP21-TNX (RCCX) modules: a comparison of Asian-Indian and European American populations. Mol Immunol (2009) 46(7):1289–303. doi:10.1016/j.molimm.2008.11.018
15. Yang Y, Chung EK, Zhou B, Blanchong CA, Yu CY, Fust G, et al. Diversity in intrinsic strengths of the human complement system: serum C4 protein concentrations correlate with C4 gene size and polygenic variations, hemolytic activities, and body mass index. J Immunol (2003) 171(5):2734–45. doi:10.4049/jimmunol.171.5.2734
16. Manzi S, Navratil JS, Ruffing MJ, Liu CC, Danchenko N, Nilson SE, et al. Measurement of erythrocyte C4d and complement receptor 1 in systemic lupus erythematosus. Arthritis Rheum (2004) 50(11):3596–604. doi:10.1002/art.20561
17. Putterman C, Furie R, Ramsey-Goldman R, Askanase A, Buyon J, Kalunian K, et al. Cell-bound complement activation products in systemic lupus erythematosus: comparison with anti-double-stranded DNA and standard complement measurements. Lupus Sci Med. (2014) 1(1):e000056. doi:10.1136/lupus-2014-000056
18. Liu CC, Manzi S, Kao AH, Navratil JS, Ahearn JM. Cell-bound complement biomarkers for systemic lupus erythematosus: from benchtop to bedside. Rheum Dis Clin North Am (2010) 36(1):161–72. doi:10.1016/j.rdc.2009.12.003
19. Liu CC, Manzi S, Kao AH, Navratil JS, Ruffing MJ, Ahearn JM. Reticulocytes bearing C4d as biomarkers of disease activity for systemic lupus erythematosus. Arthritis Rheum (2005) 52(10):3087–99. doi:10.1002/art.21305
20. Navratil JS, Manzi S, Kao AH, Krishnaswami S, Liu CC, Ruffing MJ, et al. Platelet C4d is highly specific for systemic lupus erythematosus. Arthritis Rheum (2006) 54(2):670–4. doi:10.1002/art.21627
21. Calano SJ, Shih PA, Liu CC, Kao AH, Navratil JS, Manzi S, et al. Cell-bound complement activation products (CB-CAPs) as a source of lupus biomarkers. Adv Exp Med Biol (2006) 586:381–90. doi:10.1007/0-387-34134-X_25
22. Middleton J, Crookston MC, Falk JA, Robson EB, Cook PJ, Batchelor JR, et al. Linkage of Chido and HL-A. Tissue Antigens (1974) 4(4):366–73. doi:10.1111/j.1399-0039.1974.tb00262.x
23. Giles CM, Gedde-Dahl T Jr, Robson EB, Thorsby E, Olaisen B, Arnason A, et al. Rga (Rodgers) and the HLA region: linkage and associations. Tissue Antigens (1976) 8(2):143–9. doi:10.1111/j.1399-0039.1976.tb00578.x
24. O’Neill GJ, Yang SY, Tegoli J, Berger R, Dupont B. Chido and Rodgers blood groups are distinct antigenic components of human complement C4. Nature (1978) 273(5664):668–70. doi:10.1038/273668a0
25. Mauff G, Luther B, Schneider PM, Rittner C, Stradmann-Bellinghausen B, Dawkins R, et al. Reference typing report for complement component C4. Exp Clin Immunogenet (1998) 15(4):249–60. doi:10.1159/000019079
26. Awdeh ZL, Alper CA. Inherited structural polymorphism of the fourth component of human complement. Proc Natl Acad Sci U S A (1980) 77(6):3576–80. doi:10.1073/pnas.77.6.3576
27. Sim E, Cross SJ. Phenotyping of human complement component C4, a class-III HLA antigen. Biochem J (1986) 239(3):763–7. doi:10.1042/bj2390763
28. Law SK, Dodds AW, Porter RR. A comparison of the properties of two classes, C4A and C4B, of the human complement component C4. EMBO J (1984) 3(8):1819–23.
29. Isenman DE, Young JR. The molecular basis for the difference in immune hemolysis activity of the Chido and Rodgers isotypes of human complement component C4. J Immunol (1984) 132(6):3019–27.
30. Hing SN, Giles CM, Fielder AH, Batchelor JR. HLA haplotypes with C4B5; evidence for further allelic heterogeneity. Immunogenetics (1986) 23(3):151–5. doi:10.1007/BF00373815
31. Yu CY, Belt KT, Giles CM, Campbell RD, Porter RR. Structural basis of the polymorphism of human complement components C4A and C4B: gene size, reactivity and antigenicity. EMBO J (1986) 5(11):2873–81.
32. Yu CY, Campbell RD, Porter RR. A structural model for the location of the Rodgers and the Chido antigenic determinants and their correlation with the human complement component C4A/C4B isotypes. Immunogenetics (1988) 27(6):399–405. doi:10.1007/BF00364425
33. Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, et al. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. J Exp Med (2000) 191(12):2183–96. doi:10.1084/jem.191.12.2183
34. Chung EK, Yang Y, Rennebohm RM, Lokki ML, Higgins GC, Jones KN, et al. Genetic sophistication of human complement components C4A and C4B and RP-C4-CYP21-TNX (RCCX) modules in the major histocompatibility complex. Am J Hum Genet (2002) 71(4):823–37. doi:10.1086/342777
35. Feucht HE, Felber E, Gokel MJ, Hillebrand G, Nattermann U, Brockmeyer C, et al. Vascular deposition of complement-split products in kidney allografts with cell-mediated rejection. Clin Exp Immunol (1991) 86(3):464–70. doi:10.1111/j.1365-2249.1991.tb02954.x
36. Feucht HE. Complement C4d in graft capillaries – the missing link in the recognition of humoral alloreactivity. Am J Transplant (2003) 3(6):646–52. doi:10.1034/j.1600-6143.2003.00171.x
37. Bohmig GA, Kikic Z, Wahrmann M, Eskandary F, Aliabadi AZ, Zlabinger GJ, et al. Detection of alloantibody-mediated complement activation: a diagnostic advance in monitoring kidney transplant rejection? Clin Biochem (2015). doi:10.1016/j.clinbiochem.2015.05.024
38. Walport MJ. Complement. Second of two parts. N Engl J Med (2001) 344(15):1140–4. doi:10.1056/NEJM200104123441506
39. Navratil JS, Watkins SC, Wisnieski JJ, Ahearn JM. The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. J Immunol (2001) 166(5):3231–9. doi:10.4049/jimmunol.166.5.3231
40. Lipsker D, Hauptmann G. Cutaneous manifestations of complement deficiencies. Lupus (2010) 19(9):1096–106. doi:10.1177/0961203310373370
41. Wu YL, Brookshire BP, Verani RR, Arnett FC, Yu CY. Clinical presentations and molecular basis of complement C1r deficiency in a male African-American patient with systemic lupus erythematosus. Lupus (2011) 20(11):1126–34. doi:10.1177/0961203311404914
42. Wu YL, Hauptmann G, Viguier M, Yu CY. Molecular basis of complete complement C4 deficiency in two North-African families with systemic lupus erythematosus. Genes Immun (2009) 10(5):433–45. doi:10.1038/gene.2009.10
43. Yu C, Hauptmann G, Yang Y, Wu Y, Birmingham DJ, Rovin BH, et al. Complement deficiencies in human systemic lupus erythematosus (SLE) and SLE nephritis: epidemiology and pathogenesis. In: Tsokos GC, Gordon C, Smolen J, editors. Systemic Lupus Erythematosus: A Companion to Rheumatology. Philadelphia, PA: Mosby Elsevier (2007). p. 183–93.
44. Carroll MC. A protective role for innate immunity in systemic lupus erythematosus. Nat Rev Immunol (2004) 4(10):825–31. doi:10.1038/nri1456
45. Carroll MC. The role of complement in B cell activation and tolerance. Adv Immunol (2000) 74:61–88. doi:10.1016/S0065-2776(08)60908-6
46. Prodeus AP, Goerg S, Shen LM, Pozdnyakova OO, Chu L, Alicot EM, et al. A critical role for complement in maintenance of self-tolerance. Immunity (1998) 9(5):721–31. doi:10.1016/S1074-7613(00)80669-X
47. Duncan AR, Winter G. The binding site for C1q on IgG. Nature (1988) 332(6166):738–40. doi:10.1038/332738a0
48. Gadjeva MG, Rouseva MM, Zlatarova AS, Reid KB, Kishore U, Kojouharova MS. Interaction of human C1q with IgG and IgM: revisited. Biochemistry (2008) 47(49):13093–102. doi:10.1021/bi801131h
49. Zlatarova AS, Rouseva M, Roumenina LT, Gadjeva M, Kolev M, Dobrev I, et al. Existence of different but overlapping IgG- and IgM-binding sites on the globular domain of human C1q. Biochemistry (2006) 45(33):9979–88. doi:10.1021/bi060539v
50. Stegert M, Bock M, Trendelenburg M. Clinical presentation of human C1q deficiency: how much of a lupus? Mol Immunol (2015) 67(1):3–11. doi:10.1016/j.molimm.2015.03.007
51. Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol (2000) 76:227–324. doi:10.1016/S0065-2776(01)76021-X
52. Schejbel L, Skattum L, Hagelberg S, Ahlin A, Schiller B, Berg S, et al. Molecular basis of hereditary C1q deficiency – revisited: identification of several novel disease-causing mutations. Genes Immun (2011) 12(8):626–34. doi:10.1038/gene.2011.39
53. Namjou B, Keddache M, Fletcher D, Dillon S, Kottyan L, Wiley G, et al. Identification of novel coding mutation in C1qA gene in an African-American pedigree with lupus and C1q deficiency. Lupus (2012) 21(10):1113–8. doi:10.1177/0961203312443993
55. Berkel A, Loos M, Sanal O, Mauff G, Gungen Y, Ors U, et al. Clinical and immunochemical studies in a case of selective complete C1q deficiency. Clin Exp Immunol (1979) 38:52–63.
56. Petry F, Berkel AI, Loos M. Multiple identification of a particular type of hereditary C1q deficiency in the Turkish population: review of the cases and additional genetic and functional analysis. Hum Genet (1997) 100(1):51–6. doi:10.1007/s004390050464
57. Berkel AI, Birben E, Oner C, Oner R, Loos M, Petry F. Molecular, genetic and epidemiologic studies on selective complete C1q deficiency in Turkey. Immunobiology (2000) 201(3–4):347–55. doi:10.1016/S0171-2985(00)80089-3
58. Berkel AI, Petry F, Sanal O, Tinaztepe K, Ersoy F, Bakkaloglu A, et al. Development of systemic lupus erythematosus in a patient with selective complete C1q deficiency. Eur J Pediatr (1997) 156(2):113–5. doi:10.1007/s004310050567
59. Topaloglu R, Bakkaloglu A, Slingsby JH, Mihatsch MJ, Pascual M, Norsworthy P, et al. Molecular basis of hereditary C1q deficiency associated with SLE and IgA nephropathy in a Turkish family. Kidney Int (1996) 50(2):635–42. doi:10.1038/ki.1996.359
60. Hoppenreijs EP, van Dijken PJ, Kabel PJ, Th Draaisma JM. Hereditary C1q deficiency and secondary Sjogren’s syndrome. Ann Rheum Dis (2004) 63(11):1524–5. doi:10.1136/ard.2003.016592
61. Sun-Tan C, Ozgur TT, Kilinc G, Topaloglu R, Gokoz O, Ersoy-Evans S, et al. Hereditary C1q deficiency: a new family with C1qA deficiency. Turk J Pediatr (2010) 52(2):184–6.
62. Reid KB, Thompson RA. Characterization of a non-functional form of C1q found in patients with a genetically linked deficiency of C1q activity. Mol Immunol (1983) 20(10):1117–25. doi:10.1016/0161-5890(83)90121-9
63. McAdam RA, Goundis D, Reid KB. A homozygous point mutation results in a stop codon in the C1q B-chain of a C1q-deficient individual. Immunogenetics (1988) 27(4):259–64. doi:10.1007/BF00376120
64. Chapuis RM, Hauptmann G, Grosshans E, Isliker H. Structural and functional studies in C1q deficiency. J Immunol (1982) 129(4):1509–12.
65. Petry F, Hauptmann G, Goetz J, Grosshans E, Loos M. Molecular basis of a new type of C1q-deficiency associated with a non-functional low molecular weight (LMW) C1q: parallels and differences to other known genetic C1q-defects. Immunopharmacology (1997) 38(1–2):189–201. doi:10.1016/S0162-3109(97)00065-9
66. Marquart HV, Schejbel L, Sjoholm A, Martensson U, Nielsen S, Koch A, et al. C1q deficiency in an Inuit family: identification of a new class of C1q disease-causing mutations. Clin Immunol (2007) 124(1):33–40. doi:10.1016/j.clim.2007.03.547
67. Roumenina LT, Sene D, Radanova M, Blouin J, Halbwachs-Mecarelli L, Dragon-Durey MA, et al. Functional complement C1q abnormality leads to impaired immune complexes and apoptotic cell clearance. J Immunol (2011) 187(8):4369–73. doi:10.4049/jimmunol.1101749
68. van Schaarenburg RA, Daha NA, Schonkeren JJ, Nivine Levarht EW, van Gijlswijk-Janssen DJ, Kurreeman FA, et al. Identification of a novel non-coding mutation in C1qB in a Dutch child with C1q deficiency associated with recurrent infections. Immunobiology (2015) 220(3):422–7. doi:10.1016/j.imbio.2014.10.005
69. Higuchi Y, Shimizu J, Hatanaka M, Kitano E, Kitamura H, Takada H, et al. The identification of a novel splicing mutation in C1qB in a Japanese family with C1q deficiency: a case report. Pediatr Rheumatol Online J (2013) 11(1):41. doi:10.1186/1546-0096-11-41
70. Slingsby JH, Norsworthy P, Pearce G, Vaishnaw AK, Issler H, Morley BJ, et al. Homozygous hereditary C1q deficiency and systemic lupus erythematosus. A new family and the molecular basis of C1q deficiency in three families. Arthritis Rheum (1996) 39(4):663–70. doi:10.1002/art.1780390419
71. Tsuge I, Kondo Y, Nakajima Y, Nakagawa N, Imai K, Nonoyama S, et al. Hyper IgM syndrome and complement Clq deficiency in an individual with systemic lupus erythematosus-like disease. Clin Exp Rheumatol (2010) 28(4):558–60.
72. Pickering MC, Macor P, Fish J, Durigutto P, Bossi F, Petry F, et al. Complement C1q and C8beta deficiency in an individual with recurrent bacterial meningitis and adult-onset systemic lupus erythematosus-like illness. Rheumatology (Oxford) (2008) 47(10):1588–9. doi:10.1093/rheumatology/ken289
73. Bowness P, Davies KA, Norsworthy PJ, Athanassiou P, Taylor-Wiedeman J, Borysiewicz LK, et al. Hereditary C1q deficiency and systemic lupus erythematosus. QJM (1994) 87(8):455–64.
74. Gulez N, Genel F, Atlihan F, Gullstrand B, Skattum L, Schejbel L, et al. Homozygosity for a novel mutation in the C1q C chain gene in a Turkish family with hereditary C1q deficiency. J Investig Allergol Clin Immunol (2010) 20(3):255–8.
75. Mikuska A, Stefanovic Z, Miletic V, Oxelius V-A, Sjoholm A. Systemic lupus erythematosus-like disease in two siblings with complete C1q deficiency. Period Biol (1983) 85:271–3.
76. Stone NM, Williams A, Wilkinson JD, Bird G. Systemic lupus erythematosus with C1q deficiency. Br J Dermatol (2000) 142(3):521–4. doi:10.1046/j.1365-2133.2000.03369.x
77. Mehta P, Norsworthy PJ, Hall AE, Kelly SJ, Walport MJ, Botto M, et al. SLE with C1q deficiency treated with fresh frozen plasma: a 10-year experience. Rheumatology (Oxford) (2010) 49(4):823–4. doi:10.1093/rheumatology/kep387
78. Chew CH, Chua KH, Lian LH, Puah SM, Tan SY. PCR-RFLP genotyping of C1q mutations and single nucleotide polymorphisms in Malaysian patients with systemic lupus erythematosus. Hum Biol (2008) 80(1):83–93. doi:10.3378/1534-6617(2008)80[83:PGOCMA]2.0.CO;2
79. McClellan J, King MC. Genetic heterogeneity in human disease. Cell (2010) 141(2):210–7. doi:10.1016/j.cell.2010.03.032
80. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature (2009) 461(7265):747–53. doi:10.1038/nature08494
81. Nguyen VC, Tosi M, Gross MS, Cohen-Haguenauer O, Jegou-Foubert C, de Tand MF, et al. Assignment of the complement serine protease genes C1r and C1s to chromosome 12 region 12p13. Hum Genet (1988) 78(4):363–8. doi:10.1007/BF00291737
82. Kusumoto H, Hirosawa S, Salier JP, Hagen FS, Kurachi K. Human genes for complement components C1r and C1s in a close tail-to-tail arrangement. Proc Natl Acad Sci U S A (1988) 85(19):7307–11. doi:10.1073/pnas.85.19.7307
83. Pickering RJ, Naff GB, Stroud RM, Good RA, Gewurz H. Deficiency of C1r in human serum. Effects on the structure and function of macromolecular C1. J Exp Med (1970) 131(4):803–15. doi:10.1084/jem.131.4.803
84. Moncada B, Day NK, Good RA, Windhorst DB. Lupus-erythematosus-like syndrome with a familial defect of complement. N Engl J Med (1972) 286(13):689–93. doi:10.1056/NEJM197203302861304
85. Day NK, Geiger H, Stroud R, DeBracco M, Mancaido B, Windhorst D, et al. C1r deficiency: an inborn error associated with cutaneous and renal disease. J Clin Invest (1972) 51(5):1102–8. doi:10.1172/JCI106902
86. Manderson AP, Pickering MC, Botto M, Walport MJ, Parish CR. Continual low-level activation of the classical complement pathway. J Exp Med (2001) 194(6):747–56. doi:10.1084/jem.194.6.747
87. Amano MT, Ferriani VP, Florido MP, Reis ES, Delcolli MI, Azzolini AE, et al. Genetic analysis of complement C1s deficiency associated with systemic lupus erythematosus highlights alternative splicing of normal C1s gene. Mol Immunol (2008) 45(6):1693–702. doi:10.1016/j.molimm.2007.09.034
88. Inoue N, Saito T, Masuda R, Suzuki Y, Ohtomi M, Sakiyama H. Selective complement C1s deficiency caused by homozygous four-base deletion in the C1s gene. Hum Genet (1998) 103(4):415–8. doi:10.1007/s004390050843
89. Endo Y, Kanno K, Takahashi M, Yamaguchi K, Kohno Y, Fujita T. Molecular basis of human complement C1s deficiency. J Immunol (1999) 162(4):2180–3.
90. Abe K, Endo Y, Nakazawa N, Kanno K, Okubo M, Hoshino T, et al. Unique phenotypes of C1s deficiency and abnormality caused by two compound heterozygosities in a Japanese family. J Immunol (2009) 182(3):1681–8. doi:10.4049/jimmunol.182.3.1681
91. Dragon-Durey MA, Quartier P, Fremeaux-Bacchi V, Blouin J, de Barace C, Prieur AM, et al. Molecular basis of a selective C1s deficiency associated with early onset multiple autoimmune diseases. J Immunol (2001) 166(12):7612–6. doi:10.4049/jimmunol.166.12.7612
92. Carroll MC, Campbell RD, Bentley DR, Porter RR. A molecular map of the human major histocompatibility complex class III region linking complement genes C4, C2 and factor B. Nature (1984) 307(5948):237–41. doi:10.1038/307237a0
93. Wu YL, Savelli SL, Yang Y, Zhou B, Rovin BH, Birmingham DJ, et al. Sensitive and specific real-time polymerase chain reaction assays to accurately determine copy number variations (CNVs) of human complement C4A, C4B, C4-long, C4-short, and RCCX modules: elucidation of C4 CNVs in 50 consanguineous subjects with defined HLA genotypes. J Immunol (2007) 179(5):3012–25. doi:10.4049/jimmunol.179.5.3012
94. Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular variations of the human major histocompatibility complex class III genes for serine/threonine kinase RP, complement component C4, steroid 21-hydroxylase CYP21, and tenascin TNX (the RCCX module). A mechanism for gene deletions and disease associations. J Biol Chem (1999) 274(17):12147–56.
95. Kidmose RT, Laursen NS, Dobo J, Kjaer TR, Sirotkina S, Yatime L, et al. Structural basis for activation of the complement system by component C4 cleavage. Proc Natl Acad Sci U S A (2012) 109(38):15425–30. doi:10.1073/pnas.1208031109
96. Reilly BD. Analysis of human C4A and C4B binding to an immune complex in serum. Clin Exp Immunol (1999) 117(1):12–8. doi:10.1046/j.1365-2249.1999.00940.x
97. Chung EK, Yang Y, Rupert KL, Jones KN, Rennebohm RM, Blanchong CA, et al. Determining the one, two, three, or four long and short loci of human complement C4 in a major histocompatibility complex haplotype encoding C4A or C4B proteins. Am J Hum Genet (2002) 71(4):810–22. doi:10.1086/342778
98. Yu CY. The complete exon-intron structure of a human complement component C4A gene. DNA sequences, polymorphism, and linkage to the 21-hydroxylase gene. J Immunol (1991) 146(3):1057–66.
99. Dangel AW, Mendoza AR, Baker BJ, Daniel CM, Carroll MC, Wu LC, et al. The dichotomous size variation of human complement C4 genes is mediated by a novel family of endogenous retroviruses, which also establishes species-specific genomic patterns among Old World primates. Immunogenetics (1994) 40(6):425–36. doi:10.1007/BF00177825
100. Mitchell TJ, Naughton M, Norsworthy P, Davies KA, Walport MJ, Morley BJ. IFN-gamma up-regulates expression of the complement components C3 and C4 by stabilization of mRNA. J Immunol (1996) 156(11):4429–34.
101. Mortensen S, Kidmose RT, Petersen SV, Szilagyi A, Prohaszka Z, Andersen GR. Structural basis for the function of complement component C4 within the classical and lectin pathways of complement. J Immunol (2015) 194(11):5488–96. doi:10.4049/jimmunol.1500087
102. Dodds AW, Ren XD, Willis AC, Law SK. The reaction mechanism of the internal thioester in the human complement component C4. Nature (1996) 379(6561):177–9. doi:10.1038/379177a0
103. Carroll MC, Fathallah DM, Bergamaschini L, Alicot EM, Isenman DE. Substitution of a single amino acid (aspartic acid for histidine) converts the functional activity of human complement C4B to C4A. Proc Natl Acad Sci U S A (1990) 87(17):6868–72. doi:10.1073/pnas.87.17.6868
104. Yang Y, Lhotta K, Chung EK, Eder P, Neumair F, Yu CY. Complete complement components C4A and C4B deficiencies in human kidney diseases and systemic lupus erythematosus. J Immunol (2004) 173(4):2803–14. doi:10.4049/jimmunol.173.4.2803
105. Lokki ML, Circolo A, Ahokas P, Rupert KL, Yu CY, Colten HR. Deficiency of human complement protein C4 due to identical frameshift mutations in the C4A and C4B genes. J Immunol (1999) 162(6):3687–93.
106. Rupert KL, Moulds JM, Yang Y, Arnett FC, Warren RW, Reveille JD, et al. The molecular basis of complete complement C4A and C4B deficiencies in a systemic lupus erythematosus patient with homozygous C4A and C4B mutant genes. J Immunol (2002) 169(3):1570–8. doi:10.4049/jimmunol.169.3.1570
107. Nordin Fredrikson G, Truedsson L, Sjoholm AG, Kjellman M. DNA analysis in a MHC heterozygous patient with complete C4 deficiency – homozygosity for C4 gene deletion and C4 pseudogene. Exp Clin Immunogenet (1991) 8(1):29–37.
108. Fredrikson GN, Gullstrand B, Schneider PM, Witzel-Schlomp K, Sjoholm AG, Alper CA, et al. Characterization of non-expressed C4 genes in a case of complete C4 deficiency: identification of a novel point mutation leading to a premature stop codon. Hum Immunol (1998) 59(11):713–9. doi:10.1016/S0198-8859(98)00068-8
109. Kjellman M, Laurell AB, Low B, Sjoholm AG. Homozygous deficiency of C4 in a child with a lupus erythematosus syndrome. Clin Genet (1982) 22(6):331–9. doi:10.1111/j.1399-0004.1982.tb01849.x
110. Rigby WF, Wu YL, Zan M, Zhou B, Rosengren S, Carlson C, et al. Increased frequency of complement C4B deficiency in rheumatoid arthritis. Arthritis Rheum (2012) 64(5):1338–44. doi:10.1002/art.33472
111. Hou S, Qi J, Liao D, Fang J, Chen L, Kijlstra A, et al. High C4 gene copy numbers protects against Vogt-Koyanagi-Harada syndrome in Chinese Han. Br J Ophthalmol (2014) 98(12):1733–7. doi:10.1136/bjophthalmol-2014-305596
112. Liu YH, Wan L, Chang CT, Liao WL, Chen WC, Tsai Y, et al. Association between copy number variation of complement component C4 and Graves’ disease. J Biomed Sci (2011) 18:71. doi:10.1186/1423-0127-18-71
113. Lintner KE, Patwardhan A, Rider LG, Abdul-Aziz R, Wu YL, Lundstrom E, et al. Gene copy-number variations (CNVs) of complement C4 and C4A deficiency in genetic risk and pathogenesis of juvenile dermatomyositis. Ann Rheum Dis (2015). doi:10.1136/annrheumdis-2015-207762
114. Lv Y, He S, Zhang Z, Li Y, Hu D, Zhu K, et al. Confirmation of C4 gene copy number variation and the association with systemic lupus erythematosus in Chinese Han population. Rheumatol Int (2012) 32(10):3047–53. doi:10.1007/s00296-011-2023-7
115. Kim JH, Jung SH, Bae JS, Lee HS, Yim SH, Park SY, et al. Deletion variants of RABGAP1L, 10q21.3, and C4 are associated with the risk of systemic lupus erythematosus in Korean women. Arthritis Rheum (2013) 65(4):1055–63. doi:10.1002/art.37854
116. Chen JY, Wu YL, Mok MY, Wu Y, Lintner KE, Wang C, et al. Effects of complement C4 gene copy-number variations, size dichotomy and C4A-deficiency on genetic risk and clinical presentation of East-Asian SLE. Arthritis Rheum (2016). doi:10.1002/art.39589
117. Dawkins R, Leelayuwat C, Gaudieri S, Tay G, Hui J, Cattley S, et al. Genomics of the major histocompatibility complex: haplotypes, duplication, retroviruses and disease. Immunol Rev (1999) 167:275–304. doi:10.1111/j.1600-065X.1999.tb01399.x
118. Yu CY, Whitacre CC. Sex, MHC and complement C4 in autoimmune diseases. Trends Immunol (2004) 25(12):694–9. doi:10.1016/j.it.2004.10.006
119. Boteva L, Morris DL, Cortes-Hernandez J, Martin J, Vyse TJ, Fernando MM. Genetically determined partial complement C4 deficiency states are not independent risk factors for SLE in UK and Spanish populations. Am J Hum Genet (2012) 90(3):445–56. doi:10.1016/j.ajhg.2012.01.012
120. Uejima H, Lee MP, Cui H, Feinberg AP. Hot-stop PCR: a simple and general assay for linear quantitation of allele ratios. Nat Genet (2000) 25(4):375–6. doi:10.1038/78040
121. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344(14):1058–66. doi:10.1056/NEJM200104053441406
122. Wallis R, Dodds AW, Mitchell DA, Sim RB, Reid KB, Schwaeble WJ. Molecular interactions between MASP-2, C4, and C2 and their activation fragments leading to complement activation via the lectin pathway. J Biol Chem (2007) 282(11):7844–51. doi:10.1074/jbc.M606326200
123. Johnson CA, Densen P, Hurford RK Jr, Colten HR, Wetsel RA. Type I human complement C2 deficiency. A 28-base pair gene deletion causes skipping of exon 6 during RNA splicing. J Biol Chem (1992) 267(13):9347–53.
124. Agnello V. Lupus diseases associated with hereditary and acquired deficiencies of complement. Springer Semin Immunopathol (1986) 9(2–3):161–78. doi:10.1007/BF02099020
125. Wang X, Circolo A, Lokki ML, Shackelford PG, Wetsel RA, Colten HR. Molecular heterogeneity in deficiency of complement protein C2 type I. Immunology (1998) 93(2):184–91. doi:10.1046/j.1365-2567.1998.00392.x
126. Wetsel RA, Kulics J, Lokki ML, Kiepiela P, Akama H, Johnson CA, et al. Type II human complement C2 deficiency. Allele-specific amino acid substitutions (Ser189 – > Phe; Gly444 – > Arg) cause impaired C2 secretion. J Biol Chem (1996) 271(10):5824–31. doi:10.1074/jbc.271.10.5824
127. Zhu ZB, Atkinson TP, Volanakis JE. A novel type II complement C2 deficiency allele in an African-American family. J Immunol (1998) 161(2):578–84.
128. Dragon-Durey MA, Blanc C, Roumenina LT, Poulain N, Ngo S, Bordereau P, et al. Anti-factor H autoantibodies assay. Methods Mol Biol (2014) 1100:249–56. doi:10.1007/978-1-62703-724-2_20
129. Walport MJ. Complement and systemic lupus erythematosus. Arthritis Res (2002) 4(Suppl 3):S279–93. doi:10.1186/ar586
130. Mahler M, van Schaarenburg RA, Trouw LA. Anti-C1q autoantibodies, novel tests, and clinical consequences. Front Immunol (2013) 4:117. doi:10.3389/fimmu.2013.00117
131. Fremeaux-Bacchi V, Weiss L, Demouchy C, Blouin J, Kazatchkine MD. Autoantibodies to the collagen-like region of C1q are strongly associated with classical pathway-mediated hypocomplementemia in systemic lupus erythematosus. Lupus (1996) 5(3):216–20. doi:10.1177/096120339600500309
132. Orbai AM, Truedsson L, Sturfelt G, Nived O, Fang H, Alarcon GS, et al. Anti-C1q antibodies in systemic lupus erythematosus. Lupus (2015) 24(1):42–9. doi:10.1177/0961203314547791
133. He S, Lin YL. In vitro stimulation of C1s proteolytic activities by C1s-presenting autoantibodies from patients with systemic lupus erythematosus. J Immunol (1998) 160(9):4641–7.
134. Jackson J, Sim RB, Whelan A, Feighery C. An IgG autoantibody which inactivates C1-inhibitor. Nature (1986) 323(6090):722–4. doi:10.1038/323722a0
135. Nakamura S, Yoshinari M, Saku Y, Hirakawa K, Miishima C, Murai K, et al. Acquired C1 inhibitor deficiency associated with systemic lupus erythematosus affecting the central nervous system. Ann Rheum Dis (1991) 50(10):713–6. doi:10.1136/ard.50.10.713
136. Ochonisky S, Intrator L, Wechsler J, Revuz J, Bagot M. Acquired C1 inhibitor deficiency revealing systemic lupus erythematosus. Dermatology (1993) 186(4):261–3. doi:10.1159/000247366
137. Meszaros T, Fust G, Farkas H, Jakab L, Temesszentandrasi G, Nagy G, et al. C1-inhibitor autoantibodies in SLE. Lupus (2010) 19(5):634–8. doi:10.1177/0961203309357059
138. Jozsi M, Reuter S, Nozal P, Lopez-Trascasa M, Sanchez-Corral P, Prohaszka Z, et al. Autoantibodies to complement components in C3 glomerulopathy and atypical hemolytic uremic syndrome. Immunol Lett (2014) 160(2):163–71. doi:10.1016/j.imlet.2014.01.014
139. Daha MR, van Es LA. Relative resistance of the F-42-stabilized classical pathway C3 convertase to inactivation by C4-binding protein. J Immunol (1980) 125(5):2051–4.
140. Miller EC, Chase NM, Densen P, Hintermeyer MK, Casper JT, Atkinson JP. Autoantibody stabilization of the classical pathway C3 convertase leading to C3 deficiency and Neisserial sepsis: C4 nephritic factor revisited. Clin Immunol (2012) 145(3):241–50. doi:10.1016/j.clim.2012.09.007
141. Pickering MC, Cook HT. Translational mini-review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol (2008) 151(2):210–30. doi:10.1111/j.1365-2249.2007.03574.x
142. Heinen S, Pluthero FG, van Eimeren VF, Quaggin SE, Licht C. Monitoring and modeling treatment of atypical hemolytic uremic syndrome. Mol Immunol (2013) 54(1):84–8. doi:10.1016/j.molimm.2012.10.044
143. Leah E. Experimental arthritis: Targeted anti-C5 drug neutralizes complement in the joint. Nat Rev Rheumatol (2012) 8(4):186. doi:10.1038/nrrheum.2012.31
144. Copland DA, Hussain K, Baalasubramanian S, Hughes TR, Morgan BP, Xu H, et al. Systemic and local anti-C5 therapy reduces the disease severity in experimental autoimmune uveoretinitis. Clin Exp Immunol (2010) 159(3):303–14. doi:10.1111/j.1365-2249.2009.04070.x
145. Mizuno M, Nishikawa K, Morgan BP, Matsuo S. Comparison of the suppressive effects of soluble CR1 and C5a receptor antagonist in acute arthritis induced in rats by blocking of CD59. Clin Exp Immunol (2000) 119(2):368–75. doi:10.1046/j.1365-2249.2000.01127.x
146. Ames RS, Lee D, Foley JJ, Jurewicz AJ, Tornetta MA, Bautsch W, et al. Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol (2001) 166(10):6341–8. doi:10.4049/jimmunol.166.10.6341
147. Fraser DA, Harris CL, Williams AS, Mizuno M, Gallagher S, Smith RA, et al. Generation of a recombinant, membrane-targeted form of the complement regulator CD59: activity in vitro and in vivo. J Biol Chem (2003) 278(49):48921–7. doi:10.1074/jbc.M302598200
148. Zhang Y, Nester CM, Holanda DG, Marsh HC, Hammond RA, Thomas LJ, et al. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol (2013) 24(11):1820–9. doi:10.1681/ASN.2013010045
149. Steinsson K, Erlendsson K, Valdimarsson H. Successful plasma infusion treatment of a patient with C2 deficiency and systemic lupus erythematosus: clinical experience over forty-five months. Arthritis Rheum (1989) 32(7):906–13.
150. Petry F, Botto M, Holtappels R, Walport MJ, Loos M. Reconstitution of the complement function in C1q-deficient (C1qa-/-) mice with wild-type bone marrow cells. J Immunol (2001) 167(7):4033–7. doi:10.4049/jimmunol.167.7.4033
151. Cortes-Hernandez J, Fossati-Jimack L, Petry F, Loos M, Izui S, Walport MJ, et al. Restoration of C1q levels by bone marrow transplantation attenuates autoimmune disease associated with C1q deficiency in mice. Eur J Immunol (2004) 34(12):3713–22. doi:10.1002/eji.200425616
152. Arkwright PD, Riley P, Hughes SM, Alachkar H, Wynn RF. Successful cure of C1q deficiency in human subjects treated with hematopoietic stem cell transplantation. J Allergy Clin Immunol (2014) 133(1):265–7. doi:10.1016/j.jaci.2013.07.035
153. Shamonki JM, Salmon JE, Hyjek E, Baergen RN. Excessive complement activation is associated with placental injury in patients with antiphospholipid antibodies. Am J Obstet Gynecol (2007) 196(2):.e1–5. doi:10.1016/j.ajog.2006.10.879
154. Breen KA, Seed P, Parmar K, Moore GW, Stuart-Smith SE, Hunt BJ. Complement activation in patients with isolated antiphospholipid antibodies or primary antiphospholipid syndrome. Thromb Haemost (2012) 107(3):423–9. doi:10.1160/TH11-08-0554
155. Oku K, Atsumi T, Bohgaki M, Amengual O, Kataoka H, Horita T, et al. Complement activation in patients with primary antiphospholipid syndrome. Ann Rheum Dis (2009) 68(6):1030–5. doi:10.1136/ard.2008.090670
156. Avalos I, Tsokos GC. The role of complement in the antiphospholipid syndrome-associated pathology. Clin Rev Allergy Immunol (2009) 36(2–3):141–4. doi:10.1007/s12016-008-8109-7
157. De Carolis S, Botta A, Santucci S, Salvi S, Moresi S, Di Pasquo E, et al. Complementemia and obstetric outcome in pregnancy with antiphospholipid syndrome. Lupus (2012) 21(7):776–8. doi:10.1177/0961203312444172
158. Dalakas MC. Inflammatory Muscle Diseases. N Engl J Med (2015) 373(4):393–4. doi:10.1056/NEJMc1506827
159. Whitaker JN, Engel WK. Vascular deposits of immunoglobulin and complement in idiopathic inflammatory myopathy. N Engl J Med (1972) 286(7):333–8. doi:10.1056/NEJM197202172860701
160. Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med (1986) 314(6):329–34. doi:10.1056/NEJM198602063140601
161. Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: a quantitative study. Ann Neurol (1990) 27(4):343–56. doi:10.1002/ana.410270402
162. Kissel JT, Halterman RK, Rammohan KW, Mendell JR. The relationship of complement-mediated microvasculopathy to the histologic features and clinical duration of disease in dermatomyositis. Arch Neurol (1991) 48(1):26–30. doi:10.1001/archneur.1991.00530130034016
163. Mascaro JM Jr, Hausmann G, Herrero C, Grau JM, Cid MC, Palou J, et al. Membrane attack complex deposits in cutaneous lesions of dermatomyositis. Arch Dermatol (1995) 131(12):1386–92. doi:10.1001/archderm.1995.01690240040007
164. Burgin S, Stone JH, Shenoy-Bhangle AS, McGuone D. Case records of the Massachusetts General Hospital. Case 18-2014. A 32-year-old man with a rash, myalgia, and weakness. N Engl J Med (2014) 370(24):2327–37. doi:10.1056/NEJMcpc1304161
165. Leddy JP, Griggs RC, Klemperer MR, Frank MM. Hereditary complement (C2) deficiency with dermatomyositis. Am J Med (1975) 58(1):83–91. doi:10.1016/0002-9343(75)90537-9
166. Miller FW, Cooper RG, Vencovsky J, Rider LG, Danko K, Wedderburn LR, et al. Genome-wide association study of dermatomyositis reveals genetic overlap with other autoimmune disorders. Arthritis Rheum (2013) 65(12):3239–47. doi:10.1002/art.38137
167. Miller FW, Chen W, O’Hanlon TP, Cooper RG, Vencovsky J, Rider LG, et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun (2015) 16(7):470–80. doi:10.1038/gene.2015.28
168. O’Hanlon TP, Carrick DM, Targoff IN, Arnett FC, Reveille JD, Carrington M, et al. Immunogenetic risk and protective factors for the idiopathic inflammatory myopathies: distinct HLA-A, -B, -Cw, -DRB1, and -DQA1 allelic profiles distinguish European American patients with different myositis autoantibodies. Medicine (Baltimore) (2006) 85(2):111–27. doi:10.1097/01.md.0000217525.82287.eb
169. Friedman JM, Pachman LM, Maryjowski ML, Radvany RM, Crowe WE, Hanson V, et al. Immunogenetic studies of juvenile dermatomyositis: HLA-DR antigen frequencies. Arthritis Rheum (1983) 26(2):214–6. doi:10.1002/art.1780260216
170. Rothwell S, Cooper RG, Lundberg IE, Miller FW, Gregersen PK, Bowes J, et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis (2015). doi:10.1136/annrheumdis-2015-208119
171. Horton R, Gibson R, Coggill P, Miretti M, Allcock RJ, Almeida J, et al. Variation analysis and gene annotation of eight MHC haplotypes: the MHC Haplotype Project. Immunogenetics (2008) 60(1):1–18. doi:10.1007/s00251-007-0262-2
172. Zubler RH, Nydegger U, Perrin LH, Fehr K, McCormick J, Lambert PH, et al. Circulating and intra-articular immune complexes in patients with rheumatoid arthritis. Correlation of 125I-Clq binding activity with clinical and biological features of the disease. J Clin Invest (1976) 57(5):1308–19. doi:10.1172/JCI108399
173. Hay FC, Nineham LJ, Perumal R, Roitt IM. Intra-articular and circulating immune complexes and antiglobulins (IgG and IgM) in rheumatoid arthritis; correlation with clinical features. Ann Rheum Dis (1979) 38(1):1–7. doi:10.1136/ard.38.1.1
174. Trouw LA, Haisma EM, Levarht EW, van der Woude D, Ioan-Facsinay A, Daha MR, et al. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum (2009) 60(7):1923–31. doi:10.1002/art.24622
175. Molenaar ET, Voskuyl AE, Familian A, van Mierlo GJ, Dijkmans BA, Hack CE. Complement activation in patients with rheumatoid arthritis mediated in part by C-reactive protein. Arthritis Rheum (2001) 44(5):997–1002. doi:10.1002/1529-0131(200105)44:5<997::AID-ANR178>3.3.CO;2-3
176. Sjoberg A, Onnerfjord P, Morgelin M, Heinegard D, Blom AM. The extracellular matrix and inflammation: fibromodulin activates the classical pathway of complement by directly binding C1q. J Biol Chem (2005) 280(37):32301–8. doi:10.1074/jbc.M504828200
177. Oleesky DA, Daniels RH, Williams BD, Amos N, Morgan BP. Terminal complement complexes and C1/C1 inhibitor complexes in rheumatoid arthritis and other arthritic conditions. Clin Exp Immunol (1991) 84(2):250–5.
178. Konttinen YT, Ceponis A, Meri S, Vuorikoski A, Kortekangas P, Sorsa T, et al. Complement in acute and chronic arthritides: assessment of C3c, C9, and protectin (CD59) in synovial membrane. Ann Rheum Dis (1996) 55(12):888–94. doi:10.1136/ard.55.12.888
179. Makinde VA, Senaldi G, Jawad AS, Berry H, Vergani D. Reflection of disease activity in rheumatoid arthritis by indices of activation of the classical complement pathway. Ann Rheum Dis (1989) 48(4):302–6. doi:10.1136/ard.48.4.302
180. Biro E, Nieuwland R, Tak PP, Pronk LM, Schaap MC, Sturk A, et al. Activated complement components and complement activator molecules on the surface of cell-derived microparticles in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis (2007) 66(8):1085–92. doi:10.1136/ard.2006.061309
181. Zadura AF, Theander E, Blom AM, Trouw LA. Complement inhibitor C4b-binding protein in primary Sjogren’s syndrome and its association with other disease markers. Scand J Immunol (2009) 69(4):374–80. doi:10.1111/j.1365-3083.2009.02229.x
182. Theander E, Henriksson G, Ljungberg O, Mandl T, Manthorpe R, Jacobsson LT. Lymphoma and other malignancies in primary Sjogren’s syndrome: a cohort study on cancer incidence and lymphoma predictors. Ann Rheum Dis (2006) 65(6):796–803. doi:10.1136/ard.2005.041186
183. Ioannidis JP, Vassiliou VA, Moutsopoulos HM. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjogren’s syndrome. Arthritis Rheum (2002) 46(3):741–7. doi:10.1002/art.10221
Keywords: systemic lupus erythematosus, complement C1q, complement C4, autoimmune diseases, complement C2, complement C1s, complement C1r, classical pathway
Citation: Lintner KE, Wu YL, Yang Y, Spencer CH, Hauptmann G, Hebert LA, Atkinson JP and Yu CY (2016) Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases. Front. Immunol. 7:36. doi: 10.3389/fimmu.2016.00036
Received: 04 November 2015; Accepted: 24 January 2016;
Published: 15 February 2016
Edited by:
Michael C. Carroll, Harvard Medical School, USAReviewed by:
Richard Di Paolo, Saint Louis University School of Medicine, USACopyright: © 2016 Lintner, Wu, Yang, Spencer, Hauptmann, Hebert, Atkinson and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: C. Yung Yu, eXVjQHBlZGlhdHJpY3Mub2hpby1zdGF0ZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.