 Susu Duan
Susu Duan Paul G. Thomas
Paul G. Thomas- Department of Immunology, St. Jude Children’s Research Hospital, Memphis, TN, USA
Influenza A virus (IAV) is a significant human pathogen causing annual epidemics and periodic pandemics. CD8+ cytotoxic T lymphocyte (CTL)-mediated immunity contributes to the clearance of virus-infected cells, and CTL immunity targeting the conserved internal proteins of IAVs is a key protection mechanism when neutralizing antibodies are absent during heterosubtypic IAV infection. However, CTL infiltration into the airways, its cytotoxicity, and the effects of produced proinflammatory cytokines can cause severe lung tissue injury, thereby contributing to immunopathology. Studies have discovered complicated and exquisite stimulatory and inhibitory mechanisms that regulate CTL magnitude and effector activities during IAV infection. Here, we review the state of knowledge on the roles of IAV-specific CTLs in immune protection and immunopathology during IAV infection in animal models, highlighting the key findings of various requirements and constraints regulating the balance of immune protection and pathology involved in CTL immunity. We also discuss the evidence of cross-reactive CTL immunity as a positive correlate of cross-subtype protection during secondary IAV infection in both animal and human studies. We argue that the effects of CTL immunity on protection and immunopathology depend on multiple layers of host and viral factors, including complex host mechanisms to regulate CTL magnitude and effector activity, the pathogenic nature of the IAV, the innate response milieu, and the host historical immune context of influenza infection. Future efforts are needed to further understand these key host and viral factors, especially to differentiate those that constrain optimally effective CTL antiviral immunity from those necessary to restrain CTL-mediated non-specific immunopathology in the various contexts of IAV infection, in order to develop better vaccination and therapeutic strategies for modifying protective CTL immunity.
Introduction
Influenza A virus (IAV) causes acute respiratory tract infection and is a significant human pathogen causing annual epidemics and periodic pandemics (1). Annual influenza epidemics are caused by circulating “seasonal” influenza viruses, which currently include H1N1 and H3N2 subtype IAVs and influenza B viruses. The RNA genome of IAV has high mutation rates due to the high error rate of its RNA polymerase, allowing the viruses to quickly evolve under selection pressures to develop antigenically drifted strains. Occasional influenza pandemics are caused by the introduction of novel antigenically shifted strains to human populations. These strains, including 2009 pandemic H1N1 IAV, often result from the reassortment of different zoonotic IAVs (2). Reassortment also can generate antigenically distinct new subtypes that pose a pandemic threat to the human population, such as the recent outbreak of human infections of zoonotic H7N9 IAV in China (3). Due to their ability to constantly generate new strains through mutation and reassortment, IAVs pose continuing threats to human populations.
Upon encountering IAVs, the complex host immune system senses the invasion and then mounts innate and adaptive immune responses intended to clear the virus. Multiple arms of host immunity are used for protection against influenza infection. The innate responses initiate inflammation, limit virus replication, and provide signals to activate adaptive immunity (4). The ultimate control of virus replication and clearance of virus-infected cells rely on the virus-specific adaptive immune responses, including antibodies and CD4+ and CD8+ T cells (5). IAV-specific antibodies bind and neutralize viral proteins (neutralizing antibodies) or mediate other virus clearance activities (non-neutralizing antibodies) in cooperation with immune cells, including CD8+ T cells and lung-resident phagocytes (6–10). CD4+ T cells help B cells for antibody production and CD8+ T cells for activation and proliferation. It has been long acknowledged that CD8+ cytotoxic T lymphocyte (CTL)-mediated immunity contributes to virus clearance through cytolysis of the virus-infected target cells and production of cytokines that further enhance antiviral inflammation (11). However, there is accumulating evidence that the IAV-specific CD8+ T cells and their effector mediators also contribute to immunopathology during IAV infection. A variety of mechanisms have been uncovered to regulate the magnitude and effector activities of IAV-specific CTL responses for effective virus clearance while limiting inflammation during influenza infection.
Here, we first review the historical advances in understanding protective and immunopathogenic roles of IAV-specific CTL responses using either IAV infection or non-viral infection models. Next, we focus on the current state of knowledge for how various regulatory mechanisms control immune protection and pathology by CTL responses during influenza infection. This is followed by a discussion of clinical findings about the role of CTL responses in human IAV infections, highlighting the evidence that emerged after the 2009 H1N1 pandemic and recent outbreaks of human infection by H7N9 IAVs. Finally, we summarize our current understanding of the multiple layers of host and viral factors mediating the outcome of antiviral CTL responses and suggest future key research directions.
IAV Infection Induces Antiviral CD8+ T-Cell Responses
The mammalian airways have very large mucosal surfaces for gas exchange. IAV infection begins with virus invasion and replication in the upper airway epithelium, and from there, it can spread further into the lower airways and lung, causing severe infection. Meanwhile, the lung epithelial and endothelial cells and other airway resident innate cells, including several dendritic cell (DC) subsets, alveolar macrophages, and innate lymphoid cells, sense the viruses and initiate innate inflammation, acting as the first line of defense (4). Innate and epithelial cells use specific pattern recognition receptors (PRRs) to recognize various viral products containing pathogen-associated molecular patterns (PAMPs), including endosome-bound TLR3 (dsRNA), TLR7/8 (ssRNA), cytosolic RIG-I (ssRNA), and NLRP3 (viral ssRNA) (12). Innate sensing of viruses initiates production and/or release of various inflammatory cytokines, including type I interferon (IFN), IL-1β, IL-6, and TNF-α. These cytokines induce multiple mechanisms in the infected cells to limit virus replication and also stimulate DC activation for effective antigen acquisition and presentation to initiate IAV-specific adaptive responses.
At least three subsets of lung-resident DCs have been found to play a role in responding to IAV infection in mice (13, 14). Lung plasmacytoid DCs (pDCs) are classic potent producers of type I IFN after detecting virus (15) but were initially thought to be dispensable for antiviral T-cell immunity (16). However, recent studies found that pDCs also migrate into lung-draining lymph nodes (LNs) to play a different role in certain IAV infection contexts (17, 18) that will be discussed later in this review. Two subsets of lung DCs, CD103+ and CD11b+ DCs, are migratory DCs (19, 20) that bridge innate and adaptive immunity during influenza infection. After being activated by virus infection and innate cytokines, they migrate out of the inflamed lung into the lung-draining LNs in a CCR7-mediated way (21), carrying the acquired viral antigen; in the LNs, these migratory DCs either directly present antigens to naive T cells or transfer the antigen to other specific LN DC populations for further presentation to naive T cells (19). Naive T cells in the LNs recognize the presented IAV-specific antigens using their T-cell receptors (TCRs) and receive various costimulation signals from the activated DCs, sequentially undergoing clonal selection, expansion, activation, and differentiation into IAV antigen-specific effector CD4+ and CD8+ T cells. Studies have shown that, following IAV infection, accelerated migration of DCs carrying antigens from the infected lung to LNs occurs only during the early phase of infection (the first 36 h after infection) (22, 23), and activation of the naive T cells in the LNs then occurs in an ordered, sequential fashion, in accordance with the tempo of the DC migration (23).
Influenza A virus infection drives CD4+ T-cell differentiation mainly into Th1 effector T cells. These “helper” cells provide help to B cells and CD8+ T cells in different ways via costimulatory signals and cytokines. Notably, CD4+ T-cell help is not essential for the primary effector CD8+ T-cell responses (24), although it is important for the generation of the optimal magnitude of memory CD8+ T-cell responses after IAV infection (25). Effector CD8+ T cells then migrate from the LNs into the infected and inflamed lung where the survival and proliferation of the CTLs are further shaped by interactions with various non-migratory DC populations in lung (26–28). IAV-specific effector CD8+ T cells are cytotoxic cells equipped to specifically recognize and kill the virus-infected cells and produce various cytokines.
Following IAV clearance, the generated virus-specific B cells and CD4+ and CD8+ T cells undergo a rapid contraction phase with only a small proportion surviving and differentiating into memory cells, which form the memory pool necessary to protect against future infections with the same or similar viruses. During this process, chemokine receptors CCR5 and CXCR3 are important in regulating the virus-specific CTL contraction and memory generation within the infected lung (29, 30). Studies have shown that residual IAV antigen depots in lung-draining LNs can persist for weeks after virus clearance; the prolonged presentation of the residual antigen does not prime new naive CD8+ T cells (31) but helps to maintain the large number of the memory CD8+ T cells in the draining LNs or in the lung airways possibly by regulating circulation patterns and activation states of the local memory CD8+ T cells (32–34). However, in contrast to the lung populations, this antigen is not required for maintenance and circulation of the peripheral memory CD8+ T cells (35). Recently, three major subsets of memory T cells are recognized based on their anatomical location, migration patterns, and additional phenotypic and effector markers: central memory T cells (Tcm), effector memory T cells (Tem), and tissue resident memory T cells (Trm). The biology of these different subsets is outside the scope of the current review.
It is worth noting that during the differentiation of naive CD8+ T cells into effector CTLs and then into distinct memory subsets, the cells undergo massive transcriptional programing that regulates their effector or memory potential (36). IRF4 (37, 38) and Blimp1 (39) have been found as important transcription factors that regulate CTL clonal expansion, effector differentiation, and/or effector activity during IAV infection.
In the following review, we discuss how the activation, proliferation, survival, migration, localization, and effector activity of IAV-specific CD8+ T cells are tightly regulated by antigen load, multiple cytokines, and costimulatory and/or coinhibitory signals provided by various cells in the LNs and in the infected lung. Appropriate control of the magnitude and effector activity of IAV-specific CD8+ cells is required both for an effective antigen removal and for limiting the potential immunopathology.
IAV-Specific CD8+ T Cells are Cytotoxic and Cytokine/Chemokine-Producing Cells
Influenza A virus-specific CD8+ CTLs play a vital role in eliminating IAV-infected host cells in the lung through their two well-defined effector activities: antigen-specific cytotoxicity and cytokine/chemokine production. The CTLs target only virus-infected host cells, using their TCRs to recognize specific viral peptides (p) in complex with host major histocompatibility complex (MHC) molecules (pMHC) on the cell surface. TCR recognition and engagement of pMHC is a prerequisite for subsequent CTL cytotoxicity and cytokine production. CTL cytotoxicity is mediated in three known ways: perforin/granzyme-mediated cytolysis, apoptosis mediated by FasL/Fas, and TRAIL/TRAIL-DR signaling (40–43). CTLs contain a pore-forming protein (perforin) and cytotoxic granules comprising proapoptotic proteases called granzymes. Perforin complexes form pores between the CTL and target cell, and the granules then release granzymes into the target cells through these pores, with granzyme B one of the most abundant (40). CTLs also use two membrane-bound TNF family ligands, FasL and TRAIL, for targeted cell killing. Engagement of these molecules with their cognate receptors (Fas and TRAIL-DR) on infected cells initiates an apoptotic signaling cascade (41–43).
Influenza A virus-specific CD8+ CTLs are also equipped to produce a range of cytokines and chemokines. Classically, IFN-γ and TNF-α are the most prominent effector cytokines produced by CTLs, and cells producing them are often referred to as classical “Tc1” cells. IFN-γ is a potent antiviral cytokine. It can enhance the cytotoxicity of other immune cells, promote further activation of DCs, and help B cells to promote antibody isotype switching (44). TNF-α is primarily a proinflammatory cytokine, induces non-specific death of infected cells, and regulates the function of other immune cells through TNFRs (45). CTLs are not the main source of IL-2, but a small proportion of CTLs can produce IL-2 after receiving certain costimulatory signals, which in return may provide proliferation and survival signals to themselves and other CTLs (46). CTLs producing IFN-γ, TNF-α, and IL-2 are considered more potent than those producing only one or two cytokines (47). Different subsets of IAV antigen-specific CTLs differ in their ability to produce combinations of the three cytokines (48).
IL-10 is usually produced by regulatory CD4+ T cells (Treg) and/or helper CD4+ T cells. It is commonly recognized as an immune regulatory/anti-inflammatory cytokine that serves as a brake on ongoing inflammation. IL-10 was found to be produced in large quantities in IAV-infected lungs, and surprisingly, the effector CD8+ T cells that simultaneously produced IFN-γ contributed a large fraction of IL-10 (49). Blocking the action of IL-10 during IAV infection resulted in enhanced pulmonary inflammation and lethal injury (49). Thus, CD8+ T cells produce both anti-inflammatory IL-10 and antiviral IFN-γ to fine-tune the CTL activity to an effective but restrained level.
Recently, studies found small subsets of CD8+ effector T cells in the IAV-infected lung that could produce non-classical CTL cytokines (50–53): “Tc2” cells producing IL-4, IL-5, and IFN-γ and “Tc17” cells producing IL-17 and IFN-γ. In vitro polarized Tc2 and Tc17 cells are as cytotoxic as Tc1 cells, and the adoptive transfer of Tc2 or Tc17 cells into infected mice provided different levels of survival protection after otherwise lethal IAV infection (50, 52, 53). Relative to Tc1 cells, Tc2 and Tc17 cells account for a very small proportion of effector CD8+ T cells in vivo, and the extent of their effector activity during IAV infection in vivo needs to be further defined.
The two CTL effector activities (cytotoxicity and cytokine production) are precisely regulated in the infected lung by a variety of factors, including their anatomic localization and their interactions with different antigen-presenting cells with diverse pMHC density and costimulatory signals, to achieve effective target cell killing while limiting non-specific inflammation (Figure 1). These mechanisms will be discussed in detail below.
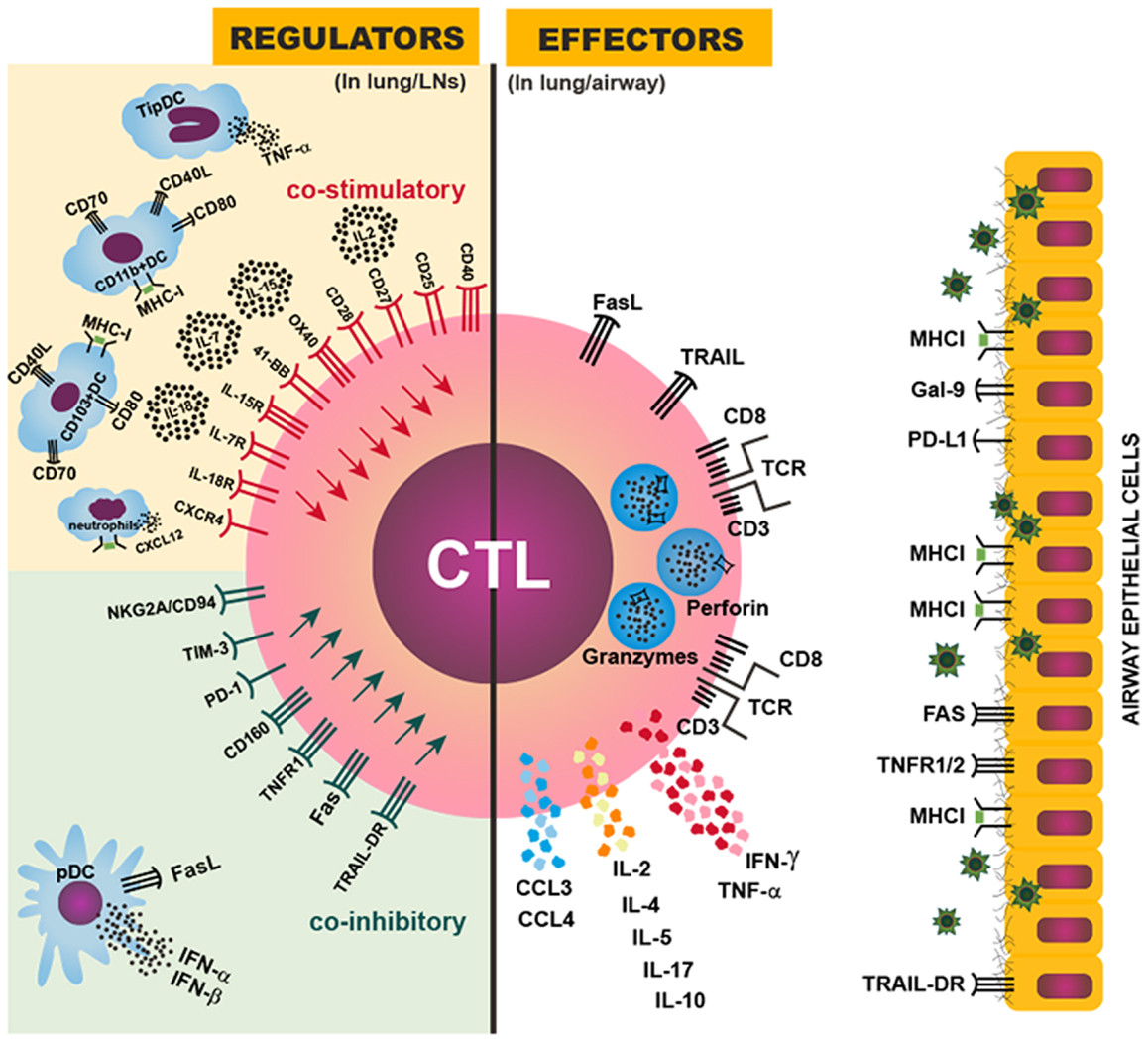
Figure 1. Regulation of CTL magnitude and effector activity. Right: CTL effector mechanisms against IAV in the infected lung or airway: the IAV-specific CTL targets IAV-infected airway epithelial cells by recognizing a viral peptide presented by MHCI molecules on the surface of infected cells; the CTL then induces cell death in the targeted cell through perforin/granzyme, FasL/Fas, and/or TRAIL/TRAIL-DR signaling; CTLs also can produce IFN-γ, TNF-α, IL-2, CCL3, CCL4, and other cytokines and chemokines to further enhance inflammation and immune activation in the infected lung. Left: various regulatory mechanisms to control the magnitude or effector activity of CTLs though costimulatory (upper) or coinhibitory (lower) signals provided in the lung-draining LNs or the infected lung. An optimal magnitude of protective CTL responses is achieved by balancing the costimulatory and coinhibitory signals, and dysregulation or imbalance among those signals can result in insufficient or exuberant CTL responses, leading to inefficient viral control or damaging immunopathology.
IAV-Specific CD8+ T Cells are Crucial for Virus Clearance and Provide Protection during IAV Infection
The role of CTLs in clearing IAV has been demonstrated in multiple studies using adoptive transfer of IAV-specific CTLs into naive recipient mice (Table 1). In these studies, after the adoptive transfers, lung virus titers and/or the time to virus clearance were reduced, leading to accelerated recovery from non-lethal infection or survival of otherwise lethal infection (54–56). The contribution of CTLs to protective anti-IAV immunity is further corroborated by studies using β2-M-deficient mice, which are defective in MHCI complex assembly and antigen presentation and thus fail to produce functional CD8+ T cells (57). The β2-M-deficient mice showed a significantly delayed pulmonary virus clearance after non-lethal IAV infection and a significantly higher mortality rate after a lethal IAV infection than the control β2-M heterozygous mice (57), showing that CD8+ T-cell immunity is important in protection against IAV infection. However, both the β2-M-deficient mice and mice depleted of CD8+ T cells were able to eventually clear the virus and recover from non-lethal IAV infection (58), suggesting that the CTL response is not the sole effector of antiviral immunity during IAV infection. IAV-specific immunity consists of multiple immune mechanisms, including CTLs, antibodies, and CD4+ T-cell responses, which promote IAV clearance and host protection.
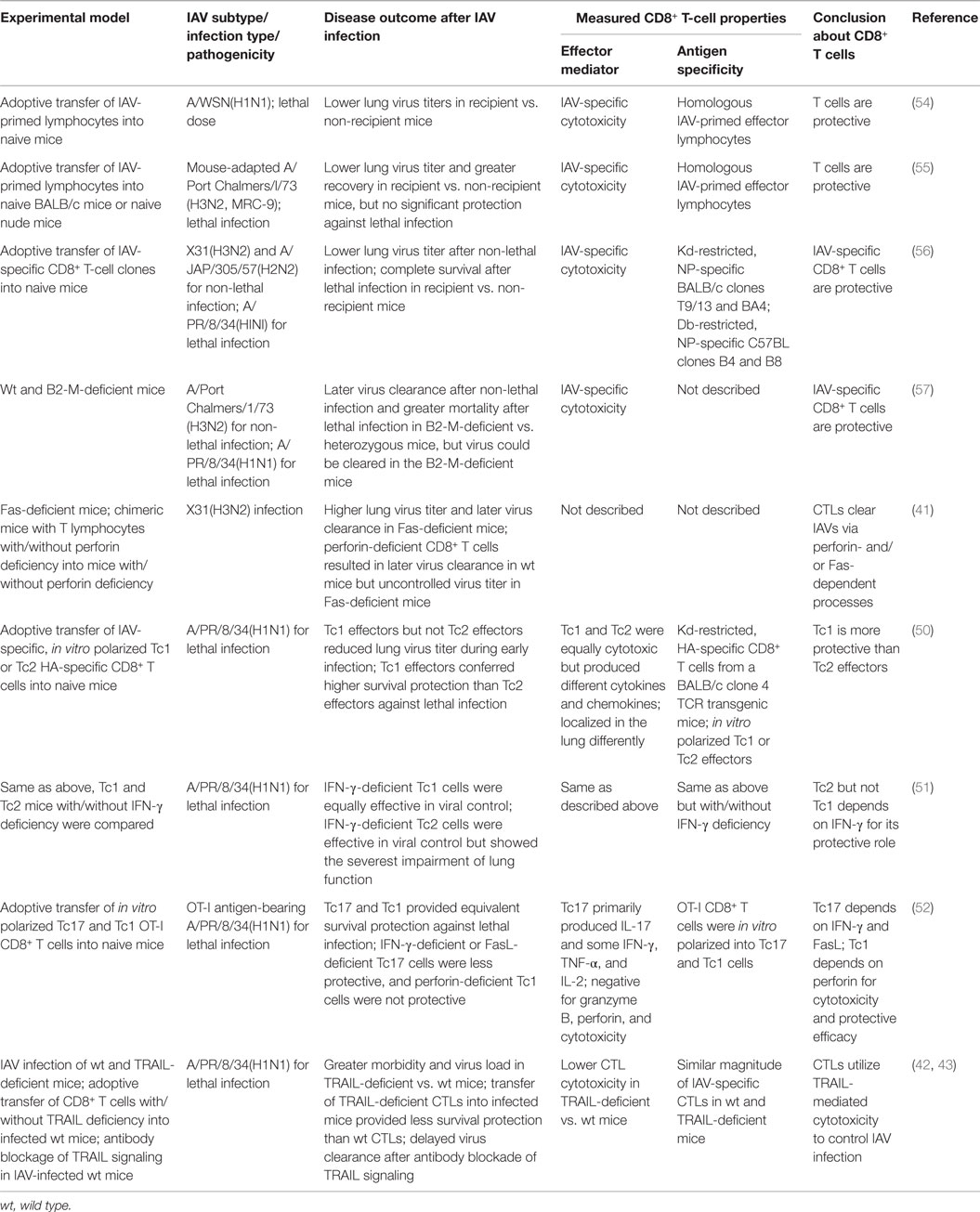
Table 1. Overview of studies demonstrating immune protection by the CD8+ T-cell responses during IAV infection.
Both CTL effector activities (cytotoxicity and cytokine production) can contribute to protective immunity, but antigen-specific target cell destruction by CTL cytotoxicity is believed to be the primary CTL activity used for IAV clearance (11). Earlier studies showed that either perforin/granzyme- or FasL/Fas signaling-mediated apoptosis provided sufficient CTL cytotoxicity for efficient virus clearance (41). Later, TRAIL/TRAIL-DR signaling was found to contribute to CTL cytotoxicity and virus clearance (42, 43). In a non-viral infection model, in the absence of perforin, the antigen-bearing alveolar epithelial cells are not sensitive to FasL/Fas-induced cell death mediated by transferred antigen-specific CTLs, suggesting that CTLs may use different cytotoxicity mechanisms depending on the lung environmental milieu. Such differences are especially evident in studies comparing Tc1, Tc2, and Tc17 cells: after in vitro polarization and adoptive transfer into IAV-infected mice, Tc1 cell efficacy depended on perforin/granzymes, while Tc17s depended on FasL/Fas signaling for cytotoxicity (50, 52) and Tc1 cells did not rely on IFN-γ, but both Tc2 and Tc17 cells required IFN-γ for their protective efficacy (50–52). Thus, CD8+ effector T cells can protect against IAV infection via a number of redundant effector mechanisms (53). The relative contribution of each mechanism to protective efficacy may depend on a variety of factors: the differentiation status of the effector CD8+ T cells; the target cell type, activation status, or receptor expression; the local lung environment in the context of IAV replication; and others.
IAV-Specific CD8+ T Cells Contribute to Immunopathology during IAV Infection
The two effector activities of IAV-specific CTLs allow effective specific killing of virus-infected cells but can also cause non-specific, tissue-destructive inflammation. In an earlier study, IAV infection of athymic nude mice, which cannot generate functional T cells, led to a longer survival and a slower progression of lung pathology than infection of wild-type mice, but the nude mice had a lower eventual survival rate, persistent lung injury, and higher lung virus titers (59). This study provided a first glimpse into the double-edged effects of antiviral T-cell immunity: while it provides necessary protective immunity, it does so at the cost of immunopathology. Since that study, immunopathology caused by IAV-specific CTLs has been further elucidated (Table 2).
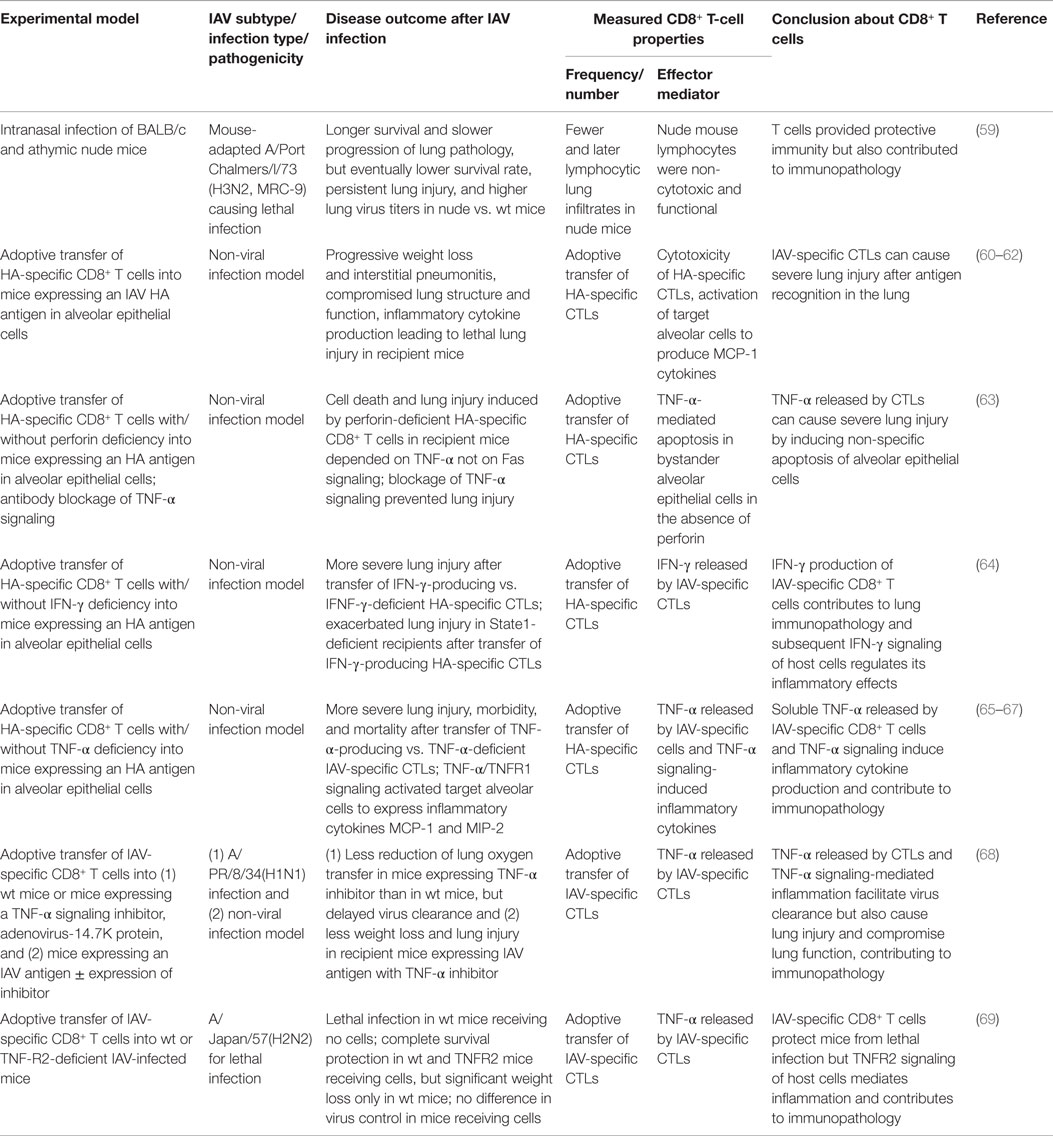
Table 2. Overview of studies demonstrating immunopathology caused by CD8+ T-cell responses during IAV infection.
One study used a non-viral infection model with CD8+ T CTLs specific for an IAV hemagglutinin (HA) antigen adoptively transferred into mice whose alveolar epithelial cells constitutively expressed the HA antigen. The CTL-mediated immune response is initiated when the transferred CTLs recognize the HA antigen (60). In this model, the contribution of the IAV-specific CTLs to lung immunopathology can be isolated from inflammation caused by virus replication, allowing detailed dissection of lung immunopathology caused by CTL cytotoxicity, TNF-α/IFN-γ release, and their subsequent effects on alveolar epithelial cells (Table 2). This non-viral infection model was able to cause lethal lung injury. The recipient mice showed progressive weight loss, interstitial pneumonitis, compromised lung structure and function, and increased inflammatory cytokine levels in the lung, demonstrating that IAV-specific CTLs can cause immunopathology (60, 61). Subsequent studies using this model revealed that the transferred CTLs were present only transiently (24–48 h) in the lung, and the targeted antigen-specific alveolar cells were stimulated by interaction with the CTLs to produce MCP-1 and other inflammatory mediators before they underwent apoptosis, causing lung inflammation (62). The soluble TNF-α released by the CTLs induced further inflammatory cytokine production (65–67, 69) and non-specific apoptosis of bystander alveolar epithelial cells (63), contributing to both lung injury and inflammation. Additionally, IFN-γ production by the CTLs and subsequent IFN-γ signaling contributed to lung immunopathology (64).
By isolating IAV-specific CTL responses, the non-viral infection model provided valuable insights into the CTLs’ contributions to lung immunopathology. It is also important to note that the sustained presence of IAV antigen in this model is not commonly found in the IAV infection model where the virus is eventually cleared; thus, the immunopathogenic effects of IAV-specific CTL responses were likely excessively amplified in this model and did not capture the protective effects that these cells provide in a natural infection. For example, a study using mice that express a TNF-α inhibitor in their alveolar epithelium demonstrates that TNF-α produced by the CTLs is a double-edged sword: it indeed facilitates virus clearance but at the same time contributes to severe lung immunopathology (68). Thus, it is this delicate balance between immune protection and pathology mediated by CTLs that determines the overall outcome in the host.
Balancing the Immune Protection and Immune Pathology of CD8+ T Cells during IAV Infection
After IAV infection, infiltration of the lung by IAV-specific CTLs and their subsequent effector activities are essential for virus clearance but also contribute substantially to lung immunopathology; therefore, IAV-specific CTL responses must be balanced to mount effective antiviral immunity but limit tissue immunopathology. Studies using mouse models have provided insights into the complex, exquisite, control, and regulation of the magnitude of IAV-specific CTLs and their effector activities. These regulatory mechanisms include various costimulatory/inhibitory signals, cytokine/chemokine signals, and different cell–cell interactions, among others, to balance the protective (Table 3) against pathological (Table 4) effects by IAV-specific CTLs (Figure 1).
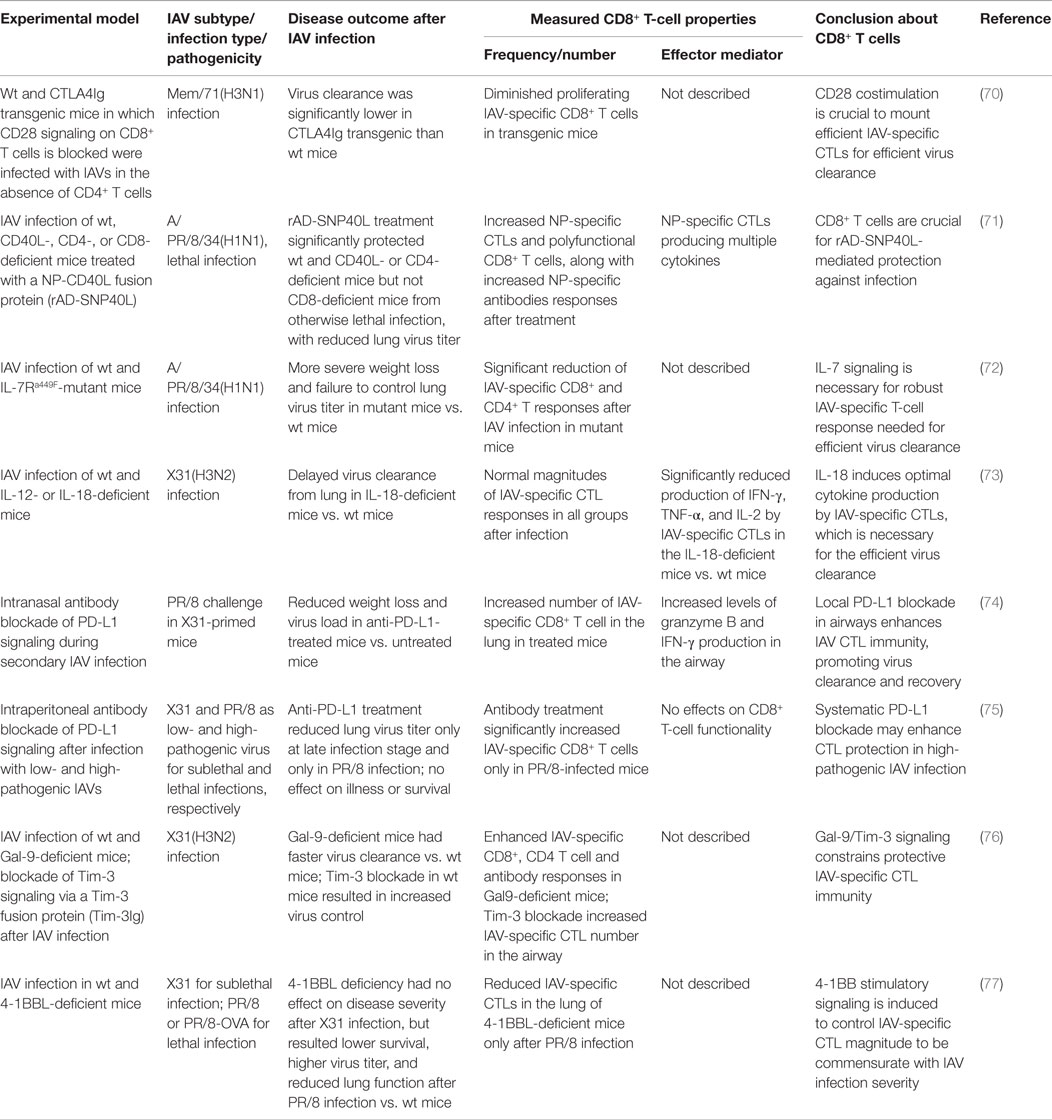
Table 3. Tight regulation of CD8+ T-cell responses provides immune protection against IAV infection.
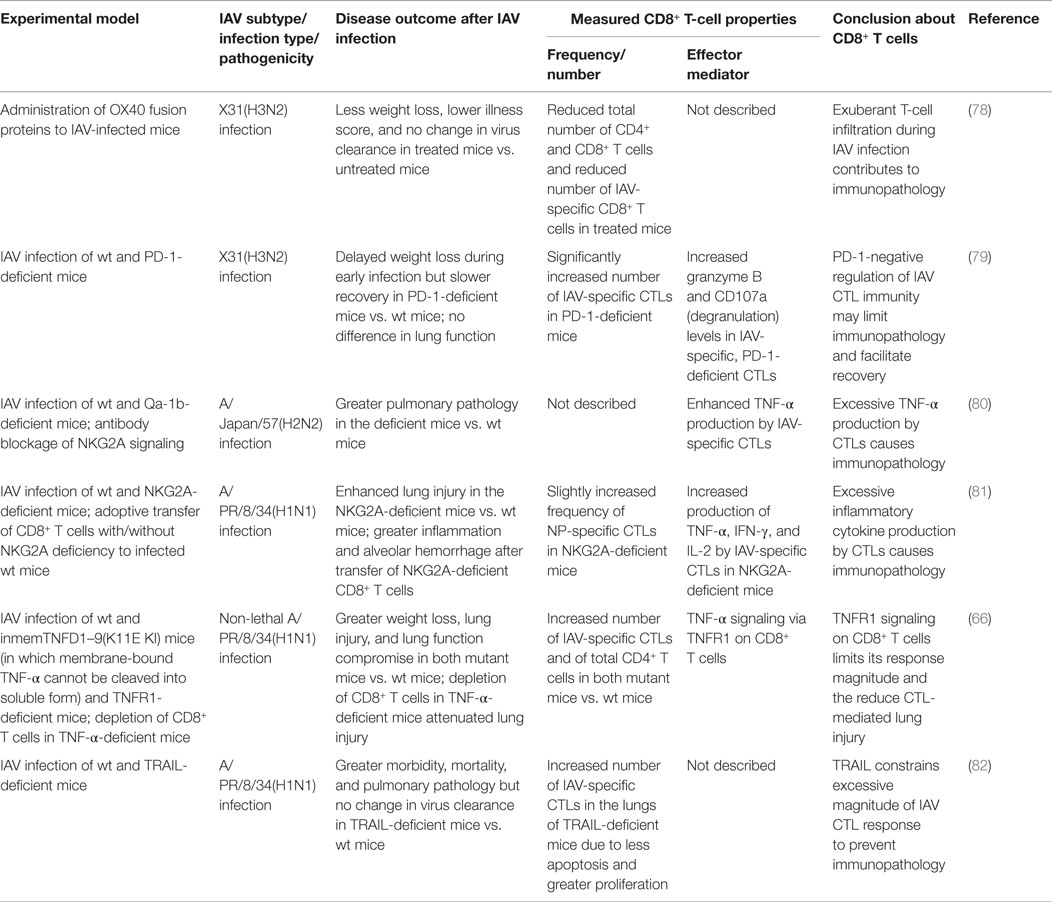
Table 4. Dysregulation of CD8+ T-cell responses contributes to immunopathology during IAV infection.
In addition to antigen stimulation, effective CD8+ T-cell responses are commonly believed to require secondary costimulatory signaling from DCs and/or proliferation signals from CD4+ helper T cells. However, during IAV infection, the primary IAV-specific CTL response is largely independent of CD4+ helper T cells (24). Thus, various costimulatory signals from DCs and other innate immune cells are particularly important in generating effective IAV-specific CTL immunity. Studies using specific receptor-deficient mice and/or signaling blockade have shown that both CD80/CD28 and CD70/27 costimulatory signaling are pivotal for initial activation and expansion of IAV-specific naive CD8+ T cells in LNs (70, 83), and CD70/27 is especially critical for accumulation of the CTLs in the infected lung by sustaining their survival (46, 84). Other costimulatory signals, including 4-1BBL/4-1BB, CD40L/CD40, and OX40L/OX40, collectively contribute to generate the optimal CTL response magnitude at late stages of infection and/or the optimal size and responsiveness of the memory CTL pool (83, 85, 86).
CD40L on the activated DC subsets provide important costimulatory signals in optimizing the magnitude of IAV-specific CTL responses in CD4+ T cell-independent immunity (71, 87). One study used an adenovirus-produced recombinant CD40L fused with IAV NP antigen (rAD-SNP40L) to target CD40 during IAV infection; this treatment successfully enhanced NP-specific CTL and antibody responses, reduced lung virus titers, and protected the mice from otherwise lethal IAV infection (71). Notably, type I IFN and IL-1 innate cytokines induced by IAV replication can activate DCs to increase the expression of these costimulatory molecules and thus promote IAV-specific CD8+ T-cell immunity (88, 89).
Another costimulatory receptor, 4-1BB, is differentially expressed on IAV-specific CTLs in lungs after infection with a low- and a high-pathogenic IAV. The higher level of 4-1BB is required to mount a higher magnitude of IAV-specific CTL responses for effective clearance of the high pathogenic IAVs (77). However, intranasal delivery of adenoviral-4-1BBL into the 4-1BBL-deficient mice after the high pathogenic IAV infection marginally improves survival at a low dose but exacerbates disease at a high dose; delivery of both doses of 4-1BBL-AdV into wild-type mice led to an increased mortality (77). This finding demonstrates an example that inducible costimulatory molecules have to be balanced to develop antiviral CTL immunity to a level commensurate with the pathogenicity of the IAVs.
Costimulatory signals can also result in exuberant T-cell inflammation that contributes to immunopathology. OX40 is a costimulatory receptor expressed only on activated T cells, and its interaction with OX40L on DCs imparts a survival signal to the T cells, preventing activation-induced cell death (90). Blockage of OX40L/OX40 signaling during IAV infection by administration of an OX40 fusion protein significantly reduced the magnitude of both IAV-specific CTL and CD4+ T-cell responses (78). However, unlike the effects of other costimulatory signals described above, reduction of T-cell responses by OX40L/OX40 blockade ameliorated disease symptoms without compromising effective virus clearance (78). Clearly, OX40L/OX40 signaling during IAV infection is not necessary for virus clearance but contributes to immunopathology.
On the other hand, there are also various coinhibitory receptors on T cells, mediating inhibitory signals to activated T cells. Coinhibitory signaling serves to prevent tissue damage by limiting the response magnitude but also may constrain the effective immunity necessary for protective efficacy. For example, tissue-expressed Galectin-9 (Gal-9) binds to its receptor Tim-3 on T cells to limit the response magnitude. Gal-9 deficiency or blockade of Tim-3 signaling resulted in more robust virus-specific CTL and antibody responses to IAV infection, leading to a more rapid virus clearance and recovery (76), suggesting that Gal-9/Tim-3 signaling constrains effective antiviral immunity. NKG2A/CD94, another inhibitory receptor originally described mainly on NK cells, was found to be expressed on IAV-specific CTLs during IAV infection. Deficiency of NKG2A or its ligand Qa-1b, or blockage of Qa-1b/NKG2A signaling, resulted in a greater pulmonary pathology accompanied by an enhanced TNF-α production by the IAV-specific CTL cells (80, 81); therefore, NKG2A-mediated negative signaling truly limits CTL immunity-mediated lung injury. Another inhibitory receptor, CD160, was also recently found on CD8+ T cells during influenza infection, and CD160-ligand/CD160 signaling reduced the proliferation capacity and perforin expression of the CD8+ T cells (91), although the role of this negative signaling in the outcome of the IAV infection is unclear.
PD-1 is another inhibitory receptor found to be unregulated on IAV-specific CTLs after primary or secondary IAV infection, and its ligand PD-L1 was found to be expressed on lung epithelial cells and innate immune cells (74, 75, 79). PD-L1/PD-1 signaling leads to severe functional impairment of CTLs. Inhibition of PD-L1/PD-1 signaling during IAV infection by blocking either PD-1 or epithelial PD-L1 with antibodies in wild-type mice enhanced CTL function and subsequent virus clearance and ameliorated disease severity (74, 75). However, in PD-1-deficient mice, CTL magnitude and function were improved after IAV infection, but recovery was delayed (79), suggesting that PD-1-negative signaling is required to limit the adverse effects of the IAV-specific CTL responses. Clearly, this negative signaling is necessary to balance CTLs’ role in virus control, speed of recovery, and immunopathology. Interestingly, another study found that only infection with highly pathogenic but not with low pathogenic IAV induced a greater PD-1 expression on the IAV-specific CTLs (75), demonstrating that the optimal magnitude of antiviral CTL immunity can be dampened as a consequence of the high pathogenicity of a particular IAV.
Plasmacytoid DCs are also found to negatively regulate the magnitude of CTL responses in LNs after lethal infection with a high dose of IAV or with a highly pathogenic H5N1 IAV but not after sublethal infection (17, 18). After the lethal infections, pDCs accumulating in the LNs showed a high expression of FasL, driving the elimination of Fas+ CD8+ cells. The dampened IAV-specific CTL responses enhanced the lethality of the infections, providing another example of how the optimal magnitude of antiviral CTL responses can be compromised in infections with IAVs of high pathogenicity, consistent with the clinical findings of lymphopenia in patients infected with highly pathogenic H5N1 IAVs (92).
Various soluble cytokines induced during IAV infection also play an important role in the trafficking, survival, and effector activity of CD8+ effector T cells required for optimal antiviral CTL responses. For example, IL-15 is chemotactic, responsible for the migration of IAV-specific CTLs to the infected lung (93) and also contributes to the survival of the CTLs in the infected lung (27, 93). Both IL-7 (72) and IL-18 (73) are necessary for the development of a robust IAV-specific CTL response required for efficient virus clearance. IL-12 was also shown to be important for CTL cytotoxicity in vitro (94).
Two signaling molecules, TNF-α and TRAIL, used in the effector mechanisms of CTLs, can also constrain the magnitude of IAV-specific CTL responses to prevent the excessive damage. TRAIL deficiency (82) and TNF-α deficiency (66) increased the number of IAV-specific CTLs accompanied with decreased apoptosis and increased proliferation but increased host morbidity and mortality after IAV infection. Thus, both TRAIL/TRAIL-DR signaling and TNF-α/TNFR1 signaling have a dual role during IAV infection depending on the cell type in which the receptors are expressed: they can kill IAV-infected cells for immune protection and also negatively regulate the magnitude of the CTL response to reduce the likelihood of immunopathology.
Various types of innate and adaptive immune cells, including IAV-specific CTLs, infiltrate the IAV-infected lung. These infiltrating cells, the lung-resident cells, and the cytokines that they produce compose the local inflammation milieu, which can further shape the antiviral CTL response. For example, maintaining an optimal magnitude of protective IAV-specific CTL responses requires interactions with multiple non-migratory DC subsets in the infected lung for essential survival and proliferation signals (26–28). An inflammatory monocyte population (sometimes referred to as TipDCs) is recruited from bone marrow to the infected lung by CCL2/CCR2-mediated chemoattraction. TipDCs and the TNF-α that they produce promote IAV-specific CTL expansion and survival in the lung, which are required for optimal protection (95). The IAV-infected lung is also characterized by a large neutrophil infiltration. Migrating neutrophils leaves behind long-lasting CXCL12-enriched trails and routes into the infected lung to facilitate efficient CD8+ T-cell migration and localization to the infection site, which is required for optimal CTL infiltration and effective virus clearance (96). In the lung interstitium, neutrophils also serve as antigen-presenting cells that promote IFN-γ production by CTLs (97). NKT cells are recruited to the lung, where they themselves have a protective role against IAV infection (98). Furthermore, NKT cells also promote the accumulation of CD103+ DCs in the LNs and promote the subsequent IAV-specific CD8+ T-cell response (99). Finally, IAV antigen-specific Treg cells are found in both primary and secondary IAV infections, negatively regulate the proliferation of IAV-specific CTLs, and limit the pulmonary inflammation during secondary IAV infection (100).
During IAV infection, not only is the magnitude of the antiviral CTL response regulated by multiple signals from various cells, but also the antiviral effector activities of the CTLs are dictated by the target cell type encountered and the costimulatory signals and antigen intensity the target cell carries. For example, CD45+ inflammatory mononuclear cells in the lung interstitium, such as CD11c-high cells, which express costimulatory ligands (CD80/86), stimulate both CTL cytotoxicity and release of inflammatory cytokines, but CD45− respiratory epithelial cells expressing few stimulatory ligands trigger only CTL cytotoxicity (14, 101). Consistent with this finding, after adoptive transfer into IAV-infected mice, highly cytotoxic Tc1 cells were found to localize near the infected airway epithelium, but Tc2 cells localized within the clusters of other inflammatory cells distant from epithelium (50), suggesting that different subsets of CD8+ T cells might interact with the subsets of innate cells resulting in diverse effector activities.
During the CTL and target cell interaction, CTL TCR recognition of its cognate pMHCI complex on the target cells determines the specificity of CTLs, while the strength of TCR/pMHCI recognition fine-tunes the extent of CTL effector activities (102–104). The requirements of TCR occupancy and signal strength to activate CTL cytotoxicity are very low to minimal, allowing rapid and specific target cell killing, while the requirement for the induction of inflammatory cytokines is much higher, limiting the extent of non-specific inflammation induced by the cytokines (102–104). Thus, the division of specialized roles among the different antigen-presenting cells and the two activation thresholds for the two CTL effector activities enable specific target cell killing while not necessarily causing excessive inflammation in the infected lung. In addition, from a temporal view of IAV replication and the antiviral CTL response, it is also reasonable to speculate that higher viral antigen load and stronger TCR signals on CTLs at the early phase of infection may promote terminal CTL effector activity, while the lower antigen load and TCR signals on CTLs at late phases of infection or after virus clearance may promote CTL memory potential or differentiation.
In summary, during IAV infection, both the magnitude and effector activities of the CTL response are exquisitely regulated by various stimulatory and inhibitory signals, cytokines, and chemokines from a variety of cell types to achieve the goal of immune protection while minimizing potential immunopathology.
CD8+ T-Cell Response is a Positive Correlate of Protective Immunity Against Heterosubtypic IAV Infection
Human populations are challenged constantly by mutated variants of circulating seasonal IAVs (currently H1N1 and H3N2) and occasionally by novel pandemic strains. An estimated 5–20% of the population worldwide is infected annually with a seasonal IAV (105). Thus, human IAV infections are almost invariably at least secondary, except in young children. Therefore, it is of great importance to understand heterosubtypic IAV immunity, which is generated by a given IAV subtype but offers some protection against challenge with another IAV subtype. In the face of a heterosubtypic IAV infection, the neutralizing antibodies generated by IAV-specific B cells can recognize only the specific IAV surface proteins from a previous infection, while IAV-specific CTLs target viral peptides that usually are derived from IAV internal proteins and are relatively conserved across different subtypes (106). Thus, cross-reactive IAV-specific CTLs generated by previous IAV infection are an essential component of heterosubtypic immunity (107, 108), as shown consistently in both animal and human studies.
Animal studies (Table 5) using a prime/challenge model with IAVs of different subtypes have shown that the cross-reactive CD8+ T cells generated by a first IAV infection are unable to prevent secondary IAV infection but clearly ameliorate morbidity and mortality, reduce the virus load, and accelerate recovery; this protection can even boost survival after an otherwise lethal challenge with H5N1 or H7N9 IAV (109–115). These studies have consistently demonstrated that CD8+ T cells mediate heterosubtypic immunity. One study found that the best predictor of protective efficacy against secondary infection was the overall size of the memory CTL pool generated by the priming infection, rather than conservation of known CD8+ epitopes between viruses (114). The size of the memory CTL pool may serve as a proxy indicator of the robustness and quality of the primary immune response and may be associated with the activation status and quality of memory CTLs. Meanwhile, this finding also illustrates our incomplete understanding of the full measure of CD8+ epitope diversity and other heterologous immune mechanisms generated by initial priming, which warrants future studies.
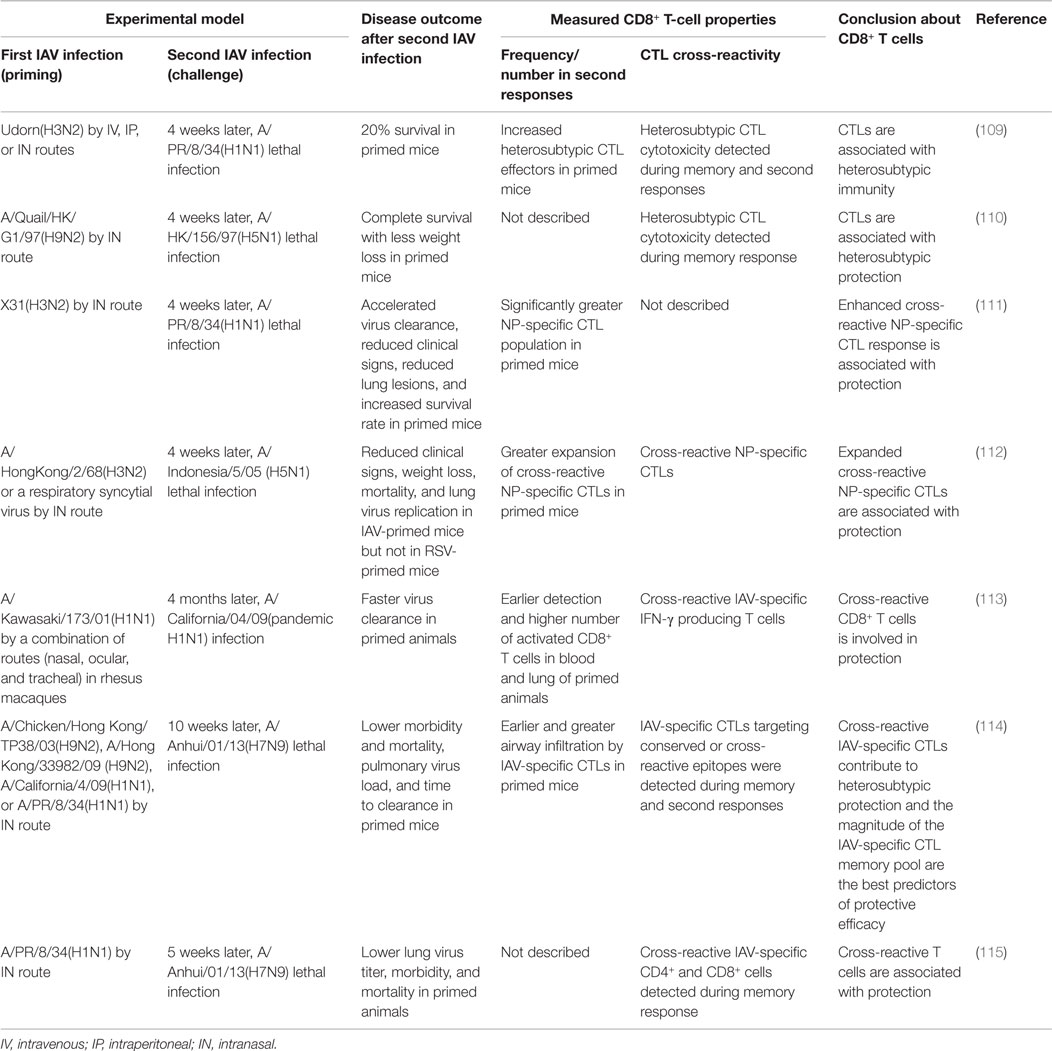
Table 5. Overview of animal studies showing that IAV-specific CD8+ T-cell responses are a correlate of protection against heterosubtypic secondary IAV infection.
Due to the many challenges posed by human studies, the role of CD8+ T cells in mediating heterosubtypic protection against illness caused by natural IAV infection is far from certain, but studies using human samples have provided some useful insights. A number of studies of human peripheral blood cells have demonstrated the existence of cross-reactive CD8+ T-cell immunity between distinct heterosubtypic IAVs, such as between seasonal IAVs and 2009 pandemic H1N1 IAV (116, 117), between the 1918 pandemic H1N1 and 2009 pandemic H1N1 IAVs (118), between seasonal IAVs and avian H5N1 IAVs (119), and between seasonal IAVs and the novel H7N9 IAVs (115, 120, 121). These studies consistently show that prior IAV infection in humans can generate a measure of cross-reactive, or “heterosubtypic,” CD8+ T-cell-mediated immunity against other serologically distinct IAVs, for potential immune recall responses upon another IAV infection.
A few studies in humans have demonstrated that the cross-reactive IAV-specific CD8+ T-cell response is a positive correlate of cross-protective immunity against secondary IAV infection (Table 6). An earlier human study using experimental H1N1 challenge showed that the extent of CTL cytotoxicity in the blood of subjects at 2 days after challenge was closely associated with less virus shedding, faster virus clearance, and less severe symptoms (122), providing the first evidence that the preexisting cross-reactive CTL response is a correlate of clinically cross-protective immunity (day 2 was too early to generate CTL responses to the challenge IAV). The 2009 H1N1 pandemic and the recent emergence of human infections with H7N9 IAV in China have provided unique opportunities to study the natural heterosubtypic IAV infection and cross-protective response in the absence of the specific neutralizing antibodies. One study followed a cohort of healthy adults through the pandemic waves in the UK (123), and another examined a cohort of hospitalized patients with severe H7N9 infection in China (124). Both studies found that the magnitude of the cross-reactive CD8+ T-cell response was positively correlated with a favorable clinical outcome in the patients. Although these findings are consistent with those obtained in mice, it is noteworthy that human studies often involve complex and uncontrollable variables, including demographic, environmental, and genetic differences. As a consequence, extreme care must be taken in interpreting the results of human studies.
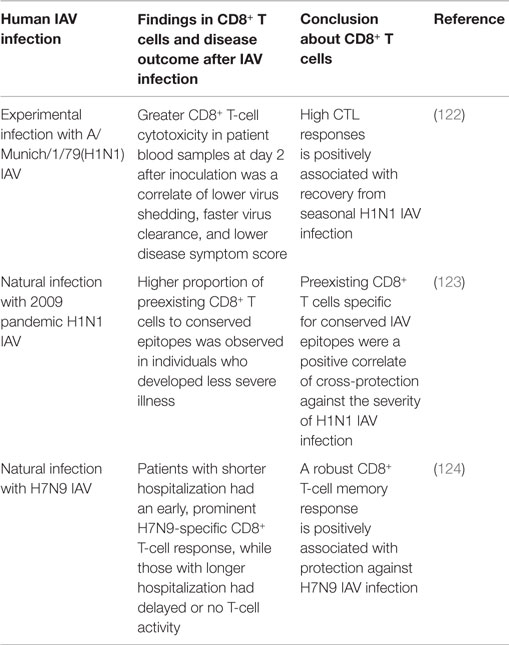
Table 6. Overview of human studies showing that cross-reactive CD8+ T-cell responses are a correlate of protective immunity against human IAV infection.
Concluding Remarks
Due to their naturally high mutation rate and their ability to generate genetic reassortants, IAVs pose a continuing threat to human populations. In order to develop better therapeutic and vaccination strategies to appropriately modify CTL immunity for protection against IAV infection, it is a great need to better understand the regulation and balance of the immune protection and pathology involved in virus-specific CTL immunity during IAV infection. We argue that the counterbalance of these effects depends on multiple layers of host and viral factors, including complex host mechanisms that regulate CTL quantity, quality, and effector activities; the pathogenic profile of the IAV, especially its ability to induce different innate host response milieus in which CTL immunity is primed and regulated; and the historical immune context of influenza infection (e.g., primary, secondary, and postvaccination). We propose that future research efforts should refine our understanding of key host and viral parameters, their points of interaction, and the effects of these interactions under different contexts of influenza occurrence (primary, secondary, postvaccination challenge, or heterologous infection), especially to differentiate those that constrain optimally effective CTL antiviral immunity from those truly necessary to restrain CTL-mediated non-specific immunopathology. Further translational research efforts can then focus on developing preventive or therapeutic means to modulate these parameters and interactions to obtain the best clinical outcomes.
Author Contributions
PT assisted in review conception, planning, and editing. SD conceived, planned, and wrote the review and designed the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by HHSN272201400006C, R01AI107625 from the National Institute of Allergy and Infectious Disease, US National Institutes of Health, and ALSAC. We thank Ashley Broussard for generating the illustration and Sharon Naron for editing the article.
References
1. Wendel I, Matrosovich M, Klenk HD. Snapshot: evolution of human influenza A viruses. Cell Host Microbe (2015) 17:416. doi:10.1016/j.chom.2015.02.001
2. Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature (2009) 459:931–9. doi:10.1038/nature08157
3. Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med (2013) 368:1888–97. doi:10.1056/NEJMoa1304459
4. Tripathi S, White MR, Hartshorn KL. The amazing innate immune response to influenza A virus infection. Innate Immun (2015) 21:73–98. doi:10.1177/1753425913508992
5. Chiu C, Openshaw PJ. Antiviral B cell and T cell immunity in the lungs. Nat Immunol (2015) 16:18–26. doi:10.1038/ni.3056
6. Lee BO, Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Makris M, Sprague F, et al. CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. J Immunol (2005) 175:5827–38. doi:10.4049/jimmunol.175.9.5827
7. Rangel-Moreno J, Carragher DM, Misra RS, Kusser K, Hartson L, Moquin A, et al. B cells promote resistance to heterosubtypic strains of influenza via multiple mechanisms. J Immunol (2008) 180:454–63. doi:10.4049/jimmunol.180.1.454
8. Carragher DM, Kaminski DA, Moquin A, Hartson L, Randall TD. A novel role for non-neutralizing antibodies against nucleoprotein in facilitating resistance to influenza virus. J Immunol (2008) 181:4168–76. doi:10.4049/jimmunol.181.6.4168
9. LaMere MW, Lam HT, Moquin A, Haynes L, Lund FE, Randall TD, et al. Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J Immunol (2011) 186:4331–9. doi:10.4049/jimmunol.1003057
10. Laidlaw BJ, Decman V, Ali MA, Abt MC, Wolf AI, Monticelli LA, et al. Cooperativity between CD8+ T cells, non-neutralizing antibodies, and alveolar macrophages is important for heterosubtypic influenza virus immunity. PLoS Pathog (2013) 9:e1003207. doi:10.1371/journal.ppat.1003207
11. Doherty PC, Turner SJ, Webby RG, Thomas PG. Influenza and the challenge for immunology. Nat Immunol (2006) 7:449–55. doi:10.1038/ni1343
12. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol (2014) 14:315–28. doi:10.1038/nri3665
13. Kim TH, Lee HK. Differential roles of lung dendritic cell subsets against respiratory virus infection. Immune Netw (2014) 14:128–37. doi:10.4110/in.2014.14.3.128
14. Hao X, Kim TS, Braciale TJ. Differential response of respiratory dendritic cell subsets to influenza virus infection. J Virol (2008) 82:4908–19. doi:10.1128/JVI.02367-07
15. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med (1999) 5:919–23. doi:10.1038/11360
16. Wolf AI, Buehler D, Hensley SE, Cavanagh LL, Wherry EJ, Kastner P, et al. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J Immunol (2009) 182:871–9. doi:10.4049/jimmunol.182.2.871
17. Boonnak K, Vogel L, Feldmann F, Feldmann H, Legge KL, Subbarao K. Lymphopenia associated with highly virulent H5N1 virus infection due to plasmacytoid dendritic cell-mediated apoptosis of T cells. J Immunol (2014) 192:5906–12. doi:10.4049/jimmunol.1302992
18. Langlois RA, Legge KL. Plasmacytoid dendritic cells enhance mortality during lethal influenza infections by eliminating virus-specific CD8 T cells. J Immunol (2010) 184:4440–6. doi:10.4049/jimmunol.0902984
19. GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J Exp Med (2008) 205:1621–34. doi:10.1084/jem.20071365
20. Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One (2009) 4:e4204. doi:10.1371/journal.pone.0004204
21. Heer AK, Harris NL, Kopf M, Marsland BJ. CD4+ and CD8+ T cells exhibit differential requirements for CCR7-mediated antigen transport during influenza infection. J Immunol (2008) 181:6984–94. doi:10.4049/jimmunol.181.10.6984
22. Legge KL, Braciale TJ. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity (2003) 18:265–77. doi:10.1016/S1074-7613(03)00023-2
23. Yoon H, Legge KL, Sung SS, Braciale TJ. Sequential activation of CD8+ T cells in the draining lymph nodes in response to pulmonary virus infection. J Immunol (2007) 179:391–9. doi:10.4049/jimmunol.179.1.391
24. Allan W, Tabi Z, Cleary A, Doherty PC. Cellular events in the lymph node and lung of mice with influenza. Consequences of depleting CD4+ T cells. J Immunol (1990) 144:3980–6.
25. Belz GT, Wodarz D, Diaz G, Nowak MA, Doherty PC. Compromised influenza virus-specific CD8(+)-T-cell memory in CD4(+)-T-cell-deficient mice. J Virol (2002) 76:12388–93. doi:10.1128/JVI.76.23.12388-12393.2002
26. McGill J, Van RN, Legge KL. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med (2008) 205:1635–46. doi:10.1084/jem.20080314
27. McGill J, Van RN, Legge KL. IL-15 trans-presentation by pulmonary dendritic cells promotes effector CD8 T cell survival during influenza virus infection. J Exp Med (2010) 207:521–34. doi:10.1084/jem.20091711
28. McGill J, Legge KL. Cutting edge: contribution of lung-resident T cell proliferation to the overall magnitude of the antigen-specific CD8 T cell response in the lungs following murine influenza virus infection. J Immunol (2009) 183:4177–81. doi:10.4049/jimmunol.0901109
29. Kohlmeier JE, Miller SC, Smith J, Lu B, Gerard C, Cookenham T, et al. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity (2008) 29:101–13. doi:10.1016/j.immuni.2008.05.011
30. Kohlmeier JE, Reiley WW, Perona-Wright G, Freeman ML, Yager EJ, Connor LM, et al. Inflammatory chemokine receptors regulate CD8(+) T cell contraction and memory generation following infection. J Exp Med (2011) 208:1621–34. doi:10.1084/jem.20102110
31. Jelley-Gibbs DM, Dibble JP, Brown DM, Strutt TM, McKinstry KK, Swain SL. Persistent depots of influenza antigen fail to induce a cytotoxic CD8 T cell response. J Immunol (2007) 178:7563–70. doi:10.4049/jimmunol.178.12.7563
32. Takamura S, Roberts AD, Jelley-Gibbs DM, Wittmer ST, Kohlmeier JE, Woodland DL. The route of priming influences the ability of respiratory virus-specific memory CD8+ T cells to be activated by residual antigen. J Exp Med (2010) 207:1153–60. doi:10.1084/jem.20090283
33. Kim TS, Hufford MM, Sun J, Fu YX, Braciale TJ. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J Exp Med (2010) 207:1161–72. doi:10.1084/jem.20092017
34. Zammit DJ, Turner DL, Klonowski KD, Lefrancois L, Cauley LS. Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity (2006) 24:439–49. doi:10.1016/j.immuni.2006.01.015
35. Kohlmeier JE, Miller SC, Woodland DL. Cutting edge: antigen is not required for the activation and maintenance of virus-specific memory CD8+ T cells in the lung airways. J Immunol (2007) 178:4721–5. doi:10.4049/jimmunol.178.8.4721
36. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12:749–61. doi:10.1038/nri3307
37. Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity (2013) 39:833–45. doi:10.1016/j.immuni.2013.10.007
38. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol (2013) 14:1155–65. doi:10.1038/ni.2710
39. Sun J, Dodd H, Moser EK, Sharma R, Braciale TJ. CD4+ T cell help and innate-derived IL-27 induce Blimp-1-dependent IL-10 production by antiviral CTLs. Nat Immunol (2011) 12:327–34. doi:10.1038/ni.1996
40. Cullen SP, Martin SJ. Mechanisms of granule-dependent killing. Cell Death Differ (2008) 15:251–62. doi:10.1038/sj.cdd.4402244
41. Topham DJ, Tripp RA, Doherty PC. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol (1997) 159:5197–200.
42. Ishikawa E, Nakazawa M, Yoshinari M, Minami M. Role of tumor necrosis factor-related apoptosis-inducing ligand in immune response to influenza virus infection in mice. J Virol (2005) 79:7658–63. doi:10.1128/JVI.79.12.7658-7663.2005
43. Brincks EL, Katewa A, Kucaba TA, Griffith TS, Legge KL. CD8 T cells utilize TRAIL to control influenza virus infection. J Immunol (2008) 181:4918–25. doi:10.4049/jimmunol.181.10.7428-a
44. Billiau A, Matthys P. Interferon-gamma: a historical perspective. Cytokine Growth Factor Rev (2009) 20:97–113. doi:10.1016/j.cytogfr.2009.02.004
45. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol (2015) 15:362–74. doi:10.1038/nri3834
46. Peperzak V, Xiao Y, Veraar EA, Borst J. CD27 sustains survival of CTLs in virus-infected nonlymphoid tissue in mice by inducing autocrine IL-2 production. J Clin Invest (2010) 120:168–78. doi:10.1172/JCI40178
47. Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol (2008) 8:247–58. doi:10.1038/nri2274
48. La Gruta NL, Turner SJ, Doherty PC. Hierarchies in cytokine expression profiles for acute and resolving influenza virus-specific CD8+ T cell responses: correlation of cytokine profile and TCR avidity. J Immunol (2004) 172:5553–60. doi:10.4049/jimmunol.172.9.5553
49. Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med (2009) 15:277–84. doi:10.1038/nm.1929
50. Cerwenka A, Morgan TM, Harmsen AG, Dutton RW. Migration kinetics and final destination of type 1 and type 2 CD8 effector cells predict protection against pulmonary virus infection. J Exp Med (1999) 189:423–34. doi:10.1084/jem.189.2.423
51. Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol (2001) 158:119–30. doi:10.1016/S0002-9440(10)63950-8
52. Hamada H, Garcia-Hernandez ML, Reome JB, Misra SK, Strutt TM, McKinstry KK, et al. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol (2009) 182:3469–81. doi:10.4049/jimmunol.0801814
53. Hamada H, Bassity E, Flies A, Strutt TM, Garcia-Hernandez ML, McKinstry KK, et al. Multiple redundant effector mechanisms of CD8+ T cells protect against influenza infection. J Immunol (2013) 190:296–306. doi:10.4049/jimmunol.1200571
54. Yap KL, Ada GL, McKenzie IF. Transfer of specific cytotoxic T lymphocytes protects mice inoculated with influenza virus. Nature (1978) 273:238–9. doi:10.1038/273238a0
55. Wells MA, Ennis FA, Albrecht P. Recovery from a viral respiratory infection. II. Passive transfer of immune spleen cells to mice with influenza pneumonia. J Immunol (1981) 126:1042–6.
56. Taylor PM, Askonas BA. Influenza nucleoprotein-specific cytotoxic T-cell clones are protective in vivo. Immunology (1986) 58:417–20.
57. Bender BS, Croghan T, Zhang L, Small PA Jr. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med (1992) 175:1143–5. doi:10.1084/jem.175.4.1143
58. Eichelberger M, Allan W, Zijlstra M, Jaenisch R, Doherty PC. Clearance of influenza virus respiratory infection in mice lacking class I major histocompatibility complex-restricted CD8+ T cells. J Exp Med (1991) 174:875–80. doi:10.1084/jem.174.4.875
59. Wells MA, Albrecht P, Ennis FA. Recovery from a viral respiratory infection. I. Influenza pneumonia in normal and T-deficient mice. J Immunol (1981) 126:1036–41.
60. Enelow RI, Mohammed AZ, Stoler MH, Liu AN, Young JS, Lou YH, et al. Structural and functional consequences of alveolar cell recognition by CD8(+) T lymphocytes in experimental lung disease. J Clin Invest (1998) 102:1653–61. doi:10.1172/JCI4174
61. Zhao MQ, Stoler MH, Liu AN, Wei B, Soguero C, Hahn YS, et al. Alveolar epithelial cell chemokine expression triggered by antigen-specific cytolytic CD8(+) T cell recognition. J Clin Invest (2000) 106:R49–58. doi:10.1172/JCI9786
62. Small BA, Dressel SA, Lawrence CW, Drake DR III, Stoler MH, Enelow RI, et al. CD8(+) T cell-mediated injury in vivo progresses in the absence of effector T cells. J Exp Med (2001) 194:1835–46. doi:10.1084/jem.194.12.1835
63. Liu AN, Mohammed AZ, Rice WR, Fiedeldey DT, Liebermann JS, Whitsett JA, et al. Perforin-independent CD8(+) T-cell-mediated cytotoxicity of alveolar epithelial cells is preferentially mediated by tumor necrosis factor-alpha: relative insensitivity to Fas ligand. Am J Respir Cell Mol Biol (1999) 20:849–58. doi:10.1165/ajrcmb.20.5.3585
64. Ramana CV, DeBerge MP, Kumar A, Alia CS, Durbin JE, Enelow RI. Inflammatory impact of IFN-gamma in CD8+ T cell-mediated lung injury is mediated by both Stat1-dependent and -independent pathways. Am J Physiol Lung Cell Mol Physiol (2015) 308:L650–7. doi:10.1152/ajplung.00360.2014
65. Xu L, Yoon H, Zhao MQ, Liu J, Ramana CV, Enelow RI. Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-alpha by antiviral CD8+ T cells. J Immunol (2004) 173:721–5. doi:10.4049/jimmunol.173.2.721
66. DeBerge MP, Ely KH, Enelow RI. Soluble, but not transmembrane, TNF-alpha is required during influenza infection to limit the magnitude of immune responses and the extent of immunopathology. J Immunol (2014) 192:5839–51. doi:10.4049/jimmunol.1302729
67. DeBerge MP, Ely KH, Cheng GS, Enelow RI. ADAM17-mediated processing of TNF-alpha expressed by antiviral effector CD8+ T cells is required for severe T-cell-mediated lung injury. PLoS One (2013) 8:e79340. doi:10.1371/journal.pone.0079340
68. Srikiatkhachorn A, Chintapalli J, Liu J, Jamaluddin M, Harrod KS, Whitsett JA, et al. Interference with intraepithelial TNF-alpha signaling inhibits CD8(+) T-cell-mediated lung injury in influenza infection. Viral Immunol (2010) 23:639–45. doi:10.1089/vim.2010.0076
69. Liu J, Zhao MQ, Xu L, Ramana CV, Declercq W, Vandenabeele P, et al. Requirement for tumor necrosis factor-receptor 2 in alveolar chemokine expression depends upon the form of the ligand. Am J Respir Cell Mol Biol (2005) 33:463–9. doi:10.1165/rcmb.2005-0204OC
70. Seah SG, Carrington EM, Ng WC, Belz GT, Brady JL, Sutherland RM, et al. Unlike CD4+ T-cell help, CD28 costimulation is necessary for effective primary CD8+ T-cell influenza-specific immunity. Eur J Immunol (2012) 42:1744–54. doi:10.1002/eji.201142211
71. Hashem AM, Gravel C, Chen Z, Yi Y, Tocchi M, Jaentschke B, et al. CD40 ligand preferentially modulates immune response and enhances protection against influenza virus. J Immunol (2014) 193:722–34. doi:10.4049/jimmunol.1300093
72. Plumb AW, Patton DT, Seo JH, Loveday EK, Jean F, Ziegler SF, et al. Interleukin-7, but not thymic stromal lymphopoietin, plays a key role in the T cell response to influenza A virus. PLoS One (2012) 7:e50199. doi:10.1371/journal.pone.0050199
73. Denton AE, Doherty PC, Turner SJ, La Gruta NL. IL-18, but not IL-12, is required for optimal cytokine production by influenza virus-specific CD8+ T cells. Eur J Immunol (2007) 37:368–75. doi:10.1002/eji.200636766
74. McNally B, Ye F, Willette M, Flano E. Local blockade of epithelial PDL-1 in the airways enhances T cell function and viral clearance during influenza virus infection. J Virol (2013) 87:12916–24. doi:10.1128/JVI.02423-13
75. Rutigliano JA, Sharma S, Morris MY, Oguin TH III, McClaren JL, Doherty PC, et al. Highly pathological influenza A virus infection is associated with augmented expression of PD-1 by functionally compromised virus-specific CD8+ T cells. J Virol (2014) 88:1636–51. doi:10.1128/JVI.02851-13
76. Sharma S, Sundararajan A, Suryawanshi A, Kumar N, Veiga-Parga T, Kuchroo VK, et al. T cell immunoglobulin and mucin protein-3 (Tim-3)/Galectin-9 interaction regulates influenza A virus-specific humoral and CD8 T-cell responses. Proc Natl Acad Sci U S A (2011) 108:19001–6. doi:10.1073/pnas.1107087108
77. Lin GH, Sedgmen BJ, Moraes TJ, Snell LM, Topham DJ, Watts TH. Endogenous 4-1BB ligand plays a critical role in protection from influenza-induced disease. J Immunol (2009) 182:934–47. doi:10.4049/jimmunol.182.2.934
78. Humphreys IR, Walzl G, Edwards L, Rae A, Hill S, Hussell T. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J Exp Med (2003) 198:1237–42. doi:10.1084/jem.20030351
79. Erickson JJ, Gilchuk P, Hastings AK, Tollefson SJ, Johnson M, Downing MB, et al. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J Clin Invest (2012) 122:2967–82. doi:10.1172/JCI62860
80. Zhou J, Matsuoka M, Cantor H, Homer R, Enelow RI. Cutting edge: engagement of NKG2A on CD8+ effector T cells limits immunopathology in influenza pneumonia. J Immunol (2008) 180:25–9. doi:10.4049/jimmunol.180.1.25
81. Ely KH, Matsuoka M, DeBerge MP, Ruby JA, Liu J, Schneider MJ, et al. Tissue-protective effects of NKG2A in immune-mediated clearance of virus infection. PLoS One (2014) 9:e108385. doi:10.1371/journal.pone.0108385
82. Brincks EL, Gurung P, Langlois RA, Hemann EA, Legge KL, Griffith TS. The magnitude of the T cell response to a clinically significant dose of influenza virus is regulated by TRAIL. J Immunol (2011) 187:4581–8. doi:10.4049/jimmunol.1002241
83. Bertram EM, Lau P, Watts TH. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol (2002) 168:3777–85. doi:10.4049/jimmunol.168.8.3777
84. Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med (2003) 198:1369–80. doi:10.1084/jem.20030916
85. Hendriks J, Xiao Y, Rossen JW, van der Sluijs KF, Sugamura K, Ishii N, et al. During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8+ memory T cells and their capacity for secondary expansion. J Immunol (2005) 175:1665–76. doi:10.4049/jimmunol.175.3.1665
86. Seah SG, Brady JL, Carrington EM, Ng WC, Sutherland RM, Hancock MS, et al. Influenza-induced, helper-independent CD8+ T cell responses use CD40 costimulation at the late phase of the primary response. J Leukoc Biol (2013) 93:145–54. doi:10.1189/jlb.0612266
87. Johnson S, Zhan Y, Sutherland RM, Mount AM, Bedoui S, Brady JL, et al. Selected toll-like receptor ligands and viruses promote helper-independent cytotoxic T cell priming by upregulating CD40L on dendritic cells. Immunity (2009) 30:218–27. doi:10.1016/j.immuni.2008.11.015
88. Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat Immunol (2013) 14:246–53. doi:10.1038/ni.2514
89. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol (2015) 15:231–42. doi:10.1038/nri3806
90. Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol (2009) 29:187–201. doi:10.1615/CritRevImmunol.v29.i3.10
91. Vigano S, Banga R, Bellanger F, Pellaton C, Farina A, Comte D, et al. CD160-associated CD8 T-cell functional impairment is independent of PD-1 expression. PLoS Pathog (2014) 10:e1004380. doi:10.1371/journal.ppat.1004380
92. Yuen KY, Chan PK, Peiris M, Tsang DN, Que TL, Shortridge KF, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet (1998) 351:467–71. doi:10.1016/S0140-6736(98)01182-9
93. Verbist KC, Cole CJ, Field MB, Klonowski KD. A role for IL-15 in the migration of effector CD8 T cells to the lung airways following influenza infection. J Immunol (2011) 186:174–82. doi:10.4049/jimmunol.1002613
94. Monteiro JM, Harvey C, Trinchieri G. Role of interleukin-12 in primary influenza virus infection. J Virol (1998) 72:4825–31.
95. Aldridge JR Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, et al. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci U S A (2009) 106:5306–11. doi:10.1073/pnas.0900655106
96. Lim K, Hyun YM, Lambert-Emo K, Capece T, Bae S, Miller R, et al. Neutrophil trails guide influenza-specific CD8(+) T cells in the airways. Science (2015) 349:aaa4352. doi:10.1126/science.aaa4352
97. Hufford MM, Richardson G, Zhou H, Manicassamy B, Garcia-Sastre A, Enelow RI, et al. Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PLoS One (2012) 7:e46581. doi:10.1371/journal.pone.0046581
98. Juno JA, Keynan Y, Fowke KR. Invariant NKT cells: regulation and function during viral infection. PLoS Pathog (2012) 8:e1002838. doi:10.1371/journal.ppat.1002838
99. Paget C, Ivanov S, Fontaine J, Blanc F, Pichavant M, Renneson J, et al. Potential role of invariant NKT cells in the control of pulmonary inflammation and CD8+ T cell response during acute influenza A virus H3N2 pneumonia. J Immunol (2011) 186:5590–602. doi:10.4049/jimmunol.1002348
100. Chappert P, Leboeuf M, Rameau P, Lalfer M, Desbois S, Liblau RS, et al. Antigen-specific Treg impair CD8(+) T-cell priming by blocking early T-cell expansion. Eur J Immunol (2010) 40:339–50. doi:10.1002/eji.200839107
101. Hufford MM, Kim TS, Sun J, Braciale TJ. Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J Exp Med (2011) 208:167–80. doi:10.1084/jem.20101850
102. Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol (2004) 5:524–30. doi:10.1038/ni0604-658a
103. Faroudi M, Utzny C, Salio M, Cerundolo V, Guiraud M, Muller S, et al. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci U S A (2003) 100:14145–50. doi:10.1073/pnas.2334336100
104. Valitutti S, Muller S, Dessing M, Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med (1996) 183:1917–21. doi:10.1084/jem.183.4.1917
105. CDC. Seasonal Influenza Q&A (2014). Available from: http://www.cdc.gov/flu/about/qa/disease.htm
106. Grant EJ, Chen L, Quinones-Parra S, Pang K, Kedzierska K, Chen W. T-cell immunity to influenza A viruses. Crit Rev Immunol (2014) 34:15–39. doi:10.1615/CritRevImmunol.2013010019
107. Grebe KM, Yewdell JW, Bennink JR. Heterosubtypic immunity to influenza A virus: where do we stand? Microbes Infect (2008) 10:1024–9. doi:10.1016/j.micinf.2008.07.002
108. Hillaire ML, Osterhaus AD, Rimmelzwaan GF. Induction of virus-specific cytotoxic T lymphocytes as a basis for the development of broadly protective influenza vaccines. J Biomed Biotechnol (2011) 2011:939860. doi:10.1155/2011/939860
109. Nguyen HH, Moldoveanu Z, Novak MJ, van Ginkel FW, Ban E, Kiyono H, et al. Heterosubtypic immunity to lethal influenza A virus infection is associated with virus-specific CD8(+) cytotoxic T lymphocyte responses induced in mucosa-associated tissues. Virology (1999) 254:50–60. doi:10.1006/viro.1998.9521
110. O’Neill E, Krauss SL, Riberdy JM, Webster RG, Woodland DL. Heterologous protection against lethal A/HongKong/156/97 (H5N1) influenza virus infection in C57BL/6 mice. J Gen Virol (2000) 81:2689–96. doi:10.1099/0022-1317-81-11-2689
111. Kreijtz JH, Bodewes R, van AG, Kuiken T, Fouchier RA, Osterhaus AD, et al. Primary influenza A virus infection induces cross-protective immunity against a lethal infection with a heterosubtypic virus strain in mice. Vaccine (2007) 25:612–20. doi:10.1016/j.vaccine.2006.08.036
112. Kreijtz JH, Bodewes R, van den Brand JM, de MG, Baas C, van AG, et al. Infection of mice with a human influenza A/H3N2 virus induces protective immunity against lethal infection with influenza A/H5N1 virus. Vaccine (2009) 27:4983–9. doi:10.1016/j.vaccine.2009.05.079
113. Weinfurter JT, Brunner K, Capuano SV III, Li C, Broman KW, Kawaoka Y, et al. Cross-reactive T cells are involved in rapid clearance of 2009 pandemic H1N1 influenza virus in nonhuman primates. PLoS Pathog (2011) 7:e1002381. doi:10.1371/journal.ppat.1002381
114. Duan S, Meliopoulos VA, McClaren JL, Guo XZ, Sanders CJ, Smallwood HS, et al. Diverse heterologous primary infections radically alter immunodominance hierarchies and clinical outcomes following H7N9 influenza challenge in mice. PLoS Pathog (2015) 11:e1004642. doi:10.1371/journal.ppat.1004642
115. McMaster SR, Gabbard JD, Koutsonanos DG, Compans RW, Tripp RA, Tompkins SM, et al. Memory T cells generated by prior exposure to influenza cross react with the novel H7N9 influenza virus and confer protective heterosubtypic immunity. PLoS One (2015) 10:e0115725. doi:10.1371/journal.pone.0115725
116. Hillaire ML, Vogelzang-van Trierum SE, Kreijtz JH, de MG, Fouchier RA, Osterhaus AD, et al. Human T-cells directed to seasonal influenza A virus cross-react with 2009 pandemic influenza A (H1N1) and swine-origin triple-reassortant H3N2 influenza viruses. J Gen Virol (2013) 94:583–92. doi:10.1099/vir.0.048652-0
117. Tu W, Mao H, Zheng J, Liu Y, Chiu SS, Qin G, et al. Cytotoxic T lymphocytes established by seasonal human influenza cross-react against 2009 pandemic H1N1 influenza virus. J Virol (2010) 84:6527–35. doi:10.1128/JVI.00519-10
118. Gras S, Kedzierski L, Valkenburg SA, Laurie K, Liu YC, Denholm JT, et al. Cross-reactive CD8+ T-cell immunity between the pandemic H1N1-2009 and H1N1-1918 influenza A viruses. Proc Natl Acad Sci U S A (2010) 107:12599–604. doi:10.1073/pnas.1007270107
119. Kreijtz JH, de MG, van Baalen CA, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. Cross-recognition of avian H5N1 influenza virus by human cytotoxic T-lymphocyte populations directed to human influenza A virus. J Virol (2008) 82:5161–6. doi:10.1128/JVI.02694-07
120. van de Sandt CE, Kreijtz JH, de MG, Geelhoed-Mieras MM, Hillaire ML, Vogelzang-van Trierum SE, et al. Human cytotoxic T lymphocytes directed to seasonal influenza A viruses cross-react with the newly emerging H7N9 virus. J Virol (2014) 88:1684–93. doi:10.1128/JVI.02843-13
121. Quinones-Parra S, Grant E, Loh L, Nguyen TH, Campbell KA, Tong SY, et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc Natl Acad Sci U S A (2014) 111:1049–54. doi:10.1073/pnas.1322229111
122. McMichael AJ, Gotch FM, Noble GR, Beare PA. Cytotoxic T-cell immunity to influenza. N Engl J Med (1983) 309:13–7. doi:10.1056/NEJM198307073090103
123. Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med (2013) 19:1305–12. doi:10.1038/nm.3350
Keywords: CD8+ T cells, influenza, human, immune regulation, immunopathology, vaccination
Citation: Duan S and Thomas PG (2016) Balancing Immune Protection and Immune Pathology by CD8+ T-Cell Responses to Influenza Infection. Front. Immunol. 7:25. doi: 10.3389/fimmu.2016.00025
Received: 09 December 2015; Accepted: 18 January 2016;
Published: 05 February 2016
Edited by:
Michael Croft, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Andrea Sant, David H. Smith Center for Vaccine Biology and Immunology, USAShahram Salek-Ardakani, University of Florida, USA
Tara Marlene Strutt, University of Central Florida, USA
Copyright: © 2016 Duan and Thomas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul G. Thomas, cGF1bC50aG9tYXNAc3RqdWRlLm9yZw==