 Nicholas van Panhuys
Nicholas van Panhuys
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol. , 25 January 2016
Sec. T Cell Biology
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00006
This article is part of the Research Topic Setting the path – How qualitative and quantitative differences in TCR-pMHC interactions regulate T cell immune responses View all 12 articles
The ability of CD4+ T cells to differentiate into effector subsets underpins their ability to shape the immune response and mediate host protection. During T cell receptor-induced activation of CD4+ T cells, both the quality and quantity of specific activatory peptide/MHC ligands have been shown to control the polarization of naive CD4+ T cells in addition to co-stimulatory and cytokine-based signals. Recently, advances in two-photon microscopy and tetramer-based cell tracking methods have allowed investigators to greatly extend the study of the role of TCR signaling in effector differentiation under in vivo conditions. In this review, we consider data from recent in vivo studies analyzing the role of TCR signal strength in controlling the outcome of CD4+ T cell differentiation and discuss the role of TCR in controlling the critical nature of CD4+ T cell interactions with dendritic cells during activation. We further propose a model whereby TCR signal strength controls the temporal aspects of T–DC interactions and the implications for this in mediating the downstream signaling events, which influence the transcriptional and epigenetic regulation of effector differentiation.
CD4+ T cells constitute a key population in the adaptive arm of the immune response, due to their ability to differentiate into various distinct effector populations such as Th1, Th2, Th17, T follicular helper (Tfh) cells, and induced regulatory (iTreg) T cells, which help to orchestrate host defense against various classes of pathogens (1, 2). The differentiation of naive CD4+ T cells into polarized effectors has been studied extensively, and two alternative mechanisms of cell fate control have been described. The predominant view espoused by the field is that following the activation of a CD4+ T cell by an antigen-presenting cell (APC) displaying cognate peptide in the context of MHCII CD4+ T cells differentiate into highly specialized subsets according to the cytokine milieu present during activation and division (1, 3, 4). However, in addition to this qualitative/cytokine-based model of differentiation, the literature also contains clear evidence for a quantitative mechanism that controls cell fate decisions. Quantitative models indicate that upon activation, CD4+ T cells are able to determine the strength of TCR signaling in terms of both the antigen load and quality of peptide presented, and in combination with co-stimulatory molecules, CD4+ T cells perform a cellular calculus to determine activatory signal strength. T-helper cells then use this qualitative signaling information to determine which program of differentiation to engage in (5).
This review aims to discuss the evidence that supports the strength of signal-based mechanisms for inducing differentiation in parallel with qualitative mechanisms and argues that TCR signaling dynamics include a temporal component that regulates the interactions between APC and CD4+ T cells, which explains much of the observed qualitative and quantitative phenomena previously described.
Cytokine-based models indicate that CD4+ T cell differentiation is controlled by a qualitative signaling mechanism in which specific combinations of cytokines produced mainly by myeloid APCs (and also by non-hematopoietic tissue elements) induce a specific pathway of differentiation following antigen-dependent activation of CD4+ T cells. Following the activation of APC by pathogen-associated molecular patterns (PAMPs), APCs begin to upregulate the levels of both MHCII and co-activation molecules allowing for the activation of CD4+ T cells in an antigen-specific manner. PAMPs additionally induce the production of specific cytokines by activated APCs, which have been shown to induce the differentiation of specialized T-helper populations (6). Cytokines have been shown to directly induce the differentiation of T-helper subsets through binding to specific receptors on the surface of CD4+ T cells, the activation of specific patterns of JAKs and STATs, which then translocate to the nucleus, and the upregulation of master transcription factors that are essential for the determination and maintenance of cell lineage and controlling downstream effector functions (1, 7). Th1 cells are induced in response to infection with bacterial or viral pathogens and are also a hallmark of autoimmune disease. Th1 differentiation can be induced following exposure to IFNγ (8) or IL-12 (9) during activation and is characterized by the expression of the master regulator of transcription Tbet (10) and production of the effector cytokine IFNγ. Th2 differentiation occurs following infection with helminthic parasites and is also responsible for much of the pathology associated with asthmatic and allergic responses. Th2 cells are characterized by the upregulation of GATA3 (11) following differentiation and the ability to produce IL-4, -5, and/or -13 upon re-stimulation. While Th2 differentiation has classically been believed to be driven by IL-4 (12), several studies have indicated that IL-4 is not required for Th2 differentiation under in vivo conditions (13, 14). Additionally, cytokines such as IL-25 and IL-33 and TSLP have also been implicated to be involved in Th2 differentiation; however, these factors were not found to be required for the induction of differentiation (15). Thus, it has proved difficult to determine a specific qualitative cytokine signal responsible for Th2 differentiation under in vivo conditions, and it has been suggested that Th2 induction may occur through a default or endogenous pathway (16). Th17 cells are believed to play a role in defending against fungal pathogens and have also been found to play a significant role in several autoimmune diseases. The Th17 lineage is characterized by the expression of the transcription factor RORγt and by the expression of the cytokines IL-17, -21, and/or -22 (17). Tfh cells perform a specialized function in that after differentiation, they localize to the B cell follicle and are critical for providing B cell help, by inducing B cell affinity maturation, differentiation, and promoting the class switching of B cell immunoglobulin isotype expression. They are generally characterized by IL-21 expression along with the transcriptional repressor Bcl-6 and the surface receptors CXCR5 and PD-1 (2). iTreg cells are functionally distinct from other groups of T-helper cells as they play a key role in the immune response by dampening down excessive CD4+ T cell activation and controlling the extent of inflammation. While iTreg cells are characterized by the expression of the transcription factor Foxp3 and production of regulatory cytokines such as TGFβ and Il-10, these factors are also present in naturally occurring or thymically derived Tregs (nTreg), making it problematic to distinguish these two subsets under in vivo conditions (18).
These studies form a large body of compelling evidence indicating that the cytokine milieu represents a highly deterministic cellular cue during the polarization process. However, it should be noted that cell fate decisions occur due to a number of factors, and as the majority of qualitative/cytokine-based studies have been conducted at single antigen concentrations in vitro or in vivo, the quantitative/TCR signal strength-based component is generally not considered giving the appearance that cytokines dominate. However, in practice, this conclusion requires a more explicit analysis to assess the relationship and possible hierarchical and temporal dominance of qualitative vs. quantitative aspects of control.
The most widely characterized example of TCR signal strength directly contributing to the alternate differentiation of CD4+ T cells has been studied looking at the comparative differences in Th1 vs. Th2 induction. It has long been established that under in vitro conditions, a strong TCR signal leads to a predominance of Th1 differentiation, whereas weak TCR signaling leads to Th2 differentiation (19, 20). Early studies using an altered peptide ligand (APL) model system compared stimulation of TCR transgenic (Tg) CD4+ T cells with either the native Moth Cytochrome C (MCC) peptide or the K99 peptide, which has a significantly weaker affinity for the Tg TCR. When APCs were loaded with an equivalent quantity of antigen, those stimulated with the higher affinity MCC peptide displayed a striking tendency to differentiate into Th1 cells, whereas those stimulated with the weaker K99 peptide differentiated to the Th2 phenotype (19, 21–23). The capacity of APCs to induce TCR signaling can be modulated by both the affinity of the peptide:MHCII (pMHCII) complex for a specific TCR and the quantity of pMHCII present on the surface of the APC. Parallel studies examining the effects of peptide load on the differentiation CD4+ T cells revealed that stimulation with APCs loaded with a high dose of peptide led to the induction of Th1 cells, whereas stimulation with a low dose of peptide resulted in the differentiation of Th2 cells (20, 24). Together, these results show that altering the initial TCR stimulus received by naive CD4+ T cells, by either pMHC affinity or density, leads to a divergent outcome in terms of the end point of differentiation, with strong TCR signaling leading to Th1 differentiation and weak TCR signaling leading to Th2 differentiation (Figure 1).
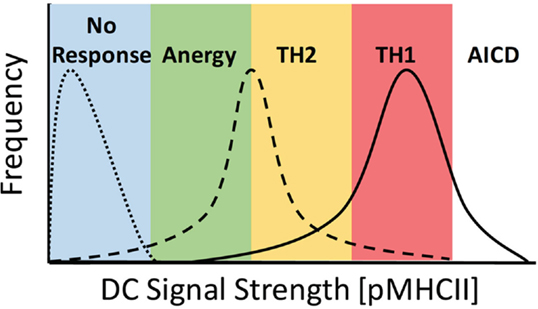
Figure 1. Ability of DCs to stimulate TCR signaling controls the activation potential of CD4+ T cells. DC priming of CD4+ T cells is dependent on the overall signal strength imparted by specific DC, where TCR signal strength = [peptide] × Ka of pMHCII. DC signaling capacity must be fitted to a distribution profile at the population level due to multiple factors, including expression levels of MHCII and co-stimulatory molecules, peptide loading efficiency, DC morphology, and availability of sites for interaction in a complex 3D environment. Here, DC lacking cognate peptide fail to stimulate a response from T cells (dotted line), whereas at low levels of signal strength, DC induce a complex response inducing anergy and Th2 and Th1 differentiation, with a predominant skewing toward TH1 differentiation (dashed line). When strong signals are present, the response is skewed toward TH1 differentiation with both Th2 differentiation and activation-induced cell death (AICD) also occurring (solid line).
Recent studies utilizing two-photon intravital microscopy and tetramer-based cell tracking methods have allowed investigators to greatly extend the study of the role of TCR signaling in effector differentiation under in vivo conditions. While the earlier studies discussed above imply that modulation of antigenic quality or quantity leads to similar outcomes at comparable levels of TCR stimulation, studies into the in vivo requirement for Treg generation by Gottschalk et al. (25, 26) indicate that TCR is able to differentiate between the two and that alteration of either of these components can affect the outcome of differentiation and/or survival, thus modulating the outcome of the immune response. Here, the induction of iTreg cell differentiation was assessed under in vivo conditions following the i.v. injection of MCC peptide or an APL to induce the activation of 5C.C7 TCR Tg CD4+ T cells. By adjusting the dose of MCC or APL peptide injected to normalize for peptide potency, comparable levels of proliferation and Foxp3 expressing iTreg cells were induced. However, those iTreg cells induced following activation with low-affinity ligands were unable to persist in vivo, illustrating the ability of TCR to distinguish between the quality and quantity of cognate peptide being presented under in vivo conditions (25). It was additionally determined that while large quantities of a weak agonist peptide and small quantities of a strong agonist peptide can provide a compensatory outcome in terms of in vivo proliferation, the stimulation of CD4+ T cells under these conditions led to divergent outcomes in terms of the gene expression profile-induced downstream. Further, intravital microscopy revealed that more stable contacts between CD4+ T cells and APC were observed in the presence of a strong agonist peptide in comparison to a weak agonist peptide, irrespective of the dose used (26), indicating antigen quality-dependent differences in T–DC interaction times may contribute to the observed divergence in gene expression profiles.
The differentiation of CD4+ T cells is influenced by TCR-mediated, co-stimulatory molecule and cytokine-induced signaling, all of which are modulated following APC activation by PAMPS or adjuvant-associated molecular patterns. The relationship between these factors in determining the outcome of differentiation is highly relevant. We recently addressed this issue by conducting a study utilizing the intravital imaging of CD4+ T cells interactions with DCs following their exposure to Th1- or Th2-polarizing adjuvants (27). Using Ca2+ flux imaging, these studies revealed a close correlation between the strength of TCR-induced signaling and the length of interaction, with Th1 differentiation being induced following increased TCR-associated signaling and longer lengths of interaction time. Under these conditions, antigen concentration was found to dominate over adjuvant in controlling differentiation. At a fixed antigen concentration, adjuvants inducing Th1 differentiation operated by increasing the co-stimulatory molecule expression on stimulatory DCs, which has previously been shown to potentiate TCR-associated signaling (28) and induce tighter T–DC interactions (29). However, by titrating the level of peptide used, we found that the concentration of antigen dominates the outcome of the response, such that, at high doses of antigen, CD4+ T cells primed with DC activated with Th2-polarizing adjuvants developed into Th1 cells and at low doses of antigen, Th2 differentiation predominated even after priming with DC activated in the presence of Th1-polarizing adjuvants. These responses showed a direct connection between alterations in Ca2+ signaling and interaction times, indicating that adjustments to TCR signal strength that affect interaction duration and signal accumulation can reverse the fate outcome, irrespective of the adjuvant used to treat the DC. These data imply that qualitative control of differentiation by cytokines reflects a secondary step in a developmental system in which the strength of initial T cell signaling regulates the capacity of the cells to respond to these mediators.
The majority of studies into the effects of TCR signal strength on CD4+ differentiation have relied upon the use of TCR Tg model systems; however, in order to more closely model responses occurring under physiological conditions, the application of tetramer-based technology has proved highly illuminating. During a polyclonal immune response, CD4+ T cells of various affinities for a given pMHCII are present in the immune repertoire; thus, it could be hypothesized from previous findings that those polyclonal cells with a high affinity for a specific pMHCII would preferentially differentiate into Th1 cells, whereas those with a lower affinity would differentiate into Th2 cells. To assess this possibility, Milner et al. (30) employed a tetramer-based staining system to deplete high-affinity CD4+ T cells and found that in the absence of such cells, stimulation of a polyclonal population with a specific pMHCII combination led to the preferential induction of a Th2 population, which was abrogated in the presence of higher affinity cells. These findings have been significantly extended by the use of an in vivo limiting-dilution system, which assessed the role of clonal differences in TCR signaling in response to a specific Listeria monocytogenes (Lm) peptide (31). Here, it was determined that single polyclonal cells responding to the Lm peptide displayed widely divergent abilities to differentiate into Th1, Tfh, and germinal center (GC) Tfh cells. However, when averaged together, these populations gave a characteristic pattern of differentiation, indicating that the diverse behaviors of individual naive CD4+ T cells with differing affinities for a specific peptide are able to account for the consistent pattern of response, which is found in a responding population of CD4+ T cells under in vivo conditions (31). Additionally, this study also indicated that the differentiation of Th1, Tfh, and GC-Tfh cells during Lm infection was dose dependent, with low-dose Lm infections leading to the predominance of Th1 and Tfh cells and high-dose infections leading to a corresponding decrease in the proportion of Th1 cells and an increase in the proportion of Tfh cells, especially GC-Tfh cells. An in vivo comparison of CD4+ T cells responding to whole PCC protein immunization further showed that precursor T cells with a higher affinity for pMHCII preferentially differentiated into Tfh cells and that LN-resident Tfh cells displayed a restricted TCR repertoire and displayed stronger pMHCII binding, (32) reinforcing the idea that Tfh cells are inducible in response to strong TCR signaling. Mechanistically, it has been shown that the generation of Tfh cells requires strong/sustained TCR signals through the analysis of the requirement for ITAM multiplicity in TCR signaling during Tfh cell generation, where it was determined that the presence of mutant ITAMs lacking their p-Tyr residues decreased proximal signaling downstream from the TCR and led to a concomitant decrease in Tfh cell generation (33). In vivo studies using 2P imaging to study the dynamic components of both B–T (34) and DC–T (35) interactions during Tfh cell responses have demonstrated that Tfh cells most frequently interact with APCs presenting strong pMHCII signaling complements and that prolonged Ag presentation was required (≥72 h) in order to most efficiently induce Tfh cell differentiation (36, 37). Together, these data indicate that very strong TCR stimuli are associated with the continued interaction of DC and CD4+ T cells for extended periods of time and that these ongoing interactions are required for Tfh cell differentiation.
Th17 differentiation has also been observed to occur in a dose-dependent manner; initial studies indicated that in vitro Th17 differentiation occurs in response to stimulation with a strong pMHCII stimulus, in conjunction with the presence of exogenous TGFβ and IL-6. Here, it was found that high doses of antigen were able to upregulate CD40L, and in combination with specific adjuvants, this increased DC IL-6 production leading to the differentiation of Th17 cells under in vitro and in vivo conditions (38). Interestingly, in the presence of IL-6 and TGFβ, the ability of naive CD4+ T cells to differentiate into Th1 or Th2 cells was abrogated, but Th17 differentiation was found to correlate with the strength of antigenic stimuli. CD4+ T cells deficient for Itk, a tyrosine kinase activated as part of the proximal response to TCR activation, have additionally been shown to have a decreased TCR-induced Ca2+ flux response (39) to antigen and have a deficiency in Th17 differentiation, even in the presence of IL-6 and TGFβ (40). Conversely, Itk-deficient cells exhibited increased Treg cell differentiation under Th17 conditions, where cells were found to be more sensitive to IL-2 signaling while exhibiting an activation profile similar to that observed in cells activated with a weak TCR stimulus (41). This observation indicates that Treg and Th17 differentiation may result from stimulation under a similar cytokine milieu, but their alternate induction is driven by TCR stimulation at opposite ends of the activation spectrum. Together, this literature clearly indicates that the predominant thesis describing CD4+ T cell differentiation by qualitative cytokine-based induction needs to be updated to better integrate the wealth of studies demonstrating a significant role for quantitative mechanisms in differentiation.
Naive CD4+ cells display an array of cytokine receptor molecules on their surfaces, such as IL-4R, IL-6R, IL-17R, IFNγR, and TGFβR, which are important for the induction of multiple differentiation programs. However, upon TCR activation, the expression of additional cytokine receptors such as the high-affinity IL-2R (CD25) (42) and the IL-12Rβ2 (43) is induced, and levels of existing cytokine receptors may be modulated according to signal strength (44). Interestingly, cytokines, such as IL-12 (45) and IFNγ (46, 47), associated with Th1 differentiation, have been shown to require the formation of mature immune synapses (IS) for their efficient delivery, with cytokine receptors localizing to the IS following formation of a mature IS, whereas IL-4 associated with Th1 differentiation (12), but not necessary for Th2 differentiation (14), is thought to act in a paracrine fashion, as does IL-2. Accordingly, these cytokines are not secreted in a directional fashion and their receptors do not display polarization even upon formation of a mature IS (47, 48).
However, responsiveness to IL-2 signaling has been shown to be regulated by TCR signal strength; activation by both weak and strong stimuli leads to an increased expression of IL-2R, but strong stimuli also cause an attenuated ability to signal via IL-2R through inhibition of phosphorylation of the transcription factor STAT5 (24), induced by the dose-dependent repression of Pten (41). The dose-dependent control of IL-2 signaling may therefore be a major factor controlling the Treg–Th17 differentiation axis. As where Th1 and Th2 differentiation is inhibited by TGFβ, Treg and Th17 differentiation predominates (38, 41, 49). However, while IL-2 is essential for Treg differentiation (18), IL-2 signaling inhibits Th17 differentiation (50); therefore, the negative feedback on STAT5 phosphorylation induced by strong TCR signaling allows for Th17 differentiation in the presence of IL-2, similar to that produced by naive T cells at early time points after activation. Further, Th17 differentiation has been shown to be dependent on the production of IL-6 by DC; thus, the potential exists that Th17 induction may require high levels of TCR stimuli in order to form a mature IS to allow for the directional secretion of IL-6. Though the directionality of IL-6 secretion at the T–DC interface has not been fully investigated, IL-6 is thought to be synaptically secreted in the central nervous system (51). The repression of IL-2R signaling may also play a significant role in the differentiation of the Tfh cell lineage. Upregulation of Bcl-6 is required for Tfh cell commitment, as opposed to the expression of Blimp-1, which is expressed by T effector lineages (52). Blimp-1 is upregulated in naive T cells upon activation and exposure to IL-2 (53) and causes the inhibition of Bcl-6 expression repressing Tfh cell differentiation in a STAT5-dependent manner (54). Additionally, the anti-apoptotic qualities of Bcl-6 may serve a protective function during Tfh cell differentiation (55) by allowing their stimulation by the very strong TCR signals over a long period of time that are required for Tfh cell differentiation, while avoiding activation-induced cell death (AICD).
In terms of Th1 differentiation, we recently showed that under both in vitro and in vivo conditions, TCR signal strength controlled downstream IL-12 receptor expression, with IL-12Rβ2 directly correlating with signal strength. Linking TCR signaling to the ability of CD4+ T cells to respond to cytokine signaling and establishing TCR-induced signaling play a dominant role in the signaling hierarchy (27). Together, these data indicate that CD4+ T cells display an ability to quantitate and integrate signals from the TCR and co-stimulatory molecules, which then regulates both the length of interaction and downstream checkpoints, such as the upregulation of cytokine receptors and their polarization to the IS, which is required for the cytokine-mediated control of effector differentiation. As the strength of signal received through TCR can alter the interaction time between T cells and DC, this indicates that it is an integral component in determining the outcome of differentiation.
T cells display a default scanning behavior under in vivo conditions possessing what could be thought of as an inbuilt “migrational momentum,” such that they will resist activatory “Stop” signals from an APC, unless a significant influence is exerted upon them. Multiple studies (26, 27, 34, 56) have shown a high degree of correlation between the length of time T cells interact with APCs and the outcome of differentiation, suggesting the need for a model of differentiation that takes into account the dynamic nature of T cell–DC interactions, the role of TCR in facilitating the nature of these interactions, and the pattern of resultant downstream signaling that regulates the outcome of differentiation.
T cells display a natural propensity for migration, with naive T cells estimated to recirculate though the secondary lymphoid organs in as little as a few hours as they search for cognate antigen (57, 58). Initial imaging studies (59, 60) assessing the behavior of naive CD4+ T cells in lymph node explant models characterized a short-lived contact duration between naive T cells and APCs of approximately 3 min. These Ag nonspecific interactions did not lead to T cell activation and were followed by a rapid dissociation and migration away from the APC, allowing the naive T cell to continue its scanning behavior. It was further noted that apart from brief pauses, naive T cells exist in a state of constant kinesis (61). The apparent random nature of in vivo migratory behavior observed in early studies led to the adoption of a random walk model, with T cells migrating as autonomous agents taking independent and stochastic paths through the secondary lymphoid tissue in search of APCs with cognate Ag (60, 61). However, more detailed studies examining the structure of secondary lymphoid organs characterized a complex matrix-like environment consisting of a network of interconnected follicular reticular cells along which T cells and APCs traffic (62). A particularly dense network of FRCs is associated near the HEVs in the paracortex, which enables the localization of tissue-derived DCs for the efficient presentation of Ag to immigrating T cells, and upon Ag administration, this site corresponds to areas of T cell clustering and early activation (63, 64). The expression of CCR7 by naive T cells and its requirement for migration into the lymphoid compartment is well documented (64, 65). However, the role it plays in guiding CD4+ T cells once inside the SLOs is less clear. FRCs in both humans and mice produce the CCR7 ligands CCL19 and CCL21 (66, 67) and likely guide the migration of naive CD4+ cells under steady-state conditions (67, 68). Upon inflammation, the potential role of chemokine gradients is yet to be fully determined, though activated dendritic cells have been observed to secrete CCL19 (69) and CCL21, and the expression can be downregulated by FRCs during an immune response (70). Ligation of CCR7 by CCL19 leads to receptor phosphorylation and internalization and a subsequent desensitization, potentially providing a mechanism for local CD4+ guidance, whereas CCL21 binding does not exhibit a reciprocal effect drawing naive CD4+ T cells toward activated DCs. The blockade (68) or disruption (71) of CCR7 ligand signaling has been shown to reduce the velocity of CD4+ migration, but failed to completely abrogate migration, indicating either the presence of additional as yet unidentified ligands or that CD4+ T cells exhibit a constitutively migrant behavior under in vivo conditions.
Upon interaction with APCs, it is vital for T cells to strictly discriminate between self and non-self ligands to avoid autoimmune disease, while at the same time maintain tolerance to nonpathogenic environmental antigens to avoid unnecessary pathology. In vitro studies have demonstrated that CD4+ T cells can be activated by less than ten cognate peptides presented by an APC in the presence of MHCII (72, 73); thus, T cell responses exhibit a very high degree of sensitivity. In order to scan the maximal number of APCs, non-productive interactions must be quickly terminated, such that in the absence of response to an agonist peptide, CD4+ T cell:APC interactions have been estimated to last between 1 and 5 min (59, 61, 74) and up to 5,000 T cells may contact and scan a single DC during an hour (74). The transition from TCR binding to signaling is highly specific, resulting in the ability to integrate TCR:pMHC interactions, which occur over a minimal range of Kd into a definitive digital on/off recognition signal (75). Thus, on the one hand, T cells must be able to rapidly and efficiently recognize their cognate Ag and, on the other, must display a rigorous intolerance to activation via the myriad of self-peptide and environmental antigens displayed by APCs.
In order to account for many of the observed details of TCR signaling and CD4+ activation, a kinetic thresholding with differential signaling feedback model of ligand discrimination has been suggested, (76) in particular, this mechanism of TCR signaling accounts for the speed, sensitivity, and specificity of signal discrimination. Kinetic thresholding models dictate that following pMHC:TCR interaction, proofreading kinases associated with the CD3 components of TCR phosphorylate the multiple tyrosine residues contained in the CD3 ITAM regions, allowing ZAP-70 association and progression of downstream signaling. Upon pMHC:TCR dissociation, the ITAMs are then dephosphorylated by constitutively active phosphatases, thereby attenuating the progression of signaling from the TCR. Thus, the duration of pMHC:TCR interaction sets the level of ITAM phosphorylation, which controls the lifetime of downstream signaling with the utilization of multiple proofreading kinases, allowing for the amplification of signaling in a nonlinear fashion (77–79). However, the initial kinetic proofreading models do not sufficiently account for all the characteristics of CD4+ activation; thus, in order to extend these models, an additional differential feedback step as identified by Stefanova et al. (80) was included. Here, Erk and SHP-1 were identified as key components of signal discrimination during the kinetic proofreading process. Following TCR engagement by both antagonistic and agonistic ligands, Lck is activated, resulting in its association and phosphorylation of both ZAP-70 and ITAMs, and Lck additionally phosphorylates the regulatory phosphatase SHP-1. In the case of TCR association by antagonistic or weak ligands, phosphorylation of SHP-1 allows binding to and inactivation of Lck and further enables a close association of SHP-1 with ZAP-70 and ITAMs, potentially resulting in the dephosphorylation of unprotected residues and cessation of signaling. However, following association with a strong agonist pMHC, the MAP kinase ERK is rapidly activated and phosphorylates Lck at Ser59, inhibiting the binding of SHP-1 and the subsequent inhibition of Lck kinase activity. Thus, ERK activation by strong agonists acts at a local level to protect Lck activity at TCR associated with agonist activation, causing the stabilization of ITAM phosphorylation and preventing desensitization through the subsequent binding of pSHP-1, which also accumulates following strong agonist binding. Under this model of TCR activation, signaling occurs in a binary fashion, with pSHP-1 inhibiting the activation unless threshold levels of agonist pMHC are encountered, whereupon, the system shifts to a fully activated state with full Erk phosphorylation, ZAP-70 activation, and phosphorylation of ITAMs.
Initial recognition of a pMHC:TCR match is amplified by Lck activation and phosphorylation of ZAP70, decreasing the activation threshold on neighboring TCR allowing for signal spreading by activation through interaction with lower affinity (self)pMHC:TCR interactions, which would be agonistic under steady-state conditions. It has been proposed that once activated ERK (76, 80) and/or Lck can act in a trans fashion, exerting effects on neighboring TCR to reduce the kinetic threshold of pMHC-TCR lifetime required for activation. Here, the transactivity of ERK and/or Lck may lead to a localized alteration in the balance of ITAM kinase vs. phosphatase activity, such that the speed of full ITAM phosphorylation occurs more rapidly than the dissociation of the pMHC:TCR complex, thereby causing TCR activation and recruitment of ZAP-70 in response to weak agonistic ligands such as self-peptides (81), resulting in a signal spreading mechanism and the formation of a local microcluster, which allows for the induction of TCR signaling from a small number of strong ligands. However, it is debatable whether this gives a better account for the stoichiometry of TCR downregulation than previously proposed models that envisage a serial engagement of pMHC by TCR with fast kinetics to account for the sustained signaling observed (82, 83). Additionally, signal spreading may provide a mechanism for the downstream calculation of TCR signal strength that cannot be accounted for by a binary mechanism of TCR activation.
Each successful pMHC:TCR interaction induces at least two major distinct pathways; the first pathway leads to the induction of a default activation program consisting of proximal MAPK phosphorylation and the second results in an increase of intracellular Ca2 + (Figure 2A). In lymphoid organs, the initial phase of T cell:APC interactions has been observed to comprise a number of brief serial interactions, with CD4+ T cell interactions lasting an average of 11–12 min (60) and CD8+ T cell interactions lasting 5–6 min (84). However, the average velocity of T cells decreases after contacting APCs loaded with cognate peptides during this first phase of interaction, and transient increases in intracellular calcium levels are observed (85). The sustained level of interaction necessary to form a mature IS does not generally occur until later in the response. However, following the kinaptic recognition (86) of cognate pMHC, T cells may receive a stimulatory signal sufficient to induce activation and maturation (87, 88). We propose that this indicates that in the absence of a sustained increase in calcium signaling sufficient to facilitate a halt in T cell migration required for the formation of a mature IS, what could be considered a default “on” pathway is sufficient to initiate TCR-driven activation of downstream transcription factors (89, 90). Thus, signaling from the TCR is able to directly turn naive T cells “on” and initiate maturation and differentiation by a primary default mechanism following the discrimination of a positive TCR signal. Second, where a sufficiently strong TCR signal is received, this may cause a halt in T cell migration by mediating a sustained increase in the intracellular calcium, thus allowing the formation of the IS and a prolonged T cell:APC interaction (91), as required for Th1 differentiation. The ability of T cells to discriminate between APCs loaded with different activatory stimuli allows T cells to perform a calculus when deciding which DC to form a synaptic relationship with (56) and indicates that T cells have specific mechanisms for determining the quantity and quality of signal they are receiving, which in turn imparts specific effector information.
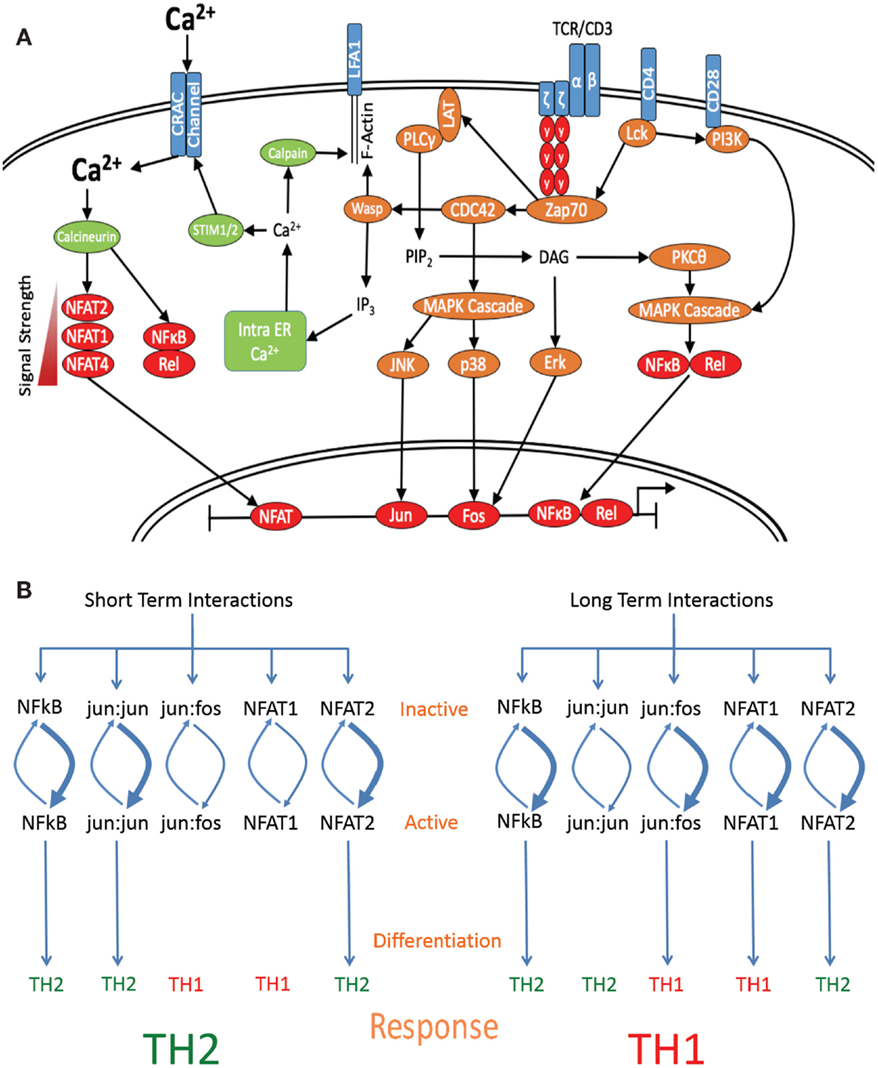
Figure 2. Length of interaction alters the activation of specific transcription factors. (A) Schematic diagram denoting the major signaling molecules involved in the T cell activation process upon stimulation of the TCR by cognate pMHCII, leading to the differential activation of downstream transcription factors based on signal strength and interaction times. (B) Short-term interactions induce a subset of default pathway of fast-acting transcription factors, which preferentially activate Th2-associated genes. Interaction for a longer period of time induces both default pathway-associated transcription factors and additionally allows for the induction of Th1-specific transcription factor activation, which are able to override the default Th2-inducing signal and skew the response toward the differentiation of a Th1 subset.
While T cells are able to calculate the level of signal imparted to them by a specific APC, a mechanism for this has not yet been fully ascertained. However, it is known that naive T cells are able to form a stable long-lasting IS within minutes when forced to interact with lipid bilayers or APCs in culture, indicating that de novo protein synthesis is not a prerequisite for IS formation (92). In vivo, T cells have been observed to progress through an initial priming phase (phase I) of activation where APC encounters consist of brief serial contacts, before transitioning to phase II where long-lasting contacts with APCs are maintained (60, 93). However, this initial priming phase does not seem to be required for induction of full T cell activation as following in vitro activation T cells may proceed directly to phase II with the formation of an IS. It is possible that in vivo, the priming phase is used as a mechanism to calculate the level of pMHC being presented, leading to a decision whether to form an IS, while under in vitro conditions, the pMHC concentrations used to study IS formation are sufficient to bypass phase I. Alternatively, as the physiological signals that normally drive naive T cell migration are not present in vitro, this may lead to a reduction in cellular momentum and thus a decreased requirement for a brake-inducing signal. To account for the observed T cell behavior, such a stopping mechanism must be both quantitative for pMHC encountered and iterative with each APC interaction. One possibility for such an integrative function is the IP3-mediated release of intracellular Ca2+, whereby receptor signal spreading leads to an exponential increase in the quantity of TCR signaling complexes available to recruit and activate PLCγ causing a concomitant increase in IP3 production and signal strength-dependent release of Ca2+ from the ER. Huse et al. (94) observed that activation at decreased [pMHC] levels or blocking of CD4 binding increased the offset time for Ca2+ release, which suggests that a threshold level of IP3 is required for Ca2+ flux. Additionally, it has been demonstrated that in vivo activation of naive T cells with a low potency APL can induce upregulation of CD69 expression without inducing a sustained Ca2+ release, indicating the ability of T cells to be activated in the absence of sufficient TCR signaling to trigger Ca2+ release (85).
Once threshold levels of intra-ER Ca2+ are reached, store-operated calcium entry is then triggered via activation of STIM1 and STIM2, which sense Ca2+ release via low-affinity Ca2+-binding EF hand domains located in the ER lumen. Translocation of STIM molecules to the plasma membrane causes opening of CRAC channels, a sustained increase in intracellular Ca2+ levels, and initiation of a cellular brake response allowing for IS formation (32), as opposed to a brief kinaptic interaction. Initially, T cell momentum dictates the overall length of T cell:APC interaction, as Ca2+ levels must reach the threshold for CRAC channel activation within a predetermined period of time in order to induce a T cell braking response; otherwise, migration will continue. As in the absence of CRAC opening, the T cell fails to arrest, and the kinaptic interaction will be broken, with ER Ca2+ slowly returning to baseline, such that, ER Ca2+ levels may form an intracellular clock based on Ca2+ reuptake vs. [pMHC] stimulation.
Several studies (73, 85, 90, 95) have reported the correlation between the arrest of naive T cell migration and an increase in Ca2+ signaling following encounter with APC, although a mechanism for this decreased motility has not been completely described (86, 89, 96, 97). What is evident from this body of work is that the spike in intracellular Ca2+ levels following TCR-induced CRAC opening marks the transition from a migratory phase to a primary signaling phase, which may better account for the T cell ability to quantitate [pMHC]. It has been reported (98) that following TCR activation, WASp-deficient T cells displayed normal IS formation initially; however, after further migration, WASp-deficient T cells were unable to reform a stable IS in the presence of PKCθ. Conversely, PKCθ-deficient T cells formed hyper-stable IS and displayed impaired migratory ability. Sims et al. (98) suggest that PKCθ is required for the periodic breaking of IS symmetry, which allows for bursts of mobility during phase I of activation, with WASp playing an opposing role in re-establishing symmetry upon encounter with subsequent APCs. Thus, the interplay between PKCθ and WASp provides a mechanism for the quantitation of [pMHC]. Large increases in intracellular [Ca2+] are associated with a decrease in T cell motility, with Ca2+ flux signaling initiating the reorganization of f-actin function, from providing the mechanical force necessary for migration (99) to one whereby f-actin stabilizes the IS (89). F-actin clustering is additionally required to stabilize activated LFA-1 at the cell surface (96); thus, f-actin-stabilized LFA-1 persistence at the cell surface may represent a further downstream mechanism for determination of antigen signal strength and may link PKCθ/WASp activity to the transition between phase I/phase II T cell migratory behavior. Additionally, the ER/CRAC channel circuit in T cell displays capacitor-like behavior, with the ability to rapidly evoke a significant increase in the intracellular Ca2+ level followed by a period of heightened intracellular [Ca2+] as Ca2+ ions are removed by specific pumps. At this time, the IP3R is unresponsive to further signaling as bound Ca2+ on the cytosolic side of the ER inhibits IP3 binding (100). During this period, the TCR-associated activation of WASp may organize f-actin stabilization of LFA-1 (and other integrins) at the kinaptic interface in response to pMHC levels, whereas upon decrease of Ca2+ to sufficient levels, PKCθ activity induces a redirection of f-actin polarization away from the T cell–APC interface, destabilizing the connection and allowing the resumption of migration.
Studies have shown that LFA-1 is required for optimal in vivo priming (101) and that LFA-1 decreases the antigen dose required for Th1 differentiation (102). Thus, an increase in activated LFA-1 at the cell surface may serve as a molecular counter for previous TCR:pMHC interactions, whereby upon the cumulative activation of sufficient LFA-1, the rate of polymerization of f-actin at the T cell:APC interface is greater than the PKCθ-induced migration-associated f-actin polymerization, enabling maintenance of the kynapse long enough for the ER to refill and induce recurrent calcium spiking. In turn, inhibiting PKCθ-induced migration for a sufficient period of time allows for the formation of a mature IS. Mechanistically, this provides an explanation for previous data indicating that the transcription of Ca2+-associated gene targets is more efficiently activated by oscillating calcium levels rather than a sustained increase in Ca2+ levels (91, 103, 104).
It is now well established that the stimulation of naive CD4+ T cells with a strong agonist or high dose of peptide leads to the induction of Th1 differentiation, while low dose or weak APL stimulation leads to the induction of Th2 differentiation (19, 21, 22, 24, 105). However, a molecular mechanism to explain these phenomena has not yet been adequately provided. Much of the data generated on the induction of Th1 differentiation have indicated that the required signal may be imparted through the formation of a mature IS. Knockout studies have revealed that many of the components, which link TCR activation with IS formation such as the calcium signaling (106–108) and actin polarization pathways (109, 110), are also required for Th1 differentiation. Thus, it is possible that transcription factors, which are regulated proximally to TCR signaling, are regulated in a temporal fashion, whereby long-term activation is required for the optimal activation and function of Th1-associated transcription factors such as Jun Kinase (JNK) (22), Erk1/2 (22, 24), AP-1 (fos:jun) (22), NFAT-1, and NFAT-4 (21). Several possible mechanisms exist that may account for the requirement for long-term signaling; Th1-associated transcription factors may take longer time to be phosphorylated and translocate to the nucleus, as suggested by Brogdon et al. (21) where a weak TCR stimulus caused translocation of NFAT-2 to the nucleus and a strong stimulation led to the nuclear recruitment of both NFAT-2 and NFAT-1. Alternately, Th1-associated gene targets may be more inaccessible, requiring greater alterations to their methylation and acetylation states before transcription is possible. Third, Th1-associated transcription factors may be subject to a greater degree of regulation by inhibitory kinases or phosphatases after activation, leading to a diminished half-life, comparatively decreasing their ability to localize to the nucleus and hence their signaling capacity.
In addition to low-dose/affinity peptide stimulation inducing Th2 differentiation, alterations to the TCR-associated signaling machinery can lead to the induction of Th2 differentiation. Inhibition of ZAP70 (111), a proximal TCR-associated kinase, which associates with phospho-ζ, was found to cause Th2 skewing, and a point mutation in the ZAP70 kinase domain has been described, which additionally induces Th2 skewing (112). As ZAP70 is thought to be required for full activation and efficient signaling, abrogation of its function is likely to lead to a decrease in the transduced signal strength and corresponding calcium flux, thus interfering with the ability of the cells to arrest and form a mature IS. Further, defects in LAT, which is phosphorylated by ZAP70 and is thought to act as a negative regulator of TCR signaling, have been described, whereby animals developed lymphoproliferative disorders with pronounced Th2 skewing (113). Here, it is likely that in the absence of sufficient negative regulation, weak signals are sufficient to activate cells but insufficient to induce full calcium flux and migratory arrest, thus leading to short-lived interactions and Th2 differentiation.
Th2-associated signaling has also been observed to be more dependent on activation of co-stimulatory pathways with defects in CD4 (23), CD28 (114), Lck (21), and PI3K (115), causing deficiencies in Th2 differentiation. As such, it is possible that as decreased overall signal strength is associated with Th2 induction, differentiation may be more highly reliant on co-stimulation in order to drive productive signaling, whereby signals that would not usually reach the threshold for activation or those that would lead to an anergic response can be modulated by effects of co-stimulatory receptor activation through Lck and its downstream effects on Zap70 induction, leading to a productive TCR engagement. Additionally, co-stimulatory signals lead to the preferential induction of the Th2-associated transcription factor NFκB1 (116, 117), through a pathway that requires PKCθ and IKKα signaling (118, 119). Thus, the reliance of Th2 induction on co-stimulation may play two roles whereby it serves to both ensure the transduction of weak TCR signaling in the presence of a strong danger signal and lead to the induction of Th2 differentiation via a default NFκB1-driven pathway in the absence of Th1 promoting transcription factors.
Other transcription factors known to affect differentiation include the AP1 binding elements fos:jun and jun:jun, which induce Th1 and Th2 differentiation, respectively (1, 22). While jun:jun homodimers are sufficient to bind DNA elements, jun:fos heterodimers are more energetically stable and thus have higher binding efficiency and target affinity, allowing binding to a wider range of targets (120, 121). However, the efficiency of dimer formation is temporally controlled through the regulation of p38 and Erk, which activate fos and JNK and then jun (120). p38, Erk, and JNK are cytosolic proteins that require phosphorylation by upstream kinases in order to migrate to the nucleus and are also subject to heavy regulation by phosphatases. Upon nuclear entry, this sets up a highly dynamic situation whereby variations in the phosphorylation: dephosphorylation rates of p38, Erk, and JNK lead to differential nuclear ratios of active fos and jun (122). The temporal model put forward here predicts that short-term signaling would lead to high levels of active jun and Th2 induction, whereas nuclear localization of p38 and Erk would occur later, increasing active fos levels and favoring jun:fos formation and Th1 induction. In support of this theory, it has been previously reported that Erk signaling is maximal at both high peptide doses (24) and where strong agonists are used (22), whereas low peptide doses and weak agonists induced low levels of Erk activation (24). Further, where weak agonists were used or Erk was inhibited, jun:jun homodimer formation was found to far exceed that of fos:jun formation and was also associated with early production of IL-4 (22). This indicates that strong TCR signals, which also induce long-term interactions, are required for full Erk activation, fos:jun formation, and Th1 induction, where as weak signals induce short-term interactions and jun:jun formation, favoring Th2 differentiation (Figure 2B).
Here, a temporally based mechanism of differentiation is suggested that accounts for many of the observed aspects of strength of signal-induced differentiation and the downstream signaling pathways associated with Th1 and Th2 differentiation. In this model, long-term interactions induce a program of Th1 differentiation and short-term interactions induce a program of Th2 differentiation. Integrating TCR signal strength through downstream signaling machinery such as calcium release allows for the influence of additional signaling systems such as co-stimulatory molecule activation and potentially cytokine signals to be combined to dictate the length of interaction between DC and CD4+ T cells and thus contributes to downstream effector differentiation (27). By varying the length of interaction time, cell fate is able to be controlled through utilizing the activation potentials of both fast-acting transcription factors such as NFAT2 and jun:jun and slow-acting transcription factors such as NFAT1 and fos:jun, with shorter interactions favoring Th2 induction and longer interactions Th1 induction. As these transcription factors are tightly regulated by both kinases and phosphatases, differentiation requires an interplay between the cycling of transcription factors between inactive/cytosolic and active/nuclear states, such that threshold concentrations of localized activated factors may be required in order to mediate the formation of epigenetic modifications and the switching on or off of the genes that control both differentiation state and effector function (3). As such, this level of control would allow for tightly regulated molecular signaling events to be transduced in a global fashion through modulation of multiple transcription factors by a temporal mechanism.
From a biological point of view, this temporal model helps explain several phenomena. As interaction time is relative to total signal strength, a model where marginal signaling such as engagement of only a few pMHC or use of very weak APL or activation in the absence of co-stimulation leads to the induction of an anergic program, and weak signaling induces Th2 differentiation and strong signaling causes Th1 induction. This provides a potential explanation for how tolerance to environmental antigens is induced, how the immune system detects a parasitic infection, and how environmental allergens induce Th2-associated allergic disease, whereas self-antigen recognition leads to Th1-associated autoimmune disease. Tolerance to common environmental antigens is generally induced as a normal response to the presentation of low levels of antigen in the absence of co-stimulation and thus induces a weak TCR response and an anergic program due to abrogated signaling and interactions (123). Nematode parasites on the other hand represent a significant pathogenic burden and secrete a large array of antigens as well as induce danger signals as they migrate through host tissue. Thus, in order to avoid host responses, parasites secrete molecules that downregulate the function of APCs and, while undergoing maturation within the host, turn over their antigenic profile, in some cases multiple times, during the course of their lifecycle. As such, parasites only secrete a specific antigen for a limited time, thus decreasing host recognition (124, 125). Further, the amount of specific antigen taken up by an individual DC is likely to be very small due to the inability of DC to phagocytize parasites, forcing reliance on endocytosis or pinocytosis for uptake of secreted Ag. Considering this, it is likely that as an evolutionary mechanism to combat nematode infection, the immune system has evolved to recognize parasites in terms of a moderate–to-low signal strength, which then allows for full activation via an endogenous pathway, in the absence of long-term signaling, which induces a program of differentiation culminating in a Th2 response. Similarly, when the immune system is stimulated by common environmental allergens, which are often capable of inducing danger signals (126), there may be a fine balance between inducing anergy and inducing a Th2 response, such that in the case of an initial encounter with an allergen in sufficient quantity or potency, a Th2 response may be induced, which when combined with chronic antigen exposure induces allergy. Conversely, autoimmune diseases tend to be predominantly Th1 or Th17 associated (127), which may be due to the thymic conditioning of the CD4+ repertoire through negative selection, such that CD4+ cells are generally non-reactive to self, except where a significant stimulus of sufficient quality is encountered (128). Such strong signaling may activate any cells remaining in the repertoire that are able to recognize self-antigen, and through recognition of a strong signal, a program of long-lasting interactions is induced leading to Th1or Th17 differentiation. During the induction of autoimmunity, strong TCR signaling may potentially occur due to the availability of a large amount of peptide via extensive presentation of self Ag or in the case of significant damage, where normally immune-privileged peptides are exposed in combination with a substantial danger signal. Evolutionarily, such signals are analogous to bacterial or viral infections, where a limited range of epitopes are presented during infection in high quantities along with the activation of specific PAMPs, leading to the delivery of high strength signals causing long-term interactions with DC and the induction of a Th1 response.
Incorporating a temporal component into our models of how CD4+ T cells interact with DC during Treg, Th17, and Tfh cell differentiation also allows for an explanation of some of the additional phenomena that have been reported to occur during differentiation, which are not fully explained by a purely qualitative model of cellular decision-making. A distinct parallel can be drawn between the differentiation of Th1–Th2 cells and Th17–Treg cells, with both Th1 and Th17 cells requiring a strong TCR signal and have been observed to interact for long periods of time with DC, whereas both Th2 and Treg cells require weak TCR signals and only interact for brief periods of time. Further, Th2 and Treg cells are able to differentiate either via endogenous signals or from cytokines that are secreted in a diffuse fashion, whereas Th1 and Th17 cells require additional cues that are delivered at the cellular synapse and require a strong and/or ongoing signal in order to regulate their ability to respond to the cytokines that direct their differentiation. Tfh cell differentiation has also been observed to have a temporal component with ongoing interactions being required for differentiation, and as such, it is appealing to hypothesize that their expression of Bcl-6 may allow for this ongoing interaction in a non-deleterious fashion, while they acquire additional interaction-based signals from DC in the form of factors such as ICOSL (52) and CD40L (38).
In summary, a temporal signaling model would explain how many of the observed alterations in TCR signaling modulate differentiation by linking TCR signal strength, co-stimulation, and cytokine inputs through a mechanism that allows for the integration of multiple signaling inputs into an overall measure of signal strength and outputs these as divergent programs of behavior during priming, which ultimately leads to alterations in cell fate and effector function.
The author confirms being the sole contributor of this work and approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Dr. Ronald Germain for helpful discussions and to Dr. Rachel Gottschalk for her critical review of the manuscript.
1. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
2. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
3. Kanno Y, Vahedi G, Hirahara K, Singleton K, O’Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol (2012) 30:707–31. doi:10.1146/annurev-immunol-020711-075058
4. Li P, Spolski R, Liao W, Leonard WJ. Complex interactions of transcription factors in mediating cytokine biology in T cells. Immunol Rev (2014) 261:141–56. doi:10.1111/imr.12199
5. Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol (1997) 15:297–322. doi:10.1146/annurev.immunol.15.1.297
6. Medzhitov R, Janeway CA Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell (1997) 91:295–8. doi:10.1016/S0092-8674(00)80412-2
7. O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity (2008) 28:477–87. doi:10.1016/j.immuni.2008.03.002
8. Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, et al. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science (2001) 292:1907–10. doi:10.1126/science.1059835
9. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (1993) 260:547–9. doi:10.1126/science.8097338
10. Grogan JL, Mohrs M, Harmon B, Lacy DA, Sedat JW, Locksley RM. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity (2001) 14:205–15. doi:10.1016/S1074-7613(01)00103-0
11. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell (1997) 89:587–96. doi:10.1016/S0092-8674(00)80240-8
12. Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, Paul WE. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J Exp Med (1990) 172:921–9. doi:10.1084/jem.172.3.921
13. Finkelman FD, Morris SC, Orekhova T, Mori M, Donaldson D, Reiner SL, et al. Stat6 regulation of in vivo IL-4 responses. J Immunol (2000) 164:2303–10. doi:10.4049/jimmunol.164.5.2303
14. van Panhuys N, Tang SC, Prout M, Camberis M, Scarlett D, Roberts J, et al. In vivo studies fail to reveal a role for IL-4 or STAT6 signaling in Th2 lymphocyte differentiation. Proc Natl Acad Sci U S A (2008) 105:12423–8. doi:10.1073/pnas.0806372105
15. Bouchery T, Kyle R, Ronchese F, Le Gros G. The differentiation of CD4(+) T-helper cell subsets in the context of helminth parasite infection. Front Immunol (2014) 5:487. doi:10.3389/fimmu.2014.00487
16. Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, et al. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity (2012) 37:660–73. doi:10.1016/j.immuni.2012.09.007
17. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
18. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
19. Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J Exp Med (1995) 182:1591–6. doi:10.1084/jem.182.5.1591
20. Hosken NA, Shibuya K, Heath AW, Murphy KM, O’Garra A. The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor-alpha beta-transgenic model. J Exp Med (1995) 182:1579–84. doi:10.1084/jem.182.5.1579
21. Brogdon JL, Leitenberg D, Bottomly K. The potency of TCR signaling differentially regulates NFATc/p activity and early IL-4 transcription in naive CD4+ T cells. J Immunol (2002) 168:3825–32. doi:10.4049/jimmunol.168.8.3825
22. Jorritsma PJ, Brogdon JL, Bottomly K. Role of TCR-induced extracellular signal-regulated kinase activation in the regulation of early IL-4 expression in naive CD4+ T cells. J Immunol (2003) 170:2427–34. doi:10.4049/jimmunol.170.5.2427
23. Leitenberg D, Boutin Y, Constant S, Bottomly K. CD4 regulation of TCR signaling and T cell differentiation following stimulation with peptides of different affinities for the TCR. J Immunol (1998) 161:1194–203.
24. Yamane H, Zhu J, Paul WE. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J Exp Med (2005) 202:793–804. doi:10.1084/jem.20051304
25. Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med (2010) 207:1701–11. doi:10.1084/jem.20091999
26. Gottschalk RA, Hathorn MM, Beuneu H, Corse E, Dustin ML, Altan-Bonnet G, et al. Distinct influences of peptide-MHC quality and quantity on in vivo T-cell responses. Proc Natl Acad Sci U S A (2012) 109:881–6. doi:10.1073/pnas.1119763109
27. van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization in vivo. Immunity (2014) 41:63–74. doi:10.1016/j.immuni.2014.06.003
28. Nurieva RI, Chuvpilo S, Wieder ED, Elkon KB, Locksley R, Serfling E, et al. A costimulation-initiated signaling pathway regulates NFATc1 transcription in T lymphocytes. J Immunol (2007) 179:1096–103. doi:10.4049/jimmunol.179.2.1096
29. Lim TS, Goh JK, Mortellaro A, Lim CT, Hammerling GJ, Ricciardi-Castagnoli P. CD80 and CD86 differentially regulate mechanical interactions of T-cells with antigen-presenting dendritic cells and B-cells. PLoS One (2012) 7:e45185. doi:10.1371/journal.pone.0045185
30. Milner JD, Fazilleau N, McHeyzer-Williams M, Paul W. Cutting edge: lack of high affinity competition for peptide in polyclonal CD4+ responses unmasks IL-4 production. J Immunol (2010) 184:6569–73. doi:10.4049/jimmunol.1000674
31. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell (2013) 153:785–96. doi:10.1016/j.cell.2013.04.007
32. Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol (2009) 10:375–84. doi:10.1038/ni.1704
33. Hwang S, Palin AC, Li L, Song KD, Lee J, Herz J, et al. TCR ITAM multiplicity is required for the generation of follicular helper T-cells. Nat Commun (2015) 6:6982. doi:10.1038/ncomms7982
34. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science (2014) 345:1058–62. doi:10.1126/science.1257861
35. Benson RA, MacLeod MK, Hale BG, Patakas A, Garside P, Brewer JM. Antigen presentation kinetics control T cell/dendritic cell interactions and follicular helper T cell generation in vivo. Elife (2015) 4:06994. doi:10.7554/eLife.06994
36. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity (2013) 38:596–605. doi:10.1016/j.immuni.2012.11.020
37. Deenick EK, Chan A, Ma CS, Gatto D, Schwartzberg PL, Brink R, et al. Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity (2010) 33:241–53. doi:10.1016/j.immuni.2010.07.015
38. Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, Kopf M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci U S A (2009) 106:876–81. doi:10.1073/pnas.0810769106
39. Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol (2005) 23:549–600. doi:10.1146/annurev.immunol.22.012703.104743
40. Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR, et al. Differential expression of interleukin-17A and -17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity (2009) 31:587–97. doi:10.1016/j.immuni.2009.07.009
41. Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, et al. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med (2014) 211:529–43. doi:10.1084/jem.20131459
42. Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol (2011) 12:551–9. doi:10.1038/ni.2030
43. Desai BB, Quinn PM, Wolitzky AG, Mongini PK, Chizzonite R, Gately MK. IL-12 receptor. II. Distribution and regulation of receptor expression. J Immunol (1992) 148:3125–32.
44. Perona-Wright G, Mohrs K, Mayer KD, Mohrs M. Differential regulation of IL-4Ralpha expression by antigen versus cytokine stimulation characterizes Th2 progression in vivo. J Immunol (2010) 184:615–23. doi:10.4049/jimmunol.0902408
45. Pulecio J, Petrovic J, Prete F, Chiaruttini G, Lennon-Dumenil AM, Desdouets C, et al. Cdc42-mediated MTOC polarization in dendritic cells controls targeted delivery of cytokines at the immune synapse. J Exp Med (2010) 207:2719–32. doi:10.1084/jem.20100007
46. Maldonado RA, Irvine DJ, Schreiber R, Glimcher LH. A role for the immunological synapse in lineage commitment of CD4 lymphocytes. Nature (2004) 431:527–32. doi:10.1038/nature02916
47. Huse M, Lillemeier BF, Kuhns MS, Chen DS, Davis MM. T cells use two directionally distinct pathways for cytokine secretion. Nat Immunol (2006) 7:247–55. doi:10.1038/ni1304
48. Perona-Wright G, Mohrs K, Mohrs M. Sustained signaling by canonical helper T cell cytokines throughout the reactive lymph node. Nat Immunol (2010) 11:520–6. doi:10.1038/ni.1866
49. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
50. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity (2007) 26:371–81. doi:10.1016/j.immuni.2007.02.009
51. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci (2012) 8:1254–66. doi:10.7150/ijbs.4679
52. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34:932–46. doi:10.1016/j.immuni.2011.03.023
53. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity (2012) 36:847–56. doi:10.1016/j.immuni.2012.02.012
54. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med (2012) 209:243–50. doi:10.1084/jem.20111174
55. Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, et al. Insights into the role of Bcl6 in follicular Th cells using a new conditional mutant mouse model. J Immunol (2013) 191:3705–11. doi:10.4049/jimmunol.1300378
56. Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat Immunol (2008) 9:282–91. doi:10.1038/ni1559
57. von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol (2003) 3:867–78. doi:10.1038/nri1222
58. Mandl JN, Liou R, Klauschen F, Vrisekoop N, Monteiro JP, Yates AJ, et al. Quantification of lymph node transit times reveals differences in antigen surveillance strategies of naive CD4+ and CD8+ T cells. Proc Natl Acad Sci U S A (2012) 109:18036–41. doi:10.1073/pnas.1211717109
59. Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science (2002) 296:1873–6. doi:10.1126/science.1071065
60. Miller MJ, Safrina O, Parker I, Cahalan MD. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J Exp Med (2004) 200:847–56. doi:10.1084/jem.20041236
61. Miller MJ, Wei SH, Cahalan MD, Parker I. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc Natl Acad Sci U S A (2003) 100:2604–9. doi:10.1073/pnas.2628040100
62. Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity (2006) 25:989–1001. doi:10.1016/j.immuni.2006.10.011
63. Bajenoff M, Granjeaud S, Guerder S. The strategy of T cell antigen-presenting cell encounter in antigen-draining lymph nodes revealed by imaging of initial T cell activation. J Exp Med (2003) 198:715–24. doi:10.1084/jem.20030167
64. Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity (2003) 19:47–57. doi:10.1016/S1074-7613(03)00175-4
65. Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol (2005) 6:895–901. doi:10.1038/ni1240
66. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol (2007) 8:1255–65. doi:10.1038/ni1513
67. Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci U S A (2000) 97:12694–9. doi:10.1073/pnas.97.23.12694
68. Germain RN, Bajenoff M, Castellino F, Chieppa M, Egen JG, Huang AY, et al. Making friends in out-of-the-way places: how cells of the immune system get together and how they conduct their business as revealed by intravital imaging. Immunol Rev (2008) 221:163–81. doi:10.1111/j.1600-065X.2008.00591.x
69. Pietila TE, Veckman V, Kyllonen P, Lahteenmaki K, Korhonen TK, Julkunen I. Activation, cytokine production, and intracellular survival of bacteria in Salmonella-infected human monocyte-derived macrophages and dendritic cells. J Leukoc Biol (2005) 78:909–20. doi:10.1189/jlb.1204721
70. Mueller SN, Hosiawa-Meagher KA, Konieczny BT, Sullivan BM, Bachmann MF, Locksley RM, et al. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science (2007) 317:670–4. doi:10.1126/science.1144830
71. Worbs T, Mempel TR, Bolter J, von Andrian UH, Forster R. CCR7 ligands stimulate the intranodal motility of T lymphocytes in vivo. J Exp Med (2007) 204:489–95. doi:10.1084/jem.20061706
72. Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature (2002) 419:845–9. doi:10.1038/nature01076
73. Ma Z, Sharp KA, Janmey PA, Finkel TH. Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLoS Biol (2008) 6:e43. doi:10.1371/journal.pbio.0060043
74. Miller MJ, Hejazi AS, Wei SH, Cahalan MD, Parker I. T cell repertoire scanning is promoted by dynamic dendritic cell behavior and random T cell motility in the lymph node. Proc Natl Acad Sci U S A (2004) 101:998–1003. doi:10.1073/pnas.0306407101
75. Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol (2005) 3:e356. doi:10.1371/journal.pbio.0030356
76. Feinerman O, Germain RN, Altan-Bonnet G. Quantitative challenges in understanding ligand discrimination by alphabeta T cells. Mol Immunol (2008) 45:619–31. doi:10.1016/j.molimm.2007.03.028
77. McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc Natl Acad Sci U S A (1995) 92:5042–6. doi:10.1073/pnas.92.11.5042
78. Lord GM, Lechler RI, George AJ. A kinetic differentiation model for the action of altered TCR ligands. Immunol Today (1999) 20:33–9. doi:10.1016/S0167-5699(98)01379-6
79. Rabinowitz JD, Beeson C, Wulfing C, Tate K, Allen PM, Davis MM, et al. Altered T cell receptor ligands trigger a subset of early T cell signals. Immunity (1996) 5:125–35. doi:10.1016/S1074-7613(00)80489-6
80. Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol (2003) 4:248–54. doi:10.1038/ni895
81. Krogsgaard M, Li QJ, Sumen C, Huppa JB, Huse M, Davis MM. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature (2005) 434:238–43. doi:10.1038/nature03391
82. Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature (1995) 375:148–51. doi:10.1038/375148a0
83. Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science (1996) 273:104–6. doi:10.1126/science.273.5271.104
84. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427:154–9. doi:10.1038/nature02238
85. Skokos D, Shakhar G, Varma R, Waite JC, Cameron TO, Lindquist RL, et al. Peptide-MHC potency governs dynamic interactions between T cells and dendritic cells in lymph nodes. Nat Immunol (2007) 8:835–44. doi:10.1038/ni1490
86. Dustin ML. T-cell activation through immunological synapses and kinapses. Immunol Rev (2008) 221:77–89. doi:10.1111/j.1600-065X.2008.00589.x
87. Gunzer M, Schafer A, Borgmann S, Grabbe S, Zanker KS, Brocker EB, et al. Antigen presentation in extracellular matrix: interactions of T cells with dendritic cells are dynamic, short lived, and sequential. Immunity (2000) 13:323–32. doi:10.1016/S1074-7613(00)00032-7
88. Gunzer M, Weishaupt C, Hillmer A, Basoglu Y, Friedl P, Dittmar KE, et al. A spectrum of biophysical interaction modes between T cells and different antigen-presenting cells during priming in 3-D collagen and in vivo. Blood (2004) 104:2801–9. doi:10.1182/blood-2004-03-1193
89. Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol (2007) 7:690–702. doi:10.1038/nri2152
90. Wei SH, Safrina O, Yu Y, Garrod KR, Cahalan MD, Parker I. Ca2+ signals in CD4+ T cells during early contacts with antigen-bearing dendritic cells in lymph node. J Immunol (2007) 179:1586–94. doi:10.4049/jimmunol.179.3.1586
91. Bhakta NR, Oh DY, Lewis RS. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat Immunol (2005) 6:143–51. doi:10.1038/ni1161
92. Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, et al. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol (2010) 28:79–105. doi:10.1146/annurev-immunol-030409-101308
93. Henrickson SE, Mempel TR, Mazo IB, Liu B, Artyomov MN, Zheng H, et al. In vivo imaging of T cell priming. Sci Signal (2008) 1:t2. doi:10.1126/stke.112pt2
94. Huse M, Klein LO, Girvin AT, Faraj JM, Li QJ, Kuhns MS, et al. Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity (2007) 27:76–88. doi:10.1016/j.immuni.2007.05.017
95. Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity (2006) 25:117–27. doi:10.1016/j.immuni.2006.04.010
96. Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nat Rev Immunol (2007) 7:131–43. doi:10.1038/nri2021
97. Gallo EM, Cante-Barrett K, Crabtree GR. Lymphocyte calcium signaling from membrane to nucleus. Nat Immunol (2006) 7:25–32. doi:10.1038/ni1295
98. Sims TN, Soos TJ, Xenias HS, Dubin-Thaler B, Hofman JM, Waite JC, et al. Opposing effects of PKCtheta and WASp on symmetry breaking and relocation of the immunological synapse. Cell (2007) 129:773–85. doi:10.1016/j.cell.2007.03.037
99. Vicente-Manzanares M, Sanchez-Madrid F. Role of the cytoskeleton during leukocyte responses. Nat Rev Immunol (2004) 4:110–22. doi:10.1038/nri1268
100. Joseph SK, Brownell S, Khan MT. Calcium regulation of inositol 1,4,5-trisphosphate receptors. Cell Calcium (2005) 38:539–46. doi:10.1016/j.ceca.2005.07.007
101. Kandula S, Abraham C. LFA-1 on CD4+ T cells is required for optimal antigen-dependent activation in vivo. J Immunol (2004) 173:4443–51. doi:10.4049/jimmunol.173.7.4443
102. Wang Y, Shibuya K, Yamashita Y, Shirakawa J, Shibata K, Kai H, et al. LFA-1 decreases the antigen dose for T cell activation in vivo. Int Immunol (2008) 20(9):1119–27. doi:10.1093/intimm/dxn070
103. Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature (1998) 392:933–6. doi:10.1038/31960
104. Montes M, McIlroy D, Hosmalin A, Trautmann A. Calcium responses elicited in human T cells and dendritic cells by cell-cell interaction and soluble ligands. Int Immunol (1999) 11:561–8. doi:10.1093/intimm/11.4.561
105. Pfeiffer C, Stein J, Southwood S, Ketelaar H, Sette A, Bottomly K. Altered peptide ligands can control CD4 T lymphocyte differentiation in vivo. J Exp Med (1995) 181:1569–74. doi:10.1084/jem.181.4.1569
106. Tahvanainen J, Kallonen T, Lahteenmaki H, Heiskanen KM, Westermarck J, Rao KV, et al. PRELI is a mitochondrial regulator of human primary T-helper cell apoptosis, STAT6, and Th2-cell differentiation. Blood (2009) 113:1268–77. doi:10.1182/blood-2008-07-166553
107. Zhang R, Yoshida A, Kumagai T, Kawaguchi H, Maruyama H, Suzuki T, et al. Vaccination with calpain induces a Th1-biased protective immune response against Schistosoma japonicum. Infect Immun (2001) 69:386–91. doi:10.1128/IAI.69.1.386-391.2001
108. Noble A, Truman JP, Vyas B, Vukmanovic-Stejic M, Hirst WJ, Kemeny DM. The balance of protein kinase C and calcium signaling directs T cell subset development. J Immunol (2000) 164:1807–13. doi:10.4049/jimmunol.164.4.1807
109. Trifari S, Sitia G, Aiuti A, Scaramuzza S, Marangoni F, Guidotti LG, et al. Defective Th1 cytokine gene transcription in CD4+ and CD8+ T cells from Wiskott-Aldrich syndrome patients. J Immunol (2006) 177:7451–61. doi:10.4049/jimmunol.177.10.7451
110. Varga G, Nippe N, Balkow S, Peters T, Wild MK, Seeliger S, et al. LFA-1 contributes to signal I of T-cell activation and to the production of T(h)1 cytokines. J Invest Dermatol (2010) 130:1005–12. doi:10.1038/jid.2009.398
111. Tanaka Y, Bi K, Kitamura R, Hong S, Altman Y, Matsumoto A, et al. SWAP-70-like adapter of T cells, an adapter protein that regulates early TCR-initiated signaling in Th2 lineage cells. Immunity (2003) 18:403–14. doi:10.1016/S1074-7613(03)00054-2
112. Siggs OM, Miosge LA, Yates AL, Kucharska EM, Sheahan D, Brdicka T, et al. Opposing functions of the T cell receptor kinase ZAP-70 in immunity and tolerance differentially titrate in response to nucleotide substitutions. Immunity (2007) 27:912–26. doi:10.1016/j.immuni.2007.11.013
113. Malissen B, Aguado E, Malissen M. Role of the LAT adaptor in T-cell development and Th2 differentiation. Adv Immunol (2005) 87:1–25. doi:10.1016/S0065-2776(05)87001-4
114. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol (2001) 1:220–8. doi:10.1038/35105024
115. Cook KD, Miller J. TCR-dependent translational control of GATA-3 enhances Th2 differentiation. J Immunol (2010) 185:3209–16. doi:10.4049/jimmunol.0902544
116. Artis D, Shapira S, Mason N, Speirs KM, Goldschmidt M, Caamano J, et al. Differential requirement for NF-kappa B family members in control of helminth infection and intestinal inflammation. J Immunol (2002) 169:4481–7. doi:10.4049/jimmunol.169.8.4481
117. Cannons JL, Yu LJ, Hill B, Mijares LA, Dombroski D, Nichols KE, et al. SAP regulates T(H)2 differentiation and PKC-theta-mediated activation of NF-kappaB1. Immunity (2004) 21:693–706. doi:10.1016/j.immuni.2004.09.012
118. Schmitz ML, Bacher S, Dienz O. NF-kappaB activation pathways induced by T cell costimulation. FASEB J (2003) 17:2187–93. doi:10.1096/fj.02-1100rev
119. Wang D, Matsumoto R, You Y, Che T, Lin XY, Gaffen SL, et al. CD3/CD28 costimulation-induced NF-kappaB activation is mediated by recruitment of protein kinase C-theta, Bcl10, and IkappaB kinase beta to the immunological synapse through CARMA1. Mol Cell Biol (2004) 24:164–71. doi:10.1128/MCB.24.1.164-171.2003
120. Smeal T, Angel P, Meek J, Karin M. Different requirements for formation of Jun: Jun and Jun: Fos complexes. Genes Dev (1989) 3:2091–100. doi:10.1101/gad.3.12b.2091
121. Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene (2001) 20:2438–52. doi:10.1038/sj.onc.1204385
122. Dunn C, Wiltshire C, MacLaren A, Gillespie DA. Molecular mechanism and biological functions of c-Jun N-terminal kinase signalling via the c-Jun transcription factor. Cell Signal (2002) 14:585–93. doi:10.1016/S0898-6568(01)00275-3
123. Sadegh-Nasseri S, Dalai SK, Korb Ferris LC, Mirshahidi S. Suboptimal engagement of the T-cell receptor by a variety of peptide-MHC ligands triggers T-cell anergy. Immunology (2010) 129:1–7. doi:10.1111/j.1365-2567.2009.03206.x
124. Hewitson JP, Grainger JR, Maizels RM. Helminth immunoregulation: the role of parasite secreted proteins in modulating host immunity. Mol Biochem Parasitol (2009) 167:1–11. doi:10.1016/j.molbiopara.2009.04.008
125. Sotillo J, Sanchez-Flores A, Cantacessi C, Harcus Y, Pickering D, Bouchery T, et al. Secreted proteomes of different developmental stages of the gastrointestinal nematode Nippostrongylus brasiliensis. Mol Cell Proteomics (2014) 13:2736–51. doi:10.1074/mcp.M114.038950
126. Platts-Mills TAE, Woodfolk JA. Allergens and their role in the allergic immune response. Immunol Rev (2011) 242:51–68. doi:10.1111/j.1600-065X.2011.01021.x
127. Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Human T helper type 1 dichotomy: origin, phenotype and biological activities. Immunology (2015) 144:343–51. doi:10.1111/imm.12399
Keywords: TCR, differentiation, T cell, signal strength, cytokine
Citation: van Panhuys N (2016) TCR Signal Strength Alters T–DC Activation and Interaction Times and Directs the Outcome of Differentiation. Front. Immunol. 7:6. doi: 10.3389/fimmu.2016.00006
Received: 16 September 2015; Accepted: 08 January 2016;
Published: 25 January 2016
Edited by:
Dietmar Zehn, Swiss Vaccine Research Institute, SwitzerlandReviewed by:
António Gil Castro, University of Minho, PortugalCopyright: © 2016 van Panhuys. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas van Panhuys, bnZhbnBhbmh1eXNAc2lkcmEub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.