 Qinghui Mu
Qinghui Mu Husen Zhang2
Husen Zhang2 Xin M. Luo
Xin M. Luo- 1Department of Biomedical Sciences and Pathobiology, Virginia Tech, Blacksburg, VA, USA
- 2Department of Civil and Environmental Engineering, Virginia Tech, Blacksburg, VA, USA
Systemic lupus erythematosus (SLE) is a multi-system autoimmune disease. Despite years of study, the etiology of SLE is still unclear. Both genetic and environmental factors have been implicated in the disease mechanisms. In the past decade, a growing body of evidence has indicated an important role of gut microbes in the development of autoimmune diseases, including type 1 diabetes, rheumatoid arthritis, and multiple sclerosis. However, such knowledge on SLE is little, though we have already known that environmental factors can trigger the development of lupus. Several recent studies have suggested that alterations of the gut microbial composition may be correlated with SLE disease manifestations, while the exact roles of either symbiotic or pathogenic microbes in this disease remain to be explored. Elucidation of the roles of gut microbes – as well as the roles of diet that can modulate the composition of gut microbes – in SLE will shed light on how this autoimmune disorder develops, and provide opportunities for improved biomarkers of the disease and the potential to probe new therapies. In this review, we aim to compile the available evidence on the contributions of diet and gut microbes to SLE occurrence and pathogenesis.
Introduction
The mammalian gut harbors trillions of microorganisms known as the microbiota (1). Increasing evidence in recent years suggest that host microbiota and immune system interact to maintain tissue homeostasis in healthy individuals (2–6). Perturbation of the host microbiota, especially in the gut, has been shown to be associated with many diseases. Among these are autoimmune disorders that include inflammatory bowel disease (IBD) (7, 8), type 1 diabetes (T1D) (9–12), rheumatoid arthritis (5, 13–15), and multiple sclerosis (16, 17). However, little is known on the role of gut microbiota in systemic lupus erythematosus (SLE) (18).
Systemic lupus erythematosus is an autoimmune disorder characterized by severe and persistent inflammation that leads to tissue damage in multiple organs. According to the Lupus Foundation of America, about two million Americans currently live with the disease. The prevalence ranges from 20 to 200 cases per 100,000 persons, with higher prevalence for people of African, Hispanic, or Asian ancestry (19, 20). Although the disease affects both males and females, women of childbearing age are diagnosed nine times more often than men.
Our research team has recently described the dynamics of gut microbiota in a classical SLE mouse model MRL/Mp-Faslpr (MRL/lpr) (21). In young, female lupus mice, we found marked depletion of Lactobacilli, and increase of Clostridial species (Lachnospiraceae) together with increased bacterial diversity compared to age-matched healthy controls. Importantly, dietary treatments that improved lupus symptoms in lupus mice also restored gut colonization of Lactobacillus and decreased that of Lachnospiraceae. In human SLE, a recent cross-sectional study has shown that a lower Firmicutes to Bacteroidetes ratio was present in women with SLE even after disease remission (22). Similarly, a higher level of Bacteroidetes was found in lupus-prone SNF1 mice with more severe disease (23), though this was not evident in MRL/lpr mice (21). These results suggest a potentially important role of gut microbiota on lupus pathogenesis, in particular a potential role of Bacteroidetes, since the relative abundance of these bacteria is increased in human SLE and at least one murine lupus model. In this review, we aim to compile the available evidence that associates gut microbes to SLE.
Environmental Factors and SLE
It is well established that genetic factors influence lupus susceptibility. However, the lack of disease concordance between genetically identical twins strongly suggests the role of non-genetic factors, most likely of environmental factors (24). The role of environmental factors in the etiology of SLE is evidenced by the dramatic difference in disease incidence between West Africans and African Americans, both derived from the same ethnic group but exposed to different environments (25). With an obviously higher burden of infections, the frequency of SLE is much lower in West Africa than Africans living in Europe or USA. The mechanism behind this observation is still unclear, but improvement in hygiene and absence of certain microbes may have contributed to the higher incidence and faster progression of lupus disease (26). In addition to microbes, a number of environmental triggering factors have been described to be associated with SLE, including UV light and cigarette smoking, some of which trigger lupus through epigenetic mechanisms (27–30).
The Hygiene Hypothesis
Increase of SLE occurrence in the developed world has been reported. Data from several regions of USA show that the incidence of SLE increased at least threefold within the second half of the twentieth century (31, 32). This increase could be related to changes of environmental factors, though better diagnostic methods and increasing awareness of the disease may partially lead to the change in SLE frequency. Similar increase has been observed in a study analyzing the incidence of SLE in Denmark (33). Genome evolution rate seems to be unpersuasive to this increase. In contrast, due to advancements on medicine and vaccination, a number of infectious agents have been gradually eliminated in developed countries, and the sanitation condition has been largely improved. Some have thus proposed that lower exposure to infections leads to the rise of allergies and some autoimmune diseases, such as T1D (34, 35). This is called “The Hygiene Hypothesis.” Considering the rise of SLE frequency in developed countries, it is reasonable to extend the hypothesis to this autoimmune disorder.
Increasing hygiene standards eliminates both pathogenic and non-pathogenic microbes from the environment. Infections from pathogenic microbes, or the lack thereof, are known to be associated with SLE occurrence. Epstein–Barr virus (EBV) and cytomegalovirus (CMV), for example, have been linked to the pathogenesis of SLE by several reports (36–40). Commensal microbes residing inside the host, in return, have been shown to maintain and expand CD8+ memory T cells during CMV infection, supporting the notion that microbiota and CMV cooperatively augment immune activation (41). While EBV and CMV are largely considered triggers of SLE, it is increasingly evident that some infections may be beneficial and the lack of them might actually facilitate SLE. In one surprising report (42), two female SLE patients with severe SLE showed improved disease after experiencing infections for a short period of time. Before the infections, both patients failed to respond to a long time of immunosuppressive therapy. Neither experienced relapse after the amelioration of SLE symptoms following the infections. One of the patients even had a successful pregnancy, which is known to trigger lupus flares. Unfortunately, the study did not identify the causing agent that ameliorated the disease. However, another study has identified hepatitis B virus (HBV) as a protective factor against SLE (43). In their study, 2.5% of SLE patients were found positive for the presence of HBV-core antibody, compared to 10.7% from normal controls, which suggests a potential benefit of HBV infection against the occurrence of SLE. In addition, in a large serologic survey, Helicobacter pylori seronegativity was found to be associated with an increased risk and earlier onset of SLE in African Americans, suggesting a protective role of H. pylori in SLE patients (44, 45). These studies suggest that, in developed countries where HBV and H. pylori infections are decreasing (46–48), the risk for developing SLE could become higher. T cell exhaustion during chronic infection may explain the ability of these pathogens to down-regulate inflammation and ameliorate SLE (49, 50).
In lupus-prone mouse models, beneficial roles of some pathogenic microbes have also been suggested. Chen et al. reported that with the infection of Toxoplasma gondii, New Zealand Black (NZB) × New Zealand White (NZW) F1 (NZB/W F1) mice had significantly decreased mortality, ameliorated proteinuria level, and reduced anti-DNA IgG in serum. IFNγ and IL-10 expression was reduced in the spleen in the presence of T. gondii, suggesting the suppression of T helper 1 (Th1) and Th2 responses, respectively, both demonstrated to be pathogenic in murine lupus (42, 51). In addition, when examining NZB/W F1 mice treated with live Plasmodium chabaudi, another prevalent parasite, several independent groups have found that the malaria-causing microbe can prevent clinical symptoms of murine lupus and protect the animals against lupus nephritis (52–54). This is perhaps due to the changed cytokine profile and redox status in both liver and kidney of the mice. Moreover, virus infection has also been found to improve murine lupus symptoms in addition to parasites. For instance, the infection of lactate dehydrogenase elevating virus (LDV) has been shown to significantly suppress the production of anti-nuclear antibody (ANA) and the development of glomerulonephritis in NZB/W F1 mice (26, 55–58). The beneficial effect is hypothesized to be associated with superoxide anion production from macrophages and modulation of prostaglandin E. While LDV and P. chabaudi do not infect humans, results from these mouse studies suggest that some infections might be associated with decreased severity of SLE.
Antibiotics and SLE
Antibiotics, which can remove gut bacteria, are known to trigger lupus flares. These include sulfa drugs such as trimethoprim–sulfamethoxazole (Septra), tetracycline-related antibiotics such as minocycline, and penicillin-related antibiotics such as amoxicillin. Increased sun sensitivity with antibiotics may be one mechanism behind the observations. However, antibiotics also cause diarrhea and remove beneficial microbes from the intestinal tract. Could it be the removal of “good” bacteria a mechanism by which antibiotics induce flares in SLE patients? In addition, bacterial metabolites produced by gut microbes can modulate immune function. Recently, several groups have found that metabolites produced by gut bacteria, especially butyrate produced by Clostridia, can promote the differentiation of regulatory T cells (Tregs) in the colon, spleen, and lymph nodes to suppress inflammation (59–62). Thus, removal of certain gut commensals with antibiotics could potentially lead to decreases of bacterial metabolites, such as homoserine lactone, N-acetylmuramic acid, and N-acetylglucosamine (63) – which could be immunosuppressive – thereby facilitating lupus progression. Incidentally, African Americans have used antibiotics much more frequently than people in West African countries (64, 65), and this may have impacted the differences in lupus prevalence and severity between the two populations.
Dietary Components and SLE
Diet, one of the main environmental factors with known effects on gut microbiota, has been studied extensively in both SLE patients and lupus-prone mice. Vitamin D (VD), vitamin A (VA), and omega-3 polyunsaturated fatty acids (PUFAs), for instance, have been found to modulate lupus onset or flares. Current knowledge suggests that dietary components can influence SLE through changing the composition and function of gut microbiota, modulating immunological pathways, and/or exerting epigenetic changes (18, 30, 66, 67). Here, we summarize the recent updates on the roles of VD, VA, and PUFAs on lupus.
Vitamin D deficiency is increasingly common, resulting in increased risks for multiple disorders (68, 69). Although VD can be synthesized by the body in sunlight, adequate VD in diet is recommended. VD plays an important role in the homeostasis of the immune system, through a nuclear receptor existing in all immune cells, VD receptor (VDR). Polymorphisms of VDR have been recently reported to be associated with SLE susceptibility (70). In SLE patients, lower VD levels are associated with higher SLE activity. Handono and colleagues found that 1,25(OH)2D3 can inhibit neutrophil extracellular trap (NET) formation in cultured cells from SLE patients with hypovitamin D (71). Inhibition of NETs prevents endothelial damage that promotes the progression of lupus disease (72), suggesting a possible benefit of supplying VD in SLE patients with suboptimal VD levels. Recently, it has been reported that VD supplementation increases the number of Treg cells and induces the shift toward Th2 response in pre-menopausal female SLE patients, although a direct efficacy toward disease activity was not observed (73, 74). Likewise, no correlation was found between SLE-associated cytokine profiles and VD levels (75). However, in juvenile-onset SLE, which is more aggressive than adult SLE, dietary intake of VD has been reported to preclude disease progression in several recent studies (75–78). It is worth noting that the doses of VD utilized in these studies were different – one was rather intensive (50,000 international units or IU/week) and the other was more standard (2,000 IU daily) – but the outcomes were similar with improvement of SLE Disease Activity Index. Further studies are required to verify these findings in juvenile-onset SLE, and to explore the mechanisms of why a lack of response to VD was seen in adult SLE.
Vitamin A has long been recognized as an immune regulator. VA exerts its effects mainly via all-trans-retinoic acid (tRA), an active metabolite of VA. For SLE, the role of VA has been revealed through oral administration of tRA to either SLE patients or lupus-prone mice. In SLE patients, some benefit of tRA to ameliorate lupus nephritis and proteinuria has been reported (79, 80). For murine lupus, several mouse models, including NZB/W F1 and MRL/lpr, showed reduced proteinuria and renal damage when supplemented with tRA (81–85). In our study (85), although tRA treatment improved lupus-like kidney disease in MRL/lpr mice, there were serious side effects: worsened inflammation in the skin, brain, and lung, as well as increased levels of circulating autoantibodies. Our findings suggest the need to monitor diverse organs in SLE patients if tRA were used as a treatment, avoiding any potential damage to organs other than the kidneys.
Polyunsaturated fatty acids, with the main representative being omega-3 fatty acid, have been studied as complementary or alternative treatments for SLE for many years. Omega-3 PUFAs cannot be synthesized by the human body or other mammals. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are two well-recognized members of omega-3 PUFAs that are found in deep sea cold water fish. Fish oil is thereby utilized in some animal studies and clinical trials to test the efficacies of omega-3 PUFAs. In 1980s, DHA and EPA were both demonstrated to ameliorate renal disease, reduce anti-dsDNA autoantibody levels, and prolong lifespan of NZB/W F1 mice (86–88). It was found that fish oil prevents murine lupus by reducing levels of various pro-inflammatory cytokines, including IL-1β, IL-6, TNFα, and TGFβ, and increasing the expression of antioxidant enzymes (89–95). In addition, Fernandes and colleagues found that DHA-enriched fish oil, compared to EPA-enriched fish oil, was better at attenuating renal disease and increasing the survival of NZB/W F1 mice (90). This suggests that the relative abundance of EPA and DHA in fish oil might impact the outcomes of experiments designed to examine the effects of fish oil on SLE. Moreover, a recent study reported that omega-6 PUFAs did not have the same beneficial effect on lupus nephritis as omega-3 PUFAs (96). The disease-ameliorating effect of omega-3 PUFA against murine lupus was further confirmed in several lupus-prone mouse models other than NZB/W F1 (86, 97, 98). Starting late 1980s, more than 10 interventional studies with omega-3 PUFAs as treatments have been done in patients with SLE. Some studies showed promising results, especially for SLE patients with cardiovascular disease, which has emerged as an important cause of death in patients with SLE (99, 100).
While VD, VA, and PUFAs are known to change the composition of gut microtioba (21, 101, 102), how different dietary components modulate the microbiota of SLE patients and subsequent disease is unclear. One recent study has described diet-mediated increases of specific microbial genera that are known to be lower in SLE (103). Further studies are necessary to determine whether the modulation of diet – likely to be less expensive and safer than immunosuppressive drugs – can be effective at establishing a healthy balance between the host and symbiotic microbiota in the gut of SLE patients. If so, diet modulation might become a cost-effective approach for the management of SLE.
Bacterial Antigens and SLE
Bacteria constitute a large part of the symbiotic microbiota living in our body. Diverse components of Gram-positive and Gram-negative bacteria have been reported to contribute to the initiation and maintenance of lupus disease through stimulating TLRs, especially TLR2 and TLR4. TLRs are pattern recognition receptors that can recognize invading microorganisms bearing pathogen-associated molecular patterns (104). Details of TLR signaling pathways and their effects on autoimmune diseases, including SLE, have been reviewed elsewhere (105). In the current review, we will focus on the roles of bacterial antigens in lupus and their possible link to the sex bias observed in SLE. We hypothesize that commensal bacteria naturally present in our microbiota might provide autoantigens that mediate the development of SLE.
Lipopolysaccharide
Lipopolysaccharide (LPS) is a Gram-negative cell wall component that can be recognized by TLR4. In SLE patients, soluble CD14 (sCD14), which is released by monocytes in response to LPS, is increased in the blood (106). The level of sCD14 is highly correlated with disease activity parameters, suggesting the involvement of LPS in lupus development. In addition, repeated injections of LPS into lupus-prone mice resulted in increased autoantibody production and development of glomerulonephritis (107–111). Activation of TLR4 also promotes lupus disease activity in transgenic mice (107, 112, 113). Lupus spontaneously develops in mice with overexpression of a molecular chaperone of TLR4 that increases its responsiveness; but when commensal bacterial flora was deleted through treatment with antibiotics, the enhanced lupus phenotype was largely ameliorated (107). This suggests that TLR4 hyperresponsiveness to gut flora (which contains LPS) plays an essential role in lupus development. Moreover, Ni and colleagues found increased levels of serum autoantibodies and more severe lung injury when challenging apolipoprotein E-deficient (ApoE−/−) mice with LPS (114). Furthermore, immunization of non-autoimmune mice (C57BL/6 or BALB/c) with phospholipid-binding proteins induced lupus-like disease, and this was facilitated by the presence of LPS (115–117). Taken together, these data suggest that enhanced TLR4 signaling by LPS stimulation is sufficient to induce SLE. LPS might do so by inducing neutrophil activation and migration (118–120), key processes that promote the development of SLE (72). Inhibition of TLR4, on the other hand, reduces autoantibody production and decreases glomerular IgG deposits in the kidney for some lupus-prone murine models (121, 122). However, in TLR4-knockout MRL/lpr mice, disease activity was not modified (123). This may be due to the different genetic backgrounds of the mice strains. Further testing of disease outcome through TLR4 knockout should be done in additional strains of lupus-prone mice to determine the role of TLR4 deficiency in lupus.
In addition to the effect of LPS on neutrophil activation (118–120), several recent studies have explored the mechanisms by which LPS induces lupus. Qin et al. reported that the interaction of TLR4 and LPS strongly induced CD40 expression in macrophages and microglia (124). It was also found that LPS had the ability to increase CD40 mRNA expression in various tissues, including liver and kidney, in NZB/W F1 mice (125). CD40 silencing reduced the glomerular deposits of IgG and C3 in these mice, revealing a possible role of LPS-TLR4-CD40 signaling in the pathogenesis of lupus. Another possible role for LPS-TLR4 in lupus is to induce autoantibody production or isotype switching toward more pathogenic immunoglobulins, like IgG (126). Both MyD88- and TRIF-mediated signaling pathways are believed to contribute to increased autoantibody levels, though TRIF may play a more important role in driving autoantigen-specific IgG response (126). Moreover, it has been found that IL-18 is induced by LPS stimulation and this cytokine may cooperate with LPS–TLR4 in breaking the tolerance in mice with lupus nephritis (127).
Systemic lupus erythematosus is a female-biased disorder. Accumulating evidences have linked TLR4 function to estrogen and estrogen receptor α (ERα). Studies by Gilkeson’s group have found that female SLE patients possess more active monocytes with enhanced TLR4 responsiveness than male SLE patients (128). In lupus-prone mice, ERα deficiency ameliorated renal damage and prolonged survival compared to ERα-sufficient controls (129). Importantly, knocking out ERα in both lupus-prone and control mice resulted in impaired TLR4 activation in immune cells, indicating that estrogen and ER signaling can influence TLR4 responsiveness (130, 131). These results suggest possible contribution of TLR4 activation to sex bias in SLE.
Other Bacterial Antigens
Lipoteichoic acid (LTA), a major component of Gram-positive bacterial wall, is also reported to be involved in lupus pathogenesis. LTA is a ligand for TLR2, whose expression is increased in T cells, B cells, and monocytes from SLE patients (132). Increased TLR2 leads to enhanced IL-17A and IL-17F production and is associated with inflammatory response of CD4+ T cells. In mice, TLR2 activation is known to trigger lupus nephritis (133). In both B6/lpr mice and pristine-induced lupus mice, TLR2 knockout resulted in decreased autoantibody levels and ameliorated lupus-like symptoms (121, 134, 135). However, like the deficiency of TLR4, in MRL/lpr mice, TLR2 deficiency did not affect lupus pathogenesis (123, 136), possibly due to mouse strain differences.
Another bacterial antigen and component of bacterial biofilms, amyloid fiber (curli), has been reported to induce autoantibody production (137). Amyloid fibers can tightly bind to extracellular DNA that exists in many bacterial biofilms. Amyloid-DNA composites have been found to be strong stimulators of both innate and adaptive responses, with the ability to promote IL-6 and TNFα production and type I interferon response in NZB/W F1 mice (138). Importantly, injection of curli-DNA composites greatly increased the autoantibody level in lupus-prone mice, and even stimulated autoantibody production in wild-type mice. Using an amyloid-induced lupus model, Cao and colleagues have recently uncovered important roles of natural killer cells and IFNγ in SLE pathogenesis downstream of type I interferon response (139).
The “SLE Microbiota”
The significance of symbiotic microbiota in the development of T1D has been shown in non-obese diabetic mice, which spontaneously develop T1D with a bias toward females (11, 12). The function of microbiota in T1D is found to be highly associated with sex hormones. Fecal transplant of male gut microbiota to female mice ameliorated the disease and increased testosterone. For SLE, although the initial comparison between lupus-prone mice in germ-free vs. conventional housing conditions showed no difference in disease severity (140), emerging evidences in both SLE patients and lupus-prone mice point to a potential link between lupus and microbiota (Figure 1).
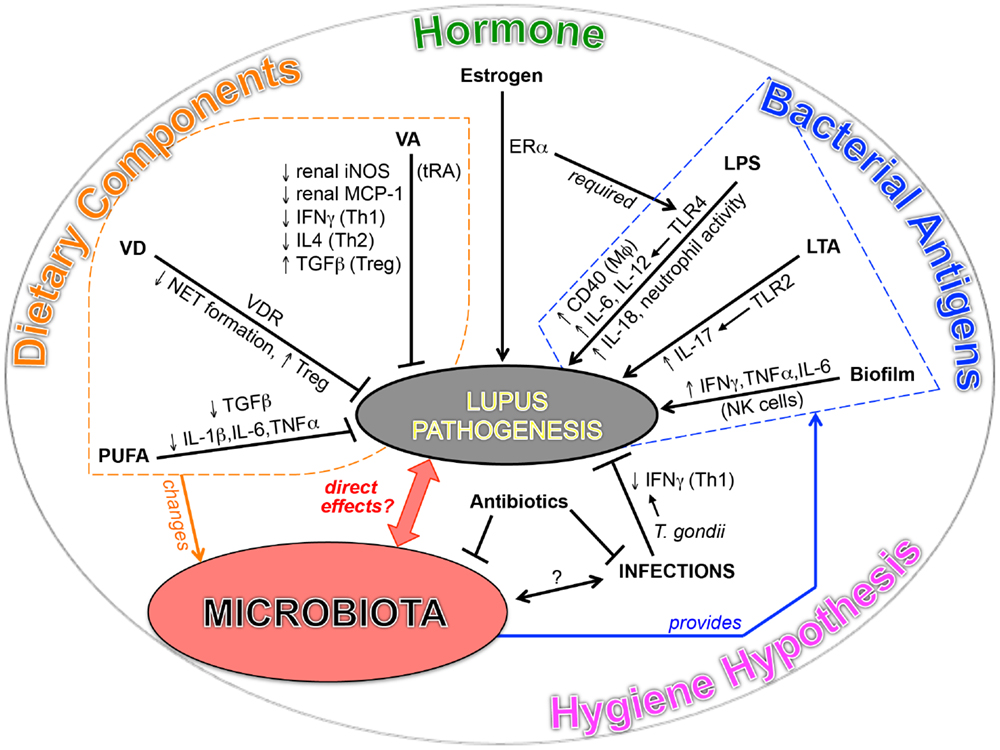
Figure 1. Emerging evidences point to a potential link between SLE and microbiota.
Intestinal dysbiosis has been reported in SLE patients. Compared to age- and sex-matched healthy controls, the fecal Firmicutes/Bacteroidetes ratio was found to be significantly lower in SLE patients even during remission (22). The same research group also described alterations in the composition and metabolic functions of gut microbiota in SLE (63). In mice, a recent study has shown that ANA production, a hallmark feature of autoimmune diseases that include SLE, is affected by neonatal colonization of gut microbiota (141). Using mice deficient of lymphotoxin-β receptor (LTβR) – the signaling of which controls development of secondary lymphoid organs – the authors found that LTβR-expressing RORγT+ innate lymphoid cells, located in the intestinal lamina propria, were important for the maintenance of immunological tolerance. Importantly, it was found that antibiotics-mediated removal of segmented filamentous bacteria (SFB) inhibited the development of ANA (141). However, in another recent study, SFB were found to be unassociated with the outcome of lupus in (SWR × NZB)-F1 (SNF1) mice (23). When given acidic pH water, SNF1 mice showed slower development of nephritis and a lower level of circulating ANA, and the improved outcome was associated with changes of gut microbiota unrelated with SFB (23). In their study, the relative abundance of Lactobacillus and the ratio of Firmicutes/Bacteroidetes were higher in mice with lower lupus severity (23). These changes were consistent, respectively, with our results in MRL/lpr mice (21) and the findings of microbiota composition in human SLE patients (22). The same authors have also reported the role of gut immune cells in female-biased development of lupus in SNF1 mice (142). Compared to male counterparts, the gut mucosa of female SNF1 mice has a higher frequency of gut-imprinted α4β7 T cells, higher expression of type I interferons, and a larger number of cells secreting IL-17, IL-22, and IL-9 (142). Altogether, the intestinal microenvironment, including microbiota, immune cells and cytokines, could contribute to the development of lupus.
Our research group has recently found that, in female lupus-prone mice, there are significant reduction of Lactobacillaceae and increase of Lachnospiraceae both prior to disease onset and in the late stage of disease with severe lupus symptoms (21). We also found that lupus-like symptoms, including nephritis, were improved with oral treatment of tRA. Importantly, the improvement was highly associated with the ability of tRA to restore Lactobacilli (21). Our work shows the potential benefits of modulating gut microbiota, especially by increasing the level of Lactobacilli, in the treatment of lupus. Lactobacilli can be introduced as probiotics, which are known to be beneficial to the host when administered in adequate amounts. Proposed health benefits provided by the consumption of Lactobacilli include prevention of constipation, hepatic disease, infections, allergies, and as recently suggested, inhibition of autoimmune diseases such as IBD and T1D (143–149). Some Lactobacillus strains have been demonstrated to exert specific effects that include modulation of host microbiota, inhibiting the formation of NETs, improving antioxidant status, or increasing the expression of genes encoding junction and adhesion proteins (150–152). This suggests an attractive prospective of utilizing certain strains of Lactobacillus in disease management for SLE.
To directly examine the potential effects of sex and gut microbiota on SLE, one approach would be to correct the imbalanced microbial composition associated with SLE with fecal transplantation – from healthy individuals to patients, or from males to females – and see if the correction ameliorates disease symptoms. This is yet to be reported for either lupus-prone mouse models or SLE patients, and remains an area that researchers actively explore.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell (2006) 124:837–48. doi: 10.1016/j.cell.2006.02.017
2. Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol (2013) 13(5):321–35. doi:10.1038/nri3430
3. Kuhn KA, Stappenbeck TS. Peripheral education of the immune system by the colonic microbiota. Semin Immunol (2013) 25(5):364–9. doi:10.1016/j.smim.2013.10.002
4. Strober W. Impact of the gut microbiome on mucosal inflammation. Trends Immunol (2013) 34(9):423–30. doi:10.1016/j.it.2013.07.001
5. Yeoh N, Burton JP, Suppiah P, Reid G, Stebbings S. The role of the microbiome in rheumatic diseases. Curr Rheumatol Rep (2013) 15(3):314. doi:10.1007/s11926-012-0314-y
6. Brown EM, Sadarangani M, Finlay BB. The role of the immune system in governing host-microbe interactions in the intestine. Nat Immunol (2013) 14(7):660–7. doi:10.1038/ni.2611
7. Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe (2010) 8:292–300. doi:10.1016/j.chom.2010.08.004
8. Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A (2007) 104:13780–5. doi:10.1073/pnas.0706625104
9. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature (2008) 455(7216):1109–13. doi:10.1038/nature07336
10. Mathis D, Benoist C. The influence of the microbiota on type-1 diabetes: on the threshold of a leap forward in our understanding. Immunol Rev (2012) 245(1):239–49. doi:10.1111/j.1600-065X.2011.01084.x
11. Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity (2013) 39(2):400–12. doi:10.1016/j.immuni.2013.08.013
12. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339(6123):1084–8. doi:10.1126/science.1233521
13. Vaahtovuo J, Munukka E, Korkeamaki M, Luukkainen R, Toivanen P. Fecal microbiota in early rheumatoid arthritis. J Rheumatol (2008) 35(8):1500–5.
14. Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest (2008) 118(1):205–16. doi:10.1172/JCI32639
15. Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity (2010) 32(6):815–27. doi:10.1016/j.immuni.2010.06.001
16. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4615–22. doi:10.1073/pnas.1000082107
17. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature (2011) 479(7374):538–41. doi:10.1038/nature10554
18. Vieira SM, Pagovich OE, Kriegel MA. Diet, microbiota and autoimmune diseases. Lupus (2014) 23(6):518–26. doi:10.1177/0961203313501401
19. Lim SS, Bayakly AR, Helmick CG, Gordon C, Easley KA, Drenkard C. The incidence and prevalence of systemic lupus erythematosus, 2002-2004: the Georgia lupus registry. Arthritis Rheumatol (2014) 66(2):357–68. doi:10.1002/art.38239
20. Somers EC, Marder W, Cagnoli P, Lewis EE, DeGuire P, Gordon C, et al. Population-based incidence and prevalence of systemic lupus erythematosus: the Michigan Lupus Epidemiology and Surveillance program. Arthritis Rheumatol (2014) 66(2):369–78. doi:10.1002/art.38238
21. Zhang H, Liao X, Sparks JB, Luo XM. Dynamics of gut microbiota in autoimmune lupus. Appl Environ Microbiol (2014) 80(24):7551–60. doi:10.1128/AEM.02676-14
22. Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio (2014) 5(5):e1548–1514. doi:10.1128/mBio.01548-14
23. Johnson BM, Gaudreau MC, Al-Gadban MM, Gudi R, Vasu C. Impact of dietary deviation on disease progression and gut microbiome composition in lupus-prone SNF1 mice. Clin Exp Immunol (2015) 181(2):323–37. doi:10.1111/cei.12609
24. Hewagama A, Richardson B. The genetics and epigenetics of autoimmune diseases. J Autoimmun (2009) 33(1):3–11. doi:10.1016/j.jaut.2009.03.007
25. Symmons DP. Frequency of lupus in people of African origin. Lupus (1995) 4(3):176–8. doi:10.1177/096120339500400303
26. Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med (2002) 347(12):911–20. doi:10.1056/NEJMra020100
27. Sawalha AH, Jeffries M, Webb R, Lu Q, Gorelik G, Ray D, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun (2008) 9(4):368–78. doi:10.1038/gene.2008.29
28. Bijl M, Kallenberg CG. Ultraviolet light and cutaneous lupus. Lupus (2006) 15(11):724–7. doi:10.1177/0961203306071705
29. Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet (2013) 4:132. doi:10.3389/fgene.2013.00132
30. Somers EC, Richardson BC. Environmental exposures, epigenetic changes and the risk of lupus. Lupus (2014) 23(6):568–76. doi:10.1177/0961203313499419
31. Uramoto KM, Michet CJ Jr, Thumboo J, Sunku J, O’Fallon WM, Gabriel SE. Trends in the incidence and mortality of systemic lupus erythematosus, 1950-1992. Arthritis Rheum (1999) 42(1):46–50. doi:10.1002/1529-0131(199901)42:1<46::AID-ANR6>3.0.CO;2-2
32. Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus (2006) 15(5):308–18. doi:10.1191/0961203306lu2305xx
33. Voss A, Green A, Junker P. Systemic lupus erythematosus in Denmark: clinical and epidemiological characterization of a county-based cohort. Scand J Rheumatol (1998) 27(2):98–105. doi:10.1080/030097498440958
34. Cooke A, Tonks P, Jones FM, O’Shea H, Hutchings P, Fulford AJ, et al. Infection with Schistosoma mansoni prevents insulin dependent diabetes mellitus in non-obese diabetic mice. Parasite Immunol (1999) 21(4):169–76. doi:10.1046/j.1365-3024.1999.00213.x
35. Strachan DP. Hay fever, hygiene, and household size. BMJ (1989) 299(6710):1259–60. doi:10.1136/bmj.299.6710.1259
36. Hayashi T, Lee S, Ogasawara H, Sekigawa I, Iida N, Tomino Y, et al. Exacerbation of systemic lupus erythematosus related to cytomegalovirus infection. Lupus (1998) 7(8):561–4. doi:10.1191/096120398678920596
37. James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest (1997) 100(12):3019–26. doi:10.1172/JCI119856
38. Nawata M, Seta N, Yamada M, Sekigawa I, Lida N, Hashimoto H. Possible triggering effect of cytomegalovirus infection on systemic lupus erythematosus. Scand J Rheumatol (2001) 30(6):360–2. doi:10.1080/030097401317148570
39. Nelson P, Rylance P, Roden D, Trela M, Tugnet N. Viruses as potential pathogenic agents in systemic lupus erythematosus. Lupus (2014) 23(6):596–605. doi:10.1177/0961203314531637
40. Rasmussen NS, Draborg AH, Nielsen CT, Jacobsen S, Houen G. Antibodies to early EBV, CMV, and HHV6 antigens in systemic lupus erythematosus patients. Scand J Rheumatol (2015) 44(2):143–9. doi:10.3109/03009742.2014.973061
41. Tanaka K, Sawamura S, Satoh T, Kobayashi K, Noda S. Role of the indigenous microbiota in maintaining the virus-specific CD8 memory T cells in the lung of mice infected with murine cytomegalovirus. J Immunol (2007) 178(8):5209–16. doi:10.4049/jimmunol.178.8.5209
42. Praprotnik S, Sodin-Semrl S, Tomsic M, Shoenfeld Y. The curiously suspicious: infectious disease may ameliorate an ongoing autoimmune destruction in systemic lupus erythematosus patients. J Autoimmun (2008) 30(1–2):37–41. doi:10.1016/j.jaut.2007.11.002
43. Ram M, Anaya JM, Barzilai O, Izhaky D, Porat Katz BS, Blank M, et al. The putative protective role of hepatitis B virus (HBV) infection from autoimmune disorders. Autoimmun Rev (2008) 7(8):621–5. doi:10.1016/j.autrev.2008.06.008
44. Esposito S, Bosis S, Semino M, Rigante D. Infections and systemic lupus erythematosus. Eur J Clin Microbiol Infect Dis (2014) 33(9):1467–75. doi:10.1007/s10096-014-2098-7
45. Sawalha AH, Schmid WR, Binder SR, Bacino DK, Harley JB. Association between systemic lupus erythematosus and Helicobacter pylori seronegativity. J Rheumatol (2004) 31(8):1546–50.
46. Wasley A, Kruszon-Moran D, Kuhnert W, Simard EP, Finelli L, McQuillan G, et al. The prevalence of hepatitis B virus infection in the United States in the era of vaccination. J Infect Dis (2010) 202(2):192–201. doi:10.1086/653622
47. Chen Y, Blaser MJ. Helicobacter pylori colonization is inversely associated with childhood asthma. J Infect Dis (2008) 198(4):553–60. doi:10.1086/590158
48. den Hoed CM, Vila AJ, Holster IL, Perez-Perez GI, Blaser MJ, de Jongste JC, et al. Helicobacter pylori and the birth cohort effect: evidence for stabilized colonization rates in childhood. Helicobacter (2011) 16(5):405–9. doi:10.1111/j.1523-5378.2011.00854.x
49. McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature (2015) 523(7562):612–6. doi:10.1038/nature14468
50. Ingram JT, Yi JS, Zajac AJ. Exhausted CD8 T cells downregulate the IL-18 receptor and become unresponsive to inflammatory cytokines and bacterial co-infections. PLoS Pathog (2011) 7(9):e1002273. doi:10.1371/journal.ppat.1002273
51. Chen M, Aosai F, Norose K, Mun HS, Ishikura H, Hirose S, et al. Toxoplasma gondii infection inhibits the development of lupus-like syndrome in autoimmune (New Zealand Black x New Zealand White) F1 mice. Int Immunol (2004) 16(7):937–46. doi:10.1093/intimm/dxh095
52. Al-Quraishy S, Abdel-Maksoud MA, El-Amir A, Abdel-Ghaffar FA, Badr G. Malarial infection of female BWF1 lupus mice alters the redox state in kidney and liver tissues and confers protection against lupus nephritis. Oxid Med Cell Longev (2013) 2013:156562. doi:10.1155/2013/156562
53. Greenwood BM, Herrick EM, Voller A. Suppression of autoimmune disease in NZB and (NZB x NZW) F1 hybrid mice by infection with malaria. Nature (1970) 226(5242):266–7. doi:10.1038/226266a0
54. Sato MN, Minoprio P, Avrameas S, Ternynck T. Changes in the cytokine profile of lupus-prone mice (NZB/NZW)F1 induced by Plasmodium chabaudi and their implications in the reversal of clinical symptoms. Clin Exp Immunol (2000) 119(2):333–9. doi:10.1046/j.1365-2249.2000.01124.x
55. Hayashi T. Effect of prostaglandin E2 on plasma lactic dehydrogenase activity in (NZB x NZW)F1 mice with a chronic infection of lactic dehydrogenase virus. J Comp Pathol (1992) 107(1):41–8. doi:10.1016/0021-9975(92)90094-B
56. Hayashi T, Mori I, Yamamoto H. Lactic dehydrogenase virus infection prevents development of anti-nuclear antibody in (NZB x NZW)F1 mice; role of prostaglandin E2 and macrophage Ia antigen expression. Int J Exp Pathol (1992) 73(5):593–601.
57. Hayashi T, Noguchi Y, Kameyama Y. Suppression of development of anti-nuclear antibody and glomerulonephritis in NZB x NZWF1 mice by persistent infection with lactic dehydrogenase virus: possible involvement of superoxide anion as a progressive effector. Int J Exp Pathol (1993) 74(6):553–60.
58. Oldstone MB, Dixon FJ. Inhibition of antibodies to nuclear antigen and to DNA in New Zealand mice infected with lactate dehydrogenase virus. Science (1972) 175(4023):784–6. doi:10.1126/science.175.4023.784
59. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341(6145):569–73. doi:10.1126/science.1241165
60. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi:10.1038/nature12726
61. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504(7480):446–50. doi:10.1038/nature12721
62. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40(1):128–39. doi:10.1016/j.immuni.2013.12.007
63. Rojo D, Hevia A, Bargiela R, Lopez P, Cuervo A, Gonzalez S, et al. Ranking the impact of human health disorders on gut metabolism: systemic lupus erythematosus and obesity as study cases. Sci Rep (2015) 5:8310. doi:10.1038/srep08310
64. Van Boeckel TP, Gandra S, Ashok A, Caudron Q, Grenfell BT, Levin SA, et al. Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect Dis (2014) 14(8):742–50. doi:10.1016/S1473-3099(14)70780-7
65. Hogberg LD, Muller A, Zorzet A, Monnet DL, Cars O. Antibiotic use worldwide. Lancet Infect Dis (2014) 14(12):1179–80. doi:10.1016/S1473-3099(14)70987-9
66. Dorrestein PC, Mazmanian SK, Knight R. Finding the missing links among metabolites, microbes, and the host. Immunity (2014) 40(6):824–32. doi:10.1016/j.immuni.2014.05.015
67. Kamen DL. Environmental influences on systemic lupus erythematosus expression. Rheum Dis Clin North Am (2014) 40(3):401–12. doi:10.1016/j.rdc.2014.05.003
68. Ginde AA, Liu MC, Camargo CA Jr. Demographic differences and trends of vitamin D insufficiency in the US population, 1988-2004. Arch Intern Med (2009) 169(6):626–32. doi:10.1001/archinternmed.2008.604
70. Carvalho C, Marinho A, Leal B, Bettencourt A, Boleixa D, Almeida I, et al. Association between vitamin D receptor (VDR) gene polymorphisms and systemic lupus erythematosus in Portuguese patients. Lupus (2015) 24(8):846–53. doi:10.1177/0961203314566636
71. Handono K, Sidarta YO, Pradana BA, Nugroho RA, Hartono IA, Kalim H, et al. Vitamin D prevents endothelial damage induced by increased neutrophil extracellular traps formation in patients with systemic lupus erythematosus. Acta Med Indones (2014) 46(3):189–98.
72. Smith CK, Kaplan MJ. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol (2015) 27(5):448–53. doi:10.1097/BOR.0000000000000197
73. Andreoli L, Dall’Ara F, Piantoni S, Zanola A, Piva N, Cutolo M, et al. A 24-month prospective study on the efficacy and safety of two different monthly regimens of vitamin D supplementation in pre-menopausal women with systemic lupus erythematosus. Lupus (2015) 24(4–5):499–506. doi:10.1177/0961203314559089
74. Piantoni S, Andreoli L, Scarsi M, Zanola A, Dall’Ara F, Pizzorni C, et al. Phenotype modifications of T-cells and their shift toward a Th2 response in patients with systemic lupus erythematosus supplemented with different monthly regimens of vitamin D. Lupus (2015) 24(4–5):490–8. doi:10.1177/0961203314559090
75. Schneider L, Colar da Silva AC, Werres Junior LC, Alegretti AP, Dos Santos AS, Santos M, et al. Vitamin D levels and cytokine profiles in patients with systemic lupus erythematosus. Lupus (2015) 24(11):1191–7. doi:10.1177/0961203315584811
76. Habibi S, Saleem MA, Ramanan AV. Juvenile systemic lupus erythematosus: review of clinical features and management. Indian Pediatr (2011) 48(11):879–87. doi:10.1007/s13312-011-0143-5
77. Hoffman IE, Lauwerys BR, De Keyser F, Huizinga TW, Isenberg D, Cebecauer L, et al. Juvenile-onset systemic lupus erythematosus: different clinical and serological pattern than adult-onset systemic lupus erythematosus. Ann Rheum Dis (2009) 68(3):412–5. doi:10.1136/ard.2008.094813
78. Lima GL, Paupitz J, Aikawa NE, Takayama L, Bonfa E, Pereira RM. A randomized double-blind placebo-controlled trial of vitamin D supplementation in adolescents and young adults with Juvenile-onset SLE: improvement in disease activity and fatigue scores. Arthritis Care Res (Hoboken) (2015). doi:10.1002/acr.22621
79. Kinoshita K, Kishimoto K, Shimazu H, Nozaki Y, Sugiyama M, Ikoma S, et al. Successful treatment with retinoids in patients with lupus nephritis. Am J Kidney Dis (2010) 55(2):344–7. doi:10.1053/j.ajkd.2009.06.012
80. Kinoshita K, Funauchi M. [Therapeutic effect of retinoic acid in lupus nephritis]. Nihon Rinsho Meneki Gakkai Kaishi (2012) 35(1):1–7. doi:10.2177/jsci.35.1
81. Kinoshita K, Yoo BS, Nozaki Y, Sugiyama M, Ikoma S, Ohno M, et al. Retinoic acid reduces autoimmune renal injury and increases survival in NZB/W F1 mice. J Immunol (2003) 170(11):5793–8. doi:10.4049/jimmunol.170.11.5793
82. Perez de Lema G, Lucio-Cazana FJ, Molina A, Luckow B, Schmid H, de Wit C, et al. Retinoic acid treatment protects MRL/lpr lupus mice from the development of glomerular disease. Kidney Int (2004) 66(3):1018–28. doi:10.1111/j.1523-1755.2004.00850.x
83. Nozaki Y, Yamagata T, Yoo BS, Sugiyama M, Ikoma S, Kinoshita K, et al. The beneficial effects of treatment with all-trans-retinoic acid plus corticosteroid on autoimmune nephritis in NZB/WF mice. Clin Exp Immunol (2005) 139(1):74–83. doi:10.1111/j.1365-2249.2005.02654.x
84. Miyabe Y, Miyabe C, Nanki T. Could retinoids be a potential treatment for rheumatic diseases? Rheumatol Int (2015) 35(1):35–41. doi:10.1007/s00296-014-3067-2
85. Liao X, Ren J, Wei CH, Ross AC, Cecere TE, Jortner BS, et al. Paradoxical effects of all-trans-retinoic acid on lupus-like disease in the MRL/lpr mouse model. PLoS One (2015) 10(3):e0118176. doi:10.1371/journal.pone.0118176
86. Robinson DR, Prickett JD, Makoul GT, Steinberg AD, Colvin RB. Dietary fish oil reduces progression of established renal disease in (NZB x NZW)F1 mice and delays renal disease in BXSB and MRL/1 strains. Arthritis Rheum (1986) 29(4):539–46. doi:10.1002/art.1780290412
87. Robinson DR, Prickett JD, Polisson R, Steinberg AD, Levine L. The protective effect of dietary fish oil on murine lupus. Prostaglandins (1985) 30(1):51–75. doi:10.1016/S0090-6980(85)80010-1
88. Prickett JD, Robinson DR, Steinberg AD. Effects of dietary enrichment with eicosapentaenoic acid upon autoimmune nephritis in female NZB X NZW/F1 mice. Arthritis Rheum (1983) 26(2):133–9. doi:10.1002/art.1780260203
89. Halade GV, Williams PJ, Veigas JM, Barnes JL, Fernandes G. Concentrated fish oil (Lovaza(R)) extends lifespan and attenuates kidney disease in lupus-prone short-lived (NZBxNZW)F1 mice. Exp Biol Med (Maywood) (2013) 238(6):610–22. doi:10.1177/1535370213489485
90. Halade GV, Rahman MM, Bhattacharya A, Barnes JL, Chandrasekar B, Fernandes G. Docosahexaenoic acid-enriched fish oil attenuates kidney disease and prolongs median and maximal life span of autoimmune lupus-prone mice. J Immunol (2010) 184(9):5280–6. doi:10.4049/jimmunol.0903282
91. Kim YJ, Kim HJ, No JK, Chung HY, Fernandes G. Anti-inflammatory action of dietary fish oil and calorie restriction. Life Sci (2006) 78(21):2523–32. doi:10.1016/j.lfs.2005.10.034
92. Bhattacharya A, Lawrence RA, Krishnan A, Zaman K, Sun D, Fernandes G. Effect of dietary n-3 and n-6 oils with and without food restriction on activity of antioxidant enzymes and lipid peroxidation in livers of cyclophosphamide treated autoimmune-prone NZB/W female mice. J Am Coll Nutr (2003) 22(5):388–99. doi:10.1080/07315724.2003.10719322
93. Chin YH, Ye MW, Cai JP, Xu XM. Differential regulation of tissue-specific lymph node high endothelial venule cell adhesion molecules by tumour necrosis factor and transforming growth factor-beta 1. Immunology (1996) 87(4):559–65. doi:10.1046/j.1365-2567.1996.490562.x
94. Chandrasekar B, Troyer DA, Venkatraman JT, Fernandes G. Dietary omega-3 lipids delay the onset and progression of autoimmune lupus nephritis by inhibiting transforming growth factor beta mRNA and protein expression. J Autoimmun (1995) 8(3):381–93. doi:10.1006/jaut.1995.0030
95. Chandrasekar B, Fernandes G. Decreased pro-inflammatory cytokines and increased antioxidant enzyme gene expression by omega-3 lipids in murine lupus nephritis. Biochem Biophys Res Commun (1994) 200(2):893–8. doi:10.1006/bbrc.1994.1534
96. Pestka JJ, Vines LL, Bates MA, He K, Langohr I. Comparative effects of n-3, n-6 and n-9 unsaturated fatty acid-rich diet consumption on lupus nephritis, autoantibody production and CD4+ T cell-related gene responses in the autoimmune NZBWF1 mouse. PLoS One (2014) 9(6):e100255. doi:10.1371/journal.pone.0100255
97. Westberg G, Tarkowski A, Svalander C. Effect of eicosapentaenoic acid rich menhaden oil and MaxEPA on the autoimmune disease of Mrl/l mice. Int Arch Allergy Appl Immunol (1989) 88(4):454–61. doi:10.1159/000234732
98. Kelley VE, Ferretti A, Izui S, Strom TB. A fish oil diet rich in eicosapentaenoic acid reduces cyclooxygenase metabolites, and suppresses lupus in MRL-lpr mice. J Immunol (1985) 134(3):1914–9.
99. Lozovoy MA, Simao AN, Morimoto HK, Scavuzzi BM, Iriyoda TV, Reiche EM, et al. Fish oil N-3 fatty acids increase adiponectin and decrease leptin levels in patients with systemic lupus erythematosus. Mar Drugs (2015) 13(2):1071–83. doi:10.3390/md13021071
100. Borges MC, Santos Fde M, Telles RW, Correia MI, Lanna CC. [Polyunsaturated omega-3 fatty acids and systemic lupus erythematosus: what do we know?]. Rev Bras Reumatol (2014) 54(6):459–66. doi:10.1016/j.rbr.2013.12.002
101. Ghosh S, Molcan E, DeCoffe D, Dai C, Gibson DL. Diets rich in n-6 PUFA induce intestinal microbial dysbiosis in aged mice. Br J Nutr (2013) 110(3):515–23. doi:10.1017/S0007114512005326
102. Cantorna MT, McDaniel K, Bora S, Chen J, James J. Vitamin D, immune regulation, the microbiota, and inflammatory bowel disease. Exp Biol Med (Maywood) (2014) 239(11):1524–30. doi:10.1177/1535370214523890
103. Cuervo A, Hevia A, Lopez P, Suarez A, Sanchez B, Margolles A, et al. Association of polyphenols from oranges and apples with specific intestinal microorganisms in systemic lupus erythematosus patients. Nutrients (2015) 7(2):1301–17. doi:10.3390/nu7021301
104. Sabroe I, Read RC, Whyte MK, Dockrell DH, Vogel SN, Dower SK. Toll-like receptors in health and disease: complex questions remain. J Immunol (2003) 171(4):1630–5. doi:10.4049/jimmunol.171.4.1630
105. Liu Y, Yin H, Zhao M, Lu Q. TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol (2014) 47(2):136–47. doi:10.1007/s12016-013-8402-y
106. Nockher WA, Wigand R, Schoeppe W, Scherberich JE. Elevated levels of soluble CD14 in serum of patients with systemic lupus erythematosus. Clin Exp Immunol (1994) 96(1):15–9. doi:10.1111/j.1365-2249.1994.tb06222.x
107. Liu B, Yang Y, Dai J, Medzhitov R, Freudenberg MA, Zhang PL, et al. TLR4 up-regulation at protein or gene level is pathogenic for lupus-like autoimmune disease. J Immunol (2006) 177(10):6880–8. doi:10.4049/jimmunol.177.10.6880
108. Zhang Y, Liu S, Yu Y, Zhang T, Liu J, Shen Q, et al. Immune complex enhances tolerogenecity of immature dendritic cells via FcgammaRIIb and promotes FcgammaRIIb-overexpressing dendritic cells to attenuate lupus. Eur J Immunol (2011) 41(4):1154–64. doi:10.1002/eji.201040767
109. Zhai JX, Zhang ZX, Feng YJ, Ding SS, Wang XH, Zou LW, et al. PDTC attenuate LPS-induced kidney injury in systemic lupus erythematosus-prone MRL/lpr mice. Mol Biol Rep (2012) 39(6):6763–71. doi:10.1007/s11033-012-1501-7
110. Granholm NA, Cavallo T. Bacterial lipopolysaccharide enhances deposition of immune complexes and exacerbates nephritis in BXSB lupus-prone mice. Clin Exp Immunol (1991) 85(2):270–7. doi:10.1111/j.1365-2249.1991.tb05717.x
111. Granholm NA, Cavallo T. Long-lasting effects of bacterial lipopolysaccharide promote progression of lupus nephritis in NZB/W mice. Lupus (1994) 3(6):507–14. doi:10.1177/096120339400300614
112. Lee TP, Tang SJ, Wu MF, Song YC, Yu CL, Sun KH. Transgenic overexpression of anti-double-stranded DNA autoantibody and activation of toll-like receptor 4 in mice induce severe systemic lupus erythematosus syndromes. J Autoimmun (2010) 35(4):358–67. doi:10.1016/j.jaut.2010.07.007
113. Lee TP, Huang JC, Liu CJ, Chen HJ, Chen YH, Tsai YT, et al. Interactions of surface-expressed TLR-4 and endosomal TLR-9 accelerate lupus progression in anti-dsDNA antibody transgenic mice. Exp Biol Med (Maywood) (2014) 239(6):715–23. doi:10.1177/1535370214525299
114. Ni JQ, Ouyang Q, Lin L, Huang Z, Lu H, Chen X, et al. Role of toll-like receptor 4 on lupus lung injury and atherosclerosis in LPS-challenge ApoE(-)/(-) mice. Clin Dev Immunol (2013) 2013:476856. doi:10.1155/2013/476856
115. Levine J, Subang R, Setty S, Cabrera J, Laplante P, Fritzler M, et al. Phospholipid-binding proteins differ in their capacity to induce autoantibodies and murine systemic lupus erythematosus. Lupus (2014) 23(8):752–68. doi:10.1177/0961203314525676
116. Levine JS, Subang R, Nasr SH, Fournier S, Lajoie G, Wither J, et al. Immunization with an apoptotic cell-binding protein recapitulates the nephritis and sequential autoantibody emergence of systemic lupus erythematosus. J Immunol (2006) 177(9):6504–16. doi:10.4049/jimmunol.177.9.6504
117. Tolomeo T, Rico De Souza A, Roter E, Dieude M, Amireault P, Subang R, et al. T cells demonstrate a Th1-biased response to native beta2-glycoprotein I in a murine model of anti-phospholipid antibody induction. Autoimmunity (2009) 42(4):292–5. doi:10.1080/08916930902828254
118. Aida Y, Pabst MJ. Neutrophil responses to lipopolysaccharide. Effect of adherence on triggering and priming of the respiratory burst. J Immunol (1991) 146(4):1271–6.
119. Doerfler ME, Danner RL, Shelhamer JH, Parrillo JE. Bacterial lipopolysaccharides prime human neutrophils for enhanced production of leukotriene B4. J Clin Invest (1989) 83(3):970–7. doi:10.1172/JCI113983
120. Guthrie LA, McPhail LC, Henson PM, Johnston RB Jr. Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide-producing enzyme. J Exp Med (1984) 160(6):1656–71. doi:10.1084/jem.160.6.1656
121. Lartigue A, Colliou N, Calbo S, Francois A, Jacquot S, Arnoult C, et al. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol (2009) 183(10):6207–16. doi:10.4049/jimmunol.0803219
122. Summers SA, Hoi A, Steinmetz OM, O’Sullivan KM, Ooi JD, Odobasic D, et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun (2010) 35(4):291–8. doi:10.1016/j.jaut.2010.05.004
123. Freeley SJ, Giorgini A, Tulone C, Popat RJ, Horsfield C, Robson MG. Toll-like receptor 2 or toll-like receptor 4 deficiency does not modify lupus in MRLlpr mice. PLoS One (2013) 8(9):e74112. doi:10.1371/journal.pone.0074112
124. Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN. LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia. Blood (2005) 106(9):3114–22. doi:10.1182/blood-2005-02-0759
125. Ripoll E, Merino A, Herrero-Fresneda I, Aran JM, Goma M, Bolanos N, et al. CD40 gene silencing reduces the progression of experimental lupus nephritis modulating local milieu and systemic mechanisms. PLoS One (2013) 8(6):e65068. doi:10.1371/journal.pone.0065068
126. Harlow L, Fernandez I, Soejima M, Ridgway WM, Ascherman DP. Characterization of TLR4-mediated auto-antibody production in a mouse model of histidyl-tRNA synthetase-induced myositis. Innate Immun (2012) 18(6):876–85. doi:10.1177/1753425912446714
127. Shui HA, Ka SM, Wu WM, Lin YF, Hou YC, Su LC, et al. LPS-evoked IL-18 expression in mesangial cells plays a role in accelerating lupus nephritis. Rheumatology (Oxford) (2007) 46(8):1277–84. doi:10.1093/rheumatology/kem136
128. Jiang W, Zhang L, Lang R, Li Z, Gilkeson G. Sex differences in monocyte activation in systemic lupus erythematosus (SLE). PLoS One (2014) 9(12):e114589. doi:10.1371/journal.pone.0114589
129. Svenson JL, EuDaly J, Ruiz P, Korach KS, Gilkeson GS. Impact of estrogen receptor deficiency on disease expression in the NZM2410 lupus prone mouse. Clin Immunol (2008) 128(2):259–68. doi:10.1016/j.clim.2008.03.508
130. Cunningham MA, Naga OS, Eudaly JG, Scott JL, Gilkeson GS. Estrogen receptor alpha modulates toll-like receptor signaling in murine lupus. Clin Immunol (2012) 144(1):1–12. doi:10.1016/j.clim.2012.04.001
131. Jiang W, Gilkeson G. Sex differences in monocytes and TLR4 associated immune responses; implications for systemic lupus erythematosus (SLE). J Immunother Appl (2014) 1:1. doi:10.7243/2055-2394-1-1
132. Liu Y, Liao J, Zhao M, Wu H, Yung S, Chan TM, et al. Increased expression of TLR2 in CD4 T cells from SLE patients enhances immune reactivity and promotes IL-17 expression through histone modifications. Eur J Immunol (2015) 45(9):2683–93. doi:10.1002/eji.201445219
133. Pawar RD, Castrezana-Lopez L, Allam R, Kulkarni OP, Segerer S, Radomska E, et al. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating toll-like receptor 2 at the glomerular filtration barrier. Immunology (2009) 128(1 Suppl):e206–21. doi:10.1111/j.1365-2567.2008.02948.x
134. Leiss H, Niederreiter B, Bandur T, Schwarzecker B, Bluml S, Steiner G, et al. Pristane-induced lupus as a model of human lupus arthritis: evolvement of autoantibodies, internal organ and joint inflammation. Lupus (2013) 22(8):778–92. doi:10.1177/0961203313492869
135. Urbonaviciute V, Starke C, Pirschel W, Pohle S, Frey S, Daniel C, et al. Toll-like receptor 2 is required for autoantibody production and development of renal disease in pristane-induced lupus. Arthritis Rheum (2013) 65(6):1612–23. doi:10.1002/art.37914
136. Ma K, Li J, Fang Y, Lu L. Roles of B cell-intrinsic TLR signals in systemic lupus erythematosus. Int J Mol Sci (2015) 16(6):13084–105. doi:10.3390/ijms160613084
137. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity (2015) 42(6):1171–84. doi:10.1016/j.immuni.2015.06.002
138. Gallucci S, Gallo P, Rapsinski G, Oppong G, Sriram U, Wilson R, et al. Bacterial amyloids promote type I interferon production and accelerate autoimmunity. AAI Annual Meeting (2015).
139. Huang X, Li J, Dorta-Estremera S, Di Domizio J, Anthony SM, Watowich SS, et al. Neutrophils regulate humoral autoimmunity by restricting interferon-gamma production via the generation of reactive oxygen species. Cell Rep (2015) 12(7):1120–32. doi:10.1016/j.celrep.2015.07.021
140. Maldonado MA, Kakkanaiah V, MacDonald GC, Chen F, Reap EA, Balish E, et al. The role of environmental antigens in the spontaneous development of autoimmunity in MRL-lpr mice. J Immunol (1999) 162(11):6322–30.
141. Van Praet JT, Donovan E, Vanassche I, Drennan MB, Windels F, Dendooven A, et al. Commensal microbiota influence systemic autoimmune responses. EMBO J (2015) 34(4):466–74. doi:10.15252/embj.201489966
142. Gaudreau MC, Johnson BM, Gudi R, Al-Gadban MM, Vasu C. Gender bias in lupus: does immune response initiated in the gut mucosa have a role? Clin Exp Immunol (2015) 180(3):393–407. doi:10.1111/cei.12587
143. Goldin BR, Gorbach SL. Clinical indications for probiotics: an overview. Clin Infect Dis (2008) 46(Suppl 2):S96–100. doi:10.1086/523333
144. Kaur IP, Kuhad A, Garg A, Chopra K. Probiotics: delineation of prophylactic and therapeutic benefits. J Med Food (2009) 12(2):219–35. doi:10.1089/jmf.2007.0544
145. Uskova MA, Kravchenko LV. [Antioxidant properties of lactic acid bacteria – probiotic and yogurt strains]. Vopr Pitan (2009) 78(2):18–23.
146. Cain AM, Karpa KD. Clinical utility of probiotics in inflammatory bowel disease. Altern Ther Health Med (2011) 17(1):72–9.
147. Lee J, Bang J, Woo HJ. Effect of orally administered Lactobacillus brevis HY7401 in a food allergy mouse model. J Microbiol Biotechnol (2013) 23(11):1636–40. doi:10.4014/jmb.1306.06047
148. Gomes AC, Bueno AA, de Souza RG, Mota JF. Gut microbiota, probiotics and diabetes. Nutr J (2014) 13:60. doi:10.1186/1475-2891-13-60
149. Cosenza L, Nocerino R, Di Scala C, di Costanzo M, Amoroso A, Leone L, et al. Bugs for atopy: the Lactobacillus rhamnosus GG strategy for food allergy prevention and treatment in children. Benef Microbes (2015) 6(2):225–32. doi:10.3920/BM2014.0158
150. Vong L, Lorentz RJ, Assa A, Glogauer M, Sherman PM. Probiotic Lactobacillus rhamnosus inhibits the formation of neutrophil extracellular traps. J Immunol (2014) 192(4):1870–7. doi:10.4049/jimmunol.1302286
151. Valladares R, Sankar D, Li N, Williams E, Lai KK, Abdelgeliel AS, et al. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP rats. PLoS One (2010) 5(5):e10507. doi:10.1371/journal.pone.0010507
Keywords: SLE, microbiota, hygiene hypothesis, bacterial antigens, diet, estrogen
Citation: Mu Q, Zhang H and Luo XM (2015) SLE: Another Autoimmune Disorder Influenced by Microbes and Diet? Front. Immunol. 6:608. doi: 10.3389/fimmu.2015.00608
Received: 22 August 2015; Accepted: 16 November 2015;
Published: 30 November 2015
Edited by:
Shohei Hori, RIKEN Center for Integrative Medical Sciences, JapanReviewed by:
Kazuhiko Yamamoto, The University of Tokyo Graduate School of Medicine, JapanHiroshi Ohno, RIKEN, Japan
Copyright: © 2015 Mu, Zhang and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin M. Luo, eGlubHVvQHZ0LmVkdQ==