 Victoria A. McGuire
Victoria A. McGuire J. Simon C. Arthur
J. Simon C. Arthur- Division of Cell Signalling and Immunology, School of Life Sciences, University of Dundee, Dundee, UK
Pathogenic bacteria are detected by pattern-recognition receptors (PRRs) expressed on innate immune cells, which activate intracellular signal transduction pathways to elicit an immune response. Toll-like receptors are, perhaps, the most studied of the PRRs and can activate the mitogen-activated protein kinase (MAPK) and Nuclear Factor-κB (NF-κB) pathways. These pathways are critical for mounting an effective immune response. In order to evade detection and promote virulence, many pathogens subvert the host immune response by targeting components of these signal transduction pathways. This mini-review highlights the diverse mechanisms that bacterial pathogens have evolved to manipulate the innate immune response, with a particular focus on those that target MAPK and NF-κB signaling pathways. Understanding the elaborate strategies that pathogens employ to subvert the immune response not only highlights the importance of these proteins in mounting effective immune responses, but may also identify novel approaches for treatment or prevention of infection.
Introduction
Innate immunity provides the first line of defense against invading pathogens. Recognition of microbial ligands, or pathogen-associated molecular patterns (PAMPs) by pattern-recognition receptors (PRRs), stimulates innate immune cells to upregulate the expression of cytokines, chemokines, and proteins that directly target microbes. Toll-like receptors (TLRs) have been well studied amongst the PRRs, with 10 described in human and 12 in mouse (1). TLRs on the cell surface recognize ligands from extracellular microbes, such as peptidoglycan by TLR1/TLR2, lipoprotein by TLR2/6, lipopolysaccharide (LPS) by TLR4, and flagellin by TLR5. TLR3, TLR7, TLR8, and TLR9 are located in intracellular vesicles where they recognize microbial nucleic acids.
Stimulation of all TLRs activates the mitogen-activated protein kinase (MAPK) and Nuclear Factor-κB (NF-κB) signaling pathways, both of which are critical for an effective immune response. The current understanding of the signaling events that trigger MAPK and NF-κB activation in response to TLR stimulation have been reviewed recently (1–4), but is summarized below and in Figure 1.
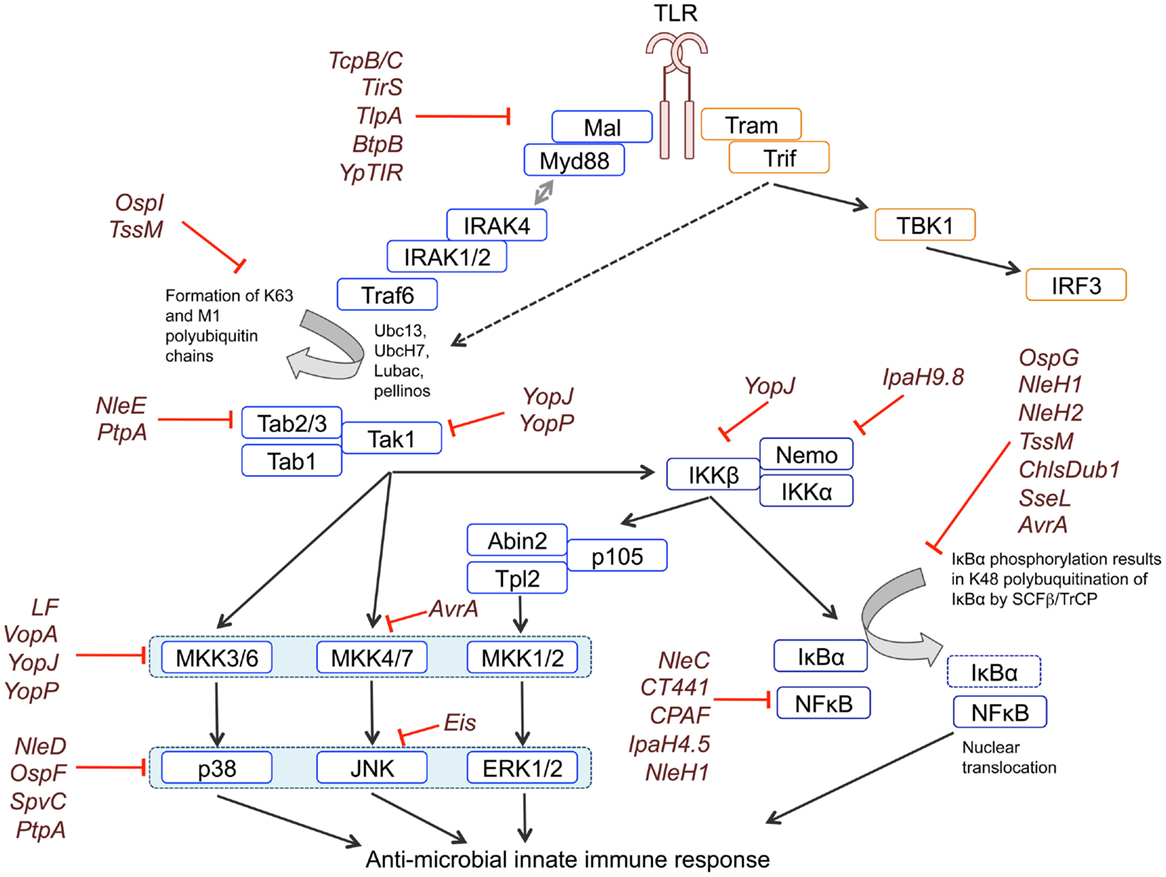
Figure 1. Blockade of MAPK and NFκB signaling by bacterial effectors. TLR signaling is initiated by the recruitment of adaptor proteins to the TIR domain of the receptor. Recruitment of MyD88 facilitates Myddosome formation through binding of IRAK4, IRAK1, and IRAK2. IRAKs bind to and recruit the E3 ubiquitin ligase TRAF6, which – perhaps with input from other E3s – generates lysine-63 (K63) linked polyubiquitin chains. K63 linked polyubiquitin chains are used as a substrate by LUBAC to form M1-K63 hybrid polyubiquitin chains. K63 and M1-K63 polyubiquitin chains are necessary for downstream signaling mediated by TAK1. TAK1 phosphorylates and activates IKKα/β, which form the IKK complex together with NEMO/IKKγ. The IKK complex phosphorylates IκBα, resulting in its K48-linked polyubiquitination and proteasomal degradation, which releases the p65 NFκB subunit from inhibition. The IKK complex also phosphorylates p105, generating the p50 NFκB subunit, and enabling the active p50-p65 NFκB dimer to translocate to the nucleus. TAK1 also controls activation of the ERK1/2, p38, and JNK MAPK pathways by acting as a MAP3K for the p38 and JNK pathways and controlling the activation of ERK1/2 via Tpl2. Phosphorylation of p105 by the IKK complex releases Tpl2 from inhibition, allowing Tpl2 to activate ERK1/2 signaling. MAPKs phosphorylate their own downstream targets including other kinases and transcription factors that regulate transcription. Activation of TLR3 and TLR4 can also recruit the TRIF adaptor, which activates NFκB and MAPK signaling via both Receptor Interacting Protein 1 (RIP1) and TRAF6 upstream of TAK1, and activates IRF3 via IKKϵ and Tank-binding kinase 1 (TBK1). Bacterial effectors block signaling by interfering with different components of the signaling cascades, as indicated in the figure.
Following detection of PAMPs by a TLR, signaling is initiated by the recruitment of adaptor proteins to the cytoplasmic Toll and IL-1 Receptor (TIR) domain of the receptor. Two main pathways of TLR signaling exist, defined on their use of either the MyD88 (myeloid differentiation primary-response protein 88) or TRIF (TIR domain-containing adaptor protein inducing interferon α/β) adaptor, with all TLRs except TLR3 able to utilize the MyD88 pathway. MyD88 recruits IL-1 receptor-associated kinase (IRAK) 4, IRAK1 and IRAK2 to form a complex known as the Myddosome, which subsequently recruits the E3 ubiquitin ligase TNF receptor-associated factor 6 (TRAF6).
TRAF6 and/or other E3 ubiquitin ligases generate lysine-63 (K63) linked polyubiquitin chains that are used by the linear ubiquitin chain assembly complex (LUBAC) to generate linear (M1)-K63 hybrid polyubiquitin chains. The formation of both K63 and M1-K63 hybrid polyubiquitin chains is required to assemble the signaling complexes that activate downstream pathways. TAK1 plays a central role in activating downstream signaling pathways. First, it phosphorylates and activates IκB kinases (IKKα/β), which form the IKK complex together with NEMO/IKKγ. The IKK complex phosphorylates IκBα, enabling its recognition by the E3 ligase complex SCF-βTrCP (SKP1–cullin-1–F-box complex containing βTrCP), resulting in its K48-linked polyubiquitination and proteasomal degradation. Loss of IκBα releases the p65 NFκB subunit allowing it to translocate to the nucleus.
TAK1 also controls activation of the ERK1/2, p38, and JNK MAPK pathways. MAPK activation requires a cascade of at least three kinases. MAPKs are activated by a MAPK Kinase (MAP2K), which itself is activated by phosphorylation by an upstream MAPK Kinase Kinase (MAP3K). TAK1 acts as a MAP3K for the p38 and JNK pathways and via IKK it controls the activation of Tpl2, the MAP3K that activates ERK1/2 downstream of TLRs. Tpl2 activity is controlled by p105, which tethers it in an inactive complex with Abin2. Phosphorylation of p105 by the IKK complex, releases this complex, allowing Tpl2 to activate its substrates. MAPKs phosphorylate their own downstream targets including other kinases and transcription factors that regulate transcription (1–4).
Pathogenic bacteria have evolved elaborate strategies to perturb intracellular signaling pathways that activate the host immune response. This review describes the mechanisms bacteria use to inhibit TLR-dependent signaling, focusing on strategies that block MAPK and NF-κB signaling and is summarised in Table 1. Interestingly, some intracellular bacteria can also activate MAPK or NF-κB pathways to their advantage at different stages of infection. For example, while within cellular vacuoles, Salmonella Typhimurium expresses the kinase SteC which phosphorylates MKK1/2 on Ser200 in the kinase domain (5). Phosphorylation of Ser200 causes MKK1/2 to autophosphorylate on Ser218 and Ser220, leading to activation of ERK1/2 and resulting in reorganization of the actin cytoskeleton, which restrains bacterial growth to control bacterial virulence (5). S. Typhimurium also uses SopE, SopE2, and SopB, which act redundantly to activate MAPK and NF-κB via Rho-family GTPases and stimulate inflammation (6). Infection of alveolar macrophages with Legionalla pneumophila causes Legionnaire’s disease. L. pneumophila translocates the kinase LegK1 into macrophages where it activates NF-κB signaling to inhibit apoptosis and promote intracellular bacterial replication (7). LegK1 phosphorylates a number of proteins in both the canonical and non-canonical NF-κB pathway, including IκBα, IκBβ, IκBϵ, p100 (NFBK2), and p105 (NFKB1) (7). Phosphorylation of IκBα on serines 32 and 36 stimulate its degradation and promote translocation of NF-κB to the nucleus, while phosphorylation of p100 on serines 866 and 870 causes its cleavage to generate the p52 subunit and induce formation of the p52/RelB non-canonical NF-κB complex.
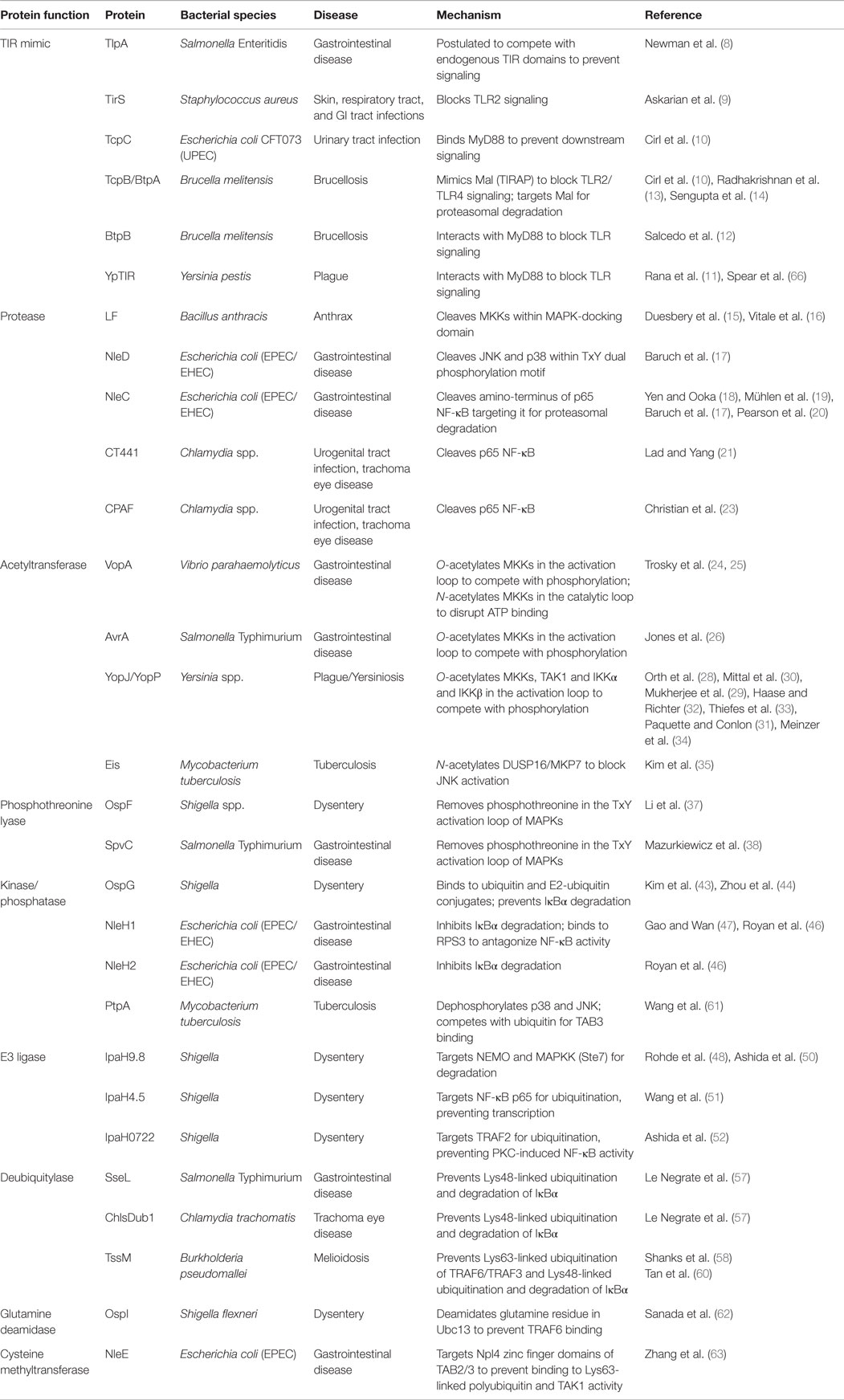
Table 1. Mechanisms used by bacteria to inhibit TLR-dependent signaling by blocking MAPKs or NFκB.
Blocking Signaling by Mimicking TIR:TIR Interactions
A number of bacteria target the initial stage of TLR activation by expressing TIR-containing proteins (Tcps) that interfere with TIR–TIR interactions. A bioinformatics screen for bacterial proteins with homology to human TIRs identified the first TIR-containing protein as TIR-like protein A (TlpA) from Salmonella enterica serovar Enteritidis (Salmonella Enteritidis), which causes food-borne gastroenteritis (8). TlpA dose-dependently suppresses TLR/IL1 induced NF-κB activity and is thought to achieve this by competing with endogenous TIR domains to block downstream signaling (8). A similar mechanism is proposed for the Staphylococcus aureus TIR domain protein TirS which blocks TLR2-induced MAPK and NF-κB signaling (9).
Other Tcps, including TcpC from the uropathogenic Escherichia coli strain CFT073, TcpB/BtpA, and BtpB from Brucella melitensis which causes the chronic and debilitating zoonotic disease Brucellosis and ypTIR from the plague-causing Yersinia pestis, are all able to bind to MyD88 and prevent downstream signaling from TLRs (10–12). Additionally, TcpB was proposed to compete with the TIR-containing adaptor protein Mal/TIRAP to prevent TLR2- and TLR4-dependent signaling (13). TcpB downregulates Mal expression by targeting phosphorylated Mal for proteasomal degradation by a mechanism similar to the cellular SOCS1-mediated degradation of Mal (14).
Bacterial Proteases
Several bacterial proteins can inhibit signaling by selectively cleaving signaling enzymes. Bacillus anthracis lethal factor (LF) is a protease that forms part of the anthrax toxin. LF specifically targets MAPK kinases (MKKs) by cleaving within the MAPK-docking domain (D-domain), which is required for binding to downstream substrates. LF-induced proteolysis disrupts or removes the D-domain to generate kinases that are unable to interact with downstream MAPKs, thereby blocking their phosphorylation and activation. Although originally described to block MKK1/2 (15), LF is capable of cleaving all MKKs except MKK5 (16), resulting in reduced kinase activity for ERK, p38, and JNK MAPK pathways.
Enteropathogenic and enterohemorrhagic E. coli (EPEC/EHEC) are closely related bacteria that cause severe food-borne gastroenteritis. Both use type III secretion systems (T3SS) to inject effector proteins into the host cell. One of these, NleD, is a zinc metalloprotease that inactivates JNK and p38 by cleaving between the dual phosphorylation sites within the kinase activation loop (17). Proteolysis as a strategy to dampen the immune response is not restricted to MAPKs as EPEC/EHEC proteases also target the NF-κB signaling pathway. NleC, another zinc protease, cleaves the p65 subunit of NF-κB at its amino-terminus to promote its proteasomal degradation (17–20) and has also been shown to target other NF-κB components including IκBα, p50, and c-Rel (19, 20). The NF-κB p65 subunit is also a target of proteolysis by the Chlamydia proteases CT441 (21, 22) and Chlamydial protease-like activity factor (CPAF) (23). CT441 inhibits NFκB activation by cleaving p65 at residue 351/2, which lies between the Rel-homologous domain and the transactivation domain (21).
Bacterial Acetyltransferases
Some bacterial effectors modify host signaling proteins to inhibit their activity. Vibrio outer protein A (VopA) is an acetyltransferase expressed by Vibrio parahaemolyticus that inhibits signaling by all MAPKs through both O-acetylation (serine and threonine acetylation) and N-acetylation (lysine acetylation) of MAP2Ks (24, 25). VopA acetylates MKK6 on three residues (Ser207, Lys210, and Thr211) in the activation loop and on Lys172 in the catalytic loop. Phosphorylation of Ser207 and Thr211 by MAP3Ks is critical for MKK6 activation, and acetylation of these resides by VopA blocks MKK6 activity. Lys172 coordinates the γ-phosphate of ATP, and its N-acetylation disrupts ATP binding to prevent phosphorylation of downstream substrates. This dual approach of preventing kinase activation and locking the kinase in an inactive state makes VopA an extremely potent inhibitor of MAPK signaling.
Salmonella Typhimurium expresses the O-actetyltranferase AvrA that modifies the threonine residue in the activation loop of MKK4 to prevent JNK activation (26). An interaction between MKK7 and AvrA was observed in a yeast-two-hybrid screen (27) suggesting that it can act on both MKKs that activate JNK. Although overexpressed AvrA inhibits both p38 and JNK phosphorylation, only JNK phosphorylation is inhibited during S. Typhimurium infection and JNK target genes are upregulated in cells infected with ΔAvrA, lending support for AvrA being targeted to the JNK signaling pathway (27).
Yersinia species deliver Yersinia outer proteins (Yops) into the host cell via a Type III secretion system. The Y. pestis/Yersinia pseudotuberculosis effector YopJ (YopP in Yersinia enterocolitica) inhibits MAPK signaling by blocking the phosphorylation and activation of MAP2Ks (28). YopJ O-actetylates critical residues in the MAP2K activation loop, as described for MKK6 and MKK2 (29–31). In addition to targeting MAP2Ks, YopJ/YopP also inhibits the MAP3K TAK1 (31–34). YopJ O-acetylates Thr184 and Thr187 in the activation loop of TAK1, preventing Thr187 autophosphorylation and thereby blocking kinase activation. Conflicting reports exist regarding the effect of YopP on the formation of the TAK1-TAB2/3 complex, with one showing that YopP interferes with TAK1-TAB2 binding (32), while a second report demonstrated that it did not affect TAK1-TAB2/3 complex formation (33). YopP may also affect ubiquitination since overexpressed YopP blocks TRAF6-dependent polyubiquitination reactions, although the authors note that they were unable to reliably detect this effect on ubiquitination in Yersinia-infected cells (32). By acetylating both TAK1 and MKKs to prevent their activation, YopJ/P targets both the MAPK and NFκB signaling pathways. This dual targeting strategy is reinforced by the demonstration that YopJ also O-acetylates IKKα and IKKβ in the activation loop to inhibit their kinase activity and prevent activation of the NFκB pathway (28, 30).
Mitogen-activated protein kinases are inactivated by a number of different phosphatases of which the dual specificity phosphatase (DUSP) family members are key regulators of MAPK dephosphorylation in immunity. The Enhanced intracellular survival (Eis) protein of Mycobacterium tuberculosis, the causative agent of tuberculosis, targets the JNK pathway. Eis N-acetylates lysine 55 of the JNK-specific DUSP16, which is also known as MAPK phosphatase 7 or MKP7 (35). Lys55 lies within the substrate-docking domain of DUSP16, and its acetylation by Eis results in reduced JNK activity in cells. Similarly, DUSP1/MKP1 that has been acetylated on Lys57 by p300 reduces p38 activity (36). Acetylated DUSP1 binds more readily to p38, resulting in higher phosphatase activity and reduced p38 activity (36). Eis acetylation of DUSP16 is thought to act in a similar manner to reduce JNK activity.
Bacterial Phosphothreonine Lyases
The OspF and SpvC proteins of Shigella and S. Typhimurium, respectively, target MAPK activation by specifically removing the phosphate group from phosphothreonine in the TxY activation loop (37, 38). Rather than acting as threonine-specific phosphatases, OspF and SpvC function as phosphothreonine lyases to irreversibly inactivate MAPKs via an eliminylation reaction whereby the threonine phosphate group is dephosphorylated by β-elimination to generate the unsaturated amino acid dehydrobutyrine (37, 39). The effect is irreversible as dehydrobutyrine lacks a hydroxyl group and cannot be phosphorylated. Although OspF and SpvC have activity against ERK, p38, and JNK (37, 38), OspF shows selectivity for ERK and p38 during Shigella infection (40, 41) and has actually been shown to potentiate JNK activity due to its phosphothreonine lyase activity on p38 disrupting a negative feedback loop between p38 and TAK1 (42).
Bacterial Kinases
Some bacteria express their own kinases. For example Shigella OspG is a serine/threonine kinase that binds to ubiquitin and E2-ubiquitin conjugates in the SCF-βTrCP complex, dampening the host immune response by reducing IκBα degradation (43, 44). The interaction between OspG and ubiquitin activates its kinase activity, which is required for it to inhibit NFκB signaling. Binding of OspG to E2-ubiquitin conjugates also represses ubiquitin transfer to E3 ligases, as it has been shown to stabilize a UbcH5b-ubiquitin complex (45).
OspG shares significant sequence homology with the NleH family of proteins in E. coli and like OspG, NleH1, and NleH2 can inhibit IκBα ubiquitination to prevent its degradation (46). However, NleH1/2 is regulated differently to OspG, since their kinase activity is not induced by ubiquitin (44). Instead, NleH1 binds to a novel subunit of NFκB, ribosomal protein S3 (RPS3), antagonizing its function of guiding p65 to specific promoters and thereby reducing its transcriptional activity (47).
Bacterial E3 Ligases
In addition to expressing kinases and phosphatases that can interfere with ubiquitin-dependent signaling, bacteria also use their own E3 ligases and deubiquitinase. IpaH proteins belong to the Novel E3 Ligase (NEL) family of ubiquitin E3 ligases of which Shigella IpaH9.8 and Salmonella SspH1 were the first described members (48). Although a large number of bacterial E3 ligases have been identified (49) many of their ubiquitination targets are unknown. Using the yeast Saccharomyces cerevisiae as a model system, it was demonstrated that IpaH9.8 acts as an E3 ligase for the MAPKK Ste7 (48), and that in human cells IpaH9.8 mediates lysine-27 polyubiquitination of NEMO/IKKγ resulting in the degradation of both proteins (50). Other IpaH family members also possess E3 ligase activity, with Shigella IpaH4.5 ubiquitinating p65 to block NF-κB transcription (51) and IpaH0722 targeting TRAF2 for ubiquitin-dependent degradation to inhibit PKC-induced NF-κB activity (52). Interestingly reports are now emerging of bacterial E3 ligases targeting other aspects of immune signaling. For example, Shigella IpaH7.8 activates the inflammasome, resulting in cell death and enhanced bacterial replication (53).
Bacterial Deubiquitinases (DUBs)
Deubiquitinases (DUBs) are proteases that remove ubiquitin from proteins. Both the AvrA and SseL proteins of S. Typhimurium and the ChlsDub1 protein of Chlamydia trachomatis possess DUB activity that inhibits K48-linked ubiquitination and degradation of IκBα, thus blocking NFκB activation (54–57). In addition to its DUB activity, ChlsDub1 has also been shown to have deNEDDylating activity (59) that may contribute to suppressing IκBα degradation by antagonizing conjugation of the ubiquitin-like NEDD8 protein to the SCF-βTrCP complex, although this has not been formally demonstrated.
Burkholderia (Pseudomonas) pseudomallei, which causes melioidosis, expresses the effector protein TssM, which possesses DUB activity against both K63 and K48-linked polyubiquitin (58, 60). TssM overexpression causes reduced K48-linked polyubiquitination of IκBα and reduced K63-linked polyubiquitination of TRAF6 to block NFκB-induced transcription (60).
Other Bacterial Effectors
In addition to the above proteins, further bacterial effectors that block MAPK and NFκB activity via different mechanisms have started to emerge. M. tuberculosis tyrosine phosphatase PtpA dephosphorylates JNK and p38 to dampen cytokine expression (61). PtpA uses a novel mechanism whereby its phosphatase activity is stimulated by binding to ubiquitin via a novel ubiquitin-interacting motif-like (UIML) region (61). Mtb PtpA also suppresses NFκB activation by competitively binding to the Npl4 zinc finger domain (NZF) of TAB3, blocking its ability to bind to ubiquitin chains and thereby reducing Tak1 activity.
The Shigella flexneri type III effector OspI blocks TRAF6 mediated signaling by selectively deamidating a glutamine residue to glutamic acid in the E2 enzyme Ubc13 (62). The deamidation of Glu100 prevents Ubc13 from binding to TRAF6, inhibiting its E3 ligase activity and thereby blocking downstream signaling.
The EPEC NleE protein also uses a unique mechanism to inhibit bacterial-induced signaling. NleE is an S-adenosyl-l-methionine (SAM)-dependent cysteine methyltransferase that targets the Npl4 zinc finger (NZF) domains in TAB2/3 (63). Modification of cysteine residues in the zinc finger domains of TAB2/3 abolishes their ability to bind to Lys63-linked polyubiquitin chains and therefore blocks downstream TAK1 activation, consistent with the observed inhibition of IκBα phosphorylation and NFκB signaling in the presence of NleE (64, 65). NleE proteins from other pathogens, such as S. flexneri protein OspZ, were shown to be functionally interchangeable with NleE in blocking NFκB signaling and may also act as a cysteine methyltransferases (65).
Summary and Future Perspectives
Bacterial pathogens have evolved diverse and elegant ways to block MAPK and NF-κB signaling downstream of TLR activation, enabling them to evade detection by the immune system and promote infection. Many bacteria employ strategies to simultaneously target a number of proteins within these, as well as other host pathways, to increase their chances of overcoming the immune response. Future discoveries in understanding how and why pathogens target particular proteins will not only demonstrate their importance in immunity, but will also help our understanding of how bacteria activate intracellular signaling pathways, and have the potential to identify new targets for the treatment or prevention of infection.
Author Contributions
Both VM and SA wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
2. Coll RC, O’Neill LA. New insights into the regulation of signalling by toll-like receptors and nod-like receptors. J Innate Immun (2010) 2(5):406–21. doi:10.1159/000315469
3. Arthur JSC, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol (2013) 13(9):679–92. doi:10.1038/nri3495
4. Cohen P. The TLR and IL-1 signalling network at a glance. J Cell Sci (2014) 127(Pt 11):2383–90. doi:10.1242/jcs.149831
5. Odendall C, Rolhion N, Förster A, Poh J, Lamont DJ, Liu M, et al. The Salmonella kinase SteC targets the MAP kinase MEK to regulate the host actin cytoskeleton. Cell Host Microbe (2012) 12(5):657–68. doi:10.1016/j.chom.2012.09.011
6. Bruno VM, Hannemann S, Lara-Tejero M, Flavell RA, Kleinstein SH, Galán JE. Salmonella Typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog (2009) 5(8):e1000538. doi:10.1371/journal.ppat.1000538
7. Ge J, Xu H, Li T, Zhou Y, Zhang Z, Li S, et al. A Legionella type IV effector activates the NF-kappaB pathway by phosphorylating the IkappaB family of inhibitors. Proc Natl Acad Sci U S A (2009) 106(33):13725–30. doi:10.1073/pnas.0907200106
8. Newman RM, Salunkhe P, Godzik A, Reed JC. Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian toll/interleukin-1 receptor family proteins. Infect Immun (2006) 74(1):594–601. doi:10.1128/IAI.74.1.594-601.2006
9. Askarian F, van Sorge NM, Sangvik M, Beasley FC, Henriksen JR, Sollid JU, et al. A Staphylococcus aureus TIR domain protein virulence factor blocks TLR2-mediated NF-κB signaling. J Innate Immun (2014) 6(4):485–98. doi:10.1159/000357618
10. Cirl C, Wieser A, Yadav M, Duerr S, Schubert S, Fischer H, et al. Subversion of toll-like receptor signaling by a unique family of bacterial toll/interleukin-1 receptor domain-containing proteins. Nat Med (2008) 14(4):399–406. doi:10.1038/nm1734
11. Rana RR, Simpson P, Zhang M, Jennions M, Ukegbu C, Spear AM, et al. Yersinia pestis TIR-domain protein forms dimers that interact with the human adaptor protein MyD88. Microb Pathog (2011) 51(3):89–95. doi:10.1016/j.micpath.2011.05.004
12. Salcedo SP, Marchesini MI, Degos C, Terwagne M, Von Bargen K, Lepidi H, et al. BtpB, a novel Brucella TIR-containing effector protein with immune modulatory functions. Front Cell Infect Microbiol (2013) 3(July):28. doi:10.3389/fcimb.2013.00028
13. Radhakrishnan GK, Yu Q, Harms JS, Splitter GA. Brucella TIR domain-containing protein mimics properties of the toll-like receptor adaptor protein TIRAP. J Biol Chem (2009) 284(15):9892–8. doi:10.1074/jbc.M805458200
14. Sengupta D, Koblansky A, Gaines J, Brown T, West AP, Zhang D, et al. Subversion of innate immune responses by Brucella through the targeted degradation of the TLR signaling adapter. J Immunol (2010) 184(2):956–64. doi:10.4049/jimmunol.0902008
15. Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science (1998) 280(5364):734–7. doi:10.1126/science.280.5364.734
16. Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. J Biochem (2000) 745:739–45. doi:10.1042/bj3520739
17. Baruch K, Gur-Arie L, Nadler C, Koby S, Yerushalmi G, Ben-Neriah Y, et al. Metalloprotease type III effectors that specifically cleave JNK and NF-κB. EMBO J (2011) 30(1):221–31. doi:10.1038/emboj.2010.297
18. Yen H, Ooka T, Iguchi A, Hayashi T, Sugimoto N, Tobe T. NleC, a type III secretion protease, compromises NF-kB activation by targeting p65/rela. PLoS Pathog (2010) 6(12):e1001231. doi:10.1371/journal.ppat.1001231
19. Mühlen S, Ruchaud-Sparagano MH, Kenny B. Proteasome-independent degradation of canonical NFκB complex components by the NleC protein of pathogenic Escherichia coli. J Biol Chem (2011) 286(7):5100–7. doi:10.1074/jbc.M110.172254
20. Pearson JS, Riedmaier P, Marchès O, Frankel G, Hartland EL. A type III effector protease NleC from enteropathogenic Escherichia coli targets NF-κB for degradation. Mol Microbiol (2011) 80(1):219–30. doi:10.1111/j.1365-2958.2011.07568.x
21. Lad SP, Yang G, Scott DA, Wang G, Nair P, Mathison J, et al. Chlamydial CT441 Is a PDZ domain-containing tail-specific protease that interferes with the NF-κB pathway of immune response. J Bacteriol (2007) 189(18):6619–25. doi:10.1128/JB.00429-07
22. Lad SP, Li J, da Silva Correia J, Pan Q, Gadwal S et al. Cleavage of p65/RelA of the NF- B pathway by Chlamydia. Proc Natl Acad Sci U S A (2007) 104(8):2933–8. doi:10.1073/pnas.0608393104
23. Christian J, Vier J, Paschen SA, Häcker G. Cleavage of the NF-κB family protein p65/RelA by the chlamydial protease-like activity factor (CPAF) impairs proinflammatory signaling in cells infected with chlamydiae. J Biol Chem (2010) 285(53):41320–7. doi:10.1074/jbc.M110.152280
24. Trosky JE, Mukherjee S, Burdette DL, Roberts M, McCarter L, Siegel RM, et al. Inhibition of MAPK signaling pathways by VopA from Vibrio parahaemolyticus. J Biol Chem (2004) 279(50):51953–7. doi:10.1074/jbc.M407001200
25. Trosky JE, Li Y, Mukherjee S, Keitany G, Ball H, Orth K. VopA inhibits ATP binding by acetylating the catalytic loop of MAPK kinases. J Biol Chem (2007) 282(47):34299–305. doi:10.1074/jbc.M706970200
26. Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, Neish AS. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe (2008) 3(4):233–44. doi:10.1016/j.chom.2008.02.016
27. Du F, Galán JE. Selective inhibition of type III secretion activated signaling by the Salmonella effector AvrA. PLoS Pathog (2009) 5(9):1–12. doi:10.1371/journal.ppat.1000595
28. Orth K, Palmer LE, Bao ZQ, Stewart S, Rudolph AE, Bliska JB, et al. Inhibition of the mitogen-activated protein kinase kinase superfamily by a Yersinia effector. Science (1999) 285(5435):1920–3. doi:10.1126/science.285.5435.1920
29. Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, et al. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science (2006) 312:1211–4. doi:10.1126/science.1126867
30. Mittal R, Peak-Chew S-Y, McMahon HT. Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc Natl Acad Sci U S A (2006) 103(49):18574–9. doi:10.1073/pnas.0608995103
31. Paquette N, Conlon J, Sweet C, Rus F, Wilson L, Pereira A, et al. Serine/threonine acetylation of TGF-β-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc Natl Acad Sci U S A (2012) 109(31):12710–5. doi:10.1073/pnas.1008203109
32. Haase R, Richter K, Pfaffinger G, Courtois G, Ruckdeschel K. Yersinia outer protein P suppresses TGF-β-activated kinase-1 activity to impair innate immune signaling in Yersinia enterocolitica-infected cells. J Immunol (2005) 175(12):8209–17. doi:10.4049/jimmunol.175.12.8209
33. Thiefes A, Wolf A, Doerrie A, Grassl GA, Matsumoto K, Autenrieth I, et al. The Yersinia enterocolitica effector YopP inhibits host cell signalling by inactivating the protein kinase TAK1 in the IL-1 signalling pathway. EMBO Rep (2006) 7(8):838–44. doi:10.1038/sj.embor.7400754
34. Meinzer U, Barreau F, Esmiol-Welterlin S, Jung C, Villard C, Léger T, et al. Yersinia pseudotuberculosis effector YopJ subverts the Nod2/RICK/TAK1 pathway and activates caspase-1 to induce intestinal barrier dysfunction. Cell Host Microbe (2012) 11(4):337–51. doi:10.1016/j.chom.2012.02.009
35. Kim KH, An DR, Song J, Yoon JY, Kim HS, Yoon HJ, et al. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci U S A (2012) 109(20):7729–34. doi:10.1073/pnas.1120251109
36. Cao W, Bao C, Padalko E, Lowenstein CJ. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J Exp Med (2008) 205(6):1491–503. doi:10.1084/jem.20071728
37. Li H, Xu H, Zhou Y, Zhang J, Long C, Li S, et al. The phosphothreonine lyase activity of a bacterial type III effector family. Science (2007) 315(5814):1000–3. doi:10.1126/science.1138960
38. Mazurkiewicz P, Thomas J, Thompson JA, Liu M, Arbibe L, Sansonetti P, et al. SpvC is a Salmonella effector with phosphothreonine lyase activity on host mitogen-activated protein kinases. Mol Microbiol (2008) 67(6):1371–83. doi:10.1111/j.1365-2958.2008.06134.x
39. Brennan DF, Barford D. Eliminylation: a post-translational modification catalyzed by phosphothreonine lyases. Trends Biochem Sci (2009) 34(3):108–14. doi:10.1016/j.tibs.2008.11.005
40. Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, et al. An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol (2007) 8(1):47–56. doi:10.1038/ni1423
41. Kramer RW, Slagowski NL, Eze NA, Giddings KS, Morrison MF, Siggers KA, et al. Yeast functional genomic screens lead to identification of a role for a bacterial effector in innate immunity regulation. PLoS Pathog (2007) 3(2):179–90. doi:10.1371/journal.ppat.0030021
42. Reiterer V, Grossniklaus L, Tschon T, Kasper CA, Sorg I, Arrieumerlou C. Shigella flexneri type III secreted effector OspF reveals new crosstalks of proinflammatory signaling pathways during bacterial infection. Cell Signal (2011) 23(7):1188–96. doi:10.1016/j.cellsig.2011.03.006
43. Kim DW, Lenzen G, Page AL, Legrain P, Sansonetti PJ, Parsot C. The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc Natl Acad Sci U S A (2005) 102(39):14046–51. doi:10.1073/pnas.0504466102
44. Zhou Y, Dong N, Hu L, Shao F. The Shigella type three secretion system effector OspG directly and specifically binds to host ubiquitin for activation. PLoS One (2013) 8(2). doi:10.1371/journal.pone.0057558
45. Pruneda JN, Smith FD, Daurie A, Swaney DL, Villén J, Scott JD, et al. E2~Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. EMBO J (2014) 33(5):437–49. doi:10.1002/embj.201386386
46. Royan SV, Jones RM, Koutsouris A, Roxas JL, Falzari K, Weflen AW, Kim A, et al. Enteropathogenic E. coli non-LEE encoded effectors NleH1 and NleH2 attenuate NF-κB activation. Mol Microbiol (2010) 78(5):1232–45. doi:10.1111/j.1365-2958.2010.07400.x
47. Gao X, Wan F, Mateo K, Callegari E, Wang D, Deng W, et al. Bacterial effector binding to ribosomal protein S3 subverts NF-κB function. PLoS Pathog (2009) 5(12):e1000708. doi:10.1371/journal.ppat.1000708
48. Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, Parsot C. Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe (2007) 1(1):77–83. doi:10.1016/j.chom.2007.02.002
49. Hicks SW, Galán JE. Hijacking the host ubiquitin pathway: structural strategies of bacterial E3 ubiquitin ligases. Curr Opin Microbiol (2010) 13(1):41–6. doi:10.1016/j.mib.2009.11.008
50. Ashida H, Kim M, Schmidt-Supprian M, Ma A, Ogawa M, Sasakawa C. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKgamma to dampen the host NF-kappaB-mediated inflammatory response. Nat Cell Biol (2010) 12(1):66–73. doi:10.1038/ncb2006
51. Wang F, Jiang Z, Li Y, He X, Zhao J, Yang X, et al. Shigella flexneri T3SS effector IpaH4.5 modulates the host inflammatory response via interaction with NF-κB p65 protein. Cell Microbiol (2013) 15(3):474–85. doi:10.1111/cmi.12052
52. Ashida H, Nakano H, Sasakawa C. Shigella IpaH0722 E3 ubiquitin Ligase effector targets TRAF2 to Inhibit PKC-NF-κB activity in invaded epithelial cells. PLoS Pathog (2013) 9(6):1–15. doi:10.1371/journal.ppat.1003409
53. Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida H, Toyotome T, et al. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc Natl Acad Sci U S A (2014) 111(40):E4254–63. doi:10.1073/pnas.1324021111
54. Collier-Hyams LS, Zeng H, Sun J, Tomlinson AD, Bao ZQ, Chen H, et al. Cutting edge: Salmonella AvrA effector inhibits the key proinflammatory, anti-apoptotic NF- B pathway. J Immunol (2002) 169(6):2846–50. doi:10.4049/jimmunol.169.6.2846
55. Ye Z, Petrof EO, Boone D, Claud EC, Sun J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am J Pathol (2007) 171(3):882–92. doi:10.2353/ajpath.2007.070220
56. Le Negrate G, Krieg A, Faustin B, Loeffler M, Godzik A, Krajewski S, et al. ChlaDub1 of chlamydia trachomatis suppresses NF-kappaB activation and inhibits IkappaBalpha ubiquitination and degradation. Cell Microbiol (2008) 10(9):1879–92. doi:10.1111/j.1462-5822.2008.01178.x
57. Le Negrate G, Faustin B, Welsh K, Loeffler M, Krajewska M, Hasegawa P, et al. Salmonella secreted factor L deubiquitinase of Salmonella typhimurium Inhibits NF-κB, suppresses IκBα ubiquitination. The J Immunol (2008) 180:5045–56. doi:10.4049/jimmunol.180.7.5045
58. Shanks J. Burtnick MN, Brett PJ, Waag DM, Spurgers KB, Ribot WJ, et al., Burkholderia mallei tssM encodes a putative deubiquitinase that is secreted and expressed inside infected RAW 264.7 murine macrophagest. Infect Immun (2009) 77(4):1636–48. doi:10.1128/IAI.01339-08
59. Misaghi S, Balsara ZR, Catic A, Spooner E, Ploegh HL, Starnbach MN. Chlamydia trachomatis-derived deubiquitinating enzymes in mammalian cells during infection. Mol Microbiol (2006) 61(1):142–50. doi:10.1111/j.1365-2958.2006.05199.x
60. Tan KS, Chen Y, Lim YC, Tan GG, Liu Y, Lim YT, et al. Suppression of host innate immune response by Burkholderia pseudomallei through the virulence factor TssM. J Immunol (2010) 184(9):5160–71. doi:10.4049/jimmunol.0902663
61. Wang J, Li BX, Ge PP, Li J, Wang Q, Gao GF, et al. Mycobacterium tuberculosis suppresses innate immunity by coopting the host ubiquitin system. Nat Immunol (2015) 16(3). doi:10.1038/ni.3096
62. Sanada T, Kim M, Mimuro H, Suzuki M, Ogawa M, Oyama A, et al. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature (2012) 483(7391):623–6. doi:10.1038/nature10894
63. Zhang L, Ding X, Cui J, Xu H, Chen J, Gong YN, et al. Cysteine methylation disrupts ubiquitin-chain sensing in NF-κB activation. Nature (2011) 481(7380):204–8. doi:10.1038/nature10690
64. Nadler C, Baruch K, Kobi S, Mills E, Haviv G, Farago M, et al. The type III secretion effector NleE inhibits NF-κB activation. PLoS Pathog (2010) 6(1):403–4. doi:10.1371/journal.ppat.1000743
65. Newton HJ, Pearson JS, Badea L, Kelly M, Lucas M, Holloway G, et al. The type III effectors NieE and NleB from enteropathogenic E. coli and Ospz from shigella block nuclear translocation of NF-κB p65. PLoS Pathog (2010) 6(5):1–16. doi:10.1371/journal.ppat.1000898
66. Spear AM, Rana RR, Jenner DC, Flick-Smith HC, Oyston PC, Simpson P, et al. A Toll/interleukin (IL)-1 receptor domain protein from Yersinia pestis interacts with mammalian IL-1/Toll-like receptor pathways but does not play a central role in the virulence of Y. pestis in a mouse model of bubonic plague. Microbiology (2012) 158(6):1593–606. doi:10.1099/mic.0.055012-0
Keywords: MAPK, NF-κB, bacterial effector, signaling, TLR, bacterial pathogen, virulence
Citation: McGuire VA and Arthur JSC (2015) Subverting Toll-Like Receptor Signaling by Bacterial Pathogens. Front. Immunol. 6:607. doi: 10.3389/fimmu.2015.00607
Received: 25 September 2015; Accepted: 13 November 2015;
Published: 01 December 2015
Edited by:
Olivier Dussurget, University Paris Diderot, FranceReviewed by:
Cammie Lesser, Harvard Medical School, USAElizabeth Hong-Geller, Los Alamos National Laboratory, USA
Sinead Corr, Trinity College Dublin, Ireland
Copyright: © 2015 McGuire and Arthur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victoria A. McGuire, v.a.mcguire@dundee.ac.uk