 Timothy A. Gottschalk
Timothy A. Gottschalk Evelyn Tsantikos
Evelyn Tsantikos Margaret L. Hibbs
Margaret L. Hibbs
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 October 2015
Sec. Immunological Tolerance and Regulation
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00550
This article is part of the Research TopicFocus on Systemic Lupus ErythematosusView all 8 articles
Systemic lupus erythematosus (SLE, lupus) is a highly complex and heterogeneous autoimmune disease that most often afflicts women in their child-bearing years. It is characterized by circulating self-reactive antibodies that deposit in tissues, including skin, kidneys, and brain, and the ensuing inflammatory response can lead to irreparable tissue damage. Over many years, clinical trials in SLE have focused on agents that control B- and T-lymphocyte activation, and, with the single exception of an agent known as belimumab which targets the B-cell survival factor BAFF, they have been disappointing. At present, standard therapy for SLE with mild disease is the agent hydroxychloroquine. During disease flares, steroids are often used, while the more severe manifestations with major organ involvement warrant potent, broad-spectrum immunosuppression with cyclophosphamide or mycophenolate. Current treatments have severe and dose-limiting toxicities and thus a more specific therapy targeting a causative factor or signaling pathway would be greatly beneficial in SLE treatment. Moreover, the ability to control inflammation alongside B-cell activation may be a superior approach for disease control. There has been a recent focus on the innate immune system and associated inflammation, which has uncovered key players in driving the pathogenesis of SLE. Delineating some of these intricate inflammatory mechanisms has been possible with studies using spontaneous mouse mutants and genetically engineered mice. These strains, to varying degrees, exhibit hallmarks of the human disease and therefore have been utilized to model human SLE and to test new drugs. Developing a better understanding of the initiation and perpetuation of disease in SLE may uncover suitable novel targets for therapeutic intervention. Here, we discuss the involvement of inflammation in SLE disease pathogenesis, with a focus on several key proinflammatory cytokines and myeloid growth factors, and review the known outcomes or the potential for targeting these factors in SLE.
Systemic lupus erythematosus (SLE, lupus) is a B-cell-mediated autoimmune disease characterized by the generation of autoantibodies against nuclear antigens and a type III hypersensitivity reaction leading to chronic systemic inflammation. The disease is polygenic and highly complex, requiring interplay between multiple immunopathogenic factors including host autoantigens and both cellular and humoral immune components that contribute to the generation of a hyperinflammatory environment resulting in organ and tissue damage (Figure 1). Deposition of circulating autoantibody–autoantigen complexes can occur in various tissues and organs of the body, resulting in a local inflammatory response and severe tissue destruction. Sites often affected include skin (cutaneous lupus), the nervous system (CNS lupus), joints and muscles (rheumatoid lupus, rhupus), and the kidney (renal lupus, lupus nephritis), which contributes most significantly to disease morbidity (1). Disease progression is non-linear and follows a relapse-remitting course, and due to its heterogeneous nature, it can vary widely from patient to patient, making diagnosis and treatment a challenge (1). The current diagnostic criteria require a patient to present with 4 out of 11 symptoms/disorders, including cutaneous rashes, inflammation of the pleura or pericardium, inflammation of joints and muscles, renal and/or neurologic disorders, hematologic and immunologic disorders, and most significantly, autoantibodies specifically targeting nuclear antigens (such as double-stranded DNA, small nuclear riboproteins, chromatin, and histone proteins), or to a lesser extent, cytoplasmic antigens (2). SLE has a strong genetic component with a high familial concordance, but environmental triggers such as UV radiation, stress, medication, or infection can contribute to disease onset. Genetic analyses, and more recently, genome-wide association studies, have uncovered various human SLE susceptibility genes that are normally responsible for maintaining immune system tolerance and homeostatic processes. These include antigen processing and presentation (HLA, TAP1/2), clearance of apoptotic debris (C1q, Dnase1), leukocyte cell surface receptors (FCGRI/II/III, ITGAM), and cell signaling and gene transcription molecules (LYN, BLK, PTPN22, STAT4, IRF5) (3, 4). SLE affects approximately one in every 2,500 individuals, although this can be highly variable based on geographical location, ethnicity, and sex [reviewed in Ref. (5, 6)]; SLE is most commonly seen in females (9:1 prevalence) with disease onset typically around child-bearing age and in those of non-Caucasian ethnicity.
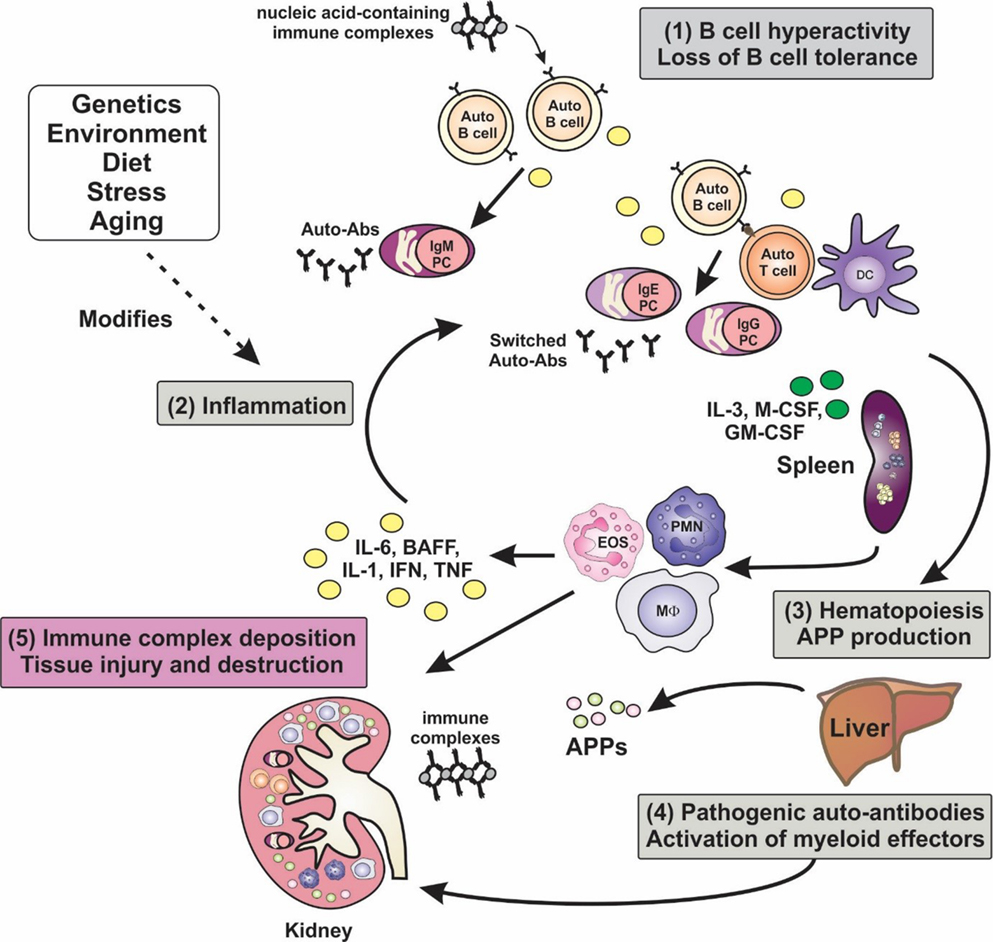
Figure 1. Inflammation is a key factor in the pathogenesis of lupus. A hallmark of lupus is the presence of hyperactive B cells and loss of B-cell tolerance. Immune complexes containing nucleic acid autoantigens can engage and activate endosomal TLRs and promote inflammation in SLE. Plasma cell expansion and the production of autoantibodies are also features, although the autoantibodies are benign unless generated in an inflammatory milieu, wherein class-switching to pathogenic isotypes occurs. Proinflammatory cytokines not only drive T-cell activation and dendritic cell maturation, but they can stimulate extramedullary hematopoiesis leading to expansion of innate immune cells, and they can induce the production of acute-phase proteins (APPs). Autoantibodies become deposited in tissues such as the glomeruli of the kidney, leading to the activation of myeloid effector cells via Fcγ and complement receptors, leading to tissue destruction. Numerous factors, including genetic make-up, environment, diet, and stress, can modify disease course and severity.
As SLE is highly complex, multifactorial, and manifests in an array of pathologies, the disease has been difficult to study in humans. Mouse models that mimic aspects of SLE pathology and pathogenesis include the naturally occurring mutants MRLlpr/lpr, NZB/W F1, congenic BSXB/Yaa, NZM2410, B6.sle1.sle2.sle3, B6.sle1.Yaa, transgenic (Tg) overexpression of BAFF in BAFF-Tg, and induced models such as pristane and ALD-DNA [reviewed in Ref. (7, 8)]. One well-studied model of SLE is the Lyn-deficient mouse (Lyn−/−) (9), which exhibits clinical, pathological, and biochemical features seen in human SLE [comprehensively reviewed in Ref. (10)]. Lyn−/− mice develop serum antinuclear antibodies (ANAs) and acquire glomerulonephritis due to immune complex deposition in kidney (9), which leads to renal dysfunction (11). The mice show hematologic disorders such as lymphopenia and thrombocytopenia (9) and develop splenomegaly and lymphadenopathy (12). Similar to SLE patients, Lyn−/− mice also have hyperactive B lymphocytes and altered signaling pathways (13–15). Lyn is a haploinsufficiency gene in autoimmunity (16), and it is implicated in human disease (10). Much of our current understanding of SLE disease pathogenesis and many preliminary therapeutic studies for SLE have come from the identification, analysis, or testing of these mouse models [reviewed in Ref. (17, 18)].
One or more mechanisms of B-cell tolerance are lost in SLE, allowing for the production of ANAs by plasma cells [reviewed in Ref. (19, 20)] (Figure 1). Upward of 90% of SLE patients have elevated titers of serum ANAs, on average 2–3 years prior to clinical onset of SLE (21), with 30–70% of SLE patients developing life-limiting renal disease (22). The temporal delay between autoantibody development and disease onset coupled with incomplete penetrance of ANA-mediated disease suggests that pathogenesis of autoantibody-driven nephritis is conditional upon other factors, such as antigen availability, a pre-established inflammatory environment, and T-cell-mediated antibody isotype switching (Figure 1). While a hallmark of inflammation is the elevation in levels of C-reactive protein (CRP), many lupus patients demonstrate normal or even reduced levels of CRP. CRP is involved in the clearance of apoptotic cells [reviewed in Ref. (23, 24)], and if they are inadequately cleared, this can expose nuclear antigens allowing for ANAs to extensively bind and form immune complexes (ICs). Such ICs can deposit in the basement membrane of the glomerular microvessels (25), resulting in activation of the alternative complement pathway and recruitment of proinflammatory macrophages and dendritic cells to the glomeruli via chemotactic signaling which upregulate inflammatory cytokine production and activate autoreactive T-cell subsets through antigen presentation and costimulation (Figure 1) (22, 26). Endosomal toll-like receptors (TLR)-7 and TLR-9 in activated B cells, plasmacytoid dendritic cells, and macrophages can respond to internalized self ICs containing nucleic acids, which can contribute to the initiation and perpetuation of the inflammatory cascade (Figure 1) [reviewed in Ref. (27)]. CD4+ T helper cells play several key roles in the pathogenesis of lupus nephritis: T helper 1 (Th1) cells are responsible for high-level production of proinflammatory cytokines, such as interferon-γ (IFN-γ), which stimulates dendritic cell and myeloid cell production of interleukin-(IL)-1, IL-6, IL-12, IL-18, TNF-α, and BAFF creating a perpetual proinflammatory loop; T helper 2 cells (Th2) produce cytokines (IL-4, IL-5), which induce antibody isotype class-switching leading to the production of high affinity, pathogenic autoantibodies [reviewed in Ref. (28, 29)]; Th17 cells also provide B-cell support, promote plasma cell differentiation and pathogenic autoantibody production and myeloid cell hyper-activation which drives systemic inflammation (30, 31); T follicular helper cells (TFH) are now also known to contribute to autoimmune germinal center reactions or autoantibody production in lupus-prone mice and SLE patients (32, 33) [reviewed in Ref. (34)]. Aside from autoantibody production (Figure 2A), autoreactive B cells contribute to the pathogenesis of lupus nephritis via two supportive mechanisms: B cells can activate autoreactive T cells through antigen presentation and costimulation (Figure 2B) and they can produce cytokines including IL-6, a proinflammatory cytokine able to drive inflammation and inhibit the generation of autoimmune suppressive regulatory T cells (Treg) (Figure 2C) (22, 29). As well as T-cell-induced antibody isotype switching within germinal centers, evidence shows ectopic germinal center-like congregations within the glomeruli of SLE patients suggesting B cells may undergo local somatic hypermutation of immunoglobulin (Ig) variable region genes generating both higher affinity autoantibodies and memory B cells (35). Inflammation and cytotoxicity caused by the immune response generated against glomerular ICs results in progressive renal tissue damage including immune cellular influx and progressive fibrotic, sclerotic, and necrotic lesions (36). As a consequence of this, patients suffer glomerular degeneration and reduced kidney function, which may result in end-stage renal failure requiring dialysis or transplantation (37). Currently, there is an incomplete understanding of the factors driving pathogenesis in lupus nephritis, which hinders the development of novel, targeted therapeutics.
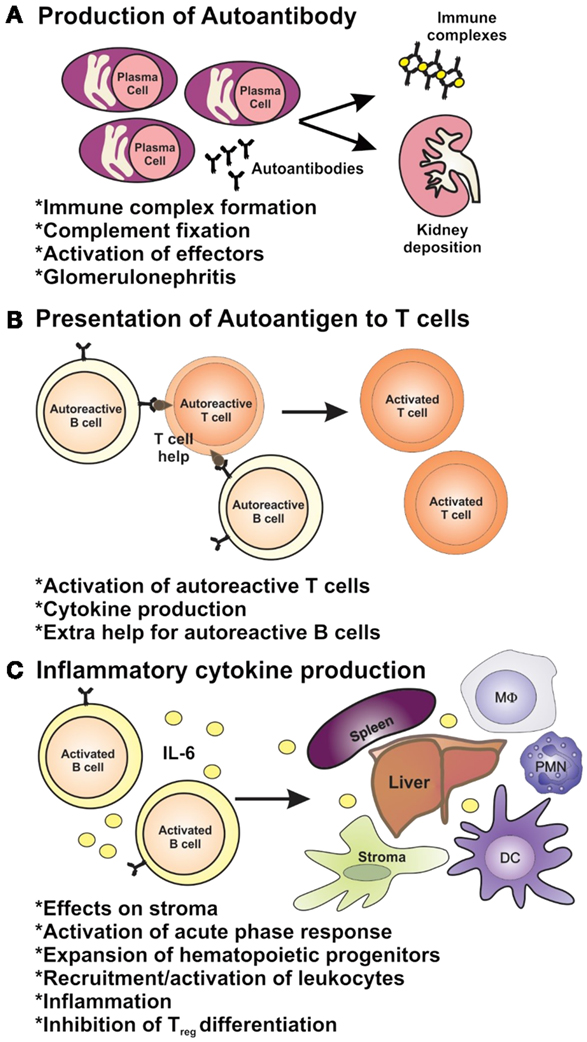
Figure 2. The roles of B cells in lupus pathogenesis. B cells have multiple roles in autoimmunity through (A) their ability to produce autoantibodies and (B) via their role as antigen-presenting cells and (C) as producers of inflammatory cytokines.
Treatment of SLE has remained fairly consistent over the past 50 years utilizing non-specific anti-inflammatory agents such as non-steroidal anti-inflammatory drugs and the immune cell modulating hydroxychloroquine for mild disease, and broad-spectrum immunosuppressants/anti-inflammatories such as corticosteroids, azathioprine, cyclophosphamide, or mycophenolate during flares or severe disease with organ involvement (38). These therapies can have severe, dose-limiting toxicities and undesirable side effects, and some patients lack an adequate response, highlighting the need for a treatment regime tailored to individual patients (38). Development and trial of novel targeted treatments for SLE have been difficult due to the complexity and limited knowledge of disease etiology, loose diagnostic criteria, various disease manifestations, and the diverse heterogeneity of patients [reviewed in Ref. (39)].
Despite promising results in animal models (40), therapeutic targeting of B cells in SLE has had minimal success, especially in patients with lupus nephritis. B-cell depletion using rituximab, a monoclonal antibody targeting the B-cell-specific receptor CD20 (anti-CD20 mAb), was unsuccessful at meeting primary and secondary endpoints in stage II/III SLE clinical trials despite showing initial promising results (41, 42). Ocrelizumab, a humanized anti-CD20 mAb, also failed in phase III clinical trials demonstrating insignificant renal improvement and increases in severe infection of patients (43). Inhibition of B-cell survival pathways by neutralizing the survival receptor TACI, using a humanized fusion protein atacicept, was initially deemed safe in phase I trials but efficacy has not been determined due to safety concerns during a phase II/III trial which led to its premature termination (44). Modulation of B-cell receptor signaling using epratuzumab (anti-CD22 mAb) has yielded some promising results in non-renal SLE patients although there is currently no data on outcomes in patients with lupus nephritis (45). Recombinant small-molecule inhibitor abatacept, which blocks T-cell co-stimulatory ligands (CD80 and CD86) on B cells and dendritic cells, also failed to meet primary and secondary endpoints in a phase II clinical trial of SLE patients which largely excluded lupus nephritis patients (46).
Despite this lack of success, belimumab, a monoclonal antibody that neutralizes the soluble B-cell activation and survival factor BAFF, has recently been approved for SLE patients with active disease in the USA and Europe by the Federal Drug Administration (FDA) and the European Medicines Agency after successful phase III clinical trials showing modest improvements in disease (47, 48). It is now also available for use in other countries, such as Australia. Belimumab is not only the first targeted therapeutic indicated for SLE but also the first new therapeutic approved for SLE in over 50 years. However, the benefits obtained with belimumab are modest and only attained in patients with mild disease who are already receiving standard therapy (47). Given the overall lack of success of lymphocyte-targeted therapeutics, coupled with the heterogeneity and diverse array of clinical manifestations of disease and the contribution of inflammatory factors to disease development in SLE, more consideration to non-B-cell targets and combination therapeutic regimes are required in order to deliver a more personalized and effective approach to treating SLE. The focus of this review will be to discuss the current understanding of proinflammatory cytokines and myeloid growth and activation factors as well as the promising outcomes and potential for the therapeutic targeting of these factors in SLE.
The inflammasome is a complex that responds to “danger signals” and induces the cleavage and release of bioactive IL-1, a cytokine upstream of many proinflammatory responses (49). The importance of inflammasome activation and the induction of IL-1 have been elucidated by several studies and have drawn an important link between the innate immune system and autoimmunity (50, 51). Increased IL-1 gene expression was observed in MRLlpr/lpr mice (52, 53), and inhibition of the NLRP3 inflammasome/IL-1β axis in MRLlpr/lpr mice attenuated proteinuria, autoantibody production, systemic proinflammatory cytokines, and kidney pathology (54, 55). When SLE was induced in mice deficient in IL-1, they showed reduced levels of autoantibody and milder disease manifestations (56). It was also found that IL-1 induced IgG production by cells from SLE patients and healthy controls (57), and increased IL-1 levels were produced by B cells from SLE patients (58) and were found in the cerebrospinal fluid (CSF) of patients with neural SLE (59). A single-nucleotide polymorphism in the IL1B gene (rs1143629) is associated with juvenile-onset SLE, implicating IL-1β in the early stages of SLE pathogenesis (60). Treatment of MRLlpr/lpr and NZB/W F1 mice with anti-IL-1R antibody reduced autoantibody titers (61, 62). A preliminary study treating patients with the IL-1R antagonist anakinra showed promising results (63); however, no further studies have since been published.
In contrast with the concept of a critical role for inflammasome activation in autoimmunity, inflammasome deficiency due to a point mutation in the NLRP3 gene has recently been identified in lupus-prone NZB/W F1 mice correlating with reduced IL-1β release (64). In addition, the NLRP3/ASC inflammasome protected against kidney damage independent of IL-1 signaling due to regulation of the anti-inflammatory activity of transforming growth factor-β (TGF-β) in C57BL6/Jlpr/lpr mice, which represent a mild spontaneous model of lupus (65). Collectively, these studies suggest that therapeutic targeting of the inflammasome in SLE may exacerbate disease in some patients or only be suitable for patients with a certain inflammatory profile. Recently, emphasis has been placed on understanding how the inflammasome contributes to proinflammatory cell death (pyroptosis) and the subsequent modification of autoantigens and generation of autoimmune responses in lupus (66).
Interferon alpha (IFN-α) is one of the most strongly implicated cytokines in the pathogenesis of SLE (67). Early studies showed that IFN-α was increased in the serum and found to be associated with increased disease activity (68–71). Correlations were also found between the levels of IFN-α and IC in the serum (69) as well as the deposition of IC in kidney sections from SLE patients (72). This may be due to IFN-α having an antagonistic effect on CRP production, leading to the elevation of available nuclear antigens (23). In addition, IFN-α was expressed at high levels in the CSF of patients with neural lupus (73, 74), and CSF from these patients had strong IFN-α-inducing ability when added to cell cultures (75). Treatment of a patient harboring a malignant tumor with IFN-α induced SLE-like symptoms (76), and it is also epidemiologically interesting that SLE patients with high IFN-α levels showed low rates of hepatitis B infections (77), highlighting the role of IFN-α not only in antitumor and antiviral responses but also in the pathogenesis of SLE. Interestingly, viral DNA and RNA as well as self nucleic acid-containing ICs stimulate IFN-α production by plasmacytoid dendritic cells via TLR-7 and TLR-9; specific inhibitors toward these TLRs potently inhibited IFN-α production (78), and treatment of lupus-prone NZB/W F1 mice with these inhibitors significantly reduced serum ANAs, glomerulonephritis, and organ damage while improving survival (79). In addition, increased TLR-7 and TLR-9 expression in PBMCs from SLE patients was correlated strongly with high levels of IFN-α mRNA (80), thus bridging innate immunity with autoimmunity. Clinical assessment of targeting IFN-α has shown safety (81). A phase II study did not reach its primary endpoint; however, exploratory analysis of the results did reveal positive reductions in disease (82). A phase I/II trial in SLE patients examining the safety of IFNα kinoid (IFN-K) that induces a host polyclonal antibody response to IFN-α is currently underway (NCT01058343). A phase I trial in lupus patients testing sifalimumab (MEDI-545), a human anti-IFN-α mAb, showed safety, tolerability, and clinical activity (83), which has led to a phase IIb trial, which has now been completed although no data have been published (NCT00299819). Studies targeting TLRs in nephritic SLE are currently lacking; however, small-molecule TLR antagonists are starting to be assessed for efficacy in animal models (84). Very interestingly, it is now apparent that hydroxychloroquine, a mainstay in lupus treatment, is a TLR-7 and TLR-9 antagonist. Thus, manipulation of TLR signaling with new agents promises to be a future growth area in the clinical management of inflammatory and autoimmune diseases such as SLE [reviewed in Ref. (85)].
Interferon gamma (IFN-γ) is a prototypic Th1 cytokine that activates a proinflammatory program in macrophages. Like IFN-α, IFN-γ is also elevated in the serum of SLE patients (86–88). Increased IFN-γ levels were also found in lymphoid organs of prediseased MRLlpr/lpr mice (52) while increased IFN-γ-producing T cells were correlated with autoantibody titers and proteinuria in aged diseased mice (89). Treatment of NZB/W F1 mice with IFN-γ accelerated disease while neutralization of IFN-γ resulted in reduced disease symptoms and improved survival (90, 91); furthermore, genetic deletion of IFN-γ receptor in these mice impaired autoantibody production and glomerulonephritis (92). Studies in the Lyn−/− mouse model showed that genetic deletion of IFN-γ led to reduced production of BAFF and decreased myeloid proliferation and T-cell hyperactivation, thereby resulting in moderation of glomerular disease (93). Analysis of PBMCs from SLE patients showed that they had significantly higher IFN-γ transcripts compared to control PBMCs (94) and that T cells from SLE patients produced more IFN-γ, which induced BAFF production by monocytes (95), while SLE NK cells produced higher IFN-γ (96). There are currently no completed trials on the effect of neutralizing IFN-γ in SLE patients (97); however, a recent single-dose study treating SLE patients with AMG811, an anti-INF-γ IgG1 mAb, was well tolerated and showed reductions in IFN-γ-mediated gene expression (98).
Tumor necrosis factor alpha (TNF-α) is an interesting and controversial cytokine in the field of SLE due to its apparent dual role (99). Similar to other proinflammatory cytokines, increased levels of TNF-α have been observed in the serum of lupus-prone animals and SLE patients (100). In particular, elevated serum levels and gene expression levels are positively associated with disease activity as well as renal involvement in SLE patients (88, 101). While TNF-α blockade has been successful as a mainstay treatment for rheumatoid arthritis (102), the assessment of this therapy in SLE patients has not been straightforward. In SLE patient studies, anti-TNF-α therapy increases the serum levels of anti-dsDNA and antiphospholipid autoantibodies (103). It would follow that a further increase in antiphospholipid antibodies in patients may lead to vascular events, which although rare, have the potential to be life-threatening. In addition, the risk of bacterial infection is increased as a result of anti-TNF-α therapy (104). Despite the risks, treatment has led to syndromes that are transient, mild in nature, have not induced flares, and have resulted in reductions in proteinuria and provided benefit to patients with lupus arthritis (105). A more recent study has demonstrated safety and efficacy of anti-TNF-α therapy in SLE (106). It is suggestive that any consideration of anti-TNF-α for the treatment of SLE patients must be for a short duration only and not recommended for patients with antiphospholipid syndrome [up to 15% of SLE patients (107, 108)]. It is still debatable whether the risks associated with therapeutic targeting of TNF-α in SLE are worth the benefits obtained.
B-cell-activating factor (BAFF) is an important B-cell survival factor with well-known pathogenic roles in SLE. It is expressed by numerous cells in the immune system but is highly expressed by innate immune cells. Mice engineered to overexpress BAFF developed autoimmune manifestations (109) while overexpression of BAFF in lupus-prone congenic strains accelerated renal pathology (110). BAFF levels are increased in the serum of patients with SLE (111, 112) and are associated with increased anti-dsDNA antibody levels (113) and disease activity (114, 115). Neutralization of BAFF in lupus-prone NZB/W F1 mice depleted B cells, prevented progressive T-cell activation and dendritic cell accumulation, and prolonged survival (116). Lyn−/− mice show excessive BAFF production by myeloid cells and treatment with anti-BAFF mAb attenuated their lupus-like disease (93). BAFF neutralization also reduced glomerulonephritis and improved survival in lupus-prone BXSB/Yaa mice (117) and NZM2410 mice (118). T cells from SLE patients produced large amounts of BAFF in culture (119) and had significantly upregulated BAFF mRNA (120) compared to control T cells. Increased BAFF expression was also found in SLE B cells and was positively associated with anti-dsDNA autoantibody and disease activity scores (121), indicating that as well as being produced by other cell types, B cells from SLE patients can produce BAFF in an autocrine manner. Especially pertinent is the FDA approval of belimumab (trade name Benlysta®), a neutralizing antibody against BAFF, for the treatment of SLE (48), spurred by the success of a randomized double-blind placebo-controlled trial that demonstrated efficacy and safety in SLE patients treated with belimumab over placebo (47). This pathway continues to dominate the focus of SLE clinical trials.
Extramedullary hematopoiesis is often a hallmark of infectious and inflammatory diseases, driven by excess production of myeloid growth factors and is evident in models of lupus such as Lyn−/−, NZB/W F1, and MRLlpr/lpr mice (Figure 1) (15, 122). This phenomenon may contribute to the splenomegaly seen in some lupus patients and lupus-prone mice (15, 122, 123). Myeloid growth factors stimulate progenitor cell release from bone marrow, myeloid cell production, and cellular activation, and this may promote enhanced inflammatory responses and tissue damage depending on tissue context. Several studies have implicated myeloid growth factors, including IL-3, M-CSF, and GM-CSF, in the inflammatory pathways and pathology in SLE.
Interleukin-3 (IL-3) is a pleiotropic, synergistic growth factor that is involved in the differentiation, activation, and support processes of many immune cells including dendritic cells (124). Although it has long been known that IL-3 can be elevated in SLE patients (125), very few studies on the role of IL-3 in SLE have since been conducted. In the Lyn-deficient mouse model of lupus, IL-3-responsive progenitor cells are elevated in spleen (15), and IL-3 induces enhanced signaling and survival of Lyn−/− plasma cells, suggesting it may play a role in the support of autoreactive plasma cells (126). Recently, a study has shown that IL-3 can drive glomerulonephritis in MRLlpr/lpr mice, hypothesized to be due to enhanced antigen presentation by dendritic cells, elevated Ig secretion, and/or basophil-mediated support functions (127). Treatment of MRLlpr/lpr mice with an anti-IL-3 mAb ameliorated nephritis, improved kidney function, and restrained production of certain autoantibodies (127). This suggests that the IL-3 axis might be an undervalued contributor to inflammation and pathology in SLE, and future studies to further our understanding of this system may benefit the development of superior treatment regimes.
Macrophage colony-stimulating factor (M-CSF) is a myeloid growth factor that induces the differentiation of myeloid precursor cells into monocytes/macrophages or dendritic cells as well as regulating macrophage functions, survival, trafficking, and proliferation, and it is associated with inflammatory pathology (128). M-CSF is elevated in the serum of SLE patients and correlates with active disease, renal pathology, and myeloid activation syndrome (129, 130). Local M-CSF production by renal mesangial cells is elevated in lupus nephritis and contributes to proteinuria, local macrophage infiltration and proliferation, and glomerular proliferation (131, 132). M-CSF is also detectable in the urine of lupus nephritis patients with levels correlating with flares in renal disease (133). Elevated M-CSF is also observed in MRLlpr/lpr mice systemically as well as in the kidney (134) and is heavily implicated in driving autoantibody production, glomerular infiltration of myeloid cells and nephritis (135). M-CSF is involved in driving a proinflammatory, immunopathogenic phenotype in MRLlpr/lpr macrophages (136), and in Lyn−/− mice, hematopoietic progenitors responsive to M-CSF are enhanced in spleen (15). Inhibition of M-CSF signaling with a selective M-CSF receptor kinase inhibitor (GW2580) prevents macrophage and T-cell accumulation in the kidney, restricts the local renal inflammatory profile, and improves kidney pathology in an induced model of lupus nephritis (137). This highlights that the M-CSF pathway may be a novel target for therapeutic trials in SLE.
Granulocyte–macrophage colony-stimulating factor (GM-CSF) is a growth factor that drives differentiation of myeloid lineage cells (granulocytes and monocyte/macrophages), and it can act on mature immune cells to upregulate an inflammatory phenotype and enhance antigen presentation and migration (138). Although there is conflicting evidence as to whether GM-CSF levels are altered in SLE (139, 140), the frequency of systemic GM-CSF-secreting immune cells is elevated and correlates with anti-dsDNA titers in SLE (141). It has also been suggested that high concentrations of GM-CSF can drive Ig secretion and leukocyte activation marker CD69 expression in lupus patients (142). GM-CSF can also be produced locally by glomerular mesangial cells and levels correlate with lupus nephritis (131). Interestingly, some studies suggest that neutrophils and dendritic cells may demonstrate resistance to GM-CSF responses in SLE (143, 144). Colony formation by splenic progenitor cells induced by GM-CSF is increased in Lyn−/− mice (15). Therapeutic targeting of the GM-CSF axis is not currently being explored in SLE, but has shown promise in rheumatoid arthritis (138). MOR103, a humanized anti-GM-CSF mAb, has recently successfully completed a phase I/II trial in rheumatoid arthritis reporting safety and preliminary efficacy (145), and a fully human anti-GM-CSF receptor mAb, mavrilimumab, is currently in phase II trial (NCT01712399). Further studies delineating the role of GM-CSF in inflammation and pathology in SLE may highlight this system as a potential target for treatment in a subset of SLE patients.
Interleukin-6 (IL-6) is a pleiotropic cytokine that acts on a range of cell types; it can influence growth and differentiation and antibody production and mediates the acute-phase inflammatory response. It is also highly implicated in the pathogenesis of many inflammatory and autoimmune diseases [reviewed in Ref. (146)]. IL-6 has consistently been shown to be elevated in the serum of SLE patients and it has been suggested that levels correlate with disease activity, making it a suitable biomarker for tracking disease activity (147–150). Similarly, IL-6 can be elevated in the urine of lupus nephritis patients, with higher levels correlating with active renal inflammation and pathology (151, 152). IL-6 has been implicated in driving autoantibody production and loss of tolerance in SLE through the upregulation of recombination-activating gene (RAG) activity (153). Studies have identified polymorphisms in the IL-6 gene, which are associated with SLE susceptibility (154). In mice, elevated levels of IL-6 are observed in numerous models, including NZB/W F1 (155), MRLlpr/lpr (156), Lyn−/− (15), ALD-DNA (157), B6.sle1.sle2.sle3 (158), and B6.sle1.Yaa (159), and it is heavily implicated in pathology. Studies in IL-6-deficient mice have shown that they are resistant to ALD-DNA-induced lupus, which ordinarily promotes anti-dsDNA autoantibody titers, proteinuria, CD4+ T-cell activation, and glomerulonephritis (157). The mechanism is thought to be due to an expansion of regulatory T cells in the absence of IL-6, which ordinarily suppresses their generation (157). Impaired Treg maturation and activity was observed in B6.sle1.sle2.sle3 mice due to overproduction of IL-6 by dendritic cells (158). IL-6 has also been shown to promote disease in Lyn−/− mice as Lyn−/−IL-6−/− show moderated B-cell hyperactivity and plasmacytosis and abrogation of T-cell hyperactivity and splenic myeloid cell expansion (15). In addition, class-switched pathogenic ANAs, glomerular IgG, and complement deposition are absent in Lyn−/−IL-6−/− mice, and glomerular structural integrity is significantly improved (15). Similarly, IL-6-deficient MRLlpr/lpr mice showed greatly improved survival with significant amelioration of renal immunopathology (160), and in B6.Sle1.Yaa mice, IL-6 deficiency eliminated autoantibody production and nephritis (161).
It has been hypothesized that classic IL-6 signaling via membrane-bound IL-6R (Figures 3A,B) does not contribute to chronic inflammation but mediates regulated pathogen clearing processes through anti-inflammatory pathways. However, IL-6 trans-signaling through soluble IL-6R/gp130 complexes (Figure 3C) is believed to be associated with chronic inflammatory conditions by driving hyperinflammatory and immunopathogenic processes (162). Specific downregulation of IL-6 trans-signaling in Lyn−/− mice through transgenic overexpression of soluble gp130-Fc fusion protein (Lyn−/−sgp130FcTg) resulted in a loss of splenomegaly, a decrease in splenic myeloid cells, and reduced systemic BAFF levels (163). Although pathogenic ANA production was sustained, as was IgG IC deposition in kidney glomeruli, renal complement deposition was significantly reduced, which suppressed renal leukocyte infiltration, thereby markedly attenuating glomerulonephritis and improving kidney function (163). These findings suggest that more emphasis should be put on examining the role of IL-6-trans-signaling in lupus nephritis, with the possibility of targeting this inflammatory pathway in disease.
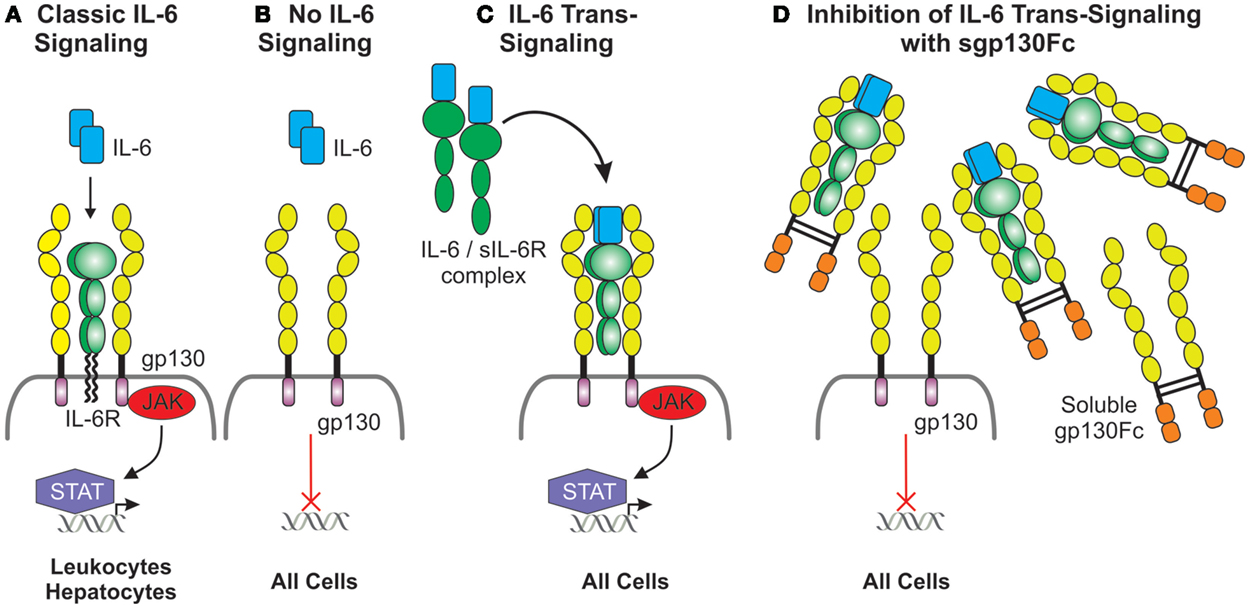
Figure 3. Duplicitous signaling roles of IL-6. (A) Classical IL-6 signaling occurs via direct interaction of IL-6 with the membrane-bound IL-6 receptor, which has a limited cellular distribution, and the ubiquitously expressed gp130. gp130 lacks intrinsic kinase activity; IL-6 signals are transduced intracellularly via the recruitment and activation of the JAK/STAT pathway. (B) Cells lacking the IL-6 receptor are not receptive to IL-6 except (C) in the presence of the IL-6/soluble IL-6 receptor complex which interacts with gp130 expressing cells and this is defined as IL-6 trans-signaling. (D) Inhibition of IL-6 trans-signaling can be achieved via the presence of excess soluble gp130Fc fusion protein.
There is much evidence to suggest that IL-6 plays a significant role in a number of inflammatory, autoimmune, and proliferative diseases, making it a strong candidate for targeted novel biological therapeutics [reviewed in Ref. (164)]. This notion is further enhanced by the improvement seen in IL-6-targeted murine SLE models. A preliminary study treating NZB/NZW F1 mice monthly with anti-IL-6 in combination with anti-IL-1α concluded that this regime only had a partial effect on disease, alleviating proteinuria (165). Young NZB/NZW F1 mice treated weekly with a rat anti-IL-6 monoclonal antibody from 3 to 9 months of age showed reduced ANA production, improved proteinuria, and increased survival, although these mice required initial tolerizing treatments of anti-CD4 to prevent a rapid-onset anti-rat Ig response (166). A more thorough study in young NZB/W F1 mice without anti-CD4 tolerance showed that anti-IL-6 mAb treatment suppressed systemic serum amyloid A (SAA) and anti-dsDNA antibody levels, suppressed hyperactivation of B and T cells, and greatly diminished the development of kidney pathology (167).
The use of murine IL-6 targeting monoclonal antibodies in human disease has been unsuccessful as IL-6/mAb immune complexes form which can further drive pathology in inflammatory settings (168). A fully humanized anti-IL6 mAb, sirukumab, has shown tolerance in humans (169, 170) and efficacy in treating rheumatoid arthritis (171). A phase I trial in cutaneous and systemic lupus patients (NCT01702740) concluded that sirukumab is generally well tolerated in SLE although mild leukopenia, neutropenia, and decreases in platelet count were observed (172). A phase II trial in lupus nephritis patients (NCT01273389) has recently been completed with official outcomes not yet reported, although unofficial accounts indicate a high frequency of serious adverse events, the majority of which were infections.
A study examined the effect of targeting IL-6R in MRLlpr/lpr mice by treating 15-week-old animals every 1–3 days for 5 weeks with a neutralizing rat anti-IL-6R mAb. These mice saw an initial reduction in anti-dsDNA autoantibodies, which rebounded and elevated in response to increasing titers of anti-rat Ig antibodies (173). The treatment was reported as successful as both kidney function and structure was improved at the endpoint compared to controls (173). Tocilizumab, a humanized murine anti-IL6-R mAb, has demonstrated efficacy in treating multiple myeloma (174) and rheumatoid arthritis (175). A phase I safety trial demonstrated that tocilizumab is well tolerated in SLE patients although similarly to sirukumab, temporary dose-dependent neutropenia was observed (176). A recent study in SLE patients showed that tocilizumab treatment can reduce B- and T-cell activation, memory B cells, and autoantibody-producing plasma cells without impacting naive B cells populations (177).
Given that anti-IL-6 and anti-IL6R inhibition may lead to unwanted neutropenia and infection, targeting IL-6 trans-signaling without disrupting classical signaling pathways may prove to be a superior approach in SLE as many anti-inflammatory and regulatory mechanisms mediated by IL-6 classic signaling will not be impacted (Figure 3D) (162, 163, 178). A fusion protein joining sgp130 to the IgG Fc region, sgp130Fc, has demonstrated effective inhibition of soluble IL-6R-mediated trans-signaling (178). Proof of concept for sgp130Fc treatment has been demonstrated in a range of inflammatory disease models, including rheumatoid arthritis (179, 180), atherosclerosis (181), and Crohn’s disease (178). Given its efficacy, it is surprising that no clinical trials utilizing this agent in lupus appear to be in the pipeline.
Components of signaling pathways that are aberrantly activated in disease are attractive drug targets. The JAK/STAT signaling pathway is activated downstream of IL-6 and gp130 (Figures 3A,C) and can be targeted by small-molecule inhibitors, which have already shown promise in cancer therapy (182). However, there are few studies on JAK/STAT inhibitor treatments in inflammatory diseases; one study demonstrated clinical efficacy of the JAK1/2 inhibitor AZD1480 in five models of experimental autoimmune encephalitis (EAE, modeling human multiple sclerosis) by inhibiting myeloid cell hyperactivation, Th1 and Th17 differentiation, and proinflammatory cytokine production as well as showing improvements in disease and clinical pathology (183). A signaling study in Lyn−/− mice showed that treatment with AZD1480 resulted in reduced splenic B cells and plasma cells as well as reduced numbers of splenocytes and thymocytes; however, effects on lupus pathology were not reported (126). Although this may be an attractive approach, inhibition of the JAK/STAT pathway in autoimmune disease should be undertaken cautiously and the use of single-specificity inhibitors may be more beneficial. Indeed, treatment using the JAK2-selective inhibitor CEP-33779 in both NZB/W F1 and MRLlpr/lpr mice saw dose-dependent improvements in lymphadenopathy and splenomegaly, reductions in systemic C3 and proinflammatory cytokines, including IL-1, IL-12, IFN-α, IL-17A, and TNF-α, reductions in autoantibody-producing plasma cells, increased survival, and significant improvements in glomerulonephritis through blockade of STAT3-mediated signaling (184, 185). These animal data lend support for trailing a JAK2 inhibitor in human disease. Interestingly, GSK recently halted development of the JAK1 inhibitor GSK2586184 following disappointing results in a phase II study in SLE (186).
Although the pathogenic pathways involved in SLE are still being defined, it is evident that a combination of deregulated processes are necessary for the disease to manifest. As pathogenesis in SLE is clearly multifactorial, the targeting of a single pathway or factor may not sufficiently alter the progression of disease. Indeed, current treatment utilizes both agents that suppress the immune response alongside those that dampen inflammation (38). One of the reasons that B-cell-targeted therapies have been largely unsuccessful in SLE trials is that they fail to target pathogenic long-lived plasma cells and memory B cells (187). Recent studies in NZB/W F1 mice treated with a B cell targeting anti-CD20 mAb alongside a plasma cell-depleting agent (188) or BAFF-blocking agent (189) resulted in superior improvements in disease compared to B-cell depletion on its own. Similarly, depletion of memory B cells using an anti-IL-15 mAb alongside TACI-Ig, a soluble form of the TACI receptor that binds to BAFF and APRIL ligands, was more efficacious than TACI-Ig alone (190). This illustrates that combining targeted therapeutics can give rise to better outcomes. As systemic inflammation is essential for disease propagation and sustenance, controlling both the proinflammatory arm alongside pathogenic plasma or memory B-cell depletion may prove to be a superior therapeutic approach to disease control in SLE.
Systemic lupus erythematosus is a highly complex autoimmune disease with multiple immunopathological disorders, the most significant being glomerulonephritis mediated by immune complex deposition in the kidney microvessels. Although autoreactive B cells are essential, disease progression relies on the establishment of a systemic inflammatory environment largely generated by myeloid cells and T lymphocytes. Current treatment utilizes broad-spectrum immunosuppressive and anti-inflammatory agents which have many off-target effects. Therapeutic targeting of the B-cell compartment in SLE has only had limited success, and a need for novel targeted therapeutics is essential for personalized, effective control of disease to improve quality of life. Many therapeutics targeting inflammatory cytokines, growth factors, and signaling pathways have shown safety and efficacy in inflammatory disorders, and preliminary studies in animal models of murine lupus and human lupus nephritis patients have demonstrated some promising disease-suppressing effects. Presently, there are no therapies for human SLE in clinical practice that target specific inflammatory pathways and thus further studies elucidating the mediators and their mode of action in generating the pathogenic inflammatory environment in lupus will greatly benefit the selection and development of therapeutics for future clinical trials.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The Guest Associate Editor, Dr. Harris, declares that, despite being affiliated with the same institution as the authors, the review process was handled objectively.
This work was supported by funding from the National Health and Medical Research Council (NH&MRC) of Australia and grants from the Central Clinical School Monash University. TAG is the recipient of an Australian Postgraduate Award and MLH is supported by a Research Fellowship from the NH&MRC Australia (APP603124).
ANAs, antinuclear antibodies; APPs, acute-phase proteins; auto-Abs, autoantibodies; BAFF, B-cell-activating factor of the TNF family; CRP, C-reactive protein; IC, immune complex; IFN, interferon; Ig, immunoglobulin; IL, interleukin; mAb, monoclonal antibody; SLE, systemic lupus erythematosus/lupus; Th, T helper; TLR, toll-like receptor; Treg, regulatory T cell.
1. Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv Immunol (2006) 92:1–69. doi: 10.1016/S0065-2776(06)92001-X
2. Petri M. Treatment of systemic lupus erythematosus: an update. Am Fam Physician (1998) 57(11):2753–60.
3. Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet (2008) 40(2):204–10. doi:10.1038/ng.81
4. Ramos PS, Brown EE, Kimberly RP, Langefeld CD. Genetic factors predisposing to systemic lupus erythematosus and lupus nephritis. Semin Nephrol (2010) 30(2):164–76. doi:10.1016/j.semnephrol.2010.01.007
5. Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus (2006) 15(5):308–18. doi:10.1191/0961203306lu2305xx
6. Lau CS, Yin G, Mok MY. Ethnic and geographical differences in systemic lupus erythematosus: an overview. Lupus (2006) 15(11):715–9. doi:10.1177/0961203306072311
7. Satoh M, Kumar A, Kanwar YS, Reeves WH. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proc Natl Acad Sci U S A (1995) 92(24):10934–8. doi:10.1073/pnas.92.24.10934
8. Du Y, Sanam S, Kate K, Mohan C. Animal models of lupus and lupus nephritis. Curr Pharm Des (2015) 21(18):2320–49. doi:10.2174/1381612821666150316115727
9. Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell (1995) 83(2):301–11. doi:10.1016/0092-8674(95)90171-X
10. Tsantikos E, Gottschalk TA, Maxwell MJ, Hibbs ML. Role of the Lyn tyrosine kinase in the development of autoimmune disease. Int J Clin Rheumatol (2014) 9(5):519–35. doi:10.2217/ijr.14.44
11. Lau M, Tsantikos E, Maxwell MJ, Tarlinton DM, Anderson GP, Hibbs ML. Loss of STAT6 promotes autoimmune disease and atopy on a susceptible genetic background. J Autoimmun (2012) 39(4):388–97. doi:10.1016/j.jaut.2012.06.003
12. Tsantikos E, Quilici C, Harder KW, Wang B, Zhu HJ, Anderson GP, et al. Perturbation of the CD4 T cell compartment and expansion of regulatory T cells in autoimmune-prone Lyn-deficient mice. J Immunol (2009) 183(4):2484–94. doi:10.4049/jimmunol.0804346
13. Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, et al. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity (1998) 8(4):497–508. doi:10.1016/S1074-7613(00)80554-3
14. Smith KGC, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by CD22: a requirement for Lyn. J Exp Med (1998) 187(5):807–11. doi:10.1084/jem.187.5.807
15. Tsantikos E, Oracki SA, Quilici C, Anderson GP, Tarlinton DM, Hibbs ML. Autoimmune disease in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. J Immunol (2010) 184(3):1348–60. doi:10.4049/jimmunol.0901878
16. Tsantikos E, Maxwell MJ, Kountouri N, Harder KW, Tarlinton DM, Hibbs ML. Genetic interdependence of Lyn and negative regulators of B cell receptor signaling in autoimmune disease development. J Immunol (2012) 189(4):1726–36. doi:10.4049/jimmunol.1103427
17. Birmingham DJ, Rovin BH, Yu CY, Hebert LA. Of mice and men: the relevance of the mouse to the study of human SLE. Immunol Res (2001) 24(2):211–24. doi:10.1385/IR:24:2:211
18. Perry D, Sang A, Yin Y, Zheng YY, Morel L. Murine models of systemic lupus erythematosus. J Biomed Biotechnol (2011) 2011:271694. doi:10.1155/2011/271694
19. Rahman A. Autoantibodies, lupus and the science of sabotage. Rheumatology (Oxford) (2004) 43(11):1326–36. doi:10.1093/rheumatology/keh354
20. Marks SD, Tullus K. Autoantibodies in systemic lupus erythematosus. Pediatr Nephrol (2012) 27(10):1855–68. doi:10.1007/s00467-011-2078-4
21. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med (2003) 349(16):1526–33. doi:10.1056/NEJMoa021933
22. Nowling TK, Gilkeson GS. Mechanisms of tissue injury in lupus nephritis. Arthritis Res Ther (2011) 13(6):250. doi:10.1186/ar3528
23. Meyer O. Anti-CRP antibodies in systemic lupus erythematosus. Joint Bone Spine (2010) 77(5):384–9. doi:10.1016/j.jbspin.2010.04.010
24. Rhodes B, Furnrohr BG, Vyse TJ. C-reactive protein in rheumatology: biology and genetics. Nat Rev (2011) 7(5):282–9. doi:10.1038/nrrheum.2011.37
25. Kramers C, Hylkema MN, van Bruggen MC, van de Lagemaat R, Dijkman HB, Assmann KJ, et al. Anti-nucleosome antibodies complexed to nucleosomal antigens show anti-DNA reactivity and bind to rat glomerular basement membrane in vivo. J Clin Invest (1994) 94(2):568–77. doi:10.1172/JCI117371
26. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med (2008) 358(9):929–39. doi:10.1056/NEJMra071297
27. Celhar T, Magalhaes R, Fairhurst AM. TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunol Res (2012) 53(1–3):58–77. doi:10.1007/s12026-012-8270-1
28. Miyake K, Akahoshi M, Nakashima H. Th subset balance in lupus nephritis. J Biomed Biotechnol (2011) 2011:980286. doi:10.1155/2011/980286
29. Iwata Y, Furuichi K, Kaneko S, Wada T. The role of cytokine in the lupus nephritis. J Biomed Biotechnol (2011) 2011:594809. doi:10.1155/2011/594809
30. Shabgah AG, Fattahi E, Shahneh FZ. Interleukin-17 in human inflammatory diseases. Postepy Dermatol Alergol (2014) 31(4):256–61. doi:10.5114/pdia.2014.40954
31. Martin JC, Baeten DL, Josien R. Emerging role of IL-17 and Th17 cells in systemic lupus erythematosus. Clin Immunol (2014) 154(1):1–12. doi:10.1016/j.clim.2014.05.004
32. Kim YU, Lim H, Jung HE, Wetsel RA, Chung Y. Regulation of autoimmune germinal center reactions in lupus-prone BXD2 mice by follicular helper T cells. PLoS One (2015) 10(3):e0120294. doi:10.1371/journal.pone.0120294
33. Zhang X, Lindwall E, Gauthier C, Lyman J, Spencer N, Alarakhia A, et al. Circulating CXCR5+CD4+helper T cells in systemic lupus erythematosus patients share phenotypic properties with germinal center follicular helper T cells and promote antibody production. Lupus (2015) 24(9):909–17. doi:10.1177/0961203314567750
34. Sweet RA, Lee SK, Vinuesa CG. Developing connections amongst key cytokines and dysregulated germinal centers in autoimmunity. Curr Opin Immunol (2012) 24(6):658–64. doi:10.1016/j.coi.2012.10.003
35. Chang A, Henderson SG, Brandt D, Liu N, Guttikonda R, Hsieh C, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol (2011) 186(3):1849–60. doi:10.4049/jimmunol.1001983
36. Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol (2004) 15(2):241–50. doi:10.1097/01.ASN.0000108969.21691.5D
37. Fiehn C, Hajjar Y, Mueller K, Waldherr R, Ho AD, Andrassy K. Improved clinical outcome of lupus nephritis during the past decade: importance of early diagnosis and treatment. Ann Rheum Dis (2003) 62(5):435–9. doi:10.1136/ard.62.5.435
38. Yildirim-Toruner C, Diamond B. Current and novel therapeutics in the treatment of systemic lupus erythematosus. J Allergy Clin Immunol (2011) 127(2):303–12. doi:10.1016/j.jaci.2010.12.1087
39. Eisenberg R. Why can’t we find a new treatment for SLE? J Autoimmun (2009) 32(3–4):223–30. doi:10.1016/j.jaut.2009.02.006
40. Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol (2007) 179(5):3351–61. doi:10.4049/jimmunol.179.5.3351
41. Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the lupus nephritis assessment with rituximab study. Arthritis Rheum (2012) 64(4):1215–26. doi:10.1002/art.34359
42. Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum (2010) 62(1):222–33. doi:10.1002/art.27233
43. Mysler EF, Spindler AJ, Guzman R, Bijl M, Jayne D, Furie RA, et al. Efficacy and safety of ocrelizumab in active proliferative lupus nephritis: results from a randomized, double-blind, phase III study. Arthritis Rheum (2013) 65(9):2368–79. doi:10.1002/art.38037
44. Ginzler EM, Wax S, Rajeswaran A, Copt S, Hillson J, Ramos E, et al. Atacicept in combination with MMF and corticosteroids in lupus nephritis: results of a prematurely terminated trial. Arthritis Res Ther (2012) 14(1):R33. doi:10.1186/ar3738
45. Rao V, Gordon C. Evaluation of epratuzumab as a biologic therapy in systemic lupus erythematosus. Immunotherapy (2014) 6(11):1165–75. doi:10.2217/imt.14.80
46. Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D’Cruz D, Wallace DJ, et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum (2010) 62(10):3077–87. doi:10.1002/art.27601
47. Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet (2011) 377(9767):721–31. doi:10.1016/S0140-6736(10)61354-2
48. Jefferson E, Liscinsky M. FDA News Release. FDA Approves Benlysta to Treat Lupus 2011 (2011). Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm246489.htm
49. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi:10.1016/S1097-2765(02)00599-3
50. Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature (2008) 452(7183):103–7. doi:10.1038/nature06664
51. Watanabe H, Gehrke S, Contassot E, Roques S, Tschopp J, Friedmann PS, et al. Danger signaling through the inflammasome acts as a master switch between tolerance and sensitization. J Immunol (2008) 180(9):5826–32. doi:10.4049/jimmunol.180.9.5826
52. Lemay S, Mao C, Singh AK. Cytokine gene expression in the MRL/lpr model of lupus nephritis. Kidney Int (1996) 50(1):85–93. doi:10.1038/ki.1996.290
53. Mao C, Singh AK. IL-1 beta gene expression in B cells derived from the murine MRL/lpr model of lupus. Autoimmunity (1996) 24(2):71–9. doi:10.3109/08916939609001949
54. Zhao J, Wang H, Huang Y, Zhang H, Wang S, Gaskin F, et al. Lupus nephritis: glycogen synthase kinase 3beta promotion of renal damage through activation of the NLRP3 inflammasome in lupus-prone mice. Arthritis Rheumatol (2015) 67(4):1036–44. doi:10.1002/art.38993
55. Zhao J, Zhang H, Huang Y, Wang H, Wang S, Zhao C, et al. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-kappaB activation. Int Immunopharmacol (2013) 17(1):116–22. doi:10.1016/j.intimp.2013.05.027
56. Voronov E, Dayan M, Zinger H, Gayvoronsky L, Lin JP, Iwakura Y, et al. IL-1 beta-deficient mice are resistant to induction of experimental SLE. Eur Cytokine Netw (2006) 17(2):109–16.
57. Jandl RC, George JL, Dinarello CA, Schur PH. The effect of interleukin 1 on IgG synthesis in systemic lupus erythematosus. Clin Immunol Immunopathol (1987) 45(3):384–94. doi:10.1016/0090-1229(87)90091-2
58. Tanaka Y, Watanabe K, Suzuki M, Saito K, Oda S, Suzuki H, et al. Spontaneous production of bone-resorbing lymphokines by B cells in patients with systemic lupus erythematosus. J Clin Immunol (1989) 9(5):415–20. doi:10.1007/BF00917107
59. Alcocer-Varela J, Aleman-Hoey D, Alarcon-Segovia D. Interleukin-1 and interleukin-6 activities are increased in the cerebrospinal fluid of patients with CNS lupus erythematosus and correlate with local late T-cell activation markers. Lupus (1992) 1(2):111–7. doi:10.1177/096120339200100209
60. Pontillo A, Reis EC, Liphaus BL, Silva CA, Carneiro-Sampaio M. Inflammasome polymorphisms in juvenile systemic lupus erythematosus. Autoimmunity (2015) 16:1–4. doi:10.3109/08916934.2015.1064399
61. Schorlemmer HU, Kanzy EJ, Langner KD, Kurrle R. Immunomodulatory activity of recombinant IL-1 receptor (IL-1-R) on models of experimental rheumatoid arthritis. Agents Actions (1993) 39:C113–6. doi:10.1007/BF01972739
62. Sun H, Liu W, Shao J, Xu H, Xiao K, Sheng G. Study on immunoregulation by interleukin-1 receptor antagonist in NZB/W F mice. J Tongji Med Univ (1997) 17(1):18–20, 24.
63. Ostendorf B, Iking-Konert C, Kurz K, Jung G, Sander O, Schneider M. Preliminary results of safety and efficacy of the interleukin 1 receptor antagonist anakinra in patients with severe lupus arthritis. Ann Rheum Dis (2005) 64(4):630–3. doi:10.1136/ard.2004.025858
64. Sester DP, Sagulenko V, Thygesen SJ, Cridland JA, Loi YS, Cridland SO, et al. Deficient NLRP3 and AIM2 inflammasome function in autoimmune NZB Mice. J Immunol (2015) 195(3):1233–41. doi:10.4049/jimmunol.1402859
65. Lech M, Lorenz G, Kulkarni OP, Grosser MO, Stigrot N, Darisipudi MN, et al. NLRP3 and ASC suppress lupus-like autoimmunity by driving the immunosuppressive effects of TGF-beta receptor signalling. Ann Rheum Dis (2014) 1–12. doi:10.1136/annrheumdis-2014-205496
66. Buyon JP, Cohen P, Merrill JT, Gilkeson G, Kaplan M, James J, et al. A highlight from the LUPUS 2014 meeting: eight great ideas. Lupus Sci Med (2015) 2(1):e000087. doi:10.1136/lupus-2015-000087
67. Obermoser G, Pascual V. The interferon-alpha signature of systemic lupus erythematosus. Lupus (2010) 19(9):1012–9. doi:10.1177/0961203310371161
68. Ytterberg SR, Schnitzer TJ. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum (1982) 25(4):401–6. doi:10.1002/art.1780250407
69. Kim T, Kanayama Y, Negoro N, Okamura M, Takeda T, Inoue T. Serum levels of interferons in patients with systemic lupus erythematosus. Clin Exp Immunol (1987) 70(3):562–9.
70. Bengtsson AA, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus (2000) 9(9):664–71. doi:10.1191/096120300674499064
71. Dall’era MC, Cardarelli PM, Preston BT, Witte A, Davis JC Jr. Type I interferon correlates with serological and clinical manifestations of SLE. Ann Rheum Dis (2005) 64(12):1692–7. doi:10.1136/ard.2004.033753
72. Panem S, Ordonez N, Vilcek J. Renal deposition of alpha interferon in systemic lupus erythematosus. Infect Immun (1983) 42(1):368–73.
73. Shiozawa S, Kuroki Y, Kim M, Hirohata S, Ogino T. Interferon-alpha in lupus psychosis. Arthritis Rheum (1992) 35(4):417–22. doi:10.1002/art.1780350410
74. Kondo M, Murakawa Y, Sumita Y, Masuda H, Kobayashi S. Lupus retinopathy associated with a high IFN-alpha level in the cerebrospinal fluid. Intern Med (2002) 41(9):754–6. doi:10.2169/internalmedicine.41.754
75. Santer DM, Yoshio T, Minota S, Moller T, Elkon KB. Potent induction of IFN-alpha and chemokines by autoantibodies in the cerebrospinal fluid of patients with neuropsychiatric lupus. J Immunol (2009) 182(2):1192–201. doi:10.4049/jimmunol.182.2.1192
76. Ronnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med (1990) 227(3):207–10. doi:10.1111/j.1365-2796.1990.tb00144.x
77. Zhao J, Qiu M, Li M, Lu C, Gu J. Low prevalence of hepatitis B virus infection in patients with systemic lupus erythematosus in southern China. Rheumatol Int (2010) 30(12):1565–70. doi:10.1007/s00296-009-1188-9
78. Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, et al. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. J Exp Med (2005) 202(8):1131–9. doi:10.1084/jem.20050914
79. Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol (2007) 37(12):3582–6. doi:10.1002/eji.200737815
80. Komatsuda A, Wakui H, Iwamoto K, Ozawa M, Togashi M, Masai R, et al. Up-regulated expression of toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Exp Immunol (2008) 152(3):482–7. doi:10.1111/j.1365-2249.2008.03646.x
81. Merrill JT, Wallace DJ, Petri M, Kirou KA, Yao Y, White WI, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann Rheum Dis (2011) 70(11):1905–13. doi:10.1136/ard.2010.144485
82. Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis (2015) 1–7. doi:10.1136/annrheumdis-2014-206090
83. Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, et al. Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum (2013) 65(4):1011–21. doi:10.1002/art.37824
84. Lamphier M, Zheng W, Latz E, Spyvee M, Hansen H, Rose J, et al. Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol (2014) 85(3):429–40. doi:10.1124/mol.113.089821
85. Hennessy EJ, Parker AE, O’Neill LA. Targeting toll-like receptors: emerging therapeutics? Nat Rev Drug Discov (2010) 9(4):293–307. doi:10.1038/nrd3203
86. al-Janadi M, al-Balla S, al-Dalaan A, Raziuddin S. Cytokine profile in systemic lupus erythematosus, rheumatoid arthritis, and other rheumatic diseases. J Clin Immunol (1993) 13(1):58–67. doi:10.1007/BF00920636
87. Robak E, Smolewski P, Wozniacka A, Sysa-Jedrzejowska A, Stepien H, Robak T. Relationship between peripheral blood dendritic cells and cytokines involved in the pathogenesis of systemic lupus erythematosus. Eur Cytokine Netw (2004) 15(3):222–30.
88. Rana A, Minz RW, Aggarwal R, Anand S, Pasricha N, Singh S. Gene expression of cytokines (TNF-alpha, IFN-gamma), serum profiles of IL-17 and IL-23 in paediatric systemic lupus erythematosus. Lupus (2012) 21(10):1105–12. doi:10.1177/0961203312451200
89. Enghard P, Langnickel D, Riemekasten G. T cell cytokine imbalance towards production of IFN-gamma and IL-10 in NZB/W F1 lupus-prone mice is associated with autoantibody levels and nephritis. Scand J Rheumatol (2006) 35(3):209–16. doi:10.1080/03009740500417791
90. Jacob CO, van der Meide PH, McDevitt HO. In vivo treatment of (NZB X NZW)F1 lupus-like nephritis with monoclonal antibody to gamma interferon. J Exp Med (1987) 166(3):798–803. doi:10.1084/jem.166.3.798
91. Schmidt T, Paust HJ, Krebs CF, Turner JE, Kaffke A, Bennstein SB, et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol (2015) 67(2):475–87. doi:10.1002/art.38955
92. Haas C, Ryffel B, Le Hir M. IFN-gamma receptor deletion prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW)F1 mice. J Immunol (1998) 160(8):3713–8.
93. Scapini P, Hu Y, Chu CL, Migone TS, Defranco AL, Cassatella MA, et al. Myeloid cells, BAFF, and IFN-gamma establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J Exp Med (2010) 207(8):1757–73. doi:10.1084/jem.20100086
94. Csiszar A, Nagy G, Gergely P, Pozsonyi T, Pocsik E. Increased interferon-gamma (IFN-gamma), IL-10 and decreased IL-4 mRNA expression in peripheral blood mononuclear cells (PBMC) from patients with systemic lupus erythematosus (SLE). Clin Exp Immunol (2000) 122(3):464–70. doi:10.1046/j.1365-2249.2000.01369.x
95. Harigai M, Kawamoto M, Hara M, Kubota T, Kamatani N, Miyasaka N. Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J Immunol (2008) 181(3):2211–9. doi:10.4049/jimmunol.181.3.2211
96. Hervier B, Beziat V, Haroche J, Mathian A, Lebon P, Ghillani-Dalbin P, et al. Phenotype and function of natural killer cells in systemic lupus erythematosus: excess interferon-gamma production in patients with active disease. Arthritis Rheum (2011) 63(6):1698–706. doi:10.1002/art.30313
97. Mathian A, Hie M, Cohen-Aubart F, Amoura Z. Targeting interferons in systemic lupus erythematosus: current and future prospects. Drugs (2015) 75(8):835–46. doi:10.1007/s40265-015-0394-x
98. Welcher AA, Boedigheimer M, Kivitz A, Amoura Z, Buyon J, Rudinskaya A, et al. Blockade of interferon-gamma (IFN-gamma) normalizes IFN regulated gene expression and serum CXCL10 (IP-10) in subjects with systemic lupus erythematosus (SLE). Arthritis Rheumatol (2015) 67(10):2713–22. doi:10.1002/art.39248
99. Aringer M, Smolen JS. Therapeutic blockade of TNF in patients with SLE-promising or crazy? Autoimmun Rev (2012) 11(5):321–5. doi:10.1016/j.autrev.2011.05.001
100. Postal M, Appenzeller S. The role of tumor necrosis factor-alpha (TNF-alpha) in the pathogenesis of systemic lupus erythematosus. Cytokine (2011) 56(3):537–43. doi:10.1016/j.cyto.2011.08.026
101. McCarthy EM, Smith S, Lee RZ, Cunnane G, Doran MF, Donnelly S, et al. The association of cytokines with disease activity and damage scores in systemic lupus erythematosus patients. Rheumatology (Oxford) (2014) 53(9):1586–94. doi:10.1093/rheumatology/ket428
102. Taylor PC, Feldmann M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat Rev (2009) 5(10):578–82. doi:10.1038/nrrheum.2009.181
103. Aringer M, Graninger WB, Steiner G, Smolen JS. Safety and efficacy of tumor necrosis factor alpha blockade in systemic lupus erythematosus: an open-label study. Arthritis Rheum (2004) 50(10):3161–9. doi:10.1002/art.20576
104. Ali T, Kaitha S, Mahmood S, Ftesi A, Stone J, Bronze MS. Clinical use of anti-TNF therapy and increased risk of infections. Drug Healthc Patient Saf (2013) 5:79–99. doi:10.2147/DHPS.S28801
105. Zhu LJ, Yang X, Yu XQ. Anti-TNF-alpha therapies in systemic lupus erythematosus. J Biomed Biotechnol (2010) 2010:465898. doi:10.1155/2010/465898
106. Uppal SS, Hayat SJ, Raghupathy R. Efficacy and safety of infliximab in active SLE: a pilot study. Lupus (2009) 18(8):690–7. doi:10.1177/0961203309102557
107. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet (2014) 384(9957):1878–88. doi:10.1016/S0140-6736(14)60128-8
108. Gezer S. Antiphospholipid syndrome. Dis Mon (2003) 49(12):696–741. doi:10.1016/j.disamonth.2003.10.001
109. MacKay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med (1999) 190(11):1697–710. doi:10.1084/jem.190.11.1697
110. Stohl W, Xu D, Kim KS, Koss MN, Jorgensen TN, Deocharan B, et al. BAFF overexpression and accelerated glomerular disease in mice with an incomplete genetic predisposition to systemic lupus erythematosus. Arthritis Rheum (2005) 52(7):2080–91. doi:10.1002/art.21138
111. Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum (2001) 44(6):1313–9. doi:10.1002/1529-0131(200106)44:6<1313::AID-ART223>3.0.CO;2-S
112. Zhang J, Roschke V, Baker KP, Wang Z, Alarcon GS, Fessler BJ, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol (2001) 166(1):6–10. doi:10.4049/jimmunol.166.1.6
113. Pers JO, Daridon C, Devauchelle V, Jousse S, Saraux A, Jamin C, et al. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann N Y Acad Sci (2005) 1050:34–9. doi:10.1196/annals.1313.004
114. Becker-Merok A, Nikolaisen C, Nossent HC. B-lymphocyte activating factor in systemic lupus erythematosus and rheumatoid arthritis in relation to autoantibody levels, disease measures and time. Lupus (2006) 15(9):570–6. doi:10.1177/0961203306071871
115. Petri M, Stohl W, Chatham W, McCune WJ, Chevrier M, Ryel J, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum (2008) 58(8):2453–9. doi:10.1002/art.23678
116. Ramanujam M, Wang X, Huang W, Liu Z, Schiffer L, Tao H, et al. Similarities and differences between selective and nonselective BAFF blockade in murine SLE. J Clin Invest (2006) 116(3):724–34. doi:10.1172/JCI26385
117. Ding H, Wang L, Wu X, Yan J, He Y, Ni B, et al. Blockade of B-cell-activating factor suppresses lupus-like syndrome in autoimmune BXSB mice. J Cell Mol Med (2010) 14(6B):1717–25. doi:10.1111/j.1582-4934.2009.00817.x
118. Ramanujam M, Bethunaickan R, Huang W, Tao H, Madaio MP, Davidson A. Selective blockade of BAFF for the prevention and treatment of systemic lupus erythematosus nephritis in NZM2410 mice. Arthritis Rheum (2010) 62(5):1457–68. doi:10.1002/art.27368
119. Yoshimoto K, Takahashi Y, Ogasawara M, Setoyama Y, Suzuki K, Tsuzaka K, et al. Aberrant expression of BAFF in T cells of systemic lupus erythematosus, which is recapitulated by a human T cell line, Loucy. Int Immunol (2006) 18(7):1189–96. doi:10.1093/intimm/dxl053
120. Morimoto S, Nakano S, Watanabe T, Tamayama Y, Mitsuo A, Nakiri Y, et al. Expression of B-cell activating factor of the tumour necrosis factor family (BAFF) in T cells in active systemic lupus erythematosus: the role of BAFF in T cell-dependent B cell pathogenic autoantibody production. Rheumatology (Oxford) (2007) 46(7):1083–6. doi:10.1093/rheumatology/kem097
121. Chu VT, Enghard P, Schurer S, Steinhauser G, Rudolph B, Riemekasten G, et al. Systemic activation of the immune system induces aberrant BAFF and APRIL expression in B cells in patients with systemic lupus erythematosus. Arthritis Rheum (2009) 60(7):2083–93. doi:10.1002/art.24628
122. Muller M, Emmendorffer A, Lohmann-Matthes ML. Expansion and high proliferative potential of the macrophage system throughout life time of lupus-prone NZB/W and MRL lpr/lpr mice. Lack of down-regulation of extramedullar macrophage proliferation in the postnatal period. Eur J Immunol (1991) 21(9):2211–7. doi:10.1002/eji.1830210932
123. Harris AA, Kamishima T, Horita T, Atsumi T, Fujita N, Omatsu T, et al. Splenic volume in systemic lupus erythematosus. Lupus (2009) 18(12):1119–20. doi:10.1177/0961203309104430
124. Lutz MB. IL-3 in dendritic cell development and function: a comparison with GM-CSF and IL-4. Immunobiology (2004) 209(1–2):79–87. doi:10.1016/j.imbio.2004.03.001
125. Fishman P, Kamashta M, Ehrenfeld M, Vianna J, Hughes GR, Sredni D, et al. Interleukin-3 immunoassay in systemic lupus erythematosus patients: preliminary data. Int Arch Allergy Immunol (1993) 100(3):215–8. doi:10.1159/000236414
126. Infantino S, Jones SA, Walker JA, Maxwell MJ, Light A, O’Donnell K, et al. The tyrosine kinase Lyn limits the cytokine responsiveness of plasma cells to restrict their accumulation in mice. Sci Signal (2014) 7(338):ra77. doi:10.1126/scisignal.2005105
127. Renner K, Hermann FJ, Schmidbauer K, Talke Y, Rodriguez Gomez M, Schiechl G, et al. IL-3 contributes to development of lupus nephritis in MRL/lpr mice. Kidney Int (2015). doi:10.1038/ki.2015.196
128. Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood (2012) 119(8):1810–20. doi:10.1182/blood-2011-09-379214
129. Yang PT, Xiao WG, Zhao LJ, Lu J, He LM, Kasai H, et al. Increase in the level of macrophage colony-stimulating factor in patients with systemic lupus erythematosus. Ann Rheum Dis (2008) 67(3):429–30. doi:10.1136/ard.2007.076117
130. Maruyama J, Inokuma S. Cytokine profiles of macrophage activation syndrome associated with rheumatic diseases. J Rheumatol (2010) 37(5):967–73. doi:10.3899/jrheum.090662
131. Matsuda M, Shikata K, Makino H, Sugimoto H, Ota Z. Glomerular expression of macrophage colony-stimulating factor and granulocyte-macrophage colony-stimulating factor in patients with various forms of glomerulonephritis. Lab Invest (1996) 75(3):403–12.
132. Isbel NM, Nikolic-Paterson DJ, Hill PA, Dowling J, Atkins RC. Local macrophage proliferation correlates with increased renal M-CSF expression in human glomerulonephritis. Nephrol Dial Transplant (2001) 16(8):1638–47. doi:10.1093/ndt/16.8.1638
133. Tian S, Li J, Wang L, Liu T, Liu H, Cheng G, et al. Urinary levels of RANTES and M-CSF are predictors of lupus nephritis flare. Inflamm Res (2007) 56(7):304–10. doi:10.1007/s00011-007-6147-x
134. Yui MA, Brissette WH, Brennan DC, Wuthrich RP, Rubin-Kelley VE. Increased macrophage colony-stimulating factor in neonatal and adult autoimmune MRL-lpr mice. Am J Pathol (1991) 139(2):255–61.
135. Lenda DM, Stanley ER, Kelley VR. Negative role of colony-stimulating factor-1 in macrophage, T cell, and B cell mediated autoimmune disease in MRL-Fas(lpr) mice. J Immunol (2004) 173(7):4744–54. doi:10.4049/jimmunol.173.7.4744
136. Menke J, Rabacal WA, Byrne KT, Iwata Y, Schwartz MM, Stanley ER, et al. Circulating CSF-1 promotes monocyte and macrophage phenotypes that enhance lupus nephritis. J Am Soc Nephrol (2009) 20(12):2581–92. doi:10.1681/ASN.2009050499
137. Chalmers SA, Chitu V, Herlitz LC, Sahu R, Stanley ER, Putterman C. Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. J Autoimmun (2015) 57:42–52. doi:10.1016/j.jaut.2014.11.007
138. van Nieuwenhuijze A, Koenders M, Roeleveld D, Sleeman MA, van den Berg W, Wicks IP. GM-CSF as a therapeutic target in inflammatory diseases. Mol Immunol (2013) 56(4):675–82. doi:10.1016/j.molimm.2013.05.002
139. Horwitz DA, Gray JD, Behrendsen SC, Kubin M, Rengaraju M, Ohtsuka K, et al. Decreased production of interleukin-12 and other Th1-type cytokines in patients with recent-onset systemic lupus erythematosus. Arthritis Rheum (1998) 41(5):838–44. doi:10.1002/1529-0131(199805)41:5<838::AID-ART10>3.0.CO;2-S
140. Fiehn C, Wermann M, Pezzutto A, Hufner M, Heilig B. [Plasma GM-CSF concentrations in rheumatoid arthritis, systemic lupus erythematosus and spondyloarthropathy]. Z Rheumatol (1992) 51(3):121–6.
141. Willeke P, Schluter B, Schotte H, Erren M, Mickholz E, Domschke W, et al. Increased frequency of GM-CSF secreting PBMC in patients with active systemic lupus erythematosus can be reduced by immunoadsorption. Lupus (2004) 13(4):257–62. doi:10.1191/0961203304lu1009oa
142. Lu C, Li J. [Effect of G-CSF and GM-CSF on expression of TNF-alpha mRNA and CD69 and secretion of IgG in peripheral blood mononuclear cells from systemic lupus erythematosus patients]. Zhongguo Shi Yan Xue Ye Xue Za Zhi (2002) 10(6):531–4.
143. Akiyama Y, Suzuki T, Imai F, Ishibashi T, Fujimaki T. Low reactivity of peripheral blood dendritic cells respond to IL-1 and GM-CSF in SLE patients. Adv Exp Med Biol (1995) 378:385–7. doi:10.1007/978-1-4615-1971-3_86
144. Armstrong DJ, Whitehead EM, Crockard AD, Bell AL. Distinctive effects of G-CSF, GM-CSF and TNFalpha on neutrophil apoptosis in systemic lupus erythematosus. Clin Exp Rheumatol (2005) 23(2):152–8.
145. Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis (2015) 74(6):1058–64. doi:10.1136/annrheumdis-2013-204816
146. Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther (2006) 8(Suppl 2):S2. doi:10.1186/ar1916
147. Chun HY, Chung JW, Kim HA, Yun JM, Jeon JY, Ye YM, et al. Cytokine IL-6 and IL-10 as biomarkers in systemic lupus erythematosus. J Clin Immunol (2007) 27(5):461–6. doi:10.1007/s10875-007-9104-0
148. Sabry A, Sheashaa H, El-Husseini A, Mahmoud K, Eldahshan KF, George SK, et al. Proinflammatory cytokines (TNF-alpha and IL-6) in Egyptian patients with SLE: its correlation with disease activity. Cytokine (2006) 35(3–4):148–53. doi:10.1016/j.cyto.2006.07.023
149. Robak E, Sysa-Jedrzejowska A, Stepien H, Robak T. Circulating interleukin-6 type cytokines in patients with systemic lupus erythematosus. Eur Cytokine Netw (1997) 8(3):281–6.
150. Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol (1991) 147(1):117–23.
151. Tsai CY, Wu TH, Yu CL, Lu JY, Tsai YY. Increased excretions of beta2-microglobulin, IL-6, and IL-8 and decreased excretion of Tamm-Horsfall glycoprotein in urine of patients with active lupus nephritis. Nephron (2000) 85(3):207–14. doi:10.1159/000045663
152. Peterson E, Robertson AD, Emlen W. Serum and urinary interleukin-6 in systemic lupus erythematosus. Lupus (1996) 5(6):571–5. doi:10.1177/096120339600500603
153. Hillion S, Garaud S, Devauchelle V, Bordron A, Berthou C, Youinou P, et al. Interleukin-6 is responsible for aberrant B-cell receptor-mediated regulation of RAG expression in systemic lupus erythematosus. Immunology (2007) 122(3):371–80. doi:10.1111/j.1365-2567.2007.02649.x
154. Cui YX, Fu CW, Jiang F, Ye LX, Meng W. Association of the interleukin-6 polymorphisms with systemic lupus erythematosus: a meta-analysis. Lupus (2015) 24(12):1308–17. doi:10.1177/0961203315588971
155. Emmendorffer A, Muller M, Wedekind D, Hartung K, Lohmann-Matthes ML. Expansion of the liver-associated macrophage system in systemic lupus erythematosus-prone NZB/W mice. J Leukoc Biol (1993) 53(3):294–300.
156. Tang B, Matsuda T, Akira S, Nagata N, Ikehara S, Hirano T, et al. Age-associated increase in interleukin 6 in MRL/lpr mice. Int Immunol (1991) 3(3):273–8. doi:10.1093/intimm/3.3.273
157. Mao X, Wu Y, Diao H, Hao J, Tian G, Jia Z, et al. Interleukin-6 promotes systemic lupus erythematosus progression with Treg suppression approach in a murine systemic lupus erythematosus model. Clin Rheumatol (2014) 33(11):1585–93. doi:10.1007/s10067-014-2717-9
158. Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J Immunol (2007) 178(1):271–9. doi:10.4049/jimmunol.178.1.271
159. Maeda K, Malykhin A, Teague-Weber BN, Sun XH, Farris AD, Coggeshall KM. Interleukin-6 aborts lymphopoiesis and elevates production of myeloid cells in systemic lupus erythematosus-prone B6.Sle1.Yaa animals. Blood (2009) 113(19):4534–40. doi:10.1182/blood-2008-12-192559
160. Cash H, Relle M, Menke J, Brochhausen C, Jones SA, Topley N, et al. Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Faslpr mice: the IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J Rheumatol (2010) 37(1):60–70. doi:10.3899/jrheum.090194
161. Maier-Moore JS, Horton CG, Mathews SA, Confer AW, Lawrence C, Pan Z, et al. Interleukin-6 deficiency corrects nephritis, lymphocyte abnormalities, and secondary Sjogren’s syndrome features in lupus-prone Sle1.Yaa mice. Arthritis Rheumatol (2014) 66(9):2521–31. doi:10.1002/art.38716
162. Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The soluble interleukin 6 receptor: generation and role in inflammation and cancer. Eur J Cell Biol (2011) 90(6–7):484–94. doi:10.1016/j.ejcb.2010.10.007
163. Tsantikos E, Maxwell MJ, Putoczki T, Ernst M, Rose-John S, Tarlinton DM, et al. Interleukin-6 trans-signaling exacerbates inflammation and renal pathology in lupus-prone mice. Arthritis Rheum (2013) 65(10):2691–702. doi:10.1002/art.38061
164. Stein B, Kung Sutherland MS. IL-6 as a drug discovery target. Drug Discov Today (1998) 3(5):202–13. doi:10.1016/S1359-6446(97)01164-1
165. Liu W, Sun H, Shao J, Xu H, Xiao K, Shen G. Immunoregulation of lupus-like NZB/W F1 mice by anti-murine IL-1 alpha, IL-6 antibodies. J Tongji Med Univ (1996) 16(4):209–11.
166. Finck BK, Chan B, Wofsy D. Interleukin 6 promotes murine lupus in NZB/NZW F1 mice. J Clin Invest (1994) 94(2):585–91. doi:10.1172/JCI117373
167. Liang B, Gardner DB, Griswold DE, Bugelski PJ, Song XY. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology (2006) 119(3):296–305. doi:10.1111/j.1365-2567.2006.02433.x
168. Lu ZY, Brailly H, Wijdenes J, Bataille R, Rossi JF, Klein B. Measurement of whole body interleukin-6 (IL-6) production: prediction of the efficacy of anti-IL-6 treatments. Blood (1995) 86(8):3123–31.
169. Zhuang Y, Xu Z, de Vries DE, Wang Q, Shishido A, Comisar C, et al. Pharmacokinetics and safety of sirukumab following a single subcutaneous administration to healthy Japanese and Caucasian subjects. Int J Clin Pharmacol Ther (2013) 51(3):187–99. doi:10.5414/CP201785
170. Xu Z, Bouman-Thio E, Comisar C, Frederick B, Van Hartingsveldt B, Marini JC, et al. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br J Clin Pharmacol (2011) 72(2):270–81. doi:10.1111/j.1365-2125.2011.03964.x
171. Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B. Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis (2014) 73(9):1616–25. doi:10.1136/annrheumdis-2013-205137
172. Szepietowski JC, Nilganuwong S, Wozniacka A, Kuhn A, Nyberg F, van Vollenhoven RF, et al. Phase I, randomized, double-blind, placebo-controlled, multiple intravenous, dose-ascending study of sirukumab in cutaneous or systemic lupus erythematosus. Arthritis Rheum (2013) 65(10):2661–71. doi:10.1002/art.38091
173. Kiberd BA. Interleukin-6 receptor blockage ameliorates murine lupus nephritis. J Am Soc Nephrol (1993) 4(1):58–61.
174. Sato K, Tsuchiya M, Saldanha J, Koishihara Y, Ohsugi Y, Kishimoto T, et al. Reshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res (1993) 53(4):851–6.
175. Navarro-Millan I, Singh JA, Curtis JR. Systematic review of tocilizumab for rheumatoid arthritis: a new biologic agent targeting the interleukin-6 receptor. Clin Ther (2012) 34(4):788.e–802.e. doi:10.1016/j.clinthera.2012.02.014
176. Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum (2010) 62(2):542–52. doi:10.1002/art.27221
177. Shirota Y, Yarboro C, Fischer R, Pham TH, Lipsky P, Illei GG. Impact of anti-interleukin-6 receptor blockade on circulating T and B cell subsets in patients with systemic lupus erythematosus. Ann Rheum Dis (2013) 72(1):118–28. doi:10.1136/annrheumdis-2012-201310
178. Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem (2001) 268(1):160–7. doi:10.1046/j.1432-1327.2001.01867.x
179. Nowell MA, Richards PJ, Horiuchi S, Yamamoto N, Rose-John S, Topley N, et al. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J Immunol (2003) 171(6):3202–9. doi:10.4049/jimmunol.171.6.3202
180. Richards PJ, Nowell MA, Horiuchi S, McLoughlin RM, Fielding CA, Grau S, et al. Functional characterization of a soluble gp130 isoform and its therapeutic capacity in an experimental model of inflammatory arthritis. Arthritis Rheum (2006) 54(5):1662–72. doi:10.1002/art.21818
181. Schuett H, Oestreich R, Waetzig GH, Annema W, Luchtefeld M, Hillmer A, et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol (2012) 32(2):281–90. doi:10.1161/ATVBAHA.111.229435
182. Zhao M, Jiang B, Gao FH. Small molecule inhibitors of STAT3 for cancer therapy. Curr Med Chem (2011) 18(26):4012–8. doi:10.2174/092986711796957284
183. Liu Y, Holdbrooks AT, De Sarno P, Rowse AL, Yanagisawa LL, McFarland BC, et al. Therapeutic efficacy of suppressing the Jak/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J Immunol (2014) 192(1):59–72. doi:10.4049/jimmunol.1301513
184. Lu LD, Stump KL, Wallace NH, Dobrzanski P, Serdikoff C, Gingrich DE, et al. Depletion of autoreactive plasma cells and treatment of lupus nephritis in mice using CEP-33779, a novel, orally active, selective inhibitor of JAK2. J Immunol (2011) 187(7):3840–53. doi:10.4049/jimmunol.1101228
185. Tagoe C, Putterman C. JAK2 inhibition in murine systemic lupus erythematosus. Immunotherapy (2012) 4(4):369–72. doi:10.2217/imt.12.20
186. van Vollenhoven RF, Layton M, Kahl L, Schifano L, Hachulla E, Machado D, et al. DRESS syndrome and reversible liver function abnormalities in patients with systemic lupus erythematosus treated with the highly selective JAK-1 inhibitor GSK2586184. Lupus (2015) 24(6):648–9. doi:10.1177/0961203315573347
187. Paran D, Naparstek Y. Is B cell-targeted therapy effective in systemic lupus erythematosus? Isr Med Assoc J (2015) 17(2):98–103.
188. Khodadadi L, Cheng Q, Alexander T, Sercan-Alp O, Klotsche J, Radbruch A, et al. Bortezomib plus continuous B cell depletion results in sustained plasma cell depletion and amelioration of lupus nephritis in NZB/W F1 mice. PLoS One (2015) 10(8):e0135081. doi:10.1371/journal.pone.0135081
189. Lin W, Seshasayee D, Lee WP, Caplazi P, McVay S, Suto E, et al. Dual B cell immunotherapy is superior to individual anti-CD20 depletion or BAFF blockade in murine models of spontaneous or accelerated lupus. Arthritis Rheumatol (2015) 67(1):215–24. doi:10.1002/art.38907
Keywords: inflammation, SLE/lupus, nephritis, immunopathology, interleukin-6, proinflammatory cytokines, lupus models, therapeutics
Citation: Gottschalk TA, Tsantikos E and Hibbs ML (2015) Pathogenic inflammation and its therapeutic targeting in systemic lupus erythematosus. Front. Immunol. 6:550. doi: 10.3389/fimmu.2015.00550
Received: 27 August 2015; Accepted: 14 October 2015;
Published: 28 October 2015
Edited by:
James Harris, Monash University, AustraliaReviewed by:
Laurence Morel, University of Florida, USACopyright: © 2015 Gottschalk, Tsantikos and Hibbs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margaret L. Hibbs, bWFyZ2FyZXQuaGliYnNAbW9uYXNoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.