 Lara Valiño-Rivas1
Lara Valiño-Rivas1 Ciro Baeza-Bermejillo1,2
Ciro Baeza-Bermejillo1,2 Laura Gonzalez-Lafuente1Ana Belen Sanz1,2
Laura Gonzalez-Lafuente1Ana Belen Sanz1,2 Alberto Ortiz1,2,3,4†
Alberto Ortiz1,2,3,4† Maria Dolores Sanchez-Niño1,2*†
Maria Dolores Sanchez-Niño1,2*†
- 1Instituto de Investigación Sanitaria de la Fundación Jiménez Díaz, Universidad Autónoma de Madrid, Madrid, Spain
- 2Red de Investigación Renal (REDINREN), Madrid, Spain
- 3School of Medicine, Universidad Autónoma de Madrid, Madrid, Spain
- 4Fundacion Renal Iñigo Alvarez de Toledo-IRSIN, Madrid, Spain
CD74 (invariant MHC class II) regulates protein trafficking and is a receptor for macrophage migration inhibitory factor (MIF) and d-dopachrome tautomerase (d-DT/MIF-2). CD74 expression is increased in tubular cells and/or glomerular podocytes and parietal cells in human metabolic nephropathies, polycystic kidney disease, graft rejection and kidney cancer and in experimental diabetic nephropathy and glomerulonephritis. Stressors like abnormal metabolite (glucose, lyso-Gb3) levels and inflammatory cytokines increase kidney cell CD74. MIF activates CD74 to increase inflammatory cytokines in podocytes and tubular cells and proliferation in glomerular parietal epithelial cells and cyst cells. MIF overexpression promotes while MIF targeting protects from experimental glomerular injury and kidney cysts, and interference with MIF/CD74 signaling or CD74 deficiency protected from crescentic glomerulonephritis. However, CD74 may protect from interstitial kidney fibrosis. Furthermore, CD74 expression by stressed kidney cells raises questions about the kidney safety of cancer therapy strategies delivering lethal immunoconjugates to CD74-expressing cells. Thus, understanding CD74 biology in kidney cells is relevant for kidney therapeutics.
CD74
CD74 (MHC class II invariant chain, Ii) is a transmembrane glycoprotein that regulates protein trafficking and is a cell surface receptor for the cytokines macrophage migration inhibitory factor (MIF) and d-dopachrome tautomerase (d-DT/MIF-2) (1). During kidney injury, leukocytes and intrinsic renal cells express CD74 (2). In the kidneys, MIF promotes experimental glomerular injury and cystogenesis, while recent reports suggest a protective role in interstitial fibrosis (2–4). However, CD74 is a multifunctional protein. Thus, the consequences of therapeutically targeting CD74 may differ from the consequences of targeting MIF. We, now, summarize the function of CD74 and review its expression and role in kidney injury, highlighting open questions. The detailed description of the role of CD74 in the immune system is beyond the scope of this review.
CD74 Functions
CD74 regulates intracellular trafficking and functions as a chaperone and a cell membrane receptor, modulating B, T, and dendritic cell responses (1, 5) and promoting tumor growth by increasing tumor cell survival or proliferation. Thus, CD74 is considered a therapeutic target in malignancy. In addition, CD74 regulates proliferation, survival, and secretion of inflammatory and fibrosis mediators in non-immune and non-tumor cells (1, 2). Thus, CD74 may modulate tissue injury and homeostasis beyond its effect on immune regulation.
Protein Trafficking
CD74 interacts with MHC class I and II proteins, contributing to antigen presentation. CD74 directs transport of MHC class II α and β chains from the endoplasmic reticulum (ER) or the cell surface to endosomes (6). As a chaperone, CD74 contributes to peptide editing in the MHC class II compartment. In endosomes, proteases degrade CD74, releasing MHC class II molecules. Prevention of CD74 degradation promotes the cell surface localization of MHC II. Extracellular CD74/cathepsin L complexes are found in human kidneys (1, 7, 8). CD74 also associates with angiotensin II type I receptors (AT1), leading to AT1 proteasomal degradation (9).
Cell Surface Receptor
Only a small amount of CD74 (2–5%) is expressed at the cell surface (10, 11). Cell surface CD74 is a receptor for MIF and MIF-2. CD74 lacks signaling motifs, but may generate fragments with transcription factor activity and binds to signaling proteins, such as CD44. Ligand binding to CD74 leads to CD74 phosphorylation, endocytosis, regulated intramembrane proteolysis (RIP), and release of CD74 intracellular domain (CD74-ICD) into the cytosol that translocates to the nucleus and activates NF-κB (10, 12–14). RIP might occur in cultured vascular smooth muscle cells since γ-secretase activity was required for MCP-1 expression induced by activating anti-CD74 antibodies. However, the pathophysiological implications remain unclear since MIF did not increase MCP-1 in these cells (15).
Migration inhibitory factor trimers activate CD74, and CD74 alone is required for MIF binding (16). In addition, MIF can also engage chemokine receptors CXCR2, CXCR4, and CXCR7 (10, 17). Cells that express two receptors, e.g., CD74 and CXCR2, may be more responsive to MIF (17). MIF-induced CD74 signaling complexes with CXCR2, CXCR4, or CXCR7 promote chemokine expression and chemotaxis (17–19). CD44 is the signaling component of the MIF-CD74 receptor complex and recruits the Src tyrosine kinase to activate ERK1/2 in macrophages and Syk, Akt, and NFκB in B cells (20, 21). Thus, the receptor complex activates kinase cascades and transcription factors. Interestingly MIF, itself increases CD44 expression (22).
Migration inhibitory factor-2 is 30% homologous to MIF, also activates CD74 (23, 24) and is responsible for residual CD74-dependent chemotactic activity in MIF−/− mice (17). MIF-2 binding to CD74/CD44 activates kinases, downstream proinflammatory pathways, and β-catenin (25). However, MIF-2 lacks the pseudo(E)LR motif present in MIF that mediates interaction with CXCR2 and CXCR4, and thus, it is a more selective CD74 agonist than MIF. Circulating MIF-2 levels correlate with severity of sepsis, a cause of acute kidney injury, and MIF or MIF-2 blockade reduced systemic inflammation, protecting mice from lethal endotoxemia.
Migration inhibitory factor or MIF-2 activation of CD74 regulates cell survival and proliferation of B cells (21) and epithelial cells, including gastric epithelial cells and type II alveolar epithelial cells (26, 27). MIF or MIF-2 binding to CD74 protects the heart and liver from injury, including from ischemia–reperfusion (28–30). In the heart, CD74 promotes phosphorylation of the AMPK catalytic alpha subunit in response to increased intracellular calcium and activation of Ca2+/calmodulin-activated kinase kinase-2 (CaMKK-2) (29). Interestingly, liver expression of CD74 protected mice from lethality induced by agonistic anti-Fas antibodies as CD74 interfered with immediate early steps in Fas signaling at the plasma membrane, and the anti-CD74 antibody milatuzumab sensitized BJAB cells to Fas-mediated apoptosis (31). Fas ligand and Fas have long been known to promote kidney injury (32). In fact, agonistic anti-Fas antibodies induced glomerular cell apoptosis in vivo (33). Thus, CD74 interference with Fas signaling should be explored in kidney cells. The MIF/CD74/AMPK pathway also protects hepatocytes in metabolic liver injury, such as non-alcoholic steatohepatitis (30). In this regard, liver fibrosis was increased in MIF−/− or CD74−/− mice suggesting an antifibrotic effect of MIF/CD74 (34). Enhanced fibrosis was thought to result from the release of MIF inhibition of PDGF-induced migration and proliferation of hepatic stellate cells. MIF/CD74 also protects the lungs. Both MIF−/− and CD74−/− mice developed spontaneous emphysema by 6 months of age (35). However, CD74 may also contribute to disease, as discussed below for glomerulonephritis and kidney cysts. In this regard, CD74 deficiency reduced atherosclerosis in low-density lipoprotein receptor-deficient LDLR−/− mice (36) and protected NOD mice from development of diabetes, probably by enhancing T regulatory cell number and impairing antigen presentation (37).
Among kidney cells, MIF induced proliferation in parietal epithelial cells but not in podocytes (4) (Figure 1). Absence of CD44 or the terminal differentiation state of podocytes may account for the differences. MIF, MIF-2, CD74, and CD44 promote clear cell renal cell carcinoma, cell proliferation, and HIF-activation (38, 39). While MIF and MIF-2 overlap in controlling cell survival and tumor formation, MIF-2 plays a dominant role in renal cancer tumor growth in vivo (40). MIF also confers resistance to senescence and cell death in mesenchymal stem cells through CD74-dependent AMPK-FOXO3a signaling and c-Met activation (41).
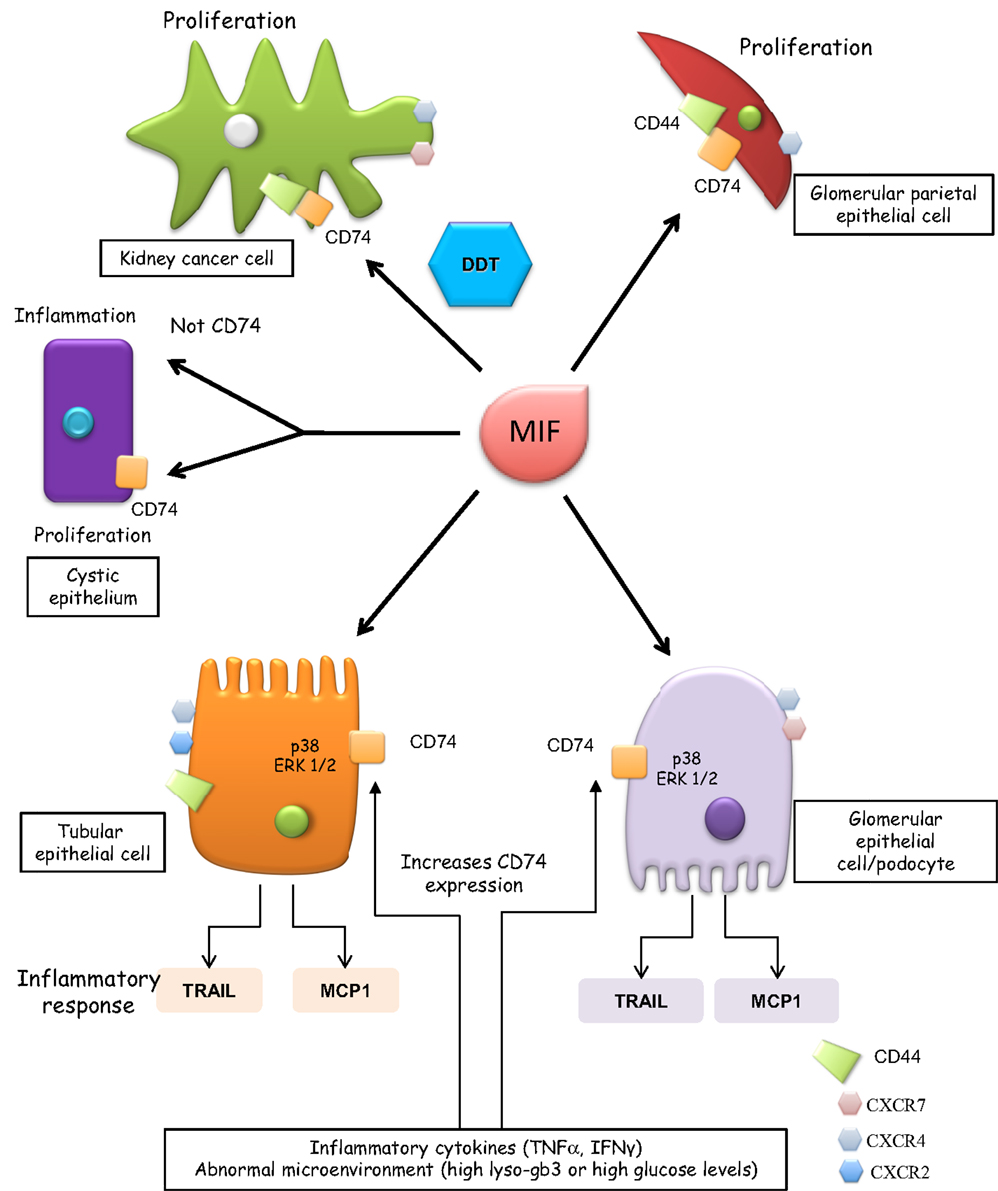
Figure 1. CD74 functions in renal cells. Glomerular parietal epithelial cells express CD44 when activated and it is thought that CD44 contributes to the proliferative response. CD44 is not expressed by podocytes and its role of CD74 signaling in tubular cells has not been characterized. Thus, in tubular cells, CD44 is not depicted as part of the CD74 signaling complex. Cells expressing CXCR2, CXCR4, and CXCR7 are also indicated, although these receptors are depicted away from MIF when in that specific cell type, there is no information on their involvement in MIF signaling. In tubular epithelium with genetic defects in PKD1, MIF promotes tubular cell proliferation and cystogenesis and a CD74 antibody blocked the MIF-induced phosphorylation of ERK but not inflammatory responses.
In renal tubular epithelial cells and podocytes, MIF binding to CD74 leads to persistent activation of p38 and ERK1/2 MAPK and expression of inflammatory mediators (e.g., TRAIL and MCP-1) (11, 42). MIF upregulation of inflammatory mediators was a late event, observed at 24 h (11). Thus, it was delayed as compared to responses elicited by the inflammatory cytokines TNF or TWEAK or metabolites, such as lyso-Gb3 (43, 44).
In summary, MIF-2 and MIF have an overlapping spectrum of activities mediated by CD74 activation and may cooperate, additively inducing chemokine secretion or survival in non-renal cells and proliferation in kidney cancer cells (45).
Regulation of CD74 Expression
CD74 expression is increased during tissue injury in diverse organs and in malignancies, including kidney cancer (2, 15, 28, 34, 46–48). There is limited information on the regulation of CD74 expression in renal cells. In normal mouse and human kidneys, tubular but not glomerular epithelium expresses low levels of CD74 (4, 11). By contrast, cultured human podocytes and proximal tubular cells and murine glomerular parietal epithelial cells express CD74 (4, 11). Abnormally high concentrations of certain metabolites (e.g., glucose and lyso-Gb3) and inflammatory cytokines, such as TNF, increase CD74 expression in podocytes and/or tubular cells (11, 49). IFN-γ increases CD74 expression in vivo in kidney tubular epithelium and in endothelial cells of larger kidney vessels (50). The factors known to upregulate CD74 expression in kidney cells may be relevant for diabetic nephropathy, Fabry disease, and inflammatory conditions.
Regulation of CD74 Interaction with MIF
Endogenous factors or drugs interfere with MIF binding to CD74 or downregulate CD74 expression and a better understanding of these interactions may provide therapeutic tools to manipulate the system. Some agents targeting MIF prevent MIF binding to CD74. These include antibodies, RPS19 (ribosomal protein S19), a component of the 40S small ribosomal subunit that binds MIF and behaves as an endogenous blocker of MIF binding to CD74 (51) and the small molecule MIF antagonist 3-(3-hydroxybenzyl)-5-methylbenzooxazol-2-one (MIF098) that decreases tautomerase activity and MIF-CD74 binding. MIF098 attenuated MIF-dependent ERK1/2 phosphorylation in human synovial fibroblasts (52) and promoted hyperoxia-induced lung injury in vivo (53), supporting the tissue-protective properties of MIF/CD74. Other compounds bind to CD74 or interfere with CD74 processing. The HLA-DRα1 domain binds to and downregulates CD74 on monocytes, directly inhibiting MIF binding to CD74 and blocking downstream inflammation in murine autoimmune encephalomyelitis. Adding a peptide extension [myelin oligodendrocyte glycoprotein (MOG)-35–55 peptide] that modified the secondary structure, enhanced the potency of the DRα1 domain in downregulating CD74 cell surface expression (54). Binding of partial MHC class II complexes comprised of linked β1α1 domains with covalently attached antigenic peptides [recombinant T-cell receptor ligands (RTLs)] to CD74 blocks the accessibility and availability of CD74 for MIF binding and downstream inflammation in monocytes (55).
The intramembrane protease presenilin homolog signal-peptide-peptidase-like 2a (SPPL2a) cleaves CD74. SPPL2a−/− mice accumulate N-terminal fragments of CD74 that impair membrane traffic within the endocytic system and alter B cell biology (56, 57). Since in SPPL2a−/− mice, CD74 signaling is inhibited, SPPL2a inhibitors may offer new pathways to inhibit CD74 signaling (58). Despite these recent advances, there is little or no information on the potential therapeutic or adverse effects of CD74 targeting strategies for kidney diseases and the available information is mainly derived from the study of CD74−/− mice, which may have developed adaptive mechanisms over development.
The role of soluble CD74 is unclear. Circulating soluble CD74 with MIF neutralizing activity was increased in primary biliary cirrhosis and was hypothesized to contribute to liver fibrosis (59). Indeed, soluble CD74 ectodomain prevents binding of MIF to cell surface receptors (10). However, MIF/sCD74 complexes were found to enhance MIF antioxidant activity (60). Autoantibodies against CD74 (anti-HLA class II-associated invariant chain peptide, CLIP) were found in 67% of ankylosing spondylitis patients, in 15% of systemic lupus erythematosus patients, and only in 0.8% of blood donors (61, 62).
CD74 and Kidney Injury
There is a growing body of evidence linking CD74 to promotion or protection from kidney injury (Figure 2).
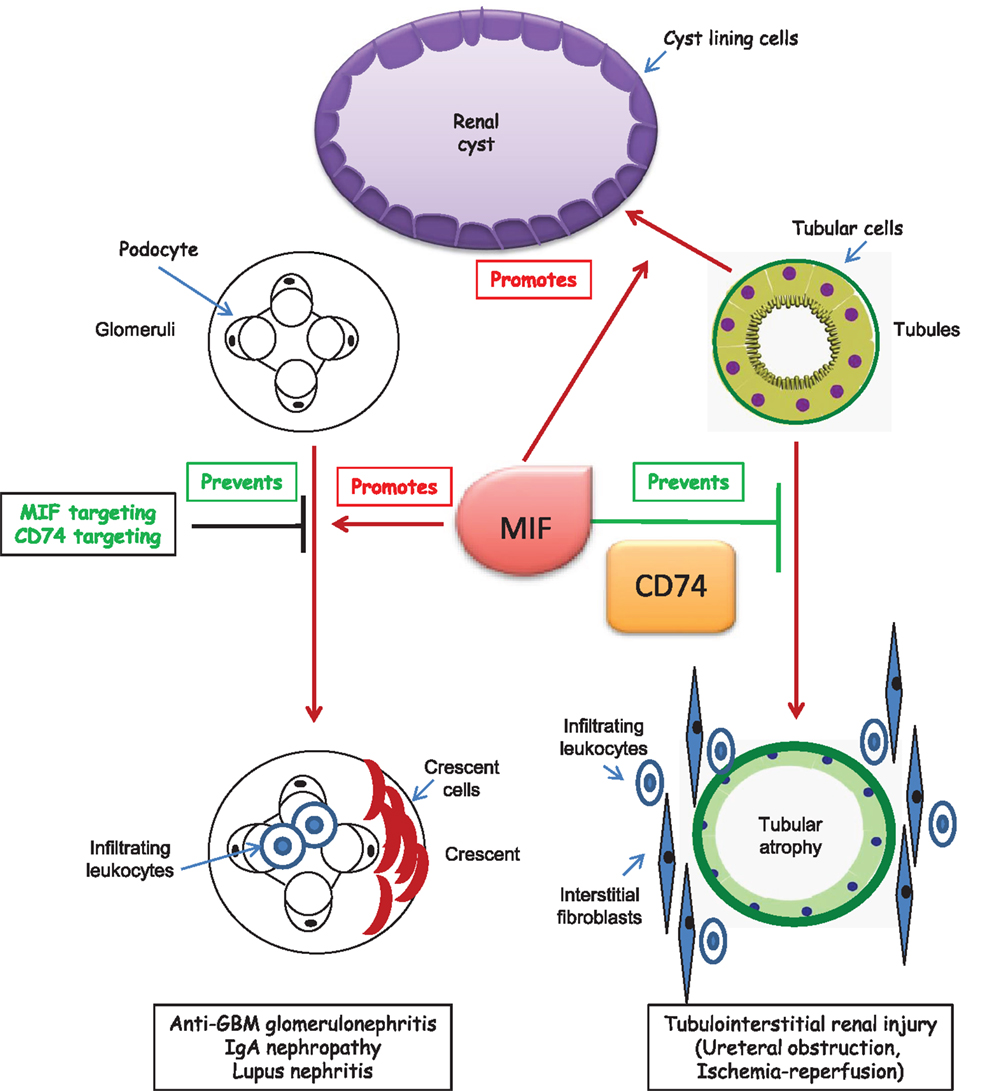
Figure 2. CD74 in kidney disease. Studies in CD74-deficient mice with kidney disease have only been published in abstract form. Thus, potential roles of CD74 in kidney disease have been mainly derived from abstracts or studies in which MIF was targeted in cultured cells or experimental animals. A putative effect of CD74 targeting on glomerular injury is only hypothetical and based on studies in which MIF was targeted. In polycystic kidney disease, MIF promotes cystogenesis. The role of CD74 is unclear, but CD74 expression is increased in cystic epithelium.
Expression of CD74 and Related Molecules in Kidney Injury
MIF has long been known to be upregulated in kidney injury (2). However, reports of increased CD74 expression are more recent and there is very little information on MIF-2.
Low CD74 expression may limit inflammatory and proliferative cell responses to MIF, and the increased renal cell CD74 expression observed during kidney injury (discussed below) may contribute, together with increased ligand availability, to elicit biological responses during kidney disease. Thus, overexpression of CD74 led to upregulation of NF-κB-dependent genes encoding cytokines in macrophages (63) and to NFκB activation and proliferation in human embryonic kidney cells (64).
CD74 is upregulated in tubular epithelial cells in at least some forms of chronic kidney disease (CKD), such as kidney graft rejection, diabetic and Fabry nephropathy and autosomal dominant polycystic kidney disease (ADPKD), and in podocytes in human diabetic nephropathy and Fabry nephropathy (11, 65, 66). A transcriptomics analysis also revealed increased kidney CD74 mRNA expression in human hypertensive nephropathy (11). CD74 is also increased in human clear cell renal cell carcinoma (67), in B cells, and kidney of lupus mice in parallel to progression of inflammation (68) and in the tubulointerstitium in anti-GBM nephritis, where it was expressed de novo in glomerular parietal epithelial cells and crescents (51). Interestingly, kidney cancer may be a complication of CKD-related acquired polycystic kidney disease (69). There is much less information on the expression of CD74 in acute kidney injury.
Migration inhibitory factor was recently identified as an important regulator of cyst growth and therapeutic target in ADPKD (3). MIF accumulated in cyst fluid of human ADPKD, promoted cystic epithelial cell proliferation and regulated apoptosis. In experimental murine polycystic kidney disease, MIF was required for renal inflammation and cyst expansion. CD74 expression was increased in Pkd1 mutant renal epithelial cells and ADPKD kidneys, suggesting that MIF actions may imply CD74 activation. However, a CD74 antibody blocked the MIF-induced phosphorylation of ERK but did not modulate the MIF-induced inflammatory response of increased MCP-1 expression in culture (3).
The CD74 signaling machinery may also be upregulated in kidney disease. CD44 is rapidly upregulated after injury in acute and chronic experimental and human kidney disease including glomerulonephritis, diabetic nephropathy, cyclosporine nephrotoxicity, urate nephropathy, and ischemia–reperfusion kidney injury (70–76). Increased CD44 expression was localized to tubules, glomerular parietal epithelial cells, mesangial cells and infiltrating macrophages, T cells, and neutrophils (70, 72, 75–78).
Finally, the expression of at least some CD74 ligands is also increased in kidney injury. MIF expression is upregulated in acute and chronic, human, and experimental kidney diseases (2). However, there is no information on MIF-2 expression and kidney disease.
CD74 Targeting and Kidney Injury
Neutralizing anti-MIF antibodies, small molecules or endogenous inhibitors or MIF−/− mice have shown that MIF aggravates anti-GBM glomerulonephritis (69–71), experimental IgAN (79), and lupus nephritis (80). More recently, the inhibitor of MIF/CD74 interactions RPS19 prevented the development of glomerular crescents, necrosis, inflammation, renal dysfunction, proteinuria, and the upregulation of MIF and CD74 in anti-GBM glomerulonephritis (51). However, MIF absence from mice did not protect from renal allograft rejection (81) or ureteral obstruction-induced kidney injury (82).
There is limited experimental and no clinical data on CD74 targeting in kidney injury. Given the multiple MIF and CD74 functions, studies are needed that explore whether targeting CD74 is therapeutic in models in which targeting MIF was beneficial. Information on the role of CD74 as a potential therapeutic target in tissue injury is mainly derived from CD74−/− mice. As discussed above, CD74 deficiency may be beneficial for some non-renal diseases, such as liver and heart disease, and deleterious in others, such as vascular injury. Specifically, liver fibrosis was increased in CD74−/− mice (34). An initial report found no protection from ureteral obstruction-induced kidney inflammation or fibrosis (82), but an abstract indicated that MIF targeting promoted interstitial fibrosis and inflammation following ureteral obstruction, whereas recombinant MIF reduced fibrosis (4). According to abstract reports, CD74 deficiency was also associated with increased interstitial fibrosis and inflammation following ureteral obstruction (day 5) and ischemia–reperfusion (day 21) (4). By contrast, CD74−/− mice are protected from glomerular injury induced by anti-GBM antiserum (Djudjaj JASN2015).
Since MIF or MIF-2 activation of CD74 is tissue protective in heart ischemia and liver injury induced by activation of the Fas receptor or metabolic disorders, a similar protective effect maybe hypothesized in ischemic acute kidney injury or metabolic kidney diseases characterized by increased CD74 expression, such as diabetic nephropathy or Fabry disease. Interventional studies should test these hypotheses and evaluate whether changes in fibrosis after kidney ischemia–reperfusion is secondary to an improved initial kidney injury or to a specific action of fibrosis mechanisms. These studies should differentiate between complete abrogation of CD74 expression (CD74−/−) and therapeutic downregulation of CD74. Incomplete blockade of the system may have different consequences than complete CD74 absence, given the potential proinflammatory effects of excess CD74 activation.
Kidney Safety of Lethal Anti-CD74 Immunoconjugates
The anti-CD74 antibody hLL1 milatuzumab, alone or as an immunoconjugate, is undergoing clinical trials to treat malignancy (83, 84). Milatuzumab binds to CD74 and promotes internalization of the antibody-CD74 complex, thus delivering conjugated antitumoral agents inside tumor cells with high CD74 expression, but not to normal cells with low CD74 levels (85). Since CD74 expression is also increased in renal cells from injured kidneys, nephrotoxicity is a potential complication of antitumoral anti-CD74 therapy, especially in patients with prior kidney disease since the active chemotherapeutic agent may be released inside already injured, CD74-expressing renal cells. This may be of special concern for one of the indications under study, multiple myeloma, which frequently causes kidney disease.
Summary and Conclusions
The role of CD74 in kidney injury has barely been explored and the scarce information available is derived from CD74-deficient mice that may not recapitulate the findings of targeting CD74 de novo in an adult. CD74 may contribute to or protect from tissue injury in a disease-specific manner. Thus, like MIF, CD74 may protect from experimental kidney interstitial fibrosis but promotes glomerular injury, while MIF (and potentially CD74) also promotes polycystic kidney disease. A few years ago, the situation was less complex. Given the longstanding preclinical evidence for a pathogenic role of MIF in glomerular kidney disease, anti-MIF strategies were tested in renal disease, although the sponsor decided to terminate a phase 1 trial of the anti-MIF monoclonal antibody imalumab in lupus nephritis (NCT01541670). However, MIF, MIF-2, and CD74 may be tissue protective or promote injury in an organ- and disease-specific manner and different forms of therapeutic manipulation of the system may be envisioned for different indications, from inhibiting to actually activating CD74 signaling. The potential impact of future intervention on CD74 for kidney disease is double. On one hand, therapeutic modulation of the system may be used to treat kidney disease. On the other, the kidney may suffer adverse effects from the therapeutic targeting on non-kidney diseases. Therapeutic approaches blocking MIF or CD74 signaling for non-renal indications may theoretically promote kidney fibrosis as an adverse effect, while therapeutic approaches activating the system may cause or aggravate glomerular injury. Thus, a better understanding of CD74 and the kidney is required for nephrologists and non-nephrologists. Future areas of research include the potential therapeutic interest of molecules aimed at increasing or decreasing CD74 activity for different forms of kidney disease, including acute and chronic kidney injury, the potential renal adverse effects of these approaches when used for non-renal indications, the impact on kidney disease of naturally occurring circulating soluble CD74 or CLIP and the kidney safety of antitumor therapeutic strategies delivering toxins into CD74-expressing cells or targeting CD74. Additional unknowns to be solved include the drivers and consequences of CD74 RIP in kidney cells, the significance for kidney disease of MIF-2 and of CD74 actions on mesenchymal stem cells, and whether AMPK is activated by CD74 in renal cells since, contrary to the heart, no difference in AMPK activation by acute ischemia was observed between MIF−/− and wild-type mice in the kidney, and this was attributed to lower CD74 expression in the kidney (86).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Grant support: ISCIII and FEDER funds CP14/00133, PI13/00047, Sociedad Española de Nefrologia, ISCIII-RETIC REDinREN/RD012/0021, Comunidad de Madrid CIFRA S2010/BMD-2378. Salary support: FIS to LV-R, Miguel Servet to MS-N. Programa Intensificación Actividad Investigadora (ISCIII/Agencia Laín-Entralgo/CM) to AO.
References
1. Borghese F, Clanchy FI. CD74: an emerging opportunity as a therapeutic target in cancer and autoimmune disease. Exp Opin Ther Targets (2011) 15:237–51. doi:10.1517/14728222.2011.550879
2. Sanchez-Nino MD, Sanz AB, Ruiz-Andres O, Poveda J, Izquierdo MC, Selgas R, et al. MIF, CD74 and other partners in kidney disease: tales of a promiscuous couple. Cytokine Growth Factor Rev (2013) 24:23–40. doi:10.1016/j.cytogfr.2012.08.001
3. Chen L, Zhou X, Fan LX, Yao Y, Swenson-Fields KI, Gadjeva M, et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest (2015) 125:2399–412. doi:10.1172/JCI80467
4. Djudjaj S, Lue H, Urzinicok T, Engel D, Martin IV, Buhl EM, et al. Macrophage migration inhibitory factor (MIF) – a novel endogeneous fibrosis limiting factor. Nephrol Dial Transplant (2013) 28(Suppl 1):i46–7. doi:10.1093/ndt/gft168
5. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol (2003) 3:791–800. doi:10.1038/nri1200
6. Roche PA, Teletski CL, Stang E, Bakke O, Long EO. Cell surface HLA-DR-invariant chain complexes are targeted to endosomes by rapid internalization. Proc Natl Acad Sci U S A (1993) 90:8581–5. doi:10.1073/pnas.90.18.8581
7. Fiebiger E, Maehr R, Villadangos J, Weber E, Erickson A, Bikoff E, et al. Invariant chain controls the activity of extracellular cathepsin L. J Exp Med (2002) 196:1263–9. doi:10.1084/jem.20020762
8. Ogrinc T, Dolenc I, Ritonja A, Turk V. Purification of the complex of cathepsin L and the MHC class II-associated invariant chain fragment from human kidney. FEBS Lett (1993) 336:555–9. doi:10.1016/0014-5793(93)80875-U
9. Szaszak M, Chen HD, Chen HC, Baukal A, Hunyady L, Catt KJ. Identification of the invariant chain (CD74) as an angiotensin AGTR1-interacting protein. J Endocrinol (2008) 199:165–76. doi:10.1677/JOE-08-0190
10. Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med (2003) 197:1467–76. doi:10.1084/jem.20030286
11. Sanchez-Nino MD, Sanz AB, Ihalmo P, Lassila M, Holthofer H, Mezzano S, et al. The MIF receptor CD74 in diabetic podocyte injury. J Am Soc Nephrol (2009) 20:353–62. doi:10.1681/ASN.2008020194
12. Starlets D, Gore Y, Binsky I, Haran M, Harpaz N, Shvidel L, et al. Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood (2006) 107:4807–16. doi:10.1182/blood-2005-11-4334
13. Becker-Herman S, Arie G, Medvedovsky H, Kerem A, Shachar I. CD74 is a member of the regulated intramembrane proteolysis-processed protein family. Mol Biol Cell (2005) 16:5061–9. doi:10.1091/mbc.E05-04-0327
14. Binsky I, Haran M, Starlets D, Gore Y, Lantner F, Harpaz N, et al. IL-8 secreted in a macrophage migration-inhibitory factor- and CD74-dependent manner regulates B cell chronic lymphocytic leukemia survival. Proc Natl Acad Sci U S A (2007) 104:13408–13. doi:10.1073/pnas.0701553104
15. Martin-Ventura JL, Madrigal-Matute J, Munoz-Garcia B, Blanco-Colio LM, Van OM, Zalba G, et al. Increased CD74 expression in human atherosclerotic plaques: contribution to inflammatory responses in vascular cells. Cardiovasc Res (2009) 83:586–94. doi:10.1093/cvr/cvp141
16. Fan C, Rajasekaran D, Syed MA, Leng L, Loria JP, Bhandari V, et al. MIF intersubunit disulfide mutant antagonist supports activation of CD74 by endogenous MIF trimer at physiologic concentrations. Proc Natl Acad Sci U S A (2013) 110:10994–9. doi:10.1073/pnas.1221817110
17. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med (2007) 13:587–96. doi:10.1038/nm1567
18. Alampour-Rajabi S, El BO, Rot A, Muller-Newen G, Bachelerie F, Gawaz M, et al. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J (2015). doi:10.1096/fj.15-273904
19. Simons D, Grieb G, Hristov M, Pallua N, Weber C, Bernhagen J, et al. Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. J Cell Mol Med (2011) 15:668–78. doi:10.1111/j.1582-4934.2010.01041.x
20. Shi X, Leng L, Wang T, Wang W, Du X, Li J, et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity (2006) 25:595–606. doi:10.1016/j.immuni.2006.08.020
21. Gore Y, Starlets D, Maharshak N, Becker-Herman S, Kaneyuki U, Leng L, et al. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J Biol Chem (2008) 283:2784–92. doi:10.1074/jbc.M703265200
22. Verschuren L, Kooistra T, Bernhagen J, Voshol PJ, Ouwens DM, van EM, et al. MIF deficiency reduces chronic inflammation in white adipose tissue and impairs the development of insulin resistance, glucose intolerance, and associated atherosclerotic disease. Circ Res (2009) 105:99–107. doi:10.1161/CIRCRESAHA.109.199166
23. Merk M, Zierow S, Leng L, Das R, Du X, Schulte W, et al. The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc Natl Acad Sci U S A (2011) 108:E577–85. doi:10.1073/pnas.1102941108
24. Merk M, Mitchell RA, Endres S, Bucala R. D-dopachrome tautomerase (D-DT or MIF-2): doubling the MIF cytokine family. Cytokine (2012) 59:10–7. doi:10.1016/j.cyto.2012.03.014
25. Xin D, Rendon BE, Zhao M, Winner M, McGhee CA, Mitchell RA. The MIF homologue D-dopachrome tautomerase promotes COX-2 expression through beta-catenin-dependent and -independent mechanisms. Mol Cancer Res (2010) 8:1601–9. doi:10.1158/1541-7786.MCR-10-0101
26. Beswick EJ I, Pinchuk V, Suarez G, Sierra JC, Reyes VE. Helicobacter pylori CagA-dependent macrophage migration inhibitory factor produced by gastric epithelial cells binds to CD74 and stimulates procarcinogenic events. J Immunol (2006) 176:6794–801. doi:10.4049/jimmunol.176.11.6794
27. Marsh LM, Cakarova L, Kwapiszewska G, von WW, Herold S, Seeger W, et al. Surface expression of CD74 by type II alveolar epithelial cells: a potential mechanism for macrophage migration inhibitory factor-induced epithelial repair. Am J Physiol Lung Cell Mol Physiol (2009) 296:L442–52. doi:10.1152/ajplung.00525.2007
28. Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature (2008) 451:578–82. doi:10.1038/nature06504
29. Qi D, Atsina K, Qu L, Hu X, Wu X, Xu B, et al. The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J Clin Invest (2014) 124:3540–50. doi:10.1172/JCI73061
30. Heinrichs D, Berres ML, Coeuru M, Knauel M, Nellen A, Fischer P, et al. Protective role of macrophage migration inhibitory factor in nonalcoholic steatohepatitis. FASEB J (2014) 28:5136–47. doi:10.1096/fj.14-256776
31. Berkova Z, Wang S, Ao X, Wise JF, Braun FK, Rezaeian AH, et al. CD74 interferes with the expression of fas receptor on the surface of lymphoma cells. J Exp Clin Cancer Res (2014) 33:80. doi:10.1186/s13046-014-0080-y
32. Ortiz A, Lorz C, Egido J. The Fas ligand/Fas system in renal injury. Nephrol Dial Transplant (1999) 14:1831–4. doi:10.1093/ndt/14.8.1831
33. Gonzalez-Cuadrado S, Lorz C, Garcia del MR, O’alle F, Alonso C, Ramiro F, et al. Agonistic anti-Fas antibodies induce glomerular cell apoptosis in mice in vivo. Kidney Int (1997) 51:1739–46. doi:10.1038/ki.1997.239
34. Heinrichs D, Knauel M, Offermanns C, Berres ML, Nellen A, Leng L, et al. Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc Natl Acad Sci U S A (2011) 108:17444–9. doi:10.1073/pnas.1107023108
35. Sauler M, Leng L, Trentalange M, Haslip M, Shan P, Piecychna M, et al. Macrophage migration inhibitory factor deficiency in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol (2014) 306:L487–96. doi:10.1152/ajplung.00284.2013
36. Sun J, Hartvigsen K, Chou MY, Zhang Y, Sukhova GK, Zhang J, et al. Deficiency of antigen-presenting cell invariant chain reduces atherosclerosis in mice. Circulation (2010) 122:808–20. doi:10.1161/CIRCULATIONAHA.109.891887
37. Mellanby RJ, Koonce CH, Monti A, Phillips JM, Cooke A, Bikoff EK. Loss of invariant chain protects nonobese diabetic mice against type 1 diabetes. J Immunol (2006) 177:7588–98. doi:10.4049/jimmunol.177.11.7588
38. Ji SQ, Su XL, Cheng WL, Zhang HJ, Zhao YQ, Han ZX. Down-regulation of CD74 inhibits growth and invasion in clear cell renal cell carcinoma through HIF-1alpha pathway. Urol Oncol (2014) 32:153–61. doi:10.1016/j.urolonc.2012.09.013
39. Du W, Wright BM, Li X, Finke J, Rini BI, Zhou M, et al. Tumor-derived macrophage migration inhibitory factor promotes an autocrine loop that enhances renal cell carcinoma. Oncogene (2013) 32:1469–74. doi:10.1038/onc.2012.143
40. Pasupuleti V, Du W, Gupta Y I, Yeh J, Montano M, Magi-Galuzzi C, et al. Dysregulated D-dopachrome tautomerase, a hypoxia-inducible factor-dependent gene, cooperates with macrophage migration inhibitory factor in renal tumorigenesis. J Biol Chem (2014) 289:3713–23. doi:10.1074/jbc.M113.500694
41. Xia W, Zhang F, Xie C, Jiang M, Hou M. Macrophage migration inhibitory factor confers resistance to senescence through CD74-dependent AMPK-FOXO3a signaling in mesenchymal stem cells. Stem Cell Res Ther (2015) 6:82. doi:10.1186/s13287-015-0076-3
42. Lorz C, Benito-Martin A, Boucherot A, Ucero AC, Rastaldi MP, Henger A, et al. The death ligand TRAIL in diabetic nephropathy. J Am Soc Nephrol (2008) 19:904–14. doi:10.1681/ASN.2007050581
43. Sanz AB, Justo P, Sanchez-Nino MD, Blanco-Colio LM, Winkles JA, Kreztler M, et al. The cytokine TWEAK modulates renal tubulointerstitial inflammation. J Am Soc Nephrol (2008) 19:695–703. doi:10.1681/ASN.2007050577
44. Sanchez-Nino MD, Poveda J, Sanz AB, Carrasco S, Ruiz-Ortega M, Selgas R, et al. 3,4-DGE is cytotoxic and decreases HSP27/HSPB1 in podocytes. Arch Toxicol (2014) 88:597–608. doi:10.1007/s00204-013-1181-7
45. Coleman AM, Rendon BE, Zhao M, Qian MW, Bucala R, Xin D, et al. Cooperative regulation of non-small cell lung carcinoma angiogenic potential by macrophage migration inhibitory factor and its homolog, D-dopachrome tautomerase. J Immunol (2008) 181:2330–7. doi:10.4049/jimmunol.181.4.2330
46. Beswick EJ, Das S I, Pinchuk V, Adegboyega P, Suarez G, Yamaoka Y, et al. Helicobacter pylori-induced IL-8 production by gastric epithelial cells up-regulates CD74 expression. J Immunol (2005) 175:171–6. doi:10.4049/jimmunol.175.1.171
47. Burton JD, Ely S, Reddy PK, Stein R, Gold DV, Cardillo TM, et al. CD74 is expressed by multiple myeloma and is a promising target for therapy. Clin Cancer Res (2004) 10:6606–11. doi:10.1158/1078-0432.CCR-04-0182
48. Lawrance IC, Fiocchi C, Chakravarti S. Ulcerative colitis and Crohn’ disease: distinctive gene expression profiles and novel susceptibility candidate genes. Hum Mol Genet (2001) 10:445–56. doi:10.1093/hmg/10.5.445
49. Sanchez-Nino MD, Sanz AB, Carrasco S, Saleem MA, Mathieson PW, Valdivielso JM, et al. Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. Nephrol Dial Transplant (2011) 26:1797–802. doi:10.1093/ndt/gfq306
50. Momburg F, Koch N, Moller P, Moldenhauer G, Butcher GW, Hammerling GJ. Differential expression of Ia and Ia-associated invariant chain in mouse tissues after in vivo treatment with IFN-gamma. J Immunol (1986) 136:940–8.
51. Lv J, Huang XR, Klug J, Frohlich S, Lacher P, Xu A, et al. Ribosomal protein S19 is a novel therapeutic agent in inflammatory kidney disease. Clin Sci (Lond) (2013) 124:627–37. doi:10.1042/CS20120526
52. Hare AA, Leng L, Gandavadi S, Du X, Cournia Z, Bucala R, et al. Optimization of N-benzyl-benzoxazol-2-ones as receptor antagonists of macrophage migration inhibitory factor (MIF). Bioorg Med Chem Lett (2010) 20:5811–4. doi:10.1016/j.bmcl.2010.07.129
53. Sauler M, Zhang Y, Min JN, Leng L, Shan P, Roberts S, et al. Endothelial CD74 mediates macrophage migration inhibitory factor protection in hyperoxic lung injury. FASEB J (2015) 29:1940–9. doi:10.1096/fj.14-260299
54. Meza-Romero R, Benedek G, Yu X, Mooney JL, Dahan R, Duvshani N, et al. HLA-DRalpha1 constructs block CD74 expression and MIF effects in experimental autoimmune encephalomyelitis. J Immunol (2014) 192:4164–73. doi:10.4049/jimmunol.1303118
55. Benedek G, Meza-Romero R, Andrew S, Leng L, Burrows GG, Bourdette D, et al. Partial MHC class II constructs inhibit MIF/CD74 binding and downstream effects. Eur J Immunol (2013) 43:1309–21. doi:10.1002/eji.201243162
56. Schneppenheim J, Dressel R, Huttl S, Lullmann-Rauch R, Engelke M, Dittmann K, et al. The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J Exp Med (2013) 210:41–58. doi:10.1084/jem.20121069
57. Beisner DR, Langerak P, Parker AE, Dahlberg C, Otero FJ, Sutton SE, et al. The intramembrane protease Sppl2a is required for B cell and DC development and survival via cleavage of the invariant chain. J Exp Med (2013) 210:23–30. doi:10.1084/jem.20121072
58. Ran Y, Ladd GZ, Ceballos-Diaz C, Jung JI, Greenbaum D, Felsenstein KM, et al. Differential inhibition of signal peptide peptidase family members by established gamma-secretase inhibitors. PLoS One (2015) 10:e0128619. doi:10.1371/journal.pone.0128619
59. Assis DN, Leng L, Du X, Zhang CK, Grieb G, Merk M, et al. The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatology (2014) 59:580–91. doi:10.1002/hep.26664
60. Stoppe C, Rex S, Goetzenich A, Kraemer S, Emontzpohl C, Soppert J, et al. Interaction of MIF family proteins in myocardial ischemia/reperfusion damage and their influence on clinical outcome of cardiac surgery patients. Antioxid Redox Signal (2015). doi:10.1089/ars.2014.6243
61. Baerlecken NT, Nothdorft S, Stummvoll GH, Sieper J, Rudwaleit M, Reuter S, et al. Autoantibodies against CD74 in spondyloarthritis. Ann Rheum Dis (2014) 73:1211–4. doi:10.1136/annrheumdis-2012-202208
62. Baraliakos X, Baerlecken N, Witte T, Heldmann F, Braun J. High prevalence of anti-CD74 antibodies specific for the HLA class II-associated invariant chain peptide (CLIP) in patients with axial spondyloarthritis. Ann Rheum Dis (2014) 73:1079–82. doi:10.1136/annrheumdis-2012-202177
63. Zhang H, Liu C, Cheng S, Wang X, Li W, Charreyre C, et al. Porcine CD74 is involved in the inflammatory response activated by nuclear factor kappa B during porcine circovirus type 2 (PCV-2) infection. Arch Virol (2013) 158:2285–95. doi:10.1007/s00705-013-1750-3
64. Liu YH, Lin CY, Lin WC, Tang SW, Lai MK, Lin JY. Up-regulation of vascular endothelial growth factor-D expression in clear cell renal cell carcinoma by CD74: a critical role in cancer cell tumorigenesis. J Immunol (2008) 181:6584–94. doi:10.4049/jimmunol.181.9.6584
65. Saleem M, Sawyer GJ, Schofield RA, Seymour ND, Gustafsson K, Fabre JW. Discordant expression of major histocompatibility complex class II antigens and invariant chain in interstitial dendritic cells. Implications for self-tolerance and immunity. Transplantation (1997) 63:1134–8. doi:10.1097/00007890-199704270-00013
66. Pagni F, Pieruzzi F, Zannella S, Di GA, Bovo G, Ferrario F, et al. Possible pathogenetic relationship between Fabry disease and renal cell carcinoma. Am J Nephrol (2012) 36:537–41. doi:10.1159/000345465
67. Young AN, Amin MB, Moreno CS, Lim SD, Cohen C, Petros JA, et al. Expression profiling of renal epithelial neoplasms: a method for tumor classification and discovery of diagnostic molecular markers. Am J Pathol (2001) 158:1639–51. doi:10.1016/S0002-9440(10)64120-X
68. Lapter S, Ben-David H, Sharabi A, Zinger H, Telerman A, Gordin M, et al. A role for the B-cell CD74/macrophage migration inhibitory factor pathway in the immunomodulation of systemic lupus erythematosus by a therapeutic tolerogenic peptide. Immunology (2011) 132:87–95. doi:10.1111/j.1365-2567.2010.03342.x
69. Bonsib SM. Renal cystic diseases and renal neoplasms: a mini-review. Clin J Am Soc Nephrol (2009) 4:1998–2007. doi:10.2215/CJN.02020309
70. Florquin S, Nunziata R, Claessen N, van den Berg FM, Pals ST, Weening JJ. CD44 expression in IgA nephropathy. Am J Kidney Dis (2002) 39:407–14. doi:10.1053/ajkd.2002.30563
71. Han DH, Song HK, Lee SY, Song JH, Piao SG, Yoon HE, et al. Upregulation of hyaluronan and its binding receptors in an experimental model of chronic cyclosporine nephropathy. Nephrology (Carlton) (2010) 15:216–24. doi:10.1111/j.1440-1797.2009.01167.x
72. Kocak B, Orug T, Turhan N, Ozcay N, Gonenc F. CD44 expression in renal ischemia-reperfusion injury in rats. Int Urol Nephrol (2009) 41:791–4. doi:10.1007/s11255-009-9542-0
73. Okada A, Yasui T, Fujii Y, Niimi K, Hamamoto S, Hirose M, et al. Renal macrophage migration and crystal phagocytosis via inflammatory-related gene expression during kidney stone formation and elimination in mice: detection by association analysis of stone-related gene expression and microstructural observation. J Bone Miner Res (2010) 25:2701–11. doi:10.1002/jbmr.158
74. Rouschop KM, Claessen N, Pals ST, Weening JJ, Florquin S. CD44 disruption prevents degeneration of the capillary network in obstructive nephropathy via reduction of TGF-beta1-induced apoptosis. J Am Soc Nephrol (2006) 17:746–53. doi:10.1681/ASN.2005080808
75. Smeets B, Uhlig S, Fuss A, Mooren F, Wetzels JF, Floege J, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol (2009) 20:2604–15. doi:10.1681/ASN.2009010122
76. Yang L, Brozovic S, Xu J, Long Y, Kralik PM, Waigel S, et al. Inflammatory gene expression in OVE26 diabetic kidney during the development of nephropathy. Nephron Exp Nephrol (2011) 119:e8–20. doi:10.1159/000324407
77. Nikolic-Paterson DJ, Jun Z, Tesch GH, Lan HY, Foti R, Atkins RC. De novo CD44 expression by proliferating mesangial cells in rat anti-Thy-1 nephritis. J Am Soc Nephrol (1996) 7:1006–14.
78. Sicking EM, Fuss A, Uhlig S, Jirak P, Dijkman H, Wetzels J, et al. Subtotal ablation of parietal epithelial cells induces crescent formation. J Am Soc Nephrol (2012) 23:629–40. doi:10.1681/ASN.2011050449
79. Leung JC, Chan LY, Tsang AW, Liu EW, Lam MF, Tang SC, et al. Anti-macrophage migration inhibitory factor reduces transforming growth factor-beta 1 expression in experimental IgA nephropathy. Nephrol Dial Transplant (2004) 19:1976–85. doi:10.1093/ndt/gfh323
80. Hoi AY, Hickey MJ, Hall P, Yamana J, O’ullivan KM, Santos LL, et al. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol (2006) 177:5687–96. doi:10.4049/jimmunol.177.8.5687
81. Jose MD, David JR, Atkins RC, Chadban SJ. Blockade of macrophage migration inhibitory factor does not prevent acute renal allograft rejection. Am J Transplant (2003) 3:1099–106. doi:10.1034/j.1600-6143.2003.00188.x
82. Rice EK, Nikolic-Paterson DJ, David JR, Bucala R, Metz CN, Atkins RC, et al. Macrophage accumulation and renal fibrosis are independent of macrophage migration inhibitory factor in mouse obstructive nephropathy. Nephrology (Carlton) (2004) 9:278–87. doi:10.1111/j.1440-1797.2004.00319.x
83. Stein R, Qu Z, Cardillo TM, Chen S, Rosario A I, Horak D, et al. Antiproliferative activity of a humanized anti-CD74 monoclonal antibody, hLL1, on B-cell malignancies. Blood (2004) 104:3705–11. doi:10.1182/blood-2004-03-0890
84. Kaufman JL, Niesvizky R, Stadtmauer EA, Chanan-Khan A, Siegel D, Horne H, et al. Phase I, multicentre, dose-escalation trial of monotherapy with milatuzumab (humanized anti-CD74 monoclonal antibody) in relapsed or refractory multiple myeloma. Br J Haematol (2013) 163:478–86. doi:10.1111/bjh.12565
85. Martin P, Furman RR, Rutherford S, Ruan J, Ely S, Greenberg J, et al. Phase I study of the anti-CD74 monoclonal antibody milatuzumab (hLL1) in patients with previously treated B-cell lymphomas. Leuk Lymphoma (2015):1–21. doi:10.3109/10428194.2014.1001987
Keywords: kidney, CD74, d-dopachrome tautomerase (d-DT/MIF-2), macrophage inhibitory factor, inflammation, Fabry, diabetes, polycystic kidney disease
Citation: Valiño-Rivas L, Baeza-Bermejillo C, Gonzalez-Lafuente L, Sanz AB, Ortiz A and Sanchez-Niño MD (2015) CD74 in kidney disease. Front. Immunol. 6:483. doi: 10.3389/fimmu.2015.00483
Received: 06 May 2015; Accepted: 05 September 2015;
Published: 23 September 2015
Edited by:
Heidi Noels, Institute for Molecular Cardiovascular Research, GermanyReviewed by:
Gabriela Godaly, Lund University, SwedenPeter Boor, Rheinisch-Westfälische Technische Hochschule Aachen, Germany
Copyright: © 2015 Valiño-Rivas, Baeza-Bermejillo, Gonzalez-Lafuente, Sanz, Ortiz and Sanchez-Niño. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Dolores Sanchez-Niño, Division of Nephrology, IIS-Fundación Jiménez Díaz, Av Reyes Católicos 2, Madrid 28040, Spain,bWRzYW5jaGV6QGZqZC5lcw==
†Alberto Ortiz and Maria Dolores Sanchez-Niño have contributed equally to this work.