 Sergey P. Morzunov1
Sergey P. Morzunov1 Svetlana F. Khaiboullina2,3
Svetlana F. Khaiboullina2,3 Stephen St. Jeor4
Stephen St. Jeor4 Albert A. Rizvanov2*
Albert A. Rizvanov2* Vincent C. Lombardi3,5*
Vincent C. Lombardi3,5*
- 1Department of Pathology, School of Medicine, University of Nevada, Reno, NV, USA
- 2Institute of Fundamental Medicine and Biology, Kazan Federal University, Kazan, Russia
- 3Whittemore Peterson Institute, Reno, NV, USA
- 4Department of Microbiology and Immunology, University of Nevada, Reno, NV, USA
- 5Department of Biochemistry, School of Medicine, University of Nevada, Reno, NV, USA
Hantavirus pulmonary syndrome (HPS) is an acute zoonotic disease transmitted primarily through inhalation of virus-contaminated aerosols. Hantavirus infection of endothelial cells leads to increased vascular permeability without a visible cytopathic effect. For this reason, it has been suggested that the pathogenesis of HPS is indirect with immune responses, such as cytokine production, playing a dominant role. In order to investigate their potential contribution to HPS pathogenesis, we analyzed the serum of hantavirus-infected subjects and healthy controls for 68 different cytokines, chemokines, angiogenic, and growth factors. Our analysis identified differential expression of cytokines that promote tissue migration of mononuclear cells including T lymphocytes, natural killer cells, and dendritic cells. Additionally, we observed a significant upregulation of cytokines known to regulate leukocyte migration and subsequent repair of lung tissue, as well as cytokines known to increase endothelial monolayer permeability and facilitate leukocyte transendothelial migration. Conversely, we observed a downregulation of cytokines associated with platelet numbers and function, consistent with the thrombocytopenia observed in subjects with HPS. This study corroborates clinical findings and extends our current knowledge regarding immunological and laboratory findings in subjects with HPS.
Introduction
Hantavirus pulmonary syndrome (HPS) is a severe life threatening disease caused by members of the genus Hantavirus. In the United States, these members include Sin Nombre virus, Bayou virus, Black Creek Canal virus, and New York virus, while South American members include Andes virus and Laguna Negra virus (1–5). Although HPS was first diagnosed as a clinical entity in 1993 in response to the four corners outbreak (6), retrospective studies have identified hantavirus-associated fatalities as early as 1978 (7). HPS cases have been reported in 34 states with the majority occurring in the Southwestern states; however, several have been reported in the Northwestern and Midwestern states. Through April 2014, the Center for Disease Control and Prevention has confirmed 639 total cases of HPS in the U.S., with the majority occurring in New Mexico (94 cases), Colorado (81 cases), and Arizona (72 cases) (8). Although the prevalence of HPS is low in the U.S., 36% of all reported HPS cases have resulted in death, underscoring the potential impact to public health.
Clinically, HPS manifests with fatigue, fever, muscle pain, headache, dizziness, nausea, and vomiting (9). Soon after onset, individuals present with bilateral diffuse interstitial edema resembling acute respiratory distress syndrome (10). Rapidly progressing pulmonary edema, myocardial depression, and hypovolemia are the leading cause of death (11). There is no specific treatment for HPS; therefore, medical care is mainly supportive with early diagnosis resulting in more successful outcomes.
Hantaviruses do not produce a visible cytopathic effect; consequently, it is believed that cytokines produced by infected cells either directly or indirectly lead to a compromised endothelial monolayer, which in turn, leads to vascular leakage. Indeed, increased numbers of cytokine-producing cells have been observed in lung and spleen tissue of HPS cases (12). We as well as others have demonstrated that endothelial cells produce the chemokines, CCL5 and CXCL10, when infected with Sin Nombre virus (13, 14). These cytokines are strong chemoattractants for mononuclear leukocytes including monocytes, lymphocytes, and natural killer (NK) cells (15, 16). Expression of these chemokines may explain the postmortem observation of monocytic interstitial pneumonia in fatal HPS cases; however, it remains to be determined whether these chemokines are expressed during active HPS. In contrast to CCL5 and CXCL10 and atypical of most viral infections, in vitro culture studies show that only a slight upregulation of type I interferon (IFN) is observed when endothelial cells are infected with hantaviruses. These data are also consistent with clinical observations that suggest that a robust IFN-α response is not characteristic of hantavirus infection (17, 18).
Although limited data exist regarding cytokine expression in subjects with HPS, a study by Borges et al. evaluated the concentrations of 11 serum analytes by ELISA. A cytokine profile was reported that defined the differential expression of a selected number of Th1 and Th2 cytokines (19). Specifically, they observed significantly elevated levels of IL-6, IFN-γ, sIL-2R, TNF-α, and decreased IL-10 when compared to controls, suggesting that activation of Th1 and Th2-type immune responses are involved. While ELISA is commonly used for such studies, it has limitations such as the necessity of a large sample volume and this issue is compounded when one wishes to analyze multiple analytes. High-throughput multiplex analysis by Luminex xMAP technology allows the simultaneous detection and quantitation of many analytes and uses a small amount of serum or plasma. In the present study, we utilized Luminex xMAP technology to conduct a comprehensive evaluation of 68 different cytokines, chemokines, angiogenic, and growth factors (hereafter referred to collectively as cytokines) in subjects with HPS, including 38 cytokines previously not investigated in association with this disease. Changes in 40 cytokines were detected in the serum of subjects with HPS when compared to healthy controls; 25 cytokines were significantly upregulated while15 were downregulated. A subset of these cytokines known to influence the migration of mononuclear effectors was upregulated, as were cytokines known to play a role in lung microbial defense and tissue repair. Another subset of cytokines associated with thrombocyte counts and function was downregulated. This study corroborates clinical findings and extends our current knowledge by providing a more comprehensive basis for the immune responses and morphology observed in laboratory and histological findings in subjects with HPS.
Materials and Methods
Subjects
Twelve clinical diagnostic serum specimens collected from 2008 to 2012 by the Nevada State Health Laboratory (NSHL) and with a confirmed diagnosis of HPS were utilized in this study. The NSHL serves as a regional reference laboratory and routinely screens subjects suspected of having HPS, by the presence of anti-hantavirus antibodies. These deidentified diagnostic specimens were deemed to be exempt from IRB approval by the University of Nevada (UNR), Research Integrity Office (Reference #616225-1) as meeting the exemption criteria defined by the Department of Health and Human Services under Human Subject Research Code 45 CFR 46.102(f). Information of each HPS case was limited to diagnosis, gender, and antibody titer range. Forty-two serum samples from healthy individuals collected under informed consent were used as controls (Human subjects protocol # B12-031). Control subjects were chosen to be consistent with published demographics of typical HPS cases regarding age and gender (male to female ratio of 54–46%, respectively, and mean age of 39.4 years) (20).
HPS Screening
Serum anti-hantavirus antibody titers were evaluated by ELISA, according to the methods described by Feldmann et al. (21). Serum dilutions (1:100–1:6400) were tested for the presence of anti-hantavirus IgG and IgM using recombinant nucleocapsid protein supplied by the United States Centers for Disease Control and Prevention (CDC, Atlanta, GA, USA). Subjects with antibody titers greater than twofold above that of negative controls were considered positive.
Multiplex Analysis
The levels of serum cytokines were analyzed using Bio-Plex (Bio-Rad, Hercules, CA, USA) multiplex magnetic bead-based antibody detection kits following the manufacturer’s instructions. The Bio-Plex Pro Human Chemokine Panel (40-Plex); Bio-Plex Pro Human Th17 Cytokine Panel; Bio-Plex Pro Human Cytokine 27-plex Panel; and Bio-Plex Human Cytokine 21-plex Panel were used for analysis of a total of 68 analytes. Fifty microliters of serum from each respective case and control was analyzed using a Luminex 200 analyzer with MasterPlex CT control software and MasterPlex QT analysis software (MiraiBio, San Bruno, CA, USA). Standard curves for each analyte were generated using standards provided by manufacturer. Serum samples from HPS cases were heat inactivated and tested for the presence of infectious virus prior to Luminex analysis. The effect of heat inactivation on cytokine stability was evaluated and those that could not be normalized were excluded from analysis.
Statistical Analysis
Mann–Whitney non-parametric analysis was utilized to identify differences in medians between HPS cases and controls. In addition, we performed classification analysis using the tree-based ensemble machine learning algorithm Random Forest (RF) (22). For this analysis, 500 random trees were built using six predictors for each node, and auto-bootstrap out-of-bag sampling was used for testing the model as previously described (23).
Results
Anti-Hantavirus Titer in HPS Serum
Twelve serum samples from subjects suspected of having hantavirus infection were tested for the presence of anti-hantavirus IgG and IgM antibodies. Antibody titers twofold greater than those of the control samples were considered diagnostic for hantavirus infection (Table 1). Previous reports suggest that anti-hantavirus IgM and IgG change with disease progression (24, 25). As reported by MacNeil and coworkers, early stage HPS is characterized by high IgM titers that peak within 11–14 days after onset whereas cases with early stage HPS often have no SNV-specific IgG titer (24). In contrast to IgM titers, median IgG titers typically displayed an increasing trend for a longer interval after the onset of disease. In light of the deidentified nature of our HPS cases, we used antibody titers to assess the stage of their illness. Six of our cases had high serum titer of IgM while IgG levels were low or undetectable, indicative of early stage disease. For the remaining six cases, high serum titers were observed for both IgG and IgM, consistent with late onset HPS.

Table 1. Antibody titer in serum from HPS cases.
Differential Expression of Serum Cytokine in HPS Cases
A total of 68 serum cytokines were measured for HPS cases and controls (Tables 2–4). To the best of our knowledge, 38 of these cytokines were previously uninvestigated in the context of HPS (indicated by an asterisk in Tables 2–4). A significant increase in the serum levels of 25 of 68 (36.7%) cytokines were observed for the HPS cases when compared to healthy controls (Table 2). The greatest difference was observed for IL-6, CXCL10, CX3CL1, MIF, and MIG, all of which were upregulated fivefold over those of controls (p < 0.001). In contrast, 15 of 68 (22.1%) cytokines were downregulated in HPS cases when compared to controls (Table 3), the greatest differences were observed for CXCL12, CCL21, CCL22, CCL27, and sCD40L (p < 0.001). Additionally, the majority of downregulated cytokines belonged to the homeostatic and inflammatory chemokine family. Of the 68 cytokines investigated, 28 (41.2%) were not statistically different when comparing cases and controls (Table 4).
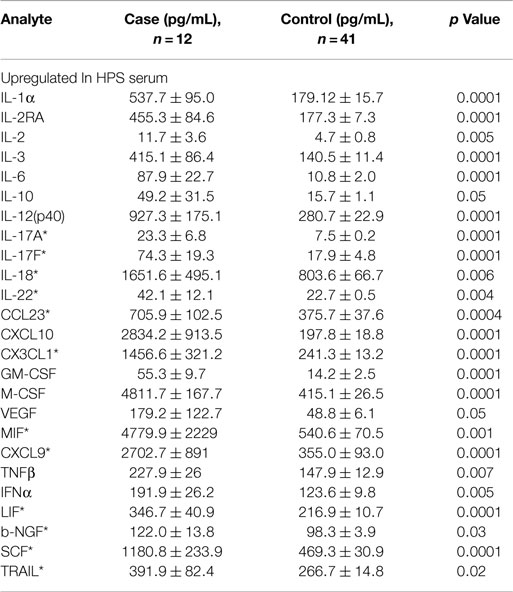
Table 2. Cytokines upregulated in HPS cases compared to healthy controls.
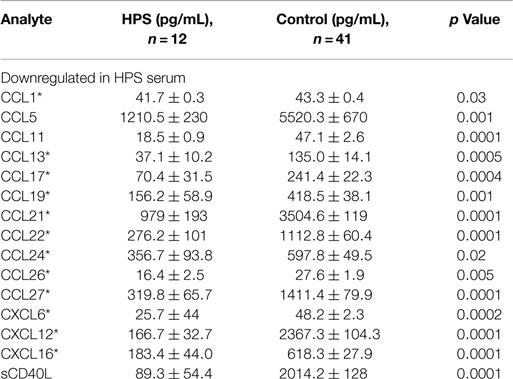
Table 3. Cytokines downregulated in HPS cases compared to healthy controls.
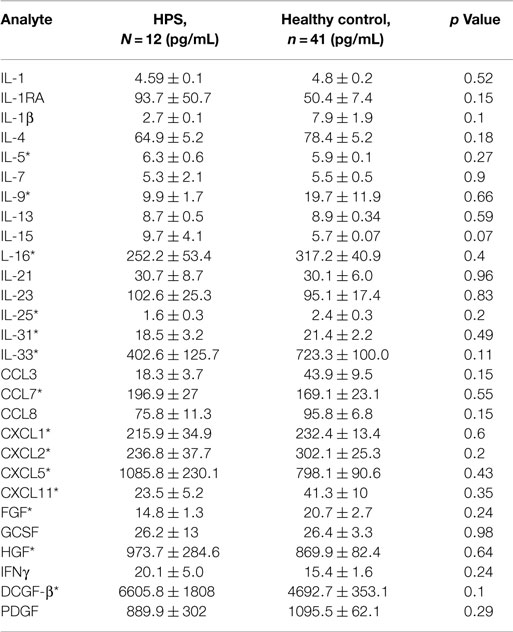
Table 4. No significant difference in cytokine expression between HPS and healthy controls.
Analysis of Serum Cytokines in Early vs. Late Stage HPS
In order to investigate the possibility that differential expression of cytokines occurs between subjects with early and late stage HPS, we compared these two subgroups with each other and to healthy controls. Surprisingly, we observed only five cytokines to be differentially expressed between the two subgroups of HPS cases (Table 5). Of these, median IL-33 and CXCL6 levels were greater in the early stage subjects whereas median CCL23, CXCL1, and TNF-β were greater in the late stage subjects. As expected, differences in cytokine expression between subgroups and controls were consistent with differences observed between total HPS cases and controls (data not shown).
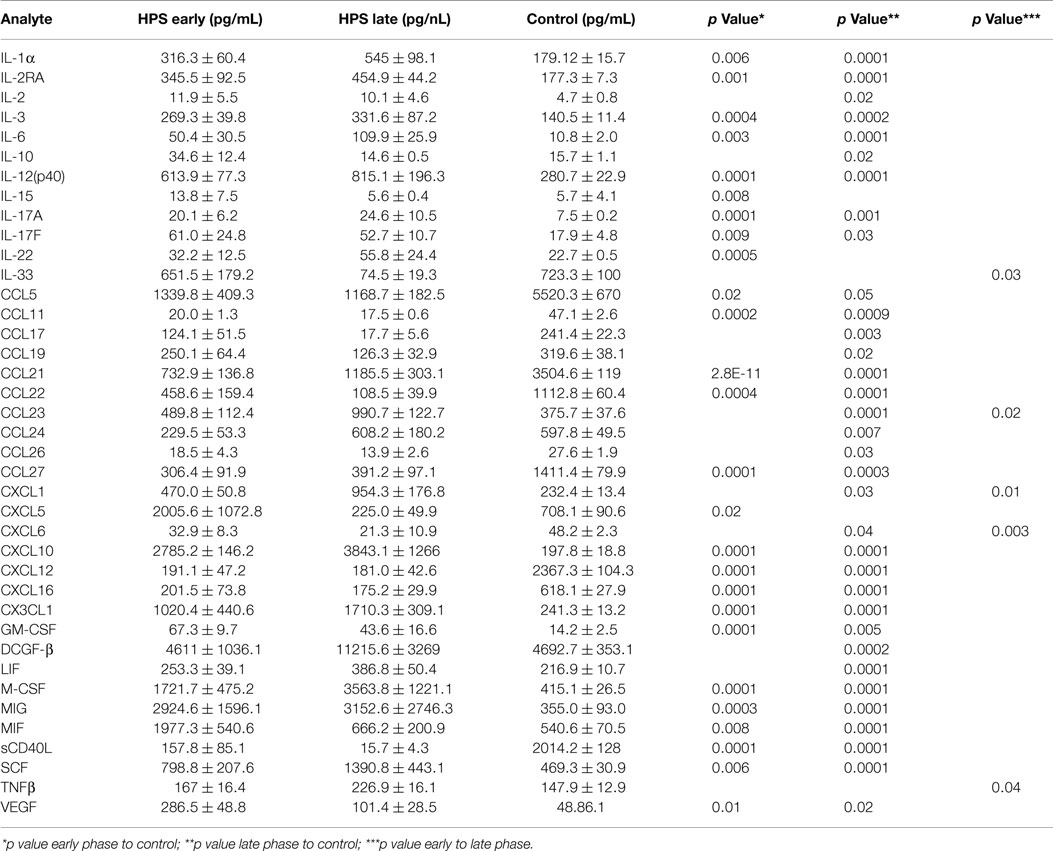
Table 5. Serum cytokine profile during early and late stages of HPS.
Classification of Cytokines by Importance
Given the complex interactions of cytokines with immune and non-immune cells, clarification of how distinct cytokines contribute to a pathological situation is often difficult to resolve. In order to provide insight into this issue, we implemented the machine logic algorithm RF to analyze our data set and potentially identify the most important cytokines that define this disease. For our analysis, 500 random decision trees were constructed with six predictors at each node, and auto-bootstrap out-of-bag sampling was implemented to test the accuracy of model. This model accurately identified HPS cases with 100% specificity and 73.81% sensitivity (Table 6). The 10 most significant cytokines for delineating HPS in decreasing order of importance are: M-CSF, CXCL16, sCD40, CXCL12, CCL22, IL-1a, CCL21, IL-12p40, CCL17, and IL-1b.
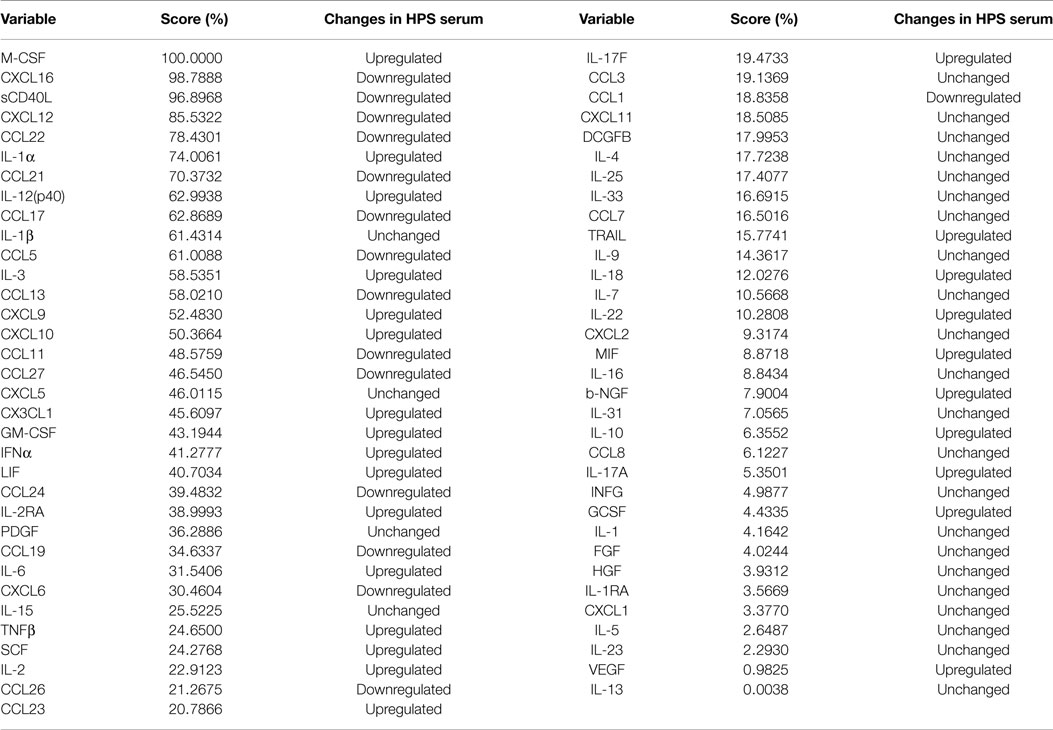
Table 6. Random forest analysis of serum cytokines in HPS vs. controls.
Discussion
The microvascular endothelium is principal target of hantavirus infection in humans and its infection in lung tissue results in significant pathology (26). Infection of endothelial cells leads to increased vascular permeability without an observable cytopathic effect; therefore, the pathogenesis of HPS is likely indirect with immune responses, such as cytokine production, playing an important role. The cytokines that we observed to be upregulated in the serum of HPS cases are involved in a number of antiviral defense mechanisms including proliferation, maturation, and activation of leukocytes, as well as survival of leukocytes, and regulation of endothelial monolayer permeability (Table 2). High levels of IL-1α, IL-6, MIF, and TNF-β suggest a strong proinflammatory milieu in the serum of HPS cases, thus promoting both inflammation and activation of immune responses. We also observed stem cell proliferation factors to be upregulated, potentially promoting the proliferation and differentiation of subsets of immune effector cells. For example, proliferation of myeloid progenitors is strongly supported by IL-3, GM-CSF, and M-CSF. Increased serum concentrations of GM-CSF and M-CSF also suggest proliferation of monocytes and granulocytes (neutrophils, eosinophils, and basophils). Upregulation of the pluripotent factor, SCF, was also observed in association with HPS, suggesting increased proliferation of T lymphocytes, NK cells, and dendritic cells.
We observed a subset of 15 serum cytokines to be downregulated in our HPS cases (Table 4). Twelve of these cytokines are involved in chemotaxis of lymphocytes, such as B cells, T cells, and NK cells, to sites of infection. Some of these cytokines, including CCL22, CXCL12, and CCL17, are associated with activation of Th2-type immunity and are potent recruiters of Th2 cells to the lungs, as well as activators of pre-B cells (27–29). A number of cytokines identified as differentially expressed in the present study are consistent with putative immune responses of lung tissue. For example, we observed the upregulation of serum IL-17F, CXCL16, and IL-22, which are involved in the regulation of leukocyte migration into lung tissue, as well as lung tissue repair (30–33). Upregulation of IL-17F has also been observed in the lung tissue of asthmatic cases and its level positively correlated with disease severity (30, 34, 35). Overexpression of IL-17F promotes neutrophil infiltration and increased airways sensitivity and thus has a significant impact on lung function (35). IL-22 is considered a key cytokine for mucosal tissue repair (36) and by activating antimicrobial responses in lung epithelial cells; it has been shown to be critical for host defense as well. Also, IL-22 promotes lung epithelial cell proliferation (37) and therefore, based on our analyses, the cytokine profile observed in our HPS cases is consistent with a pulmonary antimicrobial response and subsequent mononuclear cell migration into the lung.
The serum cytokine profile observed in our HPS subjects also suggests a mobilization of mononuclear immune effector cells (Table 2). IL-12(p40) is an autocrine chemoattractant released by activated macrophages and promotes Th1-type immunity (38, 39). Additionally, serum levels for several potent T lymphocyte and NK chemoattractants were upregulated, including CXCL10, MIG, and CCL23 (15, 16, 40, 41). MIF and VEGF, which are regulators of mononuclear cell transendothelial migration, were upregulated as well. Migration of leukocytes can also be facilitated by the upregulation of adhesion molecules on the surface of endothelial cells in response to VEGF, IL-1α, and IL-6 (42, 43). MIF and VEGF promote expression of the adhesion molecules, E-selectin, ICAM-1, and VCAM-1, and increase vascular permeability (44, 45). Additionally, VEGF can decrease tight junctions between endothelial cells enabling transmigration of immune effector cells (42, 46). The observed increased serum levels of CXCL1, which may lead to release of VEGF-A from hantavirus-activated endothelial cells, further suggests that upregulation of VEGF plays a role in HPS (47, 48).
Cytokines including CXCL10, MIF, MIG, IL-12(p40), IL-17A, and CCL23 are known to promote proliferation and migration of mononuclear immune cells, such as T lymphocytes, NK cells, monocytes, and dendritic cells (15, 49–51). Consequently, our data support the previous observations of others whereby mononuclear cell and immunoblasts are the principal cellular infiltrate in the lungs of HPS cases (12). Nevertheless, the observed cytokine expression also is consistent with the activation and migration of neutrophils. Previous studies suggest that the cytokines, IL-17F, VEGF, CXCL1, GM-CSF, and IL-22, promote neutrophil migration and lung tissue repair (52–54). These data corroborate a previous report by Mori et al., who observed low-level neutrophil infiltration in the lungs of HPS case (12). Interestingly, serum level of CXCL8, the prototype neutrophil chemoattractant, was not significantly elevated in the HPS cases in our study; however, it was identified as one of the top 10 cytokines by our RF analysis, suggesting its expression, or lack thereof, plays an important role in HPS pathology. Our data further suggest that a Th17 shift occurs in HPS (55). In the presence of IL-23, non-Th17 cells can produce IL-17 (56); however, we observed no differential expression of serum IL-23 in HPS cases. Therefore, it is likely that activated Th17 lymphocytes were the source of IL-17 in the serum of our HPS cases.
Expression of IL-17 and IL-22 in HPS suggests a developing antimicrobial state in the lung. It has been reported that IL-17 and IL-22 activate β-defensins and the S100 family of proteins (52, 57). In vivo studies using knockout mice have demonstrated that IL-17 and IL-22 are crucial for bacterial defense in the lung (58, 59). Furthermore, it has been reported that IL-17R signaling is mandatory for the establishment of an antibacterial response to M. pneumoniae, systemic fungal infection, B. fragilis, and E. coli (60–63). Consistent with this statement, a protective role for IL-22 was recently reported for experimental influenza A virus infection (64).
We also observed a subset of cytokines involved in the regulation of platelet counts and function to be downregulated in the serum of our HPS subjects, including sCD40L, CCL5, CCL22, and CXCL12 (Table 3). Consistent with our observations and the pathophysiology of HPS, CXCL12 and CCL22 act on platelets to rapidly stimulate their adhesion (65), and CCL5 and sCD40L are released by activated platelets (66–68). Wenzel and coworkers reported that serum levels of sCD40L closely correlate with platelets counts and that they are increased upon thrombocyte transfusion (69). Viallard et al. also reported a correlation between thrombocyte counts and serum sCD40L, implying that it may be used as a surrogate marker for platelet counts (66). Decreased thrombocyte counts are also well documented in association with HPS (2, 70) and our observation of downregulated sCD40L presents a potential biomarker for the thrombocytopenia. Notwithstanding, decreased serum CCL22 might also reflect the development of the thrombocytopenia observed in HPS cases. It has been shown that CCL22 is capable of aggregating platelets in the presence of low concentrations of thrombin or adenosine diphosphate (ADP), and can rapidly stimulate platelets adhesion (65). It is noteworthy that endothelial cells do not produce this cytokine; dendritic cells are the main source of CCL22 (71). Therefore, the thrombocyte aggregation and depletion observed in HPS may be the result of cytokine-driven immune responses.
Serum levels of CCL21 and CCL27 were also downregulated in the serum of our HPS subjects. These cytokines have tissue-specific activity; for example, CCL21 orchestrates dendritic cell and T cell trafficking to the lymph nodes (72–74) and CCL27 regulates migration of immune effector cells to the skin (75). Taken together, these findings suggest that the cytokines expressed during HPS promote lung tissue infiltration while reducing leukocyte trafficking to other organs and tissues.
In order to investigate the contribution of each respective cytokine to the disease process, we conducted classification analysis by RF. Of the 10 most important cytokines identified by this analysis, 3 were significantly upregulated, as determined by Mann–Whitney analysis; however, we also observed 6 to be downregulated. This observation underscores the importance of cytokine inhibition in the disease process and further suggests that depressed serum cytokine expression may be an important biomarker for monitoring disease progression.
Overall, the majority of downregulated serum cytokines were associated with Th2-type immune activation; these included CCL21, CCL17, CCL13, and CCL11. Furthermore, the cytokines significantly upregulated in HPS cases were those promoting Th1-type immunity; these included CXCL9, CXCL10, and IL-12(p40). The cytokines M-CSF, CXCL12, IL-3, LIF, GM-CSF, CCL24, which facilitate activation, differentiation, and bone marrow mobilization of myeloid progenitors, were also identified by RF analysis to differentiate HPS cases from controls. RF analysis further identified chemokines associated with platelet aggregation as important in differentiating cases from controls. Interestingly, sCD40L and CXCL12 were ranked, respectively, as the third and fourth most import cytokine in our RF analysis. Chemokines, such as sCD40L, CXCL12, and CCL17, which are stored in platelet granules, are released upon platelet aggregation, a process that is critical in HPS pathology (2, 76–78). Accordingly, nadir platelet counts in HPS may explain low serum CXCL1, CCL17, and sCD40. Taken together, RF analysis supports the supposition that HPS pathogenesis may be characterized by Th1-type immune responses and thrombocytopenia.
In summary, our data suggest that HPS is characterized by a serum cytokine profile that is consistent with putative immune responses in lung tissue. Strong activation of mononuclear immune effectors including T lymphocytes, NK cells, and dendritic cells is also suggested by this cytokine profile. Additionally, our data imply that decreased counts and increased aggregation of thrombocytes in HPS might be explained in part by the immune response to viral infection. Lastly, to the best of our knowledge, our data provide the first evidence of Th17 lymphocyte activation in association with HPS. The data presented in this study are suggestive of putative in vivo immune mechanisms and may identify the role of these cytokines in HPS pathophysiology; however, future studies using animal models would be necessary to definitively confirm their involvement.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
An award to VL from the Department of Defense (DOD) grant PR131133 supported these studies. A fellowship provided by the Program of Competitive Growth of Kazan Federal University supported the work of AR.
References
1. Duchin JS, Koster FT, Peters CJ, Simpson GL, Tempest B, Zaki SR, et al. Hantavirus pulmonary syndrome: a clinical description of 17 patients with a newly recognized disease. The Hantavirus Study Group. N Engl J Med (1994) 330(14):949–55. doi: 10.1056/NEJM199404073301401
2. Nolte KB, Feddersen RM, Foucar K, Zaki SR, Koster FT, Madar D, et al. Hantavirus pulmonary syndrome in the United States: a pathological description of a disease caused by a new agent. Hum Pathol (1995) 26(1):110–20. doi:10.1016/0046-8177(95)90123-X
3. Hjelle B, Lee SW, Song W, Torrez-Martinez N, Song JW, Yanagihara R, et al. Molecular linkage of hantavirus pulmonary syndrome to the white-footed mouse, Peromyscus leucopus: genetic characterization of the M genome of New York virus. J Virol (1995) 69(12):8137–41.
4. Khan AS, Spiropoulou CF, Morzunov S, Zaki SR, Kohn MA, Nawas SR, et al. Fatal illness associated with a new hantavirus in Louisiana. J Med Virol (1995) 46(3):281–6. doi:10.1002/jmv.1890460320
5. Lopez N, Padula P, Rossi C, Lazaro ME, Franze-Fernandez MT. Genetic identification of a new hantavirus causing severe pulmonary syndrome in Argentina. Virology (1996) 220(1):223–6. doi:10.1006/viro.1996.0305
6. Hjelle B, Jenison S, Mertz G, Koster F, Foucar K. Emergence of hantaviral disease in the southwestern United States. West J Med (1994) 161(5): 467–73.
7. Zaki SR, Khan AS, Goodman RA, Armstrong LR, Greer PW, Coffield LM, et al. Retrospective diagnosis of hantavirus pulmonary syndrome, 1978-1993: implications for emerging infectious diseases. Arch Pathol Lab Med (1996) 120(2):134–9.
8. Centers for Disease Control and Prevention. U.S. HPS Cases, by Reporting State. Atlanta, GA: Centers for Disease Control and Prevention (2014).
9. Hartline J, Mierek C, Knutson T, Kang C. Hantavirus infection in North America: a clinical review. Am J Emerg Med (2013) 31(6):978–82. doi:10.1016/j.ajem.2013.02.001
10. Warner GS. Hantavirus illness in humans: review and update. South Med J (1996) 89(3):264–71. doi:10.1097/00007611-199603000-00002
11. Hallin GW, Simpson SQ, Crowell RE, James DS, Koster FT, Mertz GJ, et al. Cardiopulmonary manifestations of hantavirus pulmonary syndrome. Crit Care Med (1996) 24(2):252–8. doi:10.1097/00003246-199602000-00012
12. Mori M, Rothman AL, Kurane I, Montoya JM, Nolte KB, Norman JE, et al. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J Infect Dis (1999) 179(2):295–302. doi:10.1086/314597
13. Khaiboullina SF, Rizvanov AA, Otteson E, Miyazato A, Maciejewski J, St Jeor S. Regulation of cellular gene expression in endothelial cells by Sin Nombre and prospect hill viruses. Viral Immunol (2004) 17(2):234–51. doi:10.1089/0882824041310504
14. Sundstrom JB, McMullan LK, Spiropoulou CF, Hooper WC, Ansari AA, Peters CJ, et al. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J Virol (2001) 75(13):6070–85. doi:10.1128/JVI.75.13.6070-6085.2001
15. Moser B, Loetscher M, Piali L, Loetscher P. Lymphocyte responses to chemokines. Int Rev Immunol (1998) 16(3–4):323–44. doi:10.3109/08830189809043000
16. Graziano FM, Cook EB, Stahl JL. Cytokines, chemokines, RANTES, and eotaxin. Allergy Asthma Proc (1999) 20(3):141–6. doi:10.2500/108854199778553055
17. Prescott J, Ye C, Sen G, Hjelle B. Induction of innate immune response genes by Sin Nombre hantavirus does not require viral replication. J Virol (2005) 79(24):15007–15. doi:10.1128/JVI.79.24.15007-15015.2005
18. Kraus AA, Raftery MJ, Giese T, Ulrich R, Zawatzky R, Hippenstiel S, et al. Differential antiviral response of endothelial cells after infection with pathogenic and nonpathogenic hantaviruses. J Virol (2004) 78(12):6143–50. doi:10.1128/JVI.78.12.6143-6150.2004
19. Borges AA, Campos GM, Moreli ML, Moro Souza RL, Saggioro FP, Figueiredo GG, et al. Role of mixed Th1 and Th2 serum cytokines on pathogenesis and prognosis of hantavirus pulmonary syndrome. Microbes Infect (2008) 10(10–11):1150–7. doi:10.1016/j.micinf.2008.06.006
20. Khan AS, Khabbaz RF, Armstrong LR, Holman RC, Bauer SP, Graber J, et al. Hantavirus pulmonary syndrome: the first 100 US cases. J Infect Dis (1996) 173(6):1297–303. doi:10.1093/infdis/173.6.1297
21. Feldmann H, Sanchez A, Morzunov S, Spiropoulou CF, Rollin PE, Ksiazek TG, et al. Utilization of autopsy RNA for the synthesis of the nucleocapsid antigen of a newly recognized virus associated with hantavirus pulmonary syndrome. Virus Res (1993) 30(3):351–67. doi:10.1016/0168-1702(93)90101-R
22. Breiman L. Classification and Regression Trees. Belmont, CA: Wadsworth International Group (1984).
23. Khaiboullina SF, DeMeirleir KL, Rawat S, Berk GS, Gaynor-Berk RS, Mijatovic T, et al. Cytokine expression provides clues to the pathophysiology of Gulf War illness and myalgic encephalomyelitis. Cytokine (2015) 72(1):1–8. doi:10.1016/j.cyto.2014.11.019
24. MacNeil A, Comer JA, Ksiazek TG, Rollin PE. Sin Nombre virus-specific immunoglobulin M and G kinetics in hantavirus pulmonary syndrome and the role played by serologic responses in predicting disease outcome. J Infect Dis (2010) 202(2):242–6. doi:10.1086/653482
25. Padula PJ, Colavecchia SB, Martinez VP, Gonzalez Della Valle MO, Edelstein A, Miguel SD, et al. Genetic diversity, distribution, and serological features of hantavirus infection in five countries in South America. J Clin Microbiol (2000) 38(8):3029–35.
26. Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, Foucar K, et al. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am J Pathol (1995) 146(3):552–79.
27. Weiss JM, Cufi P, Bismuth J, Eymard B, Fadel E, Berrih-Aknin S, et al. SDF-1/CXCL12 recruits B cells and antigen-presenting cells to the thymus of autoimmune myasthenia gravis patients. Immunobiology (2013) 218(3):373–81. doi:10.1016/j.imbio.2012.05.006
28. Nagasawa T, Tachibana K, Kishimoto T. A novel CXC chemokine PBSF/SDF-1 and its receptor CXCR4: their functions in development, hematopoiesis and HIV infection. Semin Immunol (1998) 10(3):179–85. doi:10.1006/smim.1998.0128
29. Nakayama T, Hieshima K, Nagakubo D, Sato E, Nakayama M, Kawa K, et al. Selective induction of Th2-attracting chemokines CCL17 and CCL22 in human B cells by latent membrane protein 1 of Epstein-Barr virus. J Virol (2004) 78(4):1665–74. doi:10.1128/JVI.78.4.1665-1674.2004
30. Fujie H, Niu K, Ohba M, Tomioka Y, Kitazawa H, Nagashima K, et al. A distinct regulatory role of Th17 cytokines IL-17A and IL-17F in chemokine secretion from lung microvascular endothelial cells. Inflammation (2012) 35(3):1119–31. doi:10.1007/s10753-011-9419-0
31. Al-Ramli W, Prefontaine D, Chouiali F, Martin JG, Olivenstein R, Lemiere C, et al. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol (2009) 123(5):1185–7. doi:10.1016/j.jaci.2009.02.024
32. Morgan AJ, Guillen C, Symon FA, Huynh TT, Berry MA, Entwisle JJ, et al. Expression of CXCR6 and its ligand CXCL16 in the lung in health and disease. Clin Exp Allergy (2005) 35(12):1572–80. doi:10.1111/j.1365-2222.2005.02383.x
33. Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol (2013) 87(12):6911–24. doi:10.1128/JVI.02943-12
34. Kawaguchi M, Kokubu F, Huang SK, Homma T, Odaka M, Watanabe S, et al. The IL-17F signaling pathway is involved in the induction of IFN-gamma-inducible protein 10 in bronchial epithelial cells. J Allergy Clin Immunol (2007) 119(6):1408–14. doi:10.1016/j.jaci.2007.02.036
35. Hizawa N, Kawaguchi M, Huang SK, Nishimura M. Role of interleukin-17F in chronic inflammatory and allergic lung disease. Clin Exp Allergy (2006) 36(9):1109–14. doi:10.1111/j.1365-2222.2006.02550.x
36. Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev (2013) 252(1):116–32. doi:10.1111/imr.12027
37. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14(3):275–81. doi:10.1038/nm1710
38. Bastos KR, Alvarez JM, Marinho CR, Rizzo LV, Lima MR. Macrophages from IL-12p40-deficient mice have a bias toward the M2 activation profile. J Leukoc Biol (2002) 71(2):271–8.
39. Cooper AM, Khader SA. IL-12p40: an inherently agonistic cytokine. Trends Immunol (2007) 28(1):33–8. doi:10.1016/j.it.2006.11.002
40. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol (2011) 89(2):207–15. doi:10.1038/icb.2010.158
41. Broxmeyer HE, Kim CH, Cooper SH, Hangoc G, Hromas R, Pelus LM. Effects of CC, CXC, C, and CX3C chemokines on proliferation of myeloid progenitor cells, and insights into SDF-1-induced chemotaxis of progenitors. Ann N Y Acad Sci (1999) 872:142–62.
42. Davis B, Tang J, Zhang L, Mu D, Jiang X, Biran V, et al. Role of vasodilator stimulated phosphoprotein in VEGF induced blood-brain barrier permeability in endothelial cell monolayers. Int J Dev Neurosci (2010) 28(6):423–8. doi:10.1016/j.ijdevneu.2010.06.010
43. Deissler H, Deissler H, Lang S, Lang GE. VEGF-induced effects on proliferation, migration and tight junctions are restored by ranibizumab (Lucentis) in microvascular retinal endothelial cells. Br J Ophthalmol (2008) 92(6):839–43. doi:10.1136/bjo.2007.135640
44. Cheng Q, McKeown SJ, Santos L, Santiago FS, Khachigian LM, Morand EF, et al. Macrophage migration inhibitory factor increases leukocyte-endothelial interactions in human endothelial cells via promotion of expression of adhesion molecules. J Immunol (2010) 185(2):1238–47. doi:10.4049/jimmunol.0904104
45. Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. Regulation of transendothelial migration of hematopoietic progenitor cells. Ann N Y Acad Sci (1999) 872:176–85. doi:10.1111/j.1749-6632.1999.tb08463.x
46. Deissler HL, Deissler H, Lang GE. Inhibition of vascular endothelial growth factor (VEGF) is sufficient to completely restore barrier malfunction induced by growth factors in microvascular retinal endothelial cells. Br J Ophthalmol (2011) 95(8):1151–6. doi:10.1136/bjo.2010.192229
47. Scapini P, Morini M, Tecchio C, Minghelli S, Di Carlo E, Tanghetti E, et al. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J Immunol (2004) 172(8):5034–40. doi:10.4049/jimmunol.172.8.5034
48. DiStasi MR, Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol (2009) 30(11):547–56. doi:10.1016/j.it.2009.07.012
49. Piali L, Weber C, LaRosa G, Mackay CR, Springer TA, Clark-Lewis I, et al. The chemokine receptor CXCR3 mediates rapid and shear-resistant adhesion-induction of effector T lymphocytes by the chemokines IP10 and Mig. Eur J Immunol (1998) 28(3):961–72. doi:10.1002/(SICI)1521-4141(199803)28:03<961::AID-IMMU961>3.0.CO;2-4
50. Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, et al. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med (2006) 203(7):1805–15. doi:10.1084/jem.20052545
51. Shahrara S, Pickens SR, Mandelin AM II, Karpus WJ, Huang Q, Kolls JK, et al. IL-17-mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein-1 induction. J Immunol (2010) 184(8):4479–87. doi:10.4049/jimmunol.0901942
52. Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol (2009) 2(5):403–11. doi:10.1038/mi.2009.100
53. Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol (1999) 162(4):2347–52.
54. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med (2001) 194(4):519–27. doi:10.1084/jem.194.4.519
55. Chen Z, O’Shea JJ. Th17 cells: a new fate for differentiating helper T cells. Immunol Res (2008) 41(2):87–102. doi:10.1007/s12026-007-8014-9
56. McAleer JP, Kolls JK. Mechanisms controlling Th17 cytokine expression and host defense. J Leukoc Biol (2011) 90(2):263–70. doi:10.1189/jlb.0211099
57. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203(10):2271–9. doi:10.1084/jem.20061308
58. Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A (2006) 103(6):1834–9. doi:10.1073/pnas.0510847103
59. Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature (2004) 432(7019):917–21. doi:10.1038/nature03104
60. Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect (2007) 9(1):78–86. doi:10.1016/j.micinf.2006.10.012
61. Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis (2004) 190(3):624–31. doi:10.1086/422329
62. Chung DR, Kasper DL, Panzo RJ, Chitnis T, Grusby MJ, Sayegh MH, et al. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol (2003) 170(4):1958–63. doi:10.4049/jimmunol.170.4.1958
63. Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol (2007) 178(7):4466–72. doi:10.4049/jimmunol.178.7.4466
64. Paget C, Ivanov S, Fontaine J, Renneson J, Blanc F, Pichavant M, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem (2012) 287(12):8816–29. doi:10.1074/jbc.M111.304758
65. Gear AR, Suttitanamongkol S, Viisoreanu D, Polanowska-Grabowska RK, Raha S, Camerini D. Adenosine diphosphate strongly potentiates the ability of the chemokines MDC, TARC, and SDF-1 to stimulate platelet function. Blood (2001) 97(4):937–45. doi:10.1182/blood.V97.4.937
66. Viallard JF, Solanilla A, Gauthier B, Contin C, Dechanet J, Grosset C, et al. Increased soluble and platelet-associated CD40 ligand in essential thrombocythemia and reactive thrombocytosis. Blood (2002) 99(7):2612–4. doi:10.1182/blood.V99.7.2612
67. Kameyoshi Y, Dorschner A, Mallet AI, Christophers E, Schroder JM. Cytokine RANTES released by thrombin-stimulated platelets is a potent attractant for human eosinophils. J Exp Med (1992) 176(2):587–92. doi:10.1084/jem.176.2.587
68. Antczak AJ, Singh N, Gay SR, Worth RG. IgG-complex stimulated platelets: a source of sCD40L and RANTES in initiation of inflammatory cascade. Cell Immunol (2010) 263(1):129–33. doi:10.1016/j.cellimm.2010.03.009
69. Wenzel F, Gunther W, Baertl A, Gruber W, Sorg RV, Haas R, et al. Platelet transfusion alters CD40L blood level and release capacity in patients suffering from thrombocytopenia. Transfusion (2012) 52(6):1213–20. doi:10.1111/j.1537-2995.2011.03438.x
70. Sargianou M, Watson DC, Chra P, Papa A, Starakis I, Gogos C, et al. Hantavirus infections for the clinician: from case presentation to diagnosis and treatment. Crit Rev Microbiol (2012) 38(4):317–29. doi:10.3109/1040841X.2012.673553
71. Vulcano M, Albanesi C, Stoppacciaro A, Bagnati R, D’Amico G, Struyf S, et al. Dendritic cells as a major source of macrophage-derived chemokine/CCL22 in vitro and in vivo. Eur J Immunol (2001) 31(3):812–22. doi:10.1002/1521-4141(200103)31:3<812::AID-IMMU812>3.0.CO;2-L
72. Foo SY, Phipps S. Regulation of inducible BALT formation and contribution to immunity and pathology. Mucosal Immunol (2010) 3(6):537–44. doi:10.1038/mi.2010.52
73. Johnson LA, Jackson DG. Cell traffic and the lymphatic endothelium. Ann N Y Acad Sci (2008) 1131:119–33. doi:10.1196/annals.1413.011
74. Johnson LA, Jackson DG. Control of dendritic cell trafficking in lymphatics by chemokines. Angiogenesis (2014) 17(2):335–45. doi:10.1007/s10456-013-9407-0
75. Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity (2002) 16(1):1–4. doi:10.1016/S1074-7613(01)00261-8
76. von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res (2007) 100(1):27–40. doi:10.1161/01.RES.0000252802.25497.b7
77. Andre P, Nannizzi-Alaimo L, Prasad SK, Phillips DR. Platelet-derived CD40L: the switch-hitting player of cardiovascular disease. Circulation (2002) 106(8):896–9. doi:10.1161/01.CIR.0000028962.04520.01
Keywords: hantavirus pulmonary syndrome, serum, cytokines, chemokines, growth factors, immune response, hantaviruses
Citation: Morzunov SP, Khaiboullina SF, St. Jeor S, Rizvanov AA and Lombardi VC (2015) Multiplex analysis of serum cytokines in humans with hantavirus pulmonary syndrome. Front. Immunol. 6:432. doi: 10.3389/fimmu.2015.00432
Received: 03 June 2015; Accepted: 09 August 2015;
Published: 31 August 2015
Edited by:
Gayane Manukyan, National Academy of Sciences of the Republic of Armenia, ArmeniaReviewed by:
Joshua J. Obar, Montana State University, USARudra Bhowmick, Oklahoma State University, USA
Levon Abrahamyan, Université de Montréal, Canada
Copyright: © 2015 Morzunov, Khaiboullina, St. Jeor, Rizvanov and Lombardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Albert A. Rizvanov, Institute of Fundamental Medicine and Biology, Kazan (Volga Region) Federal University, 18 Kremlevskaya Street, Kazan 420008, Tatarstan, Russia,YWxiZXJ0LnJpenZhbm92QGtwZnUucnU=;
Vincent C. Lombardi, Department of Biochemistry and Molecular Biology, University of Nevada, 1664 N Virginia Street, Reno, NV MS 0330, USA,dmxvbWJhcmRpQG1lZGljaW5lLm5ldmFkYS5lZHU=