 Ali Alawieh
Ali Alawieh Andrew Elvington
Andrew Elvington Stephen Tomlinson
Stephen Tomlinson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 August 2015
Sec. Multiple Sclerosis and Neuroimmunology
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00417
The complement system is a component of the immune system involved in both recognition and response to pathogens, and it is implicated in an increasing number of homeostatic and disease processes. It is well documented that reperfusion of ischemic tissue results in complement activation and an inflammatory response that causes post-reperfusion injury. This occurs following cerebral ischemia and reperfusion and triggers secondary damage that extends beyond the initial infarcted area, an outcome that has rationalized the use of complement inhibitors as candidate therapeutics after stroke. In the central nervous system, however, recent studies have revealed that complement also has essential roles in synaptic pruning, neurogenesis, and neuronal migration. In the context of recovery after stroke, these apparent divergent functions of complement may account for findings that the protective effect of complement inhibition in the acute phase after stroke is not always maintained in the subacute and chronic phases. The development of effective stroke therapies based on modulation of the complement system will require a detailed understanding of complement-dependent processes in both early neurodegenerative events and delayed neuro-reparatory processes. Here, we review the role of complement in normal brain physiology, the events initiating complement activation after cerebral ischemia-reperfusion injury, and the contribution of complement to both injury and recovery. We also discuss how the design of future experiments may better characterize the dual role of complement in recovery after ischemic stroke.
Following cerebral ischemia, most patients reperfuse at least part of the ischemic area (1), and the restoration of blood flow initiates an inflammatory cascade that causes secondary neuronal injury which can have a significant impact on functional recovery. Reperfusion can occur spontaneously, or can be achieved by surgical or pharmacological means (2). Once reperfusion occurs, there is an inflammatory reaction in which resident cells, neutrophils, macrophages, platelets, cytokines, molecular oxygen, and complement play important roles, and which culminates in necrotic and apoptotic cell death (3). The only approved pharmacological agent for the treatment of ischemic stroke is recombinant tissue-type plasminogen activator (tPA), which promotes reperfusion by enhancing the dissolution of blood clots. However, tPA must be administered with 3 h of symptom onset, and since there is a risk of uncontrollable intracranial hemorrhage, physicians are often reluctant to use this drug, with the result that only about 5% of stroke patients are treated with tPA (4, 5). There is thus a significant need for new and effective approaches to treat stroke, and reducing inflammation and secondary injury in the area surrounding the ischemic core (i.e., the penumbra) is a major therapeutic goal. Nevertheless, it is becoming increasingly clear that inflammation also has homeostatic functions within the brain, and post-ischemic inflammation is also associated with neural protection and regeneration. Therefore, understanding how inflammation is involved in the balance between neurodegenerative and neuroprotective mechanisms will be important for the development of therapeutics that optimally promote long-term functional recovery after stroke. Here, we review the role of complement in neuroinflammatory processes during brain homeostasis and in the post-ischemic brain.
In the CNS, as in other organs and tissues, complement plays a key role in the inflammatory reaction following ischemia and reperfusion. Disruption of the blood–brain barrier following ischemic stroke allows access of complement proteins to the brain, although proteins of the complement system can be locally produced by the cells of the CNS (6–8). Complement is implicated in human ischemic stroke by studies demonstrating complement activation in patients. Complement activation and deposition in areas of cerebral ischemia has also been shown in rodent models of focal cerebral ischemia, together with the upregulation of various complement proteins within the CNS. Studies utilizing complement inhibited mice or mice genetically deficient in various complement proteins have further helped elucidate the role of complement in the pathogenesis of ischemic stroke and functional recovery. However, as with other inflammatory processes, more recent studies have also documented a role for complement in homeostatic functions within the brain, and complement is also associated with post-ischemic neural protection, repair, and regeneration.
In addition to its well-documented role in host defense, complement plays important roles in various other physiologic and homeostatic functions, such as immune complex catabolism, the clearance of dead and dying cells, and the modulation of adaptive immune responses. Complement is also involved in various developmental and regenerative processes, and is integrated with multiple other biological systems and pathways [reviewed in Ref. (9)]. Complement activation products consisting of opsonins, anaphylatoxins, and a terminal cytolytic complex mediate the effector functions of complement. The complement cascade is activated by one of three pathways, referred to as the classical, alternative, and lectin pathways, and all pathways converge at the cleavage and activation of the complement protein C3 (Figure 1). In general, the classical pathway is activated by antibodies and apoptotic cells via recognition by C1q, and the lectin pathway is activated via recognition of carbohydrate patterns by mannose-binding lectin (MBL) or ficolins. The alternative pathway is constitutively active, and spontaneously hydrolyzed C3 can become deposited on any surface to take part in further C3 activation on that surface. An alternative pathway-activating surface is one that is unable to regulate further C3 activation and amplification. The alternative pathway also functions to amplify the classical and lectin pathways. There are, however, additional means by which complement can be activated. Extrinsic protease pathways can bypass the early pathways of complement activation and directly cleave C3 or C5 proteins; for example, thrombin can directly cleave C3 and act as a C5 convertase (10, 11). The initial products of C3 cleavage are soluble C3a and cell-bound C3b. C3b is further cleaved to yield membrane-bound iC3b, C3dg, and C3d opsonins that are recognized by receptors on immune cells. C3 cleavage also leads to C5 cleavage to yield soluble C5a and membrane-bound C5b. C5b initiates the terminal pathway and formation of the membrane attack complex (MAC) that can cause direct cell lysis, but that can also stimulate cells to release inflammatory molecules. The anaphylatoxins C3a and C5a have multiple physiologic and inflammatory activities, including the recruitment and activation leukocytes. Complement is regulated by various fluid-phase and membrane-bound inhibitors [reviewed in Ref. (12)].
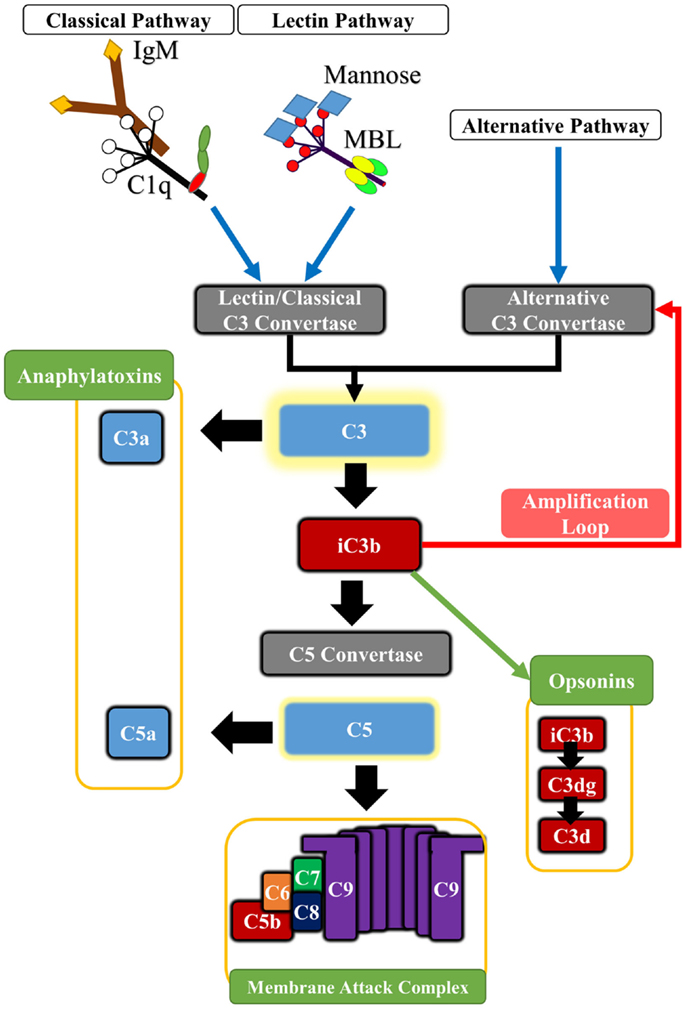
Figure 1. Basic outline of the principle pathways of complement activation and complement effector molecules. The classical pathway is triggered by the binding of C1q to antibody Fc regions, pentraxins or certain cell surface determinants. The lectin pathway is triggered by Mannose-binding lectin (MBL) or ficolins which bind to carbohydrate patterns, including those found on some IgM antibodies. Both pathways lead to cleavage of C2 and C4 components, forming the classical pathway C3 convertase (C4b2a) that cleaves C3. The alternative pathway is spontaneously activated forming the alternative pathway C3 convertase (C3bBb), and it also serves to amplify classical and lectin pathway activation. Cleavage of C3 leads to the generation of C3a and C3b. Activated C3b deposits on cell surfaces and is further degraded to iC3b, C3dg, and C3d, which serve as opsonins for receptors on immune cells. In addition, C3b associates with preformed C3 convertase, forming C5 convertase that in turn cleaves C5 into C5a and C5b. Deposition of C5b on cell surface initiates assembly of the cytolytic membrane attack complex (MAC or C5b-9). The C5a and C3a anaphylatoxins are potent pro-inflammatory molecules, and also modulate various homeostatic effects through G-protein signaling.
In contrast to systemic complement proteins that are produced predominantly by the liver, complement proteins are also be synthesized locally in the CNS. Primary studies using astrocyte and glial cell cultures demonstrated that these cells express nearly all components of the complement system (6–8). It was also shown that bacterial infection or inflammation increased expression of complement mRNA in the CNS (13, 14). Early studies focused on the protective role that the complement system played in the CNS, including sensing and response to infection, and the clearance of debris, apoptotic cells, and plaques (15–18). Further Research has also uncovered an essential role for complement in key aspects of brain function such as in synaptic pruning during CNS development, as well as other forms of synaptic plasticity (19–21). Evidence for the role of complement in elimination of synapses came from studies on the development of the retinogeniculate pathway in mice (19–21). C1q and C3 proteins produced by astrocytes were found to tag weaker synapses for removal by microglia, which drives specific innervation. This process reduces excessive connectivity between brain areas that may seed epileptogenesis as reported in C1q−/− mice (22). In addition, the anaphylatoxins, C3a and C5a, have also been implicated in cerebellar development in neonatal rats. C3a and C5a receptors (C3aR and C5aR1) were transiently elevated in rat cerebellar granule neurons peaking on the 12th postnatal day (23), and agonists to these receptors transiently altered the thickness of cerebellar cortical levels. A C5a agonist caused enlargement of the external granule layer and promoted granule cell survival by inhibiting caspase-9 activity and maintaining mitochondrial integrity. A C3a agonist increased the thickness of the internal granule layer while reducing the thickness of the external granule layer, suggesting a putative role of C3a in accelerating the migration of granule cells from the external to internal granule layer (23, 24).
Components of classical and terminal complement pathways, along with the complement anaphylatoxins, have also been shown to play a role in neurogenesis and neuroprotection. The addition of C1q to rat primary neuronal cultures resulted in alteration in microRNA and mRNA expression in a direction promoting neuronal survival (25). Pathways implicated in this effect include cholesterol metabolism, synaptic function, and neuronal growth factor production. A similar neuroprotective role for C1q was reported in response to β-amyloid and serum amyloid P-induced neurotoxicity (26). Interestingly, the MAC also demonstrated a pro-survival effect on oligodendrocyte progenitor cells at sublytic concentrations. Different pathways were thought to mediate the survival and proliferative effects of sublytic MAC on oligodendrocyte progenitor cells in vitro, including the inhibition of caspase-3 and caspase-8 activation, upregulation of bcl-2 expression, cleavage of Bid, increase in cellular FLIP long isoform, and downregulation of FasL expression (27, 28). The use of PI3K inhibitor (LY294002) partly reversed the effects of MAC on oligodendrocyte survival, suggesting that sublytic MAC concentrations may promote downstream signaling through the PI3K pathway (28). On the other hand, it has been shown that complement receptor 2 (CR2) is expressed in adult neural progenitor cells of the dentate gyrus, and that the CR2 ligands C3d and interferon-α inhibit neural progenitor cell proliferation. Furthermore, CR2-deficient mice displayed increased basal neurogenesis, indicating that CR2 may regulate neurogenesis and that complement activation (via C3d generation) may inhibit neurogenesis (29).
The C3a and C5a anaphylatoxins signal through the C3aR and C5aR1 G-protein coupled receptors, but we lack a clear understanding of their function and downstream effector mechanisms in the CNS. Certainly both anaphylatoxins can promote the production of cytokines and inflammatory mediators, and can direct and activate leukocytes, but they can also affect neuronal survival and synaptic plasticity. For example, C5aR1 is expressed on pre-synaptic terminals of mossy fibers within the hippocampus, suggesting a possible role for this receptor in synaptic/cellular plasticity (30). In addition, expression of C3aR on both neurons and glial cells suggests that C3a may play a role beyond immunological protection (31). Several studies have investigated common and different neuroprotective mechanisms of C3a and C5a. C3a was found to be involved in basal and ischemia-induced neurogenesis that was inhibited in C3-deficient mice and in mice treated with C3a receptor antagonist (C3aRA) (32). In murine primary neuronal cultures, C3a reversibly bound to neuronal cells promoting migration and differentiation through the recruitment of the intracellular ERK1/2 signaling pathway (33). C5a also recruited the ERK1/2 signaling pathway in response to glutamate-induced neurotoxicity (34). In this latter study, C5a was found to inhibit DNA fragmentation and pro-caspase-3 activation in neuronal and hippocampal cultures. Both C3a and C5a were found to increase the gene expression of neuronal growth factor in microglia, an effect mediated by C3aR and C5aR1 and inhibited by pertussis toxin (that blocks G-protein (Gi) coupled receptors) (35, 36). The increase in neuronal growth factor mRNA levels was not always associated with significant protein upregulation, but a significant synergistic effect of both anaphylatoxins with IL-1β on neuronal growth factor protein upregulation has been reported (36). Both C3a and C5a contributed to neuroprotection from glutamate-induced excitotoxicity, although each peptide had a different specific protective pattern. C3a was only protective against N-methyl-d-aspartate (NMDA) in the presence of astrocytes, and did not contribute to neuroprotection against Kainaite-induced excitotoxicity (37). By contrast, C5a exhibited a neuroprotective role in Kainaite rather than NMDA neurotoxicity. Also, intraventricular infusion of both Kainic acid and C5a in mice reduced caspase-3 activation and neuronal apoptosis (38), and C5aR1-deficient mice were shown to have increased susceptibility to Kainaite-induced excitotoxicity compared to wild type; treatment of wild-type mice with C5a reversed the glutamate-induced decrease in GluR2 receptor and reduced neuronal apoptosis (39). More recently, C5a was shown to reduce extracellular glutamate accumulation through the upregulation of glutamate transporter GLT-1 in microglia, suggesting a distinct mechanism of protection against excitotoxicity (40). Figure 2 summarizes the interplay between the complement system and other immune components in brain physiology and response to pathology.
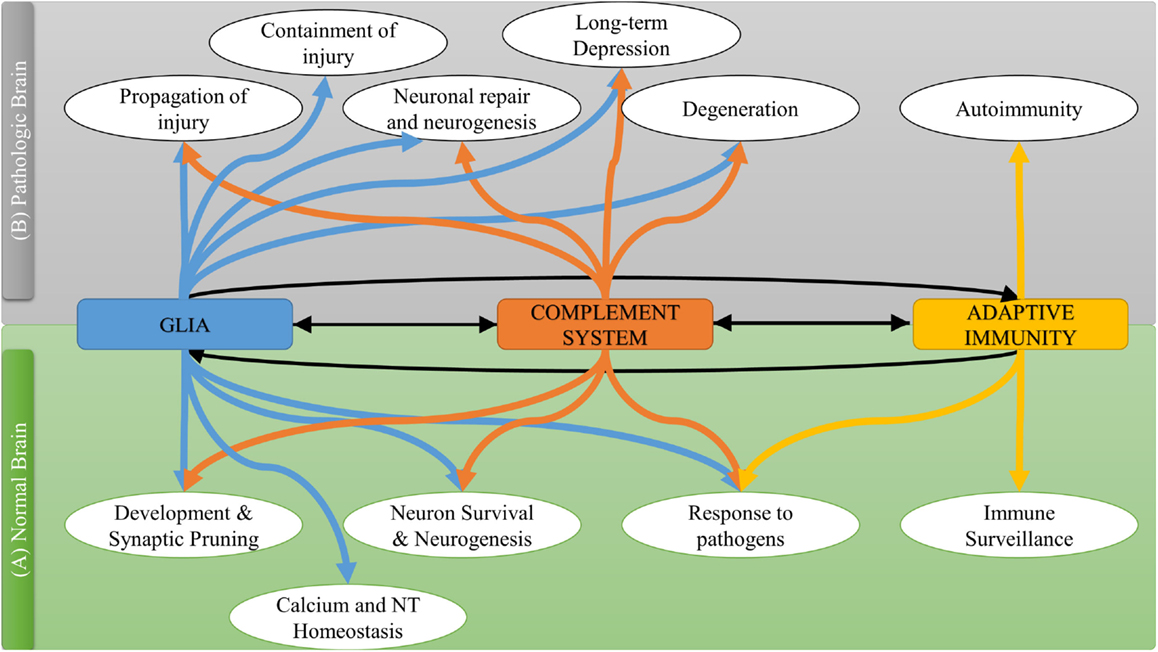
Figure 2. The interplay between complement system and other immune components in normal and pathological brain.
The complement system has long been recognized as a potential therapeutic target for the reduction of secondary damage and improvement of outcome after stroke. Research has focused on identifying the role of complement in brain ischemia-reperfusion injury (IRI) and investigating how complement inhibition affects outcome in models of stroke. There follows a summary of studies that have been performed in vitro, in animal models of ischemic and hemorrhagic stroke, and in human stroke.
Oxygen-glucose deprivation of cultured neuronal cells is a widely used in vitro model for cerebral ischemia, a procedure that results in both apoptotic and necrotic cell death. Upon hypoxic insult, neuronal cultures have been shown to overexpress several complement proteins. Both mRNA and protein levels of C1q were elevated in rat neuronal cells exposed to hypoxia, and newly produced C1q preferentially deposited on hypoxic neurons, serving as both a primary opsonin and an activator of the complement cascade (41). Similarly, mouse and rat neuronal cell cultures showed increased C3 expression in response to hypoxia, a response that was shown to be associated with activation of caspase-3, a marker for apoptosis. Both C3 expression and caspase-3 activation were reduced with intravenous immunoglobulin (IVIG) treatment, suggesting that IVIG may represent an interventional therapy for stroke (42, 43). In addition, blocking C5a signaling by the use of C5aR1 antagonist or the use of neurons from C5aR1-deficient mice reduced ischemia-induced apoptosis in murine neuronal cultures indicating a pathogenic role for C5a (44, 45). The neuroprotective effect of C5aR1 antagonism could be enhanced with hypothermia without alteration in C5aR1 levels, suggesting a putative therapeutic advantage of coupling both treatments (45). On the other hand, human neurons were found to express the complement inhibitors CD59, CD46 (membrane cofactor protein) and CD55 (decay accelerating factor), and hypoxic insult neither altered inhibitor expression nor the deposition of C3d, suggesting that human neurons are protected from the effects of C3 opsonization and the MAC (46). Table 1 shows a brief summary of the different in vitro studies on complement involvement in experimental stroke.
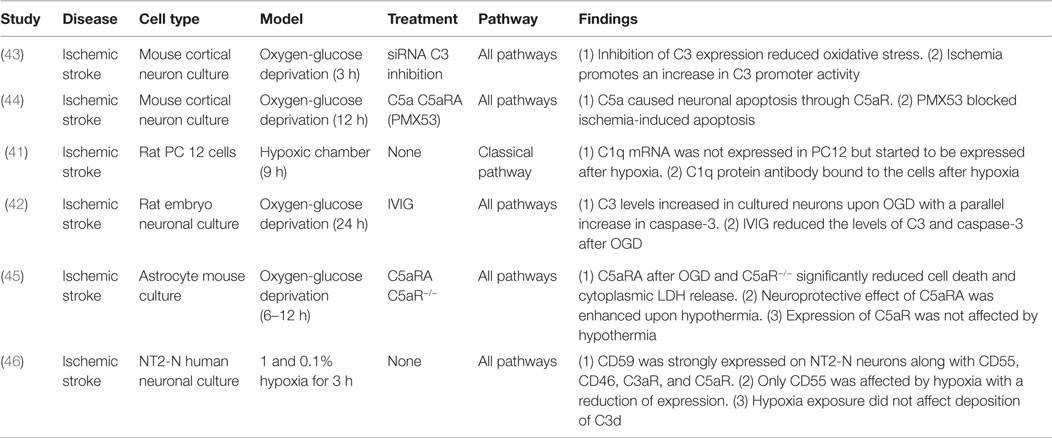
Table 1. Summary of in vitro studies on the role of complement in cerebral I/R.
Animal models of ischemic stroke involve transient or permanent occlusion of the middle cerebral artery or common carotid artery, or cerebral clot embolization. Notably, the advantage of the cerebral embolization model, although more difficult and less commonly utilized, is that it better allows the evaluation of the effect of potential adjuvant therapies to tissue plasminogen activator (t-PA), the only approved treatment for acute stroke. As a plasma protease, t-PA is capable of proteolytically activating components of the complement system via the recently recognized extrinsic pathway. In support of this, an early study reported that after cerebral embolization rabbits treated with t-PA had higher levels of C3 and C5 compared to vehicle (47). Interestingly, complement depletion in the same model using cobra venom factor (CVF) did not have any effect on infarct size in the presence or absence of t-PA treatment (48). However, this study did not investigate other outcome measures that complete complement depletion may affect, and no subsequent studies have further investigated the crosstalk between t-PA and the complement system in the context of acute stroke treatment (Table 2). The use of CVF in rodent models of transient ischemia consistently demonstrates a protective effect of complement depletion. Rats subjected to bilateral transient common carotid artery occlusion and pretreated with CVF had a better outcome compared to control treated rats in terms of somatosensory evoked potentials (49). CVF also reduced infarct volume and neuronal atrophy after rat transient middle cerebral artery occlusion (MCAO), as well as after neonatal rat hypoxia (50, 51). However, in permanent ischemia rat model, CVF did not effectively reduce infract volume (52). The prominent role of reperfusion in the activation of plasma complement proteins near ischemic tissue may explain why complement depletion did not alter outcomes in models utilizing permanent ischemia.
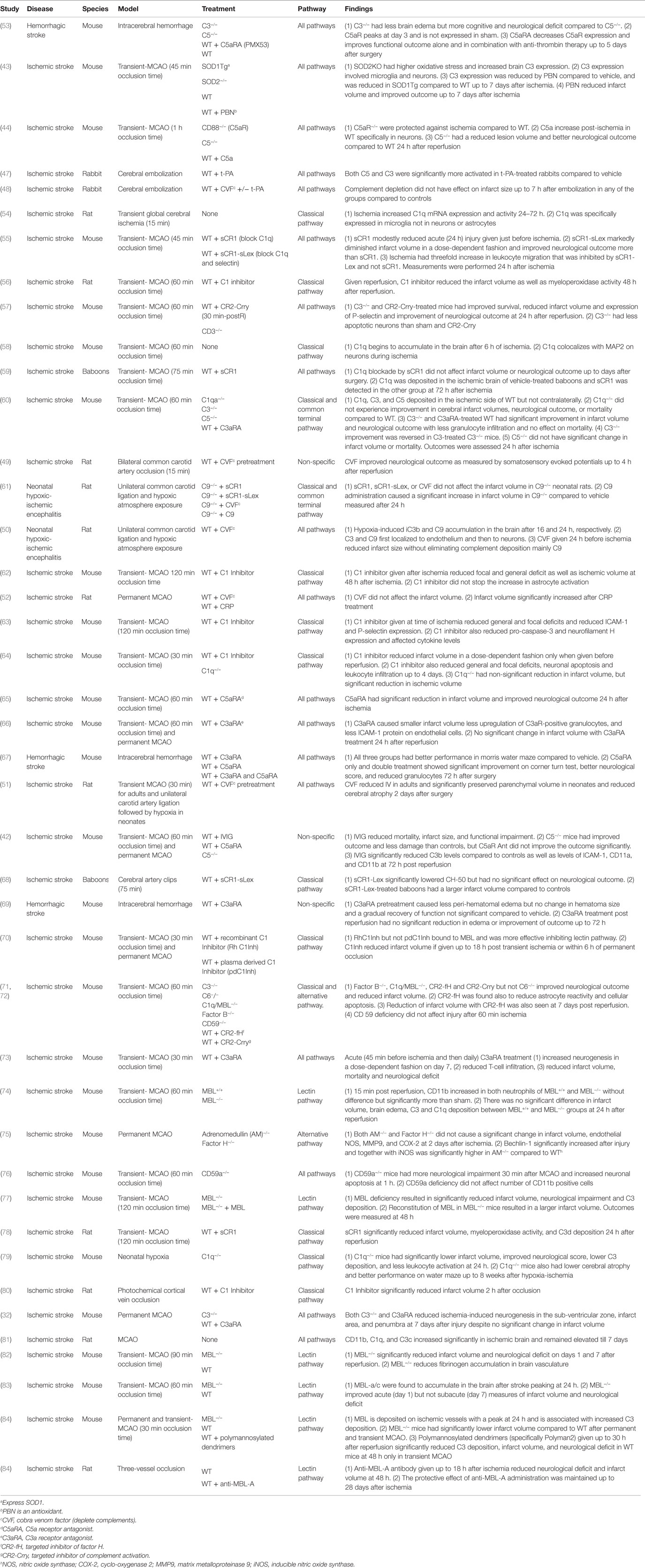
Table 2. Summary of studies investigating the role of complement in cerebral I/R using animal models.
Following stroke, damage to the blood–brain barrier will allow access of hematogenous complement proteins to the CNS, but there is evidence that increased local expression of complement proteins contribute to secondary injury after ischemia. For example, expression of C1q, C3, and CD11b (a component of complement receptor 3 expressed on phagocytes) is upregulated in response to ischemia in models of transient MCAO (54, 58, 60, 81). Interestingly, the increase in C1q mRNA expression was specific to microglia and did not occur in neurons or astrocytes (54), but C1q protein in the ischemic brain co-localized with the neuronal marker MAP2, suggesting that C1q is produced by microglia in response to ischemia and accumulates on neuronal cells (58). However, despite increased expression of C1q, the role of the classical pathway in post-ischemic injury is not completely clear since whereas C1q deficiency protects against murine neonatal hypoxic-ischemic brain injury (79, 85), it does not protect against murine transient MCAO (60). In neurodegenerative diseases, C1q and or C3 opsonization of synapses stimulates microglial phagocytosis, and complement-mediated synaptic clearance precedes neuronal loss (21, 86). During development, however, C1q and C3 opsonins have been shown to play an essential role in synaptic pruning and neuron remodeling (21). A feature of cerebral IRI is that it carries characteristics of both neurodegenerative disease and developmental mechanisms. Activation of inflammatory cascades after stroke causes significant secondary injury resulting in loss of both synapses and neurons in the penumbra area. Following this early insult, stroke is characterized by a window of neuroplastic response that mimics early developmental mechanisms including axonal growth and sprouting, and formation of new synapses and intra-cortical projections (87–89) [see reviews (90, 91)]. An important, yet unexplored question, is whether inhibition of the complement system after cerebral ischemia and reperfusion reduces loss of synaptic connections in the ischemic penumbra or whether this inhibition will prevent adequate synaptic pruning during rehabilitation induced cortical re-organization.
In an influential paper on the application of complement inhibition in stroke, Huang et al. described the use of soluble complement receptor 1 (sCR1) and its sialyl Lewis × glycosylated form (sCR1-sLex) to study the effect of systemic complement inhibition and selectin-targeted complement inhibition on outcomes after murine stroke. Soluble CR1 inhibits all complement pathways at the C3 activation step, and the sLex carbohydrate moiety binds to both P and E selectin, adhesion molecules that are upregulated on activated endothelium. Following MCAO and reperfusion, sCR1 modestly reduced ischemic injury while sCR1-sLex markedly diminished infarct volume, improved neurological outcome, and inhibited leukocyte migration (55). Thus, decoration sCR1 with the sLex moiety increased the efficacy of complement inhibition by targeting the inhibitor to selectin-expressing activated brain endothelial surfaces. A more recent study demonstrated that a truncated form of sCR1 also significantly improved outcome in a rat model of transient MCAO (78). Nevertheless, studies in a non-human primate stroke model failed to reproduce the earlier findings of sCR1 and sCR1-sLex treatment that was reported in mice (59). Similarly, the therapeutic value of sCR1-sLex failed to translate to baboons in a study by Ducruet et al. (66). In this latter study, sCR1-sLex reduced complement hemolytic activity, but failed to improve neurological outcome; in fact, treatment increased infarct volume in baboons. One possible explanation for this outcome is that sCR1-sLex has multivalent selectin-targeting sites and may promote homotypic platelet–platelet aggregation, and potentially thrombosis.
Another strategy used to target complement inhibition to an anatomical site is to link a complement inhibitor to a recombinant portion of complement receptor type 2 (CR2). Ligands for CR2 are C3 activation products that become covalently attached at sites of complement activation (57). Complement receptor 1-related gene/protein-y (Crry) is a rodent structural and functional analog of CR1, and a CR2-Crry construct administered 90 min after ischemia in a murine MCAO model was shown to improve survival, reduce infarct volume, reduce P-selectin expression and neutrophil recruitment, and improve neurological outcome. Acute outcomes after a single injection of CR2-Crry were comparable to those in C3-deficient mice (57). It has also been shown that targeted complement inhibition with CR2-Crry does not impair host susceptibility to infection, unlike systemic inhibition of C3 deficiency, a potentially important consideration for stroke patients who are at increased risk of infection (57).
The sCR1 and Crry inhibitors described above are pan complement inhibitors. Interventional studies in experimental models of stroke have also been performed with C1-inhibitor (C1-inh) that inhibits only the classical and lectin pathways. In various ischemic stroke models, C1-inh has been shown to decrease infarct volume (56, 62, 64, 70, 80), reduce focal and general deficits (62–64), inhibit leukocyte infiltration and myeloperoxidase activity (56, 63, 64), and diminish neuronal apoptosis (63, 64) (summarized in Table 2). Taken together with the data reported above that C1q deficiency is not protective in murine models of ischemic stroke, these data suggest a role for the lectin pathway in promoting cerebral IRI. Of note, however, all of the studies with C1-inh evaluated acute outcome after stroke, and it is possible that C1q-mediated uptake of apoptotic cells, an important anti-inflammatory and reparatory mechanism, may play a role in outcome in the subacute phase after stroke.
Further investigation has indeed demonstrated an important role for the lectin pathway in murine models of stroke. MBL deficiency resulted in significantly reduced infarct volumes, neurological impairment, and C3 deposition, whereas reconstitution of MBL-deficient mice with MBL resulted in larger infarct volumes (77). Two additional studies have also since demonstrated that MBL deficiency is protective against ischemic stroke (82, 84), and it has also been shown that an inhibitor of the lectin pathway, Polyman2, is protective when given up to 24 h after ischemia in mice (84). The same study demonstrated that an anti-MBL antibody is also protective in a rat model of stroke when given up to 18 h after ischemia, and as assessed 28 days after stroke. Thus, lectin pathway inhibition appears to be a therapeutic target with a wide therapeutic window. It should be noted, nevertheless, that Morrison et al. [(74) p. 52] found no significant difference in infarct volume, brain edema, or complement deposition between wild-type and MBL-deficient mice, and that Ducruet et al. (83) found that acute-phase protection in MBL-deficient mice was not sustained in the subacute phase. With regard to the different subacute outcomes in lectin pathway deficient vs. lectin pathway inhibited mice, an explanation could be that while acute complement activation is injurious, it also plays a role in repair and regenerative mechanisms in the subacute phase. Unlike deficiency, acute temporary lectin pathway inhibition would have minimal impact on the lectin pathway in the subacute phase.
The alternative complement pathway can be spontaneously activated, but it also serves as an amplification loop for the classical or lectin pathways. Whichever way complement is activated, it has been estimated that 80% of complement activation products can be the result of alternative pathway amplification (92). This is likely the reason that although the classical and lectin pathways can variously play essential roles in autoimmune, inflammatory, and ischemic disease models, the alternative pathway is nearly always required for full in vivo expression of injury (93, 94). And cerebral IRI is no exception, since although the studies reported above indicate a key role for the lectin pathway in ischemic stroke, mice deficient in factor B (alternative pathway protein) or treated with CR2-fH (targeted alternative pathway inhibitor) were protected to a similar extent as C1q/MBL-deficient mice in terms of several injury and neurological outcome measures (72). Together these data indicate that the alternative pathway is not alone sufficient to initiate complement activation, but that it acts to propagate cerebral injury via amplification of the cascade. Of note, a single CR2-fH treatment administered 90 min post ischemia resulted in significantly reduced astrocyte reactivity, cellular apoptosis and infarct volume for up to 7 days after reperfusion (72). In unpublished work, we have shown that unlike CR2-fH, CR2-Crry does not provide sustained protection into the subacute phase after stroke. This may reflect an advantage of not blocking all complement pathways in order to maintain some level of complement activation that may contribute to homeostatic or repair processes.
With regard to the terminal complement pathway, data indicate that the MAC is an important mediator of IRI in several organs and tissues, including spinal cord injury (95) and traumatic brain injury (96). In the case of cerebral IRI, however, the role of the MAC is less clear. Mice deficient in C6, a component of the MAC, are not protected from cerebral injury after 60 min MCAO and 24 h reperfusion, indicating that the MAC does not play a role in cerebral IRI (72). In agreement with this conclusion, mice deficient in CD59, a membrane-bound inhibitor of the MAC, do not display exacerbated cerebral injury in the same model (72). However, CD59-deficient mice show worse outcomes in the MCAO model with shorter ischemic times (30 min as opposed to 60 min), and thus the terminal pathway may contribute to cerebral IRI under conditions of less severe injury or ischemia (76).
Studies on the anaphylatoxins, C3a and C5a, have indicated important roles for these peptides in cerebral IRI. In a transient MCAO model, C3-deficient mice and wild-type mice treated with a C3a receptor antagonist (C3aRA) showed significant improvements in infarct volume and neurological outcomes, with less granulocyte infiltration. Furthermore, the effect was reversed when C3-deficient mice were reconstituted with C3 (60). A similar effect of C3aRA was shown in a transient MCAO model (66), but not in a model with permanent occlusion (73). The different effects of C3aRA in the two models may be related to inhibition of C3a-dependent activation of blood-derived immune cells following reperfusion. C5aR1-deficient mice are also protected against cerebral IRI following transient MCAO (44), but pretreatment of wild-type mice with C5aRA did not consistently improve neurological outcome or infarct volume (42, 65). Interestingly, C5aRA was also found to improve outcome in animal models of hemorrhagic stroke, either alone or in combination with C3aRA (53, 67, 69). These studies on the role of C3a and C5a in stroke pathogenesis were performed by analyzing acute outcomes, and it remains unclear what effect interfering with C3a- and C5a-mediated signaling would have on subacute and chronic outcomes. A concern with inhibiting C3a and C5a signaling, especially at later time points after stroke, is the effect on neurogenesis and cognitive performance, since both anaphylatoxins have implicated roles in basal and stress-induced neurogenesis (32–34). In this regard, a study using C3aRA treatment after MCAO indicated that the effect of C3a inhibition on neurogenesis is dose dependent. High-dose treatment with C3aRA (twice daily for 10 days) reduced ischemia-induced neurogenesis in the sub-ventricular zone, infarct area, and penumbra at 7 days after permanent MCAO (32). However, low dose C3aRA treatment increased neurogenesis in a dose-dependent fashion on day 7 in a transient MCAO model with 30 min ischemia (73). This may indicate that a titrated reduction of C3a-dependent signaling may limit the pro-inflammatory effects of C3a (especially in models that include reperfusion), while at the same time maintain the homeostatic effects of C3a on neurogenesis.
So what is the complement-activating event after stroke? Seminal studies by Williams et al. (97) and Zhang et al. (98, 99) demonstrated that following intestinal ischemia and reperfusion, complement is activated by natural self-reactive IgM antibodies that recognize post-ischemic neoepitopes. By screening IgM-producing hybridomas, an IgM monoclonal antibody was isolated that could restore pathogenic injury in otherwise protected antibody-deficient (Rag1−/−) mice. The intestinal target of this mAb was identified as non-muscle myosin (98), and antibodies to this post-ischemic neoepitope have additionally been shown to play a role in hind-limb (100) and myocardial IRI (101). Although the early studies attributed IgM-mediated activation of the classical pathway as an initiating event in IRI, more recent studies have demonstrated that IRI is driven by IgM-mediated activation of the lectin pathway, at least in the organ systems that have been investigated (99, 102, 103). The animal studies described above, and the human studies described below, indicate that the lectin pathway also initiates cerebral IRI, and recent studies have demonstrated that pathogenic natural IgM also plays a role in propagating injury in murine ischemic stroke (71) (Figure 3). As with intestinal IRI, Rag1-deficient mice are protected from cerebral IRI, and two IgM monoclonal antibodies were identified that reconstituted cerebral IRI in Rag1-deficient mice; these monoclonal antibodies recognized annexin IV and a subset of phospholipids, and thus identified danger-associated molecular determinants expressed after stroke in mice (71). Post-ischemic blockade of annexin IV or phospholipid neoepitopes thus represents a potential therapeutic strategy for inhibiting complement activation and reducing cerebral IRI. The same two post-ischemic neoepitopes are also expressed in the intestine (104) and heart [(57), Circulation, in press] after ischemia and reperfusion, indicating that similar pathophysiologically important epitopes recognized by IgM natural antibodies are expressed in different organs and tissues. Furthermore, it has been shown that human endothelial cells (HUVEC) express these two neoepitopes after exposure to hypoxia (71).
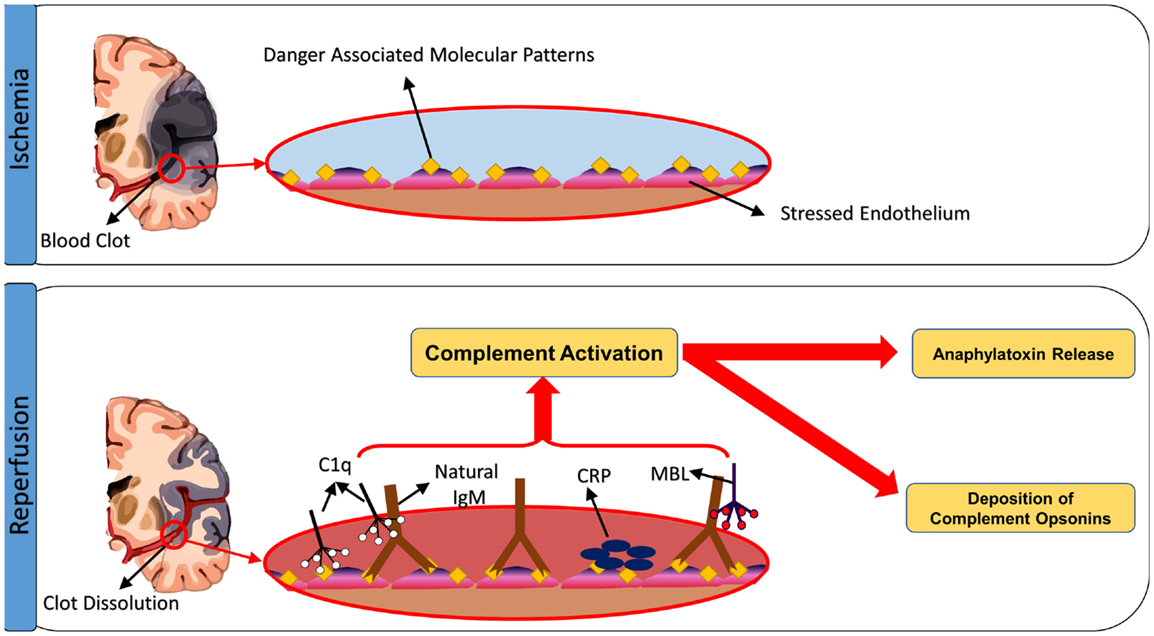
Figure 3. Triggers of complement activation after cerebral ischemia-reperfusion injury. Ischemic insult induces expression of neoepitopes or danger-associated molecular patterns (DAMPs) on the surface of stressed endothelial cells. The exposed DAMPs are recognized by circulating natural self-reactive antibodies, principally IgM, which triggers complement activation. Although IgM binds C1q, it appears to be the binding of MBL and activation of the lectin pathway that drives ischemia and reperfusion injury in the organs systems examined, including the brain. Complement can be also activated through direct binding of C1q to apoptotic cells, as well as through C-reactive protein-induced complement activation.
There are no reported clinical trials on the use of complement inhibition in stroke, a direct consequence of the aforementioned conflicting pre-clinical data. Before an intervention can enter a clinical trial, stringent criteria recommended by the stroke therapy academic industry roundtable (STAIR) must be met. In brief, there should be consistent pre-clinical data in different animal species and in animal cohorts that represent the human stroke predisposed population. Such populations include both sexes, adult and elderly age groups, and patients with comorbidities such as hypertension and diabetes (105). Thus, studies on the role of complement in human stroke are correlational.
Studies on gene polymorphisms at complement gene loci, associated polymorphisms at the C5 and factor H gene loci with increased incidence of cerebrovascular accidents (106, 107). Among 16 single nucleotide polymorphisms (SNPs) at the C3 gene locus, two SNPs (rs2277984 and rs3745565) were associated with ischemic stroke (108). MBL genotype is also associated with stroke severity. In two cohorts of stroke patients, MBL-sufficient phenotypes were associated with larger infarct and less favorable outcome compared to MBL-deficient phenotypes (77, 109). Prospective studies on men with advanced atherosclerosis, higher levels of C4 and C5 are associated with the incidence of stroke (110). Similarly, a prospective study of 5850 healthy men showed that C4 and C3 levels correlated with other cardiovascular risk factors, and that very high C4 levels may predict the incidence of stroke (111). C4 was also shown to be a stroke predictor in men with known or suspected coronary artery disease (112). Compared to healthy volunteers, stroke patients were found to have higher plasma levels of C3a, C3, C4, C5, factor B, and the terminal complement complex (113–118). Serum levels of C3, C3c, and C4 were also associated with increased stroke severity in cardio-embolic stroke patients (118, 119). Despite these human studies that report alterations in serum complement levels in stroke patients and the association with outcome, the studies remain correlative and cannot demonstrate a definitive role for complement in the pathogenesis of stroke. Table 3 summarizes studies investigating complement system changes in patients with ischemic or hemorrhagic stroke.
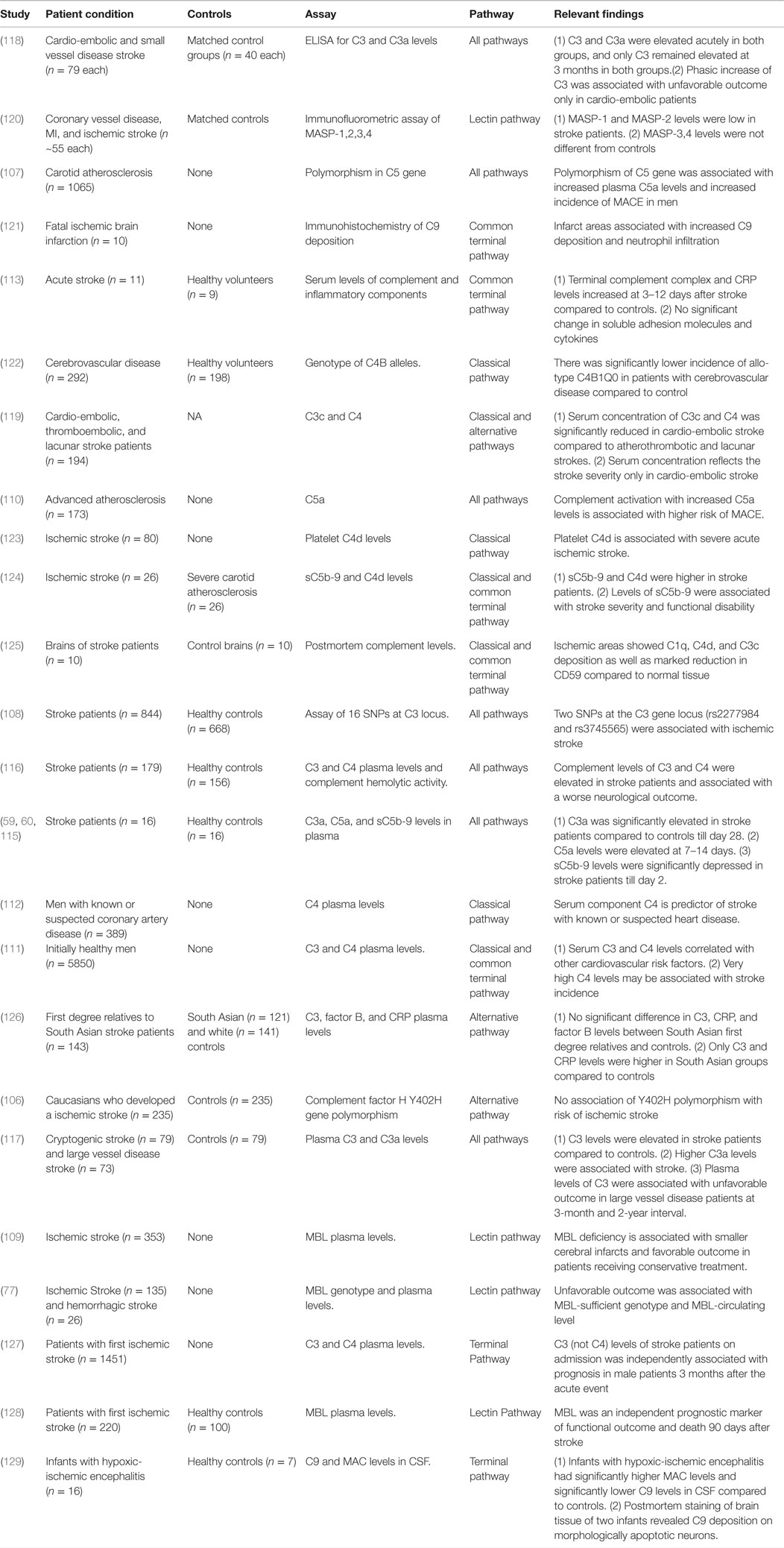
Table 3. Summary of studies performed in human patients investigating the role of complement in cerebral I/R.
The predominant focus of studies on complement and stroke has been on the role of complement in perpetuating the inflammatory cascade and contributing to neuronal death. By comparison, very few studies have investigated the putative protective role that complement may play in ischemic rescue and ischemia-induced neurogenesis. The great majority of reports investigate acute-phase outcomes where complement blockade prevents activation of pro-inflammatory mechanisms. However, as indicated by a few studies, the same favorable outcomes may not occur in the subacute and chronic phases after stroke, when complement opsonins may be important for mediating anti-inflammatory events such as removal of debris, and when anaphylatoxins may contribute to homeostatic pro-survival and neuro-regenerative effects. It will be important to investigate how manipulation of the complement system impacts subacute and chronic outcomes, as well as to evaluate areas away from the ischemic core. Investigation of chronic outcomes may reveal a role for complement in maintaining neuronal survival after acute damage has been resolved, and possible mechanisms may include stimulation of neuronal growth factor production by astrocytes and microglia (35, 36), or reduction of glutamate-induced excitotoxicity (37–40). Stroke and brain injury are associated with a window of synaptic plasticity after injury that contributes to neurological recovery (88, 90). While various mechanisms have been implicated in this process, a role for complement has not been investigated. Based on the essential role of complement in synaptic pruning during CNS development, complement may also play a role in synaptic pruning and hippocampal plasticity after stroke, and may thus contribute to improved neurological outcome and facilitate rehabilitation.
Going forward, the design of experimental models to assess longer endpoints and multiple outcome measures after stroke may reveal a more diverse role for complement in ischemic injury and rescue. Future studies should also investigate the role of different complement components in early and delayed pathophysiological events. One area of investigation that has received little attention is the role of complement opsonins in early neuronal and synaptic loss after stroke, as well as the role of these opsonins in the resolution of inflammation and synaptic re-wiring that can occur later. Future studies should also address the interplay between complement opsonins and immune cells bearing receptors for these opsonins, especially infiltrating and resident macrophages/microglia; these opsonins may also play a role in driving macrophage polarization toward an anti-inflammatory and pro-regenerative phenotype. We also have an incomplete understanding of the role of the anaphylatoxins in stroke. They are implicated in recruiting and activating immune cells acutely, as well as stimulating neuronal genesis and migration in delayed phases after stroke. It is also not clear whether C3a and C5a have redundant, overlapping, or possibly divergent effects. Tools to address these questions are available in the form of genetically deficient mice and inhibitors that block the generation of specific complement activation products or that antagonize complement receptors, and it will be important for future studies to better evaluate long-term effects and outcomes after stroke.
As mentioned in Section “Studies in Human Patients,” polymorphism in complement genes is associated with ischemic stroke, and that acute stroke patients have higher serum levels of complement proteins compared to controls (Table 3). In addition, postmortem studies have identified both complement and IgM deposition in the human brain after stroke (125). This suggests that complement activation and deposition occurs in the human brain similarly to that in experimental models. Thus, given the correlation between complement activity and stroke outcome in human patients, the use of complement inhibition holds promise as a potential therapeutic intervention. Optimally, a complement inhibitor should effectively inhibit early pathogenic complement activation without depleting systemic complement activity in order to maintain the homeostatic and protective role of complement during recovery. A clear understanding of the role of different complement activation products and their spatial and temporal effects will be important to guide the development of an optimal anti-complement therapeutic strategy. As a final note, thrombolytic therapy using t-PA also results in complement activation (130), and it is possible that complement inhibition in the context of t-PA administration could provide a new adjuvant therapy to t-PA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ST is supported by grants from Veterans Affairs (RX001141 and BX001201) and the NIH (P20GM109040).
1. Barber PA, Davis SM, Infeld B, Baird AE, Donnan GA, Jolley D, et al. Spontaneous reperfusion after ischemic stroke is associated with improved outcome. Stroke (1998) 29:2522–8. doi:10.1161/01.STR.29.12.2522
2. Patel RD, Saver JL. Evolution of reperfusion therapies for acute brain and acute myocardial ischemia: a systematic, comparative analysis. Stroke (2013) 44:94–8. doi:10.1161/STROKEAHA.112.666925
3. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med (2011) 17:796–808. doi:10.1038/nm.2399
4. Barber PA, Zhang J, Demchuk AM, Hill MD, Buchan AM. Why are stroke patients excluded from TPA therapy? An analysis of patient eligibility. Neurology (2001) 56:1015–20. doi:10.1212/WNL.56.8.1015
5. Katzan IL, Hammer MD, Hixson ED, Furlan AJ, Abou-Chebl A, Nadzam DM, et al. Utilization of intravenous tissue plasminogen activator for acute ischemic stroke. Arch Neurol (2004) 61:346–50. doi:10.1001/archneur.61.3.346
6. Levi-Strauss M, Mallat M. Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. J Immunol (1987) 139:2361–6.
7. Gasque P, Fontaine M, Morgan BP. Complement expression in human brain. Biosynthesis of terminal pathway components and regulators in human glial cells and cell lines. J Immunol (1995) 154:4726–33.
8. Gasque P, Dean Y, McGreal E, Vanbeek J, Morgan B. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology (2000) 49:171–86. doi:10.1016/S0162-3109(00)80302-1
9. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11:785–97. doi:10.1038/ni.1923
10. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med (2006) 12:682–7. doi:10.1038/nm1419
11. Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol (2008) 632:71–9. doi:10.1007/978-0-387-78952-1_6
12. Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol (2009) 9:729–40. doi:10.1038/nri2620
13. Stahel PF, Bamum SR. Bacterial meningitis: complement gene expression in the central nervous system. Immunopharmacology (1997) 38:65–72. doi:10.1016/S0162-3109(97)80150-6
14. Beek J, Elward K, Gasque P. Activation of complement in the central nervous system. Ann N Y Acad Sci (2003) 992:56–71. doi:10.1111/j.1749-6632.2003.tb03138.x
15. Rogers J, Strohmeyer R, Kovelowski CJ, Li R. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia (2002) 40:260–9. doi:10.1002/glia.10153
16. Boos L, Szalai AJ, Barnum SR. C3a expressed in the central nervous system protects against LPS-induced shock. Neurosci Lett (2005) 387:68–71. doi:10.1016/j.neulet.2005.07.015
17. Rupprecht TA, Angele B, Klein M, Heesemann J, Pfister HW, Botto M, et al. Complement C1q and C3 are critical for the innate immune response to Streptococcus pneumoniae in the central nervous system. J Immunol (2007) 178:1861–9. doi:10.4049/jimmunol.178.3.1861
18. Ramaglia V, Daha M, Baas F. The complement system in the peripheral nerve: friend or foe? Mol Immunol (2008) 45:3865–77. doi:10.1016/j.molimm.2008.06.018
19. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131:1164–78. doi:10.1016/j.cell.2007.10.036
20. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron (2012) 74:691–705. doi:10.1016/j.neuron.2012.03.026
21. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci (2012) 35:369–89. doi:10.1146/annurev-neuro-061010-113810
22. Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A (2010) 107:7975–80. doi:10.1073/pnas.0913449107
23. Benard M, Gonzalez BJ, Schouft MT, Falluel-Morel A, Vaudry D, Chan P, et al. Characterization of C3a and C5a receptors in rat cerebellar granule neurons during maturation. Neuroprotective effect of C5a against apoptotic cell death. J Biol Chem (2004) 279:43487–96. doi:10.1074/jbc.M404124200
24. Benard M, Raoult E, Vaudry D, Leprince J, Falluel-Morel A, Gonzalez BJ, et al. Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Mol Immunol (2008) 45:3767–74. doi:10.1016/j.molimm.2008.05.027
25. Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci (2011) 31:3459–69. doi:10.1523/JNEUROSCI.3932-10.2011
26. Pisalyaput K, Tenner AJ. Complement component C1q inhibits beta-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J Neurochem (2008) 104:696–707. doi:10.1111/j.1471-4159.2007.05012.x
27. Soane L, Rus H, Niculescu F, Shin ML. Inhibition of oligodendrocyte apoptosis by sublytic C5b-9 is associated with enhanced synthesis of bcl-2 and mediated by inhibition of caspase-3 activation. J Immunol (1999) 163:6132–8.
28. Cudrici C, Niculescu F, Jensen T, Zafranskaia E, Fosbrink M, Rus V, et al. C5b-9 terminal complex protects oligodendrocytes from apoptotic cell death by inhibiting caspase-8 processing and up-regulating FLIP. J Immunol (2006) 176:3173–80. doi:10.4049/jimmunol.176.11.7131
29. Moriyama M, Fukuhara T, Britschgi M, He Y, Narasimhan R, Villeda S, et al. Complement receptor 2 is expressed in neural progenitor cells and regulates adult hippocampal neurogenesis. J Neurosci (2011) 31:3981–9. doi:10.1523/JNEUROSCI.3617-10.2011
30. Crane JW, Baiquni GP, Sullivan RK, Lee JD, Sah P, Taylor SM, et al. The C5a anaphylatoxin receptor CD88 is expressed in presynaptic terminals of hippocampal mossy fibres. J Neuroinflammation (2009) 6:34. doi:10.1186/1742-2094-6-34
31. Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia (1999) 26:201–11. doi:10.1002/(SICI)1098-1136(199905)26:3<201::AID-GLIA2>3.0.CO;2-M
32. Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J (2006) 25:1364–74. doi:10.1038/sj.emboj.7601004
33. Shinjyo N, Stahlberg A, Dragunow M, Pekny M, Pekna M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells (2009) 27:2824–32. doi:10.1002/stem.225
34. Mukherjee P, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through mitogen-activated protein kinase-dependent inhibition of caspase 3. J Neurochem (2001) 77:43–9. doi:10.1046/j.1471-4159.2001.00167.x
35. Heese K, Hock C, Otten U. Inflammatory signals induce neurotrophin expression in human microglial cells. J Neurochem (1998) 70:699–707. doi:10.1046/j.1471-4159.1998.70020699.x
36. Jauneau AC, Ischenko A, Chatagner A, Benard M, Chan P, Schouft MT, et al. Interleukin-1beta and anaphylatoxins exert a synergistic effect on NGF expression by astrocytes. J Neuroinflammation (2006) 3:8. doi:10.1186/1742-2094-3-8
37. Van Beek J, Nicole O, Ali C, Ischenko A, Mackenzie ET, Buisson A, et al. Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. Neuroreport (2001) 12:289–93. doi:10.1097/00001756-200102120-00022
38. Osaka H, Mukherjee P, Aisen PS, Pasinetti GM. Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. J Cell Biochem (1999) 73:303–11.10.1002/(SICI)1097-4644(19990601) 73:3<303::AID-JCB2>3.0.CO;2-2
39. Mukherjee P, Thomas S, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. J Neuroinflammation (2008) 5:5. doi:10.1186/1742-2094-5-5
40. Persson M, Pekna M, Hansson E, Ronnback L. The complement-derived anaphylatoxin C5a increases microglial GLT-1 expression and glutamate uptake in a TNF-alpha-independent manner. Eur J Neurosci (2009) 29:267–74. doi:10.1111/j.1460-9568.2008.06575.x
41. Tohgi H, Utsugisawa K, Nagane Y. Hypoxia-induced expression of C1q, a subcomponent of the complement system, in cultured rat PC12 cells. Neurosci Lett (2000) 291:151–4. doi:10.1016/S0304-3940(00)01399-9
42. Arumugam TV, Tang SC, Lathia JD, Cheng A, Mughal MR, Chigurupati S, et al. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc Natl Acad Sci U S A (2007) 104:14104–9. doi:10.1073/pnas.0700506104
43. Yang J, Ahn HN, Chang M, Narasimhan P, Chan PH, Song YS. Complement component 3 inhibition by an antioxidant is neuroprotective after cerebral ischemia and reperfusion in mice. J Neurochem (2013) 124:523–35. doi:10.1111/jnc.12111
44. Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM. Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J (2012) 26:3680–90. doi:10.1096/fj.11-202382
45. Thundyil J, Pavlovski D, Hsieh YH, Gelderblom M, Magnus T, Fairlie DP, et al. C5a receptor (CD88) inhibition improves hypothermia-induced neuroprotection in an in vitro ischemic model. Neuromolecular Med (2012) 14:30–9. doi:10.1007/s12017-012-8167-0
46. Pedersen ED, Froyland E, Kvissel AK, Pharo AM, Skalhegg BS, Rootwelt T, et al. Expression of complement regulators and receptors on human NT2-N neurons – effect of hypoxia and reoxygenation. Mol Immunol (2007) 44:2459–68. doi:10.1016/j.molimm.2006.10.022
47. Bednar MM, Gross CE, Russell SR, Short D, Giclas PC. Activation of complement by tissue plasminogen activator, but not acute cerebral ischemia, in a rabbit model of thromboembolic stroke. J Neurosurg (1997) 86:139–42. doi:10.3171/jns.1997.86.1.0139
48. Lew SM, Gross CE, Bednar MM, Russell SJ, Fuller SP, Ellenberger CL, et al. Complement depletion does not reduce brain injury in a rabbit model of thromboembolic stroke. Brain Res Bull (1999) 48:325–31. doi:10.1016/S0361-9230(99)00004-0
49. Vasthare US, Barone FC, Sarau HM, Rosenwasser RH, Dimartino M, Young WF, et al. Complement depletion improves neurological function in cerebral ischemia. Brain Res Bull (1998) 45:413–9. doi:10.1016/S0361-9230(97)00408-5
50. Cowell RM, Plane JM, Silverstein FS. Complement activation contributes to hypoxic-ischemic brain injury in neonatal rats. J Neurosci (2003) 23:9459–68.
51. Figueroa E, Gordon LE, Feldhoff PW, Lassiter HA. The administration of cobra venom factor reduces post-ischemic cerebral injury in adult and neonatal rats. Neurosci Lett (2005) 380:48–53. doi:10.1016/j.neulet.2005.01.027
52. Gill R, Kemp JA, Sabin C, Pepys MB. Human C-reactive protein increases cerebral infarct size after middle cerebral artery occlusion in adult rats. J Cereb Blood Flow Metab (2004) 24:1214–8. doi:10.1097/01.WCB.0000136517.61642.99
53. Li G, Fan RM, Chen JL, Wang CM, Zeng YC, Han C, et al. Neuroprotective effects of argatroban and C5a receptor antagonist (PMX53) following intracerebral hemorrhage. Clin Exp Immunol (2013) 175:285–95. doi:10.1111/cei.12220
54. Schafer MK, Schwaeble WJ, Post C, Salvati P, Calabresi M, Sim RB, et al. Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J Immunol (2000) 164:5446–52. doi:10.4049/jimmunol.164.10.5446
55. Huang J, Kim LJ, Mealey R, Marsh HC Jr, Zhang Y, Tenner AJ, et al. Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science (1999) 285:595–9. doi:10.1126/science.285.5427.595
56. Akita N, Nakase H, Kanemoto Y, Kaido T, Nishioka T, Sakaki T. The effect of C 1 esterase inhibitor on ischemia: reperfusion injury in the rat brain]. No To Shinkei (2001) 53:641.
57. Atkinson C, Song H, Lu B, Qiao F, Burns TA, Holers VM, et al. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J Clin Invest (2005) 115:2444–53. doi:10.1172/JCI25208
58. Mack WJ, Sughrue ME, Ducruet AF, Mocco J, Sosunov SA, Hassid BG, et al. Temporal pattern of C1q deposition after transient focal cerebral ischemia. J Neurosci Res (2006) 83:883–9. doi:10.1002/jnr.20775
59. Mocco J, Mack WJ, Ducruet AF, King RG, Sughrue ME, Coon AL, et al. Preclinical evaluation of the neuroprotective effect of soluble complement receptor type 1 in a nonhuman primate model of reperfused stroke. J Neurosurg (2006) 105:595–601. doi:10.3171/jns.2006.105.4.595
60. Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res (2006) 99:209–17. doi:10.1161/01.RES.0000232544.90675.42
61. Imm MD, Feldhoff PW, Feldhoff RC, Lassiter HA. The administration of complement component C9 augments post-ischemic cerebral infarction volume in neonatal rats. Neurosci Lett (2002) 325:175–8. doi:10.1016/S0304-3940(02)00271-9
62. De Simoni MG, Storini C, Barba M, Catapano L, Arabia AM, Rossi E, et al. Neuroprotection by complement (C1) inhibitor in mouse transient brain ischemia. J Cereb Blood Flow Metab (2003) 23:232–9. doi:10.1097/00004647-200302000-00010
63. Storini C, Rossi E, Marrella V, Distaso M, Veerhuis R, Vergani C, et al. C1-inhibitor protects against brain ischemia-reperfusion injury via inhibition of cell recruitment and inflammation. Neurobiol Dis (2005) 19:10–7. doi:10.1016/j.nbd.2004.11.001
64. De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol (2004) 164:1857–63. doi:10.1016/S0002-9440(10)63744-3
65. Kim GH, Mocco J, Hahn DK, Kellner CP, Komotar RJ, Ducruet AF, et al. Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery (2008) 63:122–5. doi:10.1227/01.NEU.0000335079.70222.8D
66. Ducruet AF, Hassid BG, Mack WJ, Sosunov SA, Otten ML, Fusco DJ, et al. C3a receptor modulation of granulocyte infiltration after murine focal cerebral ischemia is reperfusion dependent. J Cereb Blood Flow Metab (2008) 28:1048–58. doi:10.1038/sj.jcbfm.9600608
67. Garrett MC, Otten ML, Starke RM, Komotar RJ, Magotti P, Lambris JD, et al. Synergistic neuroprotective effects of C3a and C5a receptor blockade following intracerebral hemorrhage. Brain Res (2009) 1298:171–7. doi:10.1016/j.brainres.2009.04.047
68. Ducruet AF, Mocco J, Mack WJ, Coon AL, Marsh HC, Pinsky DJ, et al. Pre-clinical evaluation of an sLe x-glycosylated complement inhibitory protein in a non-human primate model of reperfused stroke. J Med Primatol (2007) 36:375–80. doi:10.1111/j.1600-0684.2007.00213.x
69. Rynkowski MA, Kim GH, Garrett MC, Zacharia BE, Otten ML, Sosunov SA, et al. C3a receptor antagonist attenuates brain injury after intracerebral hemorrhage. J Cereb Blood Flow Metab (2008) 29:98–107. doi:10.1038/jcbfm.2008.95
70. Gesuete R, Storini C, Fantin A, Stravalaci M, Zanier ER, Orsini F, et al. Recombinant C1 inhibitor in brain ischemic injury. Ann Neurol (2009) 66:332–42. doi:10.1002/ana.21740
71. Elvington A, Atkinson C, Kulik L, Zhu H, Yu J, Kindy MS, et al. Pathogenic natural antibodies propagate cerebral injury following ischemic stroke in mice. J Immunol (2012) 188:1460–8. doi:10.4049/jimmunol.1102132
72. Elvington A, Atkinson C, Zhu H, Yu J, Takahashi K, Stahl GL, et al. The alternative complement pathway propagates inflammation and injury in murine ischemic stroke. J Immunol (2012) 189:4640–7. doi:10.4049/jimmunol.1201904
73. Ducruet AF, Zacharia BE, Sosunov SA, Gigante PR, Yeh ML, Gorski JW, et al. Complement inhibition promotes endogenous neurogenesis and sustained anti-inflammatory neuroprotection following reperfused stroke. PLoS One (2012) 7:e38664. doi:10.1371/journal.pone.0038664
74. Morrison H, Frye J, Davis-Gorman G, Funk J, McDonagh P, Stahl G, et al. The contribution of mannose binding lectin to reperfusion injury after ischemic stroke. Curr Neurovasc Res (2011) 8:52–63. doi:10.2174/156720211794520260
75. Hurtado O, Serrano J, Sobrado M, Fernández A, Lizasoain I, Martínez-Murillo R, et al. Lack of adrenomedullin, but not complement factor H, results in larger infarct size and more extensive brain damage in a focal ischemia model. Neuroscience (2010) 171:885–92. doi:10.1016/j.neuroscience.2010.09.021
76. Harhausen D, Khojasteh U, Stahel PF, Morgan BP, Nietfeld W, Dirnagl U, et al. Membrane attack complex inhibitor CD59a protects against focal cerebral ischemia in mice. J Neuroinflammation (2010) 7:15. doi:10.1186/1742-2094-7-15
77. Cervera A, Planas AM, Justicia C, Urra X, Jensenius JC, Torres F, et al. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS One (2010) 5:e8433. doi:10.1371/journal.pone.0008433
78. Li S, Xian J, He L, Luo X, Tan B, Yang Y, et al. The protective effect of SCR(15-18) on cerebral ischemia-reperfusion injury. Neurol Res (2011) 33:866–74. doi:10.1179/1743132811Y.0000000016
79. Ten VS, Sosunov SA, Mazer SP, Stark RI, Caspersen C, Sughrue ME, et al. C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke (2005) 36:2244–50. doi:10.1161/01.STR.0000182237.20807.d0
80. Heimann A, Takeshima T, Horstick G, Kempski O. C1-esterase inhibitor reduces infarct volume after cortical vein occlusion. Brain Res (1999) 838:210–3. doi:10.1016/S0006-8993(99)01740-0
81. Luo H, Li W, Yang F, Zhou L, Wen P, Zhou J. Expressions of complement C1q and C3c in rat brain tissues with cerebral ischemia/reperfusion injury. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2013) 29:897–900.
82. De La Rosa X, Cervera A, Kristoffersen AK, Valdes CP, Varma HM, Justicia C, et al. Mannose-binding lectin promotes local microvascular thrombosis after transient brain ischemia in mice. Stroke (2014) 45:1453–9. doi:10.1161/STROKEAHA.113.004111
83. Ducruet AF, Sosunov SA, Zacharia BE, Gorski J, Yeh ML, Derosa P, et al. The neuroprotective effect of genetic mannose-binding lectin deficiency is not sustained in the sub-acute phase of stroke. Transl Stroke Res (2011) 2:588–99. doi:10.1007/s12975-011-0104-2
84. Orsini F, Villa P, Parrella S, Zangari R, Zanier ER, Gesuete R, et al. Targeting mannose-binding lectin confers long-lasting protection with a surprisingly wide therapeutic window in cerebral ischemia. Circulation (2012) 126:1484–94. doi:10.1161/CIRCULATIONAHA.112.103051
85. Ten VS, Yao J, Ratner V, Sosunov S, Fraser DA, Botto M, et al. Complement component c1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J Neurosci (2010) 30:2077–87. doi:10.1523/JNEUROSCI.5249-09.2010
86. Alexander JJ, Anderson AJ, Barnum SR, Stevens B, Tenner AJ. The complement cascade: Yin-Yang in neuroinflammation – neuro-protection and -degeneration. J Neurochem (2008) 107:1169–87. doi:10.1111/j.1471-4159.2008.05668.x
87. Carmichael ST, Wei L, Rovainen CM, Woolsey TA. New patterns of intracortical projections after focal cortical stroke. Neurobiol Dis (2001) 8:910–22. doi:10.1006/nbdi.2001.0425
88. Carmichael ST. Plasticity of cortical projections after stroke. Neuroscientist (2003) 9:64–75. doi:10.1177/1073858402239592
89. Carmichael ST, Archibeque I, Luke L, Nolan T, Momiy J, Li S. Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol (2005) 193:291–311. doi:10.1016/j.expneurol.2005.01.004
90. Wieloch T, Nikolich K. Mechanisms of neural plasticity following brain injury. Curr Opin Neurobiol (2006) 16:258–64. doi:10.1016/j.conb.2006.05.011
91. Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci (2009) 10:861–72. doi:10.1038/nrn2735
92. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol (2004) 138:439–46. doi:10.1111/j.1365-2249.2004.02627.x
93. Holers VM, Thurman JM. The alternative pathway of complement in disease: opportunities for therapeutic targeting. Mol Immunol (2004) 41:147–52. doi:10.1016/j.molimm.2004.03.012
94. Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol (2006) 176:1305–10. doi:10.4049/jimmunol.176.3.1305
95. Mead RJ, Singhrao SK, Neal JW, Lassmann H, Morgan BP. The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol (2002) 168:458–65. doi:10.4049/jimmunol.168.1.458
96. Bellander BM, Singhrao SK, Ohlsson M, Mattsson P, Svensson M. Complement activation in the human brain after traumatic head injury. J Neurotrauma (2001) 18:1295–311. doi:10.1089/08977150152725605
97. Williams JP, Pechet TT, Weiser MR, Reid R, Kobzik L, Moore FD Jr, et al. Intestinal reperfusion injury is mediated by IgM and complement. J Appl Physiol (1985) (1999) 86:938–42.
98. Zhang M, Austen WG Jr, Chiu I, Alicot EM, Hung R, Ma M, et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A (2004) 101:3886–91. doi:10.1073/pnas.0400347101
99. Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, et al. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol (2006) 177:4727–34. doi:10.4049/jimmunol.177.7.4727
100. Austen WG Jr, Zhang M, Chan R, Friend D, Hechtman HB, Carroll MC, et al. Murine hindlimb reperfusion injury can be initiated by a self-reactive monoclonal IgM. Surgery (2004) 136:401–6. doi:10.1016/j.surg.2004.05.016
101. Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, et al. Identification of the target self-antigens in reperfusion injury. J Exp Med (2006) 203:141–52. doi:10.1084/jem.20050390
102. McMullen ME, Hart ML, Walsh MC, Buras J, Takahashi K, Stahl GL. Mannose-binding lectin binds IgM to activate the lectin complement pathway in vitro and in vivo. Immunobiology (2006) 211:759–66. doi:10.1016/j.imbio.2006.06.011
103. Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am J Physiol Heart Circ Physiol (2009) 297:H1853–9. doi:10.1152/ajpheart.00049.2009
104. Kulik L, Fleming SD, Moratz C, Reuter JW, Novikov A, Chen K, et al. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. J Immunol (2009) 182:5363–73. doi:10.4049/jimmunol.0803980
105. Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke (2009) 40:2244–50. doi:10.1161/STROKEAHA.108.541128
106. Zee RY, Diehl KA, Ridker PM. Complement factor H Y402H gene polymorphism, C-reactive protein, and risk of incident myocardial infarction, ischaemic stroke, and venous thromboembolism: a nested case-control study. Atherosclerosis (2006) 187:332–5. doi:10.1016/j.atherosclerosis.2005.09.009
107. Hoke M, Speidl W, Schillinger M, Minar E, Zehetmayer S, Schonherr M, et al. Polymorphism of the complement 5 gene and cardiovascular outcome in patients with atherosclerosis. Eur J Clin Invest (2012) 42:921–6. doi:10.1111/j.1365-2362.2012.02669.x
108. Olsson S, Stokowska A, Holmegaard L, Jood K, Blomstrand C, Pekna M, et al. Genetic variation in complement component C3 shows association with ischaemic stroke. Eur J Neurol (2011) 18:1272–4. doi:10.1111/j.1468-1331.2011.03377.x
109. Osthoff M, Katan M, Fluri F, Schuetz P, Bingisser R, Kappos L, et al. Mannose-binding lectin deficiency is associated with smaller infarction size and favorable outcome in ischemic stroke patients. PLoS One (2011) 6:e21338. doi:10.1371/journal.pone.0021338
110. Speidl WS, Exner M, Amighi J, Kastl SP, Zorn G, Maurer G, et al. Complement component C5a predicts future cardiovascular events in patients with advanced atherosclerosis. Eur Heart J (2005) 26:2294–9. doi:10.1093/eurheartj/ehi339
111. Engstrom G, Hedblad B, Janzon L, Lindgarde F. Complement C3 and C4 in plasma and incidence of myocardial infarction and stroke: a population-based cohort study. Eur J Cardiovasc Prev Rehabil (2007) 14:392–7. doi:10.1097/01.hjr.0000244582.30421.b2
112. Cavusoglu E, Eng C, Chopra V, Ruwende C, Yanamadala S, Clark LT, et al. Usefulness of the serum complement component C4 as a predictor of stroke in patients with known or suspected coronary artery disease referred for coronary angiography. Am J Cardiol (2007) 100:164–8. doi:10.1016/j.amjcard.2007.02.075
113. Pedersen ED, Waje-Andreassen U, Vedeler CA, Aamodt G, Mollnes TE. Systemic complement activation following human acute ischaemic stroke. Clin Exp Immunol (2004) 137:117–22. doi:10.1111/j.1365-2249.2004.02489.x
114. Aivazian VA, Boiadzhian AS, Manukian LA, Avetisian GV, Grigorian GS. [Complement componenets, C3 and factors B, in the blood of patients with acute ischemic stroke]. Zh Nevrol Psikhiatr Im S S Korsakova (2005) Suppl 15:57–60.
115. Mocco J, Wilson DA, Komotar RJ, Sughrue ME, Coates K, Sacco RL, et al. Alterations in plasma complement levels after human ischemic stroke. Neurosurgery (2006) 59:28–33. doi:10.1227/01.NEU.0000219221.14280.65
116. Cojocaru IM, Cojocaru M, Tanasescu R, Burcin C, Atanasiu AN, Petrescu AM, et al. Changes in plasma levels of complement in patients with acute ischemic stroke. Rom J Intern Med (2008) 46:77–80.
117. Stokowska A, Olsson S, Holmegaard L, Jood K, Blomstrand C, Jern C, et al. Plasma C3 and C3a levels in cryptogenic and large-vessel disease stroke: associations with outcome. Cerebrovasc Dis (2011) 32:114–22. doi:10.1159/000328238
118. Stokowska A, Olsson S, Holmegaard L, Jood K, Blomstrand C, Jern C, et al. Cardioembolic and small vessel disease stroke show differences in associations between systemic C3 levels and outcome. PLoS One (2013) 8:e72133. doi:10.1371/journal.pone.0072133
119. Di Napoli M. Systemic complement activation in ischemic stroke. Stroke (2001) 32:1443–8. doi:10.1161/01.STR.32.6.1443-a
120. Frauenknecht V, Thiel S, Storm L, Meier N, Arnold M, Schmid JP, et al. Plasma levels of mannan-binding lectin (MBL)-associated serine proteases (MASPs) and MBL-associated protein in cardio- and cerebrovascular diseases. Clin Exp Immunol (2013) 173:112–20. doi:10.1111/cei.12093
121. Lindsberg PJ, Ohman J, Lehto T, Karjalainen-Lindsberg ML, Paetau A, Wuorimaa T, et al. Complement activation in the central nervous system following blood-brain barrier damage in man. Ann Neurol (1996) 40:587–96. doi:10.1002/ana.410400408
122. Kramer J, Harcos P, Prohaszka Z, Horvath L, Karadi I, Singh M, et al. Frequencies of certain complement protein alleles and serum levels of anti-heat-shock protein antibodies in cerebrovascular diseases. Stroke (2000) 31:2648–52. doi:10.1161/01.STR.31.11.2648
123. Mehta N, Uchino K, Fakhran S, Sattar MA, Branstetter BFT, Au K, et al. Platelet C4d is associated with acute ischemic stroke and stroke severity. Stroke (2008) 39:3236–41. doi:10.1161/STROKEAHA.108.514687
124. Széplaki G, Szegedi R, Hirschberg K, Gombos T, Varga L, Karádi I, et al. Strong complement activation after acute ischemic stroke is associated with unfavorable outcomes. Atherosclerosis (2009) 204:315–20. doi:10.1016/j.atherosclerosis.2008.07.044
125. Pedersen ED, Loberg EM, Vege E, Daha MR, Maehlen J, Mollnes TE. In situ deposition of complement in human acute brain ischaemia. Scand J Immunol (2009) 69:555–62. doi:10.1111/j.1365-3083.2009.02253.x
126. Somani R, Grant PJ, Kain K, Catto AJ, Carter AM. Complement C3 and C-reactive protein are elevated in South Asians independent of a family history of stroke. Stroke (2006) 37:2001–6. doi:10.1161/01.STR.0000231649.56080.6d
127. Zhang B, Yang N, Gao C. Is plasma C3 and C4 levels useful in young cerebral ischemic stroke patients? Associations with prognosis at 3 months. J Thromb Thrombolysis (2014) 39:209–14. doi:10.1007/s11239-014-1100-7
128. Zhang ZG, Wang C, Wang J, Zhang Z, Yang YL, Gao L, et al. Prognostic value of mannose-binding lectin: 90-day outcome in patients with acute ischemic stroke. Mol Neurobiol (2014) 51:230–9. doi:10.1007/s12035-014-8682-0
129. Schultz SJ, Aly H, Hasanen BM, Khashaba MT, Lear SC, Bendon RW, et al. Complement component 9 activation, consumption, and neuronal deposition in the post-hypoxic-ischemic central nervous system of human newborn infants. Neurosci Lett (2005) 378:1–6. doi:10.1016/j.neulet.2004.12.008
Keywords: complement, stroke, innate immunity, neuroprotection, brain ischemia, reperfusion injury
Citation: Alawieh A, Elvington A and Tomlinson S (2015) Complement in the homeostatic and ischemic brain. Front. Immunol. 6:417. doi: 10.3389/fimmu.2015.00417
Received: 26 March 2015; Accepted: 30 July 2015;
Published: 12 August 2015
Edited by:
Hans-Peter Hartung, Heinrich-Heine University Düsseldorf, GermanyReviewed by:
Tanja Kuhlmann, Universitätsklinikum Münster, GermanyCopyright: © 2015 Alawieh, Elvington and Tomlinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen Tomlinson, Department of Microbiology and Immunology, Medical University of South Carolina, 173 Ashley Avenue, BSB 203, MSC 504, Charleston, SC 29425, USA,dG9tbGluc3NAbXVzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.