 Nicole von Burg
Nicole von Burg Gleb Turchinovich1,2
Gleb Turchinovich1,2 Daniela Finke
Daniela Finke
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 13 August 2015
Sec. Immunological Memory
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00416
This article is part of the Research TopicInnate immune cell determinants of T cell immunity: from basic mechanisms to clinical implications.View all 14 articles
Innate lymphoid cells (ILCs) have emerged as a new family of immune cells with crucial functions in innate and adaptive immunity. ILC subsets mirror the cytokine and transcriptional profile of CD4+ T helper (TH) cell subsets. Hence, group 1 (ILC1), group 2 (ILC2), and group 3 (ILC3) ILCs can be distinguished by the production of TH1, TH2, and TH17-type cytokines, respectively. Cytokine release by ILCs not only shapes early innate immunity but can also orchestrate TH immune responses to microbial or allergen exposure. Recent studies have identified an unexpected effector function of ILCs as antigen presenting cells. Both ILC2s and ILC3s are able to process and present foreign antigens (Ags) via major histocompatibility complex class II, and to induce cognate CD4+ T cell responses. In addition, Ag-stimulated T cells promote ILC activation and effector functions indicating a reciprocal interaction between the adaptive and innate immune system. A fundamental puzzle in ILC function is how ILC/T cell interactions promote host protection and prevent autoimmune diseases. Furthermore, the way in which microenvironmental and inflammatory signals determine the outcome of ILC/T cell immune responses in various tissues is not yet understood. This review focuses on recent advances in understanding the mechanisms that coordinate the collaboration between ILCs and T cells under homeostatic and inflammatory conditions. We also discuss the potential roles of T cells and other immune cells to regulate ILC functions and to maintain homeostasis in mucosal tissues.
Adaptive immune responses are tightly controlled by the selection of the T and B cell receptor repertoire and by transcriptional networks regulating commitment, expansion, and contraction of the responses. Upon cognate antigen (Ag)–peptide–major histocompatibility complex (MHC) recognition Ag-specific T helper (TH) cells proliferate and differentiate into effector TH cell subsets with distinguishable cytokine profiles. Almost 30 years ago, interferon (IFN)-γ-secreting TH1 cells were discriminated from TH2 cells, whose cytokine profile includes interleukin (IL)-4, IL-5, and IL-13 (1). Additional subsets of TH cells, such as TH17 (2), regulatory T (Treg) cells (3), TH9 (4), T follicular helper cells (5), and more recently granulocyte-macrophage colony-stimulating factor (GM-CSF) producing TH cells (6–8), were described.
In the past 5 years, new subsets of innate immune cells have emerged as a first line of defense at mucosal barriers. Like conventional natural killer (cNK) cells, they belong to the lymphoid lineage and develop from common lymphoid progenitor (CLP) cells but unlike T and B cells, they lack rearranged Ag-receptors. Hence, they were termed innate lymphoid cells (ILCs). ILCs are found in various tissues including mucosa, lymphoid tissue, liver, skin, and fat. They depend on the expression of the common cytokine receptor γ chain (γc chain) and the transcriptional repressor inhibitor of DNA binding 2 (ID2) for their development (9–11). The factors involved in regulating different stages of ILC commitment from CLPs have been recently reviewed in Ref. (12). ILCs resemble TH cells in their developmental requirements, transcriptional regulation, and in their cytokine secretion pattern. Thus, they were classified into three groups, which are able to immediately react to microbial and inflammatory challenge with cytokine production thereby limiting pathogen spread and tissue injury (9). Group 1 ILCs consist of cNK cells and so-called helper ILC1s; both secrete the TH1-type cytokine IFN-γ. Group 2 ILCs are characterized by the production of TH2-type cytokines IL-4, IL-5, and/or IL-13. Group 3 ILCs include fetal lymphoid tissue-inducer (LTi) cells, as well as adult ILC3s either expressing the natural cytotoxicity receptor (NCR) NKp46 (NCR+ILC3s) or lacking this molecule (NCR−ILC3s). Cells within this group produce the TH17-type cytokines, IL-17 and/or IL-22 (9). The classification into ILC1, 2, and 3 is sometimes unhelpfully restrictive because ILCs have the potential to modulate their phenotypic and transcriptional signature upon activation and inflammation. When exposed to inflammatory conditions, NCR−ILC3s can produce IFNγ (13, 14), and NCR+ILC3s are able to convert into IFNγ-producing ILC1-like cells (15, 16). Moreover, in multiple sclerosis patients, blockade of CD25 (IL-2Rα) induces phenotypic changes of ILC3s toward cNK cells (17). Additional evidence for heterogeneity among ILC subsets comes from clonal analysis in humans demonstrating that the spectrum of cytokines produced by ILC3s is diverse (18) and in some cases, both ILC2 and ILC3 cytokines are produced (19). Finally, environmental factors, such as retinoic acid, short chain fatty acids, vitamins, aryl hydrocarbon receptor (AHR) ligands, stearyl sulfate, and probably bacterial metabolites, can shape ILC phenotypes and functions (20–24). Together, these data now provide convincing evidence that, similar to TH cells, ILCs have a degree of plasticity in their cytokine profile. As for TH cell commitment, cytokine-mediated conditioning, as well as epigenetic (25, 26) and transcriptional regulation (27) may account for changes of ILC subset-determining transcription factors and cytokines.
The biological relevance of ILCs is based on their capacity to sense environmental and inflammatory signals, and to respond with the secretion of cytokines important for immune defense, allergic reactions, and tissue repair. Recent data provide additional evidence that ILCs can condition T cell responses, either through cytokines, direct cell–cell contact, or through effects on accessory cells. This review will focus on the effects of ILC–T cell interactions for maintaining immune homeostasis. We will highlight major questions on how ILCs may cooperate with T cells thereby regulating T cell responses.
Dendritic cells (DCs) are professional Ag-presenting cells (APCs) known for their robust capacity to activate naïve T cells and to modulate innate and adaptive immune responses (28). Distinct DC subsets have decisive roles in engaging pathways responsible for skewing the type of effector TH cell response (29, 30). Moreover, DCs can suppress immune responses in order to maintain peripheral immune homeostasis and tolerance to self-Ags (31). As a key step in shaping the type of TH cell response, cytokines secreted by innate immune cells including APCs can account for the expression of TH subset-specific transcription factors (32). For example, IL-12 activates signal transducer and activator of transcription (STAT)-4 and induces the expression of the T-box transcription factor T-bet, which is critical for TH1 cell commitment (33, 34). T-bet expression and TH1 cell differentiation are further promoted by IL-2 (35). IL-4 induces STAT6 activation, which enhances Gata3 expression thereby initiating differentiation into TH2 cell lineage (36). Additionally, IL-2 signaling followed by STAT5 activation plays a crucial role in TH2 cell commitment by the induction of IL-4 transcription (37, 38). IL-6 signaling through STAT3, together with transforming growth factor (TGF)-β, induces retinoic acid-related orphan receptor (ROR)-γt expression and consequently the differentiation of pathogenic TH17 cells from naïve TH cells (39). A key issue in establishing immune homeostasis is the induction of Treg cells that prevent immunopathology by maintaining tolerance. In addition, active suppression of inappropriate T cell responses is mediated by the induction of immune-regulatory cytokines, such as IL-10 (40), the expression of inhibitory receptors including cytotoxic T-lymphocyte-associated protein (CTLA)-4 or programed cell death (PD)-1 or the lack of co-stimulation and bystander signals. Altogether, cytokines and activating or inhibiting receptors of innate immune cells are pivotal for generating and conditioning TH cell responses.
The group 1 ILCs comprised cNK cells and helper ILC1s. Both subsets secrete IFNγ and express the transcription factor T-bet (15, 16, 41–43). The expression of Eomesodermin (Eomes) is considered as a key factor for distinguishing cNK cells (Eomes+) from ILC1s (Eomes−) (43). However, splenic NK1.1+ CD127 (IL-7Rα)+ cells, which are in some studies referred to as ILC1s, express considerable levels of Eomes (44). Nfil3, another transcription factor, has been attributed a role in specifying cNK cells versus ILC1s. Although important for the development of all ILC lineages, studies of Nfil3-deficient mice (42, 45, 46) revealed that cNK cells have greater dependency on Nfil3 than ILC1s (47, 48). This is probably due to direct transcriptional control of Eomes expression by Nfil3 (49). Thus, NK cells resident in the salivary gland appear to be a prototype of ILC1s, as they also do not require Nfil3 for their development (48). Cells defined as ILC1s in the intestinal epithelium in humans and mice express the epithelial homing marker CD103 and readily produce IFNγ upon stimulation (41). CD103+ intraepithelial ILC1s, similar to cNK cells, express Eomes and T-bet, and are Nfil3-dependent, but in contrast to cNK cells do not require IL-15 for their development. Phenotypically, cNK cells express DX5 and, unlike most ILC1s, lack Trail or CD127 expression (43, 47, 48). Some ILC1-like cells derive from RORγt+ ILC3s by a process that is accompanied by the loss of RORγt expression and the upregulation of T-bet in both mice and humans (15, 16, 50). Future research on T-bet+ IFNγ-secreting subsets will help to clarify the developmental and functional relationship of group 1 ILCs.
Unlike group 2 and group 3 ILCs, murine cNK cells and ILC1s do not express MHC class II (MHC II) molecules, thus being incapable of direct Ag-dependent interaction with CD4+ TH cells (Table 1). Nevertheless, in recent years, a number of reports described new aspects of a direct crosstalk between T and cNK/ILC1 cells. Several studies defined a regulatory role for cNK cells in controlling T cell-dependent immune responses by direct cytotoxic activity toward CD4+ and CD8+ T cells (51–53), as well as toward APCs required for T cell priming. Two recent publications demonstrated that type 1 IFN confer the resistance to cNK cell-mediated lysis of activated CD8+ T cells (54, 55). CD8+ T cells isolated from IFN-α-receptor-1-deficient (Ifnar1−/−) mice were preferentially targeted by cNK cells resulting in the elimination of cytotoxic CD8+ T cells in response to viral infection through a perforin-dependent pathway. Another study proposed a role for NKp46 in limiting graft versus host disease (GVHD) (56), although it has remained obscure whether NKp46 is required for the direct killing of host-reactive T cells, or if it operates via targeting of accessory APCs. More recently, Schuster et al. reported that cNK cells specifically limit the number of virus-reactive CD4+ T cells in a model of chronic murine cytomegalovirus (MCMV) infection in the salivary gland (57). Intriguingly, this process is dependent on the TNF-superfamily ligand Trail, which is, in addition to NKp46 also expressed by ILC1s. This suggests a possible contribution of ILC1s to the processes described above. Additionally, in humans, activated cNK cells could be shown to positively regulate CD4+ TH cell activity (58). cNK cells stimulated by cytokines or through activating receptors were shown to upregulate the co-stimulatory molecules, OX40L and members of B7 family (CD80/CD86). Interaction with such cNK cells led to augmented IFNγ production and enhanced T cell receptor-dependent proliferation of autologous CD4+ TH cells.
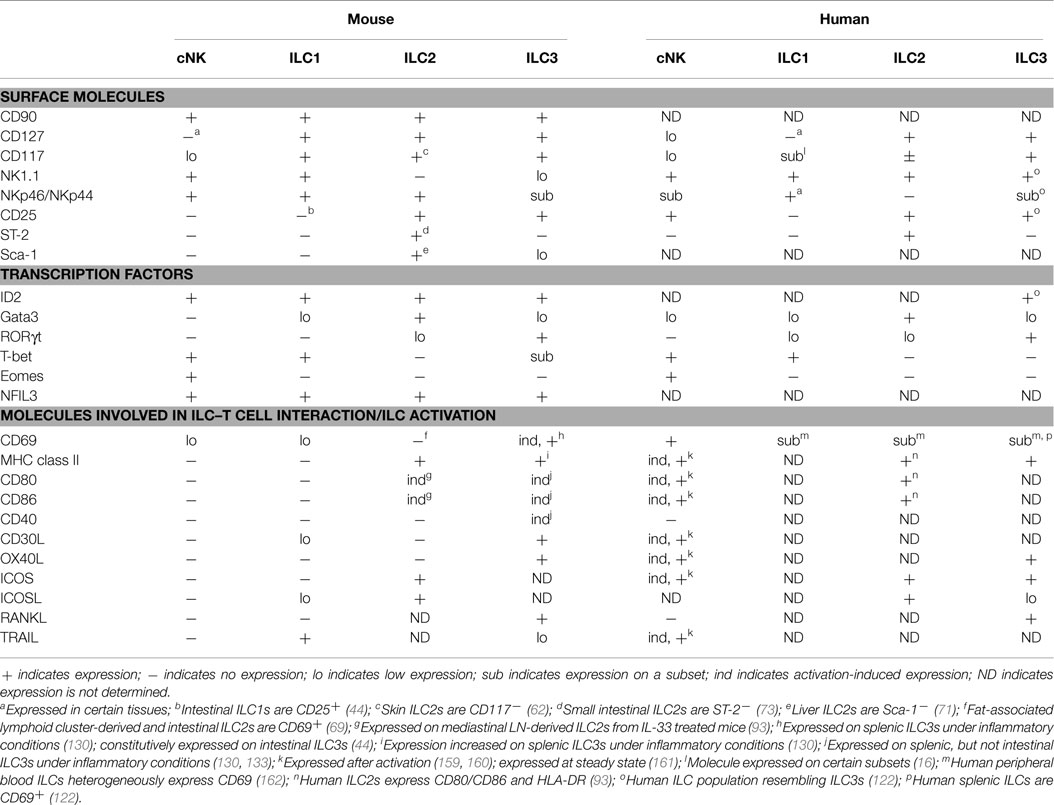
Table 1. Phenotype of mouse and human ILCs.
Conventional natural killer/ILC1 and T cell crosstalk operates in a reverse direction as well. Two studies showed that Treg cells play an important role in keeping cNK cell activity in check (59, 60). Gasteiger et al. demonstrated that upon depletion of Treg cells, cNK cells become hyper-responsive toward MHC I-deficient target cells that are recognized via missing-self mechanism. This was attributed to the increased availability of IL-2 produced by activated CD4+ T cells (59). Another report demonstrated in a genetic model of type 1 diabetes that the acute removal of Treg cells leads to the accumulation of activated cNK cells in pancreatic islets (60). On the contrary, in this experimental setting, depletion of Treg cells did not result in an increase of IL-2 secretion by CD4+ TH cells, but more likely increased the availability of IL-2 to cNK cells by decreasing IL-2 consumption by Treg cells. Interestingly, the accumulating cNK cells express CD127 (61) and might therefore constitute an “ILC1-like” subset. These studies provide the first example of Treg cell-dependent control of cNK cell and possibly ILC1 activity. Given the importance of IL-2 for the expansion of other ILC subsets (45, 62), Treg cells might also be involved in controlling their activity. Taken together, these findings illustrate the reciprocal immuno-regulatory relationship between group 1 ILCs and T cells.
ILC2s are the most homogenous ILC subset albeit with a specific phenotypic signature in the lung and intestine (44, 63). They express CD127, CD90.2 (Thy1), various levels of CD25, and the IL-33-receptor subunit ST2 (Table 1). The development of ILC2s depends on the transcription factors, ROR-α, Gata3, and T cell factor (TCF)-1 (64–67). ILC2s in both humans and mice secrete TH2-type cytokines IL-4, IL-5, and/or IL-13 in response to IL-9, IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), as well as during pulmonary inflammation or infection with Nippostrongylus brasiliensis, a helminth controlled by TH2-type cytokine responses (63, 68–78). In addition to ILC2s, another cell type, the multipotent progenitor type 2 (MPPtype2) is described. MPPtype2 cells exhibit similar phenotypic and functional characteristics with ILC2s (79), but do not produce TH2-type cytokines in response to IL-33 (80). The release of TH2-type cytokines by ILC2s is not only involved in N. brasiliensis expulsion (81) but can also trigger airway inflammation and allergic responses in humans (82–84). Together, ILC2s share developmental and inducible cytokine signatures with TH2 cells suggesting a role in type 2 immune responses.
Type 2 immune responses are severely impaired in IL-4-receptor-α-deficient (Il4Rα−/−) and IL-4-deficient (Il4−/−) mice indicating that IL-4 has a role in TH2 cell differentiation (85, 86). Further, the accumulation of TH2 cells after N. brasiliensis/ovalbumin challenge is dramatically reduced in IL-4 and IL-13-double-deficient (Il4−/−Il13−/−) mice as compared to wild type (WT) mice (87). TH2 cell differentiation is most likely initiated by innate immune cells, which become activated in the early phase of immune responses. Beside basophils and mast cells (88–90), it is now well established that ILC2s can secrete IL-4 suggesting a role for these cells in the induction of TH2 cell differentiation and type 2 immune responses. Indeed, several reports provide evidence that ILC2s and CD4+ T cells cooperate at multiple levels (91–97). In mice, which either have dramatically reduced numbers or a complete lack of ILC2s, the generation of type 2 immune responses upon N. brasiliensis infection, challenge with house dust mite Ag or with protease-allergen papain is impaired indicating a contribution of ILC2s to TH2 cell responses (91, 93, 95). The addition of ILC2s to cultures of naïve CD4+ T cells promotes the differentiation into TH2 cells, while inhibiting the differentiation into TH1 cells even in the presence of IL-12, a cytokine that drives TH1 differentiation (33, 34, 92). In line with this finding, type 2 cytokines are not detectable when TH cells are co-cultured with ILC2s unable to secrete IL-4 (94). On the other hand, in vivo differentiation of TH1/TH17 cells occurs independently of ILC2s, since mice, which lack ILC2s, show normal responses when exposed to Saccharopolyspora rectivirgula, a bacterium inducing TH1/TH17 inflammatory responses (95). Together, there is evidence that ILC2-derived IL-4 contributes to type 2 cytokine production of TH cells, although an IL-4-independent pathway for ILC2-driven type 2 immune responses may also occur (91). Beside the direct effect of ILC2s on TH2 differentiation, TH2-type cytokines secreted by ILC2s can also affect CD4+ T cells indirectly via DCs. Evidence for this comes from the finding that ILC2-derived IL-13 promotes migration of DCs into lung-draining lymph nodes (LNs), where activated DCs induce the differentiation of CD4+ T cells into TH2 cells (91).
Interleukin-33, a pro-inflammatory cytokine expressed by a variety of cell types can trigger the generation of inducible regulatory T (iTreg) cells (98) and the activation of ILC2s to produce type 2 cytokines and amphiregulin (AREG). AREG is an epithelial growth factor that promotes restoration of airway epithelial integrity following influenza virus-induced damage (63). Importantly, analysis of ILC2-depleted, influenza virus-infected mice revealed a strong reduction in AREG mRNA suggesting that ILC2s are the main source of AREG under such inflammatory conditions. In other inflammatory models, mast cells were thought to be the major source of AREG and importantly, in these models, AREG was found to be critical for efficient Treg cell function (99). In view of their abundance in the skin, lung, and colon, their strong responsiveness to IL-33, and early inflammatory signals, AREG-secreting ILC2s may have a function in tissue repair and likely also in triggering Treg cell responses.
Another mechanism through which ILC2s have an influence on CD4+ TH cells is by their ability to serve as APCs. Co-stimulatory signals via OX40 are crucial for effector/memory T cell responses and for initiating TH2 differentiation (100, 101). OX40-ligand (OX40L) is detectable on ILC2s, and the production of TH2-type cytokines in ILC2-T cell co-cultures is significantly inhibited when anti-OX40L antibodies (Abs) are added, suggesting that ILC2s promote TH2-responses via OX40/OX40L interactions (94). Further evidence for cell–cell interactions between ILC2s and CD4+ T cells is provided by the finding that human and mouse ILC2s express both inducible T cell co-stimulator (ICOS) and ICOS-ligand (ICOSL) (70, 102), a co-stimulatory receptor/ligand pair known for its function for survival, proliferation, and cytokine secretion of TH cell subsets (103). Moreover, ILC2s can process Ags and present peptides on MHC II. They express the co-stimulatory molecules, CD80 and CD86, and induce proliferation of TH2 cells, albeit to a lesser extent than professional APCs (92, 93). Interestingly, the expression of MHC II is higher on LN-, spleen-, and Peyer’s Patch (PP)-derived ILC2s than on peritoneal lavage-, bronchoalveolar lavage-, and lung-derived ILC2s. Therefore, lymphoid tissue-specific factors might be responsible for sustained MHC II expression.
Together with the finding that ILC2s can express MHC II and co-stimulatory molecules, the direct ILC2–T cell interaction not only promotes TH responses but also extends to cytokine-mediated help from activated TH cells for ILC2 effector functions. During the acute phase of N. brasiliensis infection, Rag2-deficient (Rag2−/−) mice show a similar expansion of ILC2s as WT mice. However, adaptive immune cells are required for prolonged ILC2 expansion and complete clearance of the infection (70). In a papain-induced inflammation model, IL-9 production by ILC2s is severely reduced in Rag2−/− mice suggesting that cytokine secretion by ILC2s is also dependent on the adaptive immune system (68). In vitro co-culture of CD4+ T cells and ILC2s results in the upregulation of IL-4 mRNA in ILC2s, suggesting that TH cells induce type 2 cytokine production by ILC2s (94). Additionally, activated CD4+ T cells in co-culture with ILC2s can directly induce ILC2 proliferation and IL-5/IL-13 secretion (92). This effect is partially impaired by adding anti-IL-2-neutralizing Abs but not by separating CD4+ T cells from ILC2s in transwell assays, suggesting an IL-2-driven feedback mechanism from activated CD4+ T cells to ILC2s (92). In line with this, treatment of mice with IL-2/anti-IL-2 complexes results in increased in vivo proliferation of ILC2s (62) and expansion of ILC2 progenitors in the bone marrow (BM) (45). IL-2 can also promote IL-9 release by ILC2s, whereas IL-33 induces the upregulation of the IL-2-receptor subunit CD25 on ILC2s (104). The induction of CD25 expression may help ILC2s to become more sensitive to T cell-derived IL-2. It is currently unclear to what extent ILC2s and Treg cells, which express high levels of CD25, or other TH subsets, compete for IL-2. Hence, the expression of CD25 by ILC2s may also reduce the availability of IL-2 for T cells. Based on these observations, we propose the following model (Figure 1): ILC2s can be rapidly activated by various alarm signals leading to the release of TH2-type cytokines, which help to induce TH2 cell responses and DC migration into LNs toward T cell zones. Further, activated ILC2s secrete AREG, and it remains to be investigated whether this can trigger Treg cell responses. The cognate interaction between ILC2s and CD4+ T cells via MHC II–Ag presentation, co-stimulatory signals, and cytokines helps to amplify both ILC2 and CD4+ T cell responses.
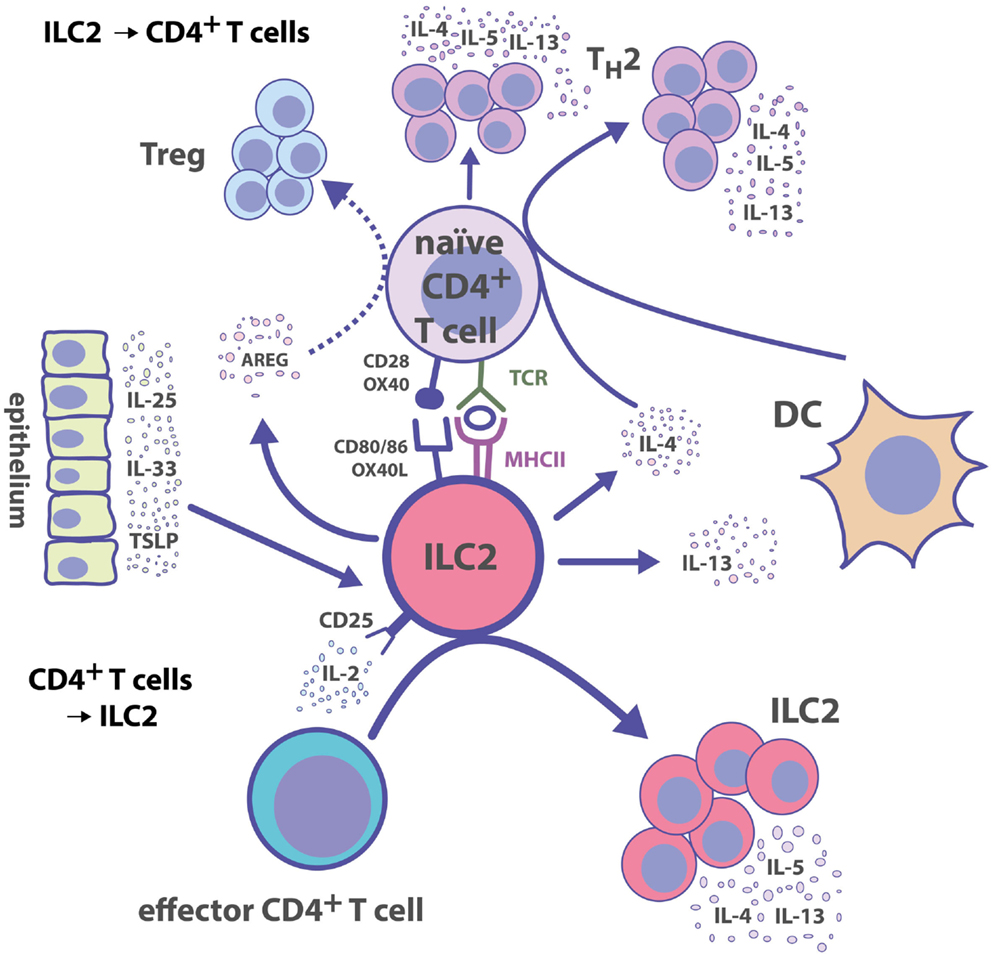
Figure 1. Group 2 ILC–CD4+ T cell interactions. ILC2s polarize CD4+ T cell responses toward TH2 immunity directly by presenting cognate Ag and by secreting TH2-inducing cytokines. Reciprocally, activated CD4+ T cells produce IL-2, which serves as a growth factor leading to the expansion of ILC2s.
All ILC3 subsets depend on the transcription factor RORγt for their development (105–107), and produce the TH17-type cytokine IL-22 (107–111). IL-22 has a major role in protecting intestinal epithelial cells from bacterial infections and in promoting tissue repair through induction of epithelial cell proliferation and production of antimicrobial peptides (112). Group 3 ILCs can be phenotypically classified into a subset of fetal RORγt+ CD127+ CD117+ LTi cells (106, 113–116), and adult NCR+ or NCR−RORγt+ ILC3s (107, 108, 111, 117).
ILC3s can modulate TH cell immune responses in several ways. One pathway involves the development of lymphoid tissue and T cell zone stroma. Already before birth, the cellular crosstalk of fetal lymphotoxin (LT)α1β2-expressing LTi cells with mesenchymal stromal cells (MSCs) plays a pivotal role in the formation of LNs and PPs, in which immune responses are generated. Adult ILC3s retain the capacity to induce lymphoid tissue formation (118, 119). Following lymphocytic choriomeningitis virus (LCMV) infection in mice, the crosstalk between LTα1β2-expressing ILC3s and T cell zone fibroblastic reticular cells helps to restore the disrupted T-zone compartment and hence the structure to generate proper immune responses (120). Similarly, LTα1β2+ ILC3s can restore lymphoid follicle organization in the colon of mice infected with Citrobacter rodentium (121). The interaction of ILC3s with MSCs is also reciprocal. In humans, the crosstalk between LTα1β2+ ILC3s and marginal reticular cells (MRCs), a subset of marginal zone stromal cells, induces the production of MRC-derived survival factors for ILC3s, such as IL-7 (122). A second pathway, by which ILC3s can modulate TH cell immune responses, is through altering the recruitment of CD4+ TH cells. ILC3s are able to release soluble LTα3, which promotes the homing of CD4+ TH cells to the gut lamina propria where they differentiate into functional TH cell subsets (Figure 2) (123). In a model of airway inflammation, ILC3-derived IL-22 reduces CCL17 production by epithelial cells thereby limiting TH2 cell recruitment and immune responses to allergens in the lung (124). These data show that ILC3s have an impact on generating functional T cell compartments and recruitment of CD4+ TH cells to mucosal sites.
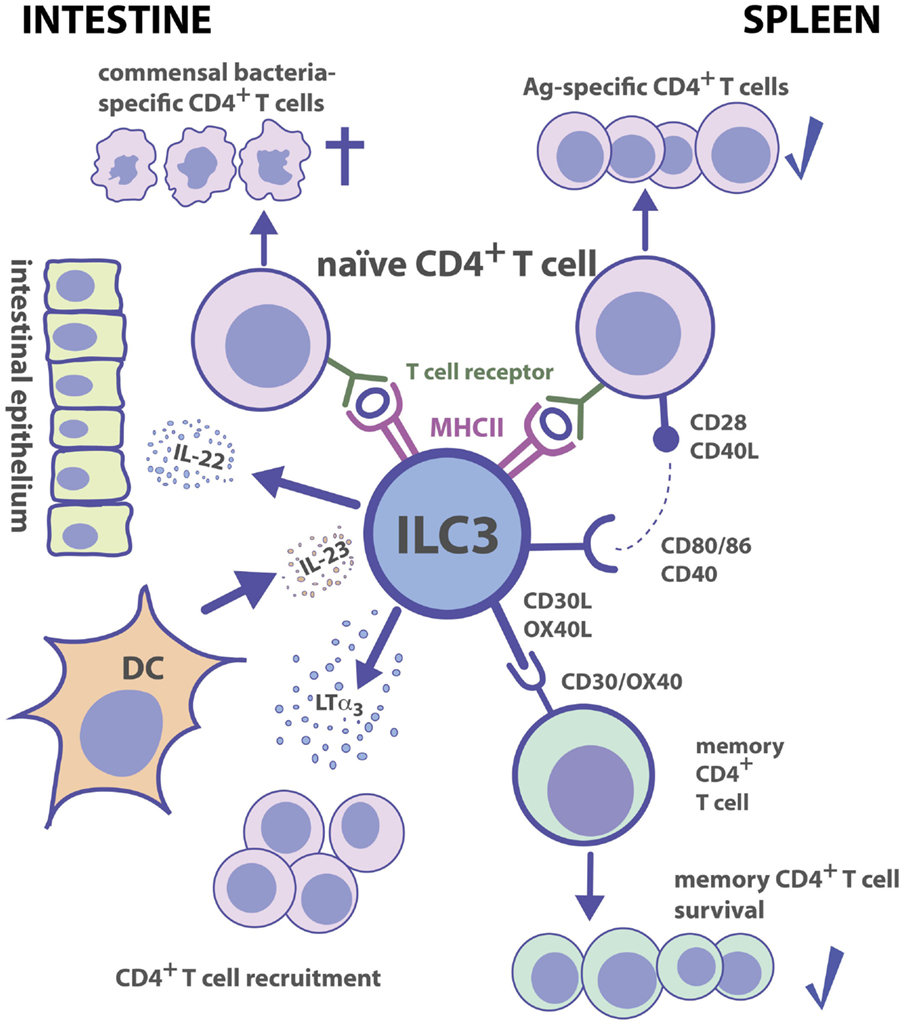
Figure 2. Group 3 ILC–CD4+ T cell interactions. Tissue localization greatly affects the outcome of Ag-dependent T cell–ILC3 interaction. Intestinal ILC3s maintain tolerance toward commensal microbiota, while splenic ILC3s are efficient in the induction of Ag-specific CD4+ T cell responses and memory CD4+ T cell survival.
In the adult spleen, ILC3s are localized in the marginal zone and around the central arterioles, and in LNs in proximity to high endothelial venules and interfollicular areas (122, 125–127). Because of the close association of splenic ILC3s to Ag-entry sites and T cells as well as their expression of the co-stimulatory molecules, CD30-ligand (CD30L) and OX40L, it has been assumed that they may directly interact with T cells during adaptive immune responses (125). Mice with a deficiency in CD30 and OX40 (CD30−/−OX40−/− mice) lack proper memory Ab responses due to a failure in survival of primed CD4+ TH cells (128). In vitro, ILC3s can promote survival of memory CD4+ TH cells from WT but not from CD30−/−OX40−/− mice suggesting that both CD30L and OX40L molecules expressed by ILC3s are essential for CD4+ TH memory responses (128). This possibility was supported by an in vivo study, which identified ILC3s as the key players in the maintenance of CD4+ memory TH cells (Figure 2) (129).
A third mechanism by which ILC3s interact with CD4+ TH cells is through receptors required for immune recognition. ILC3s isolated from various tissues of fetal, neonatal, and adult mice express MHC II and MHC II-associated gene transcripts (44, 113, 130–132). NCR−ILC3s are able to internalize, process, and present foreign Ags to CD4+ TH cells (130, 131). Under non-inflammatory conditions, ILC3s express neither CD40 and CD80 nor CD86 (130, 131). However, following stimulation with IL-1β splenic but not intestinal, NCR−ILC3s can upregulate co-stimulatory molecules (130). A recent study confirmed that even after toll-like receptor ligand (TLRL) or pro-inflammatory cytokine exposure, intestinal ILC3s do not upregulate co-stimulatory molecules (133). The finding that mLN-derived ILC3s are as well unable to express co-stimulatory molecules upon stimulation is likely due to the fact that ILC3s found in the mLNs are originally intestinal ILC3s, which were trafficking from the intestine to the mLNs (127). It is noteworthy that genome-wide transcriptional profiling of splenic ILC3s reveals an enrichment for genes involved in cell activation and immune responses (63). In contrast to splenic ILC3s, intestinal ILC3s express the activation marker, CD69 (44), a glycoprotein involved in establishing oral tolerance (134) and limiting dextran sodium sulfate (DSS)-induced inflammation (135). Moreover, ILC3s present in the small intestine express neuropilin-1 (Nrp1) (44), which promotes Treg cell survival and functional activity (136–138). It is therefore conceivable that ILC3s exert tissue-specific immune functions with immunogenic versus tolerogenic activity in the spleen and intestine, respectively. This hypothesis is further supported by the notion that splenic NCR−ILC3s promote CD4+ TH cell responses in vitro and in vivo, whereas intestinal ILC3s fail to efficiently stimulate CD4+ TH cells (Figure 2) (130). In mice, intestinal ILC3s express lower levels of MHC II as compared to ILC3s identified in other tissues (130, 131, 133). Together with the observation that intestinal ILC3s lack co-stimulatory molecules, this may contribute to maintaining intestinal T cell tolerance, similar to immature DCs expressing low surface levels of MHC II and co-stimulatory molecules (139).
Hepworth et al. reported the development of spontaneous intestinal inflammation in mice lacking MHC II exclusively on ILC3s (ILC3ΔMHCII mice) and found a role for intestinal ILC3s in limiting commensal bacteria-specific pro-inflammatory colonic CD4+ TH cell responses through induction of PD (131, 133). Since other laboratories failed to detect spontaneous signs of inflammation in ILC3ΔMHCII mice (130, 132), it is possible that the development of immunopathology is triggered by microbial co-factors. In the intestine, ILC3s can inhibit TH17 cell-mediated inflammation through AHR signaling, release of IL-22, and by preventing the expansion of aberrant segmented filamentous bacteria (SFB) (140). In pediatric Crohn’s disease (CD) patients, MHC II levels on intestinal ILC3s are significantly reduced, and such low expression correlates with increased frequencies of colonic TH17 cells and circulating commensal bacteria-specific IgG (133). This study is the first to describe an association of ILC3-mediated Ag presentation and control of commensal bacteria-specific adaptive immunity in humans. It remains unclear which are the mechanisms that underlie loss of MHC II in CD patients and whether this is sufficient to trigger inflammatory bowel disease. Together, these findings suggest that intestinal ILC3s can inhibit expansion of TH17 cells and immunopathology after exposure to pro-inflammatory stimuli.
Analogously to ILC2–T cell interactions, the crosstalk between ILC3s and CD4+ TH cells might be bidirectional and depends on cytokines. This is further supported by the findings that the presence of the adaptive immune system has an effect on the number and IL-22 production of intestinal ILC3s, most likely through competition for growth factors (141, 142). Human and activated mouse ILC3s produce IL-2 (19, 130), and conversely, TLR2-driven proliferation of human ILC3s is partially dependent on IL-2 (19). Availability of IL-2 alone or in combination with Pam3Cys promotes increased CD25 expression in human ILC3s suggesting that CD25 expression might help ILC3s to win the competition for IL-2 against T cells (19). Moreover, there is some evidence that mouse ILC3s have a higher capacity to bind IL-2 than activated CD4+ TH cells (133). Therefore, the availability of IL-2 can restrict ILC3 and TH responses as a result of receptor density, efficiency of binding, and kinetics of IL-2 consumption.
The critical question regarding maintenance of immune homeostasis is where, when, and how immune responses prevent tissue injury. The intestine is a prime example that has been extensively studied with respect to cellular networks and pathways patrolling tissue integrity and regulating inflammation. Treg and TH17 cells are the most abundant CD4+ TH cells in the intestinal mucosa under steady state (143–145). The balance between the two subsets is crucial for the outcome of mucosal immune responses (146). Commensal bacteria have a specific impact on the number of both TH subsets (147) and on the capacity of ILC3s to regulate TH subset responses (148). On the other hand, ILC3s contribute to maintenance of intestinal epithelial barrier function thereby limiting microbes entry and inflammatory TH cell responses (108, 109, 117, 141, 148). Whereas under steady-state conditions, intestinal ILC3s produce high levels of IL-22, the production of IL-17 is rather low (44). Importantly, TH17 cells are induced by SFB (149, 150) by a mechanism that requires SFB presentation by DCs (132, 151). In contrast, ILC3 presentation of Ag prevents amplification of SFB-independent TH17 cells (132). In line with this, the expansion of SFB and pathogenic TH17 cells inversely correlates with the number of intestinal ILC3s (140). In an IL-17-dependent autoimmune mouse model, it was recently shown that SFB colonization was associated with enhanced auto-Ab titers (152). The increase in IL-17-producing cells, as observed in CD patients (153), is probably not sufficient per se to induce immunopathology. Specificity of inflammatory TH cells, intestinal infections, pro-inflammatory bystander cells, and loss of functional Treg cells might be required to trigger intestinal inflammation.
All these studies published in recent years raised the question of whether and how ILC–T cell interactions regulate pro- or anti-inflammatory responses in the gut. Since ILC3s can prevent dissemination of commensal bacteria in the gut and commensal bacteria-specific TH cell responses (123, 131, 132, 148), they probably promote an immunological tolerogenic state in the gut. In addition, the production of GM-CSF by ILC3s has the potential to enhance iTreg cell numbers and function thereby promoting intestinal homeostasis (154). In some colitis models, however, ILC3s were reported to enhance intestinal inflammation (13, 15), and pathogenic ILC1 numbers were increased in patients with CD (16, 41). The functional polarization toward IFNγ-producing ILC1s or IL-22-producing ILC3s appears to depend on tissue-specific and pro-inflammatory conditions. Environmental changes may immediately affect the ratio and/or polarization of ILC and T cell subsets. For example, induction of pro-inflammatory cytokines, such as IL-23, was shown to counteract the responsiveness toward IL-33, and the generation of iTregs in the intestine (98). As for TH cell differentiation, it is likely that the amount of cytokines determines ILC cytokine polarization. Under homeostatic conditions, the intestine provides a microenvironment enriched of cytokines with inhibitory effects, such as TGF-β. At high dose, TGF-β inhibits TH17 responses, whereas low-dose TGF-β promotes TH17-differentiation (155–157). A similar impact of cytokine concentrations for immune homeostasis has also been discussed for IL-22 (158). Therefore, excessive release of cytokines by ILCs may contribute to immunopathology, whereas under steady-state conditions, ILCs rather promote epithelial tissue integrity and tolerogenic T cell responses. During inflammation, ILC3s can switch off RORγt expression, which may eventually be regained at later time points. The modulation of cytokine receptors during a critical time window of ILC activation and ILC-T cell interaction might also contribute to prevent excessive immunopathology. This has been shown for a number of receptors controlling growth and survival of both ILCs and T cells. Finally, the polarization toward protective versus inflammatory response in the gut likely requires a tight balance between temporal regulation, amount, and combination of cytokines co-expressed by individual ILCs.
Our understanding of immune homeostasis has been challenged by the notion that environmental factors, including commensal bacteria and nutritional components, as well as cholinergic and metabolic signals can regulate immune functions and pro-inflammatory processes. ILCs are important “early sentinel” cells, which connect innate and adaptive immunity by sensing environmental changes, such as infections and inflammation and by the release of immuno-regulatory cytokines. They not only contribute to T cell immune homeostasis by promoting TH cell differentiation and effector functions but can also directly interact with CD4+ TH cells. Both ILC2s and ILC3s internalize and present Ag to TH cells. Considering the fact that the number of ILCs in most tissues is rather low as compared to other immune cells, they appear to have a surprising in vivo impact on immune homeostasis. The localization of ILCs in relatively high density at Ag-entry sites and T cell areas as well as bystander effects on classical DCs might explain this effect. In addition, advances in two-photon microscopy have shown that several CD4+ TH cells are often clustering with the same APC, a fact that may increase local cytokine concentrations for optimal cell–cell interactions. The capacity to elicit cognate TH cell proliferation or rather prevent TH cell responses strongly depends on environmental factors and the nature of Ag, and it will be important to further investigate the mechanisms by which ILCs prevent or promote T cell responses in various tissues. For example, it will be interesting to unravel whether ILCs can express inhibitory receptors and/or collaborate with Treg cells. Finally, there are clearly cytokine-driven reciprocal effects between ILCs and T cells, which might help to coordinate and/or limit immune responses. Taken together, a better understanding of the regulation of cytokine expression by ILCs and their interaction with T cells will help to develop new strategies to treat inflammatory diseases in humans.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors sincerely apologize to all colleagues whose work has been omitted due to space limitations. We would like to thank A. Baerenwaldt, T. Rolink, E. Palmer, G. de Libero, and all members of the lab for comments on the manuscript. DF is supported by the Swiss National Science Foundation (SNF) grant 310030-153247/1. NB is supported by the Horton foundation. GT is supported by the Basel University Research Fund Junior Researcher.
1. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136(7):2348–57.
2. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol (1995) 155(12):5483–6.
3. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
4. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol (2008) 9(12):1341–6. doi: 10.1038/ni.1659
5. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192(11):1545–52. doi:10.1084/jem.192.11.1545
6. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12(6):568–75. doi:10.1038/ni.2031
7. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol (2011) 12(6):560–7. doi:10.1038/ni.2027
8. Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res (2014) 24(12):1387–402. doi:10.1038/cr.2014.154
9. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi:10.1038/nri3365
10. Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature (1999) 397(6721):702–6. doi:10.1038/17812
11. Boos MD, Yokota Y, Eberl G, Kee BL. Mature natural killer cell and lymphoid tissue-inducing cell development requires Id2-mediated suppression of E protein activity. J Exp Med (2007) 204(5):1119–30. doi:10.1084/jem.20061959
12. Lim AW, McKenzie AN. Deciphering the transcriptional switches of innate lymphoid cell programming: the right factors at the right time. Genes Immun (2015) 16(3):177–86. doi:10.1038/gene.2014.83
13. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464(7293):1371–5. doi:10.1038/nature08949
14. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature (2013) 494(7436):261–5. doi:10.1038/nature11813
15. Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity (2010) 33(5):736–51. doi:10.1016/j.immuni.2010.10.017
16. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14(3):221–9. doi:10.1038/ni.2534
17. Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med (2012) 4(145):145ra106. doi:10.1126/scitranslmed.3004140
18. Crellin NK, Trifari S, Kaplan CD, Cupedo T, Spits H. Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. J Exp Med (2010) 207(2):281–90. doi:10.1084/jem.20091509
19. Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, Di Santo JP, Spits H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by toll-like receptor 2. Immunity (2010) 33(5):752–64. doi:10.1016/j.immuni.2010.10.012
20. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science (2014) 343(6169):432–7. doi:10.1126/science.1247606
21. van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature (2014) 508(7494):123–7. doi:10.1038/nature13158
22. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334(6062):1561–5. doi:10.1126/science.1214914
23. Hu X, Wang Y, Hao LY, Liu X, Lesch CA, Sanchez BM, et al. Sterol metabolism controls T(H)17 differentiation by generating endogenous RORgamma agonists. Nat Chem Biol (2015) 11(2):141–7. doi:10.1038/nchembio.1714
24. Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity (2012) 36(1):92–104. doi:10.1016/j.immuni.2011.11.011
25. Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity (2009) 30(5):646–55. doi:10.1016/j.immuni.2009.05.001
26. Shih HY, Sciumè G, Poholek AC, Vahedi G, Hirahara K, Villarino AV, et al. Transcriptional and epigenetic networks of helper T and innate lymphoid cells. Immunol Rev (2014) 261(1):23–49. doi:10.1111/imr.12208
27. Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol (2010) 11(8):674–80. doi:10.1038/ni.1899
28. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med (1973) 137(5):1142–62. doi:10.1084/jem.137.5.1142
29. Maldonado-López R, De Smedt T, Michel P, Godfroid J, Pajak B, Heirman C, et al. CD8alpha+ and CD8alpha- subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med (1999) 189(3):587–92. doi:10.1084/jem.189.3.587
30. Feili-Hariri M, Falkner DH, Morel PA. Polarization of naive T cells into Th1 or Th2 by distinct cytokine-driven murine dendritic cell populations: implications for immunotherapy. J Leukoc Biol (2005) 78(3):656–64. doi:10.1189/jlb.1104631
31. Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol (2010) 11(8):647–55. doi:10.1038/ni.1894
32. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
33. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100(6):655–69. doi:10.1016/S0092-8674(00)80702-3
34. Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature (1996) 382(6587):171–4. doi:10.1038/382171a0
35. Liao W, Lin JX, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nat Immunol (2011) 12(6):551–9. doi:10.1038/ni.2030
36. Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity (1996) 4(3):313–9. doi:10.1016/S1074-7613(00)80439-2
37. Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, et al. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci U S A (2004) 101(11):3880–5. doi:10.1073/pnas.0400339101
38. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity (2003) 19(5):739–48. doi:10.1016/S1074-7613(03)00292-9
39. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi:10.1038/nature04753
40. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. doi:10.1146/annurev.immunol.19.1.683
41. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity (2013) 38(4):769–81. doi:10.1016/j.immuni.2013.02.010
42. Klose CS, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157(2):340–56. doi:10.1016/j.cell.2014.03.030
43. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med (2014) 211(3):563–77. doi:10.1084/jem.20131560
44. Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol (2015) 16(3):306–17. doi:10.1038/ni.3094
45. Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med (2014) 211(9):1733–40. doi:10.1084/jem.20140145
46. Geiger TL, Abt MC, Gasteiger G, Firth MA, O’Connor MH, Geary CD, et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med (2014) 211(9):1723–31. doi:10.1084/jem.20140212
47. Seillet C, Huntington ND, Gangatirkar P, Axelsson E, Minnich M, Brady HJ, et al. Differential requirement for Nfil3 during NK cell development. J Immunol (2014) 192(6):2667–76. doi:10.4049/jimmunol.1302605
48. Cortez VS, Fuchs A, Cella M, Gilfillan S, Colonna M. Cutting edge: salivary gland NK cells develop independently of Nfil3 in steady-state. J Immunol (2014) 192(10):4487–91. doi:10.4049/jimmunol.1303469
49. Male V, Nisoli I, Kostrzewski T, Allan DS, Carlyle JR, Lord GM, et al. The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med (2014) 211(4):635–42. doi:10.1084/jem.20132398
50. Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, et al. The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat Immunol (2013) 14(4):389–95. doi:10.1038/ni.2545
51. Lu L, Ikizawa K, Hu D, Werneck MB, Wucherpfennig KW, Cantor H. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity (2007) 26(5):593–604. doi:10.1016/j.immuni.2007.03.017
52. Soderquest K, Walzer T, Zafirova B, Klavinskis LS, Polić B, Vivier E, et al. Cutting edge: CD8+ T cell priming in the absence of NK cells leads to enhanced memory responses. J Immunol (2011) 186(6):3304–8. doi:10.4049/jimmunol.1004122
53. Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature (2012) 481(7381):394–8. doi:10.1038/nature10624
54. Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, et al. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity (2014) 40(6):961–73. doi:10.1016/j.immuni.2014.05.003
55. Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, et al. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity (2014) 40(6):949–60. doi:10.1016/j.immuni.2014.05.004
56. Ghadially H, Ohana M, Elboim M, Gazit R, Gur C, Nagler A, et al. NK cell receptor NKp46 regulates graft-versus-host disease. Cell Rep (2014) 7(6):1809–14. doi:10.1016/j.celrep.2014.05.011
57. Schuster IS, Wikstrom ME, Brizard G, Coudert JD, Estcourt MJ, Manzur M, et al. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity (2014) 41(4):646–56. doi:10.1016/j.immuni.2014.09.013
58. Zingoni A, Sornasse T, Cocks BG, Tanaka Y, Santoni A, Lanier LL. Cross-talk between activated human NK cells and CD4+ T cells via OX40-OX40 ligand interactions. J Immunol (2004) 173(6):3716–24. doi:10.4049/jimmunol.173.6.3716
59. Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med (2013) 210(6):1167–78. doi:10.1084/jem.20122462
60. Sitrin J, Ring A, Garcia KC, Benoist C, Mathis D. Regulatory T cells control NK cells in an insulitic lesion by depriving them of IL-2. J Exp Med (2013) 210(6):1153–65. doi:10.1084/jem.20122248
61. Gasteiger G, Hemmers S, Bos PD, Sun JC, Rudensky AY. IL-2-dependent adaptive control of NK cell homeostasis. J Exp Med (2013) 210(6):1179–87. doi:10.1084/jem.20122571
62. Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol (2013) 14(6):564–73. doi:10.1038/ni.2584
63. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12(11):1045–54. doi:10.1031/ni.2131
64. Wong SH, Walker JA, Jolin HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol (2012) 13(3):229–36. doi:10.1038/ni.2208
65. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity (2012) 37(4):634–48. doi:10.1016/j.immuni.2012.06.020
66. Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity (2012) 37(3):463–74. doi:10.1016/j.immuni.2012.06.012
67. Yang Q, Monticelli LA, Saenz SA, Chi AW, Sonnenberg GF, Tang J, et al. T cell factor 1 is required for group 2 innate lymphoid cell generation. Immunity (2013) 38(4):694–704. doi:10.1016/j.immuni.2012.12.003
68. Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat Immunol (2011) 12(11):1071–7. doi:10.1038/ni.2133
69. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature (2010) 463(7280):540–4. doi:10.1038/nature08636
70. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464(7293):1367–70. doi:10.1038/nature08900
71. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A (2010) 107(25):11489–94. doi:10.1073/pnas.1003988107
72. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013) 502(7470):245–8. doi:10.1038/nature12526
73. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity (2012) 36(3):451–63. doi:10.1016/j.immuni.2011.12.020
74. Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol (2011) 12(11):1055–62. doi:10.1038/ni.2104
75. Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol (2002) 169(1):443–53. doi:10.4049/jimmunol.169.1.443
76. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol (2011) 12(7):631–8. doi:10.1038/ni.2045
77. Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med (2013) 5(170):170ra116. doi:10.1126/scitranslmed.3005374
78. Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci U S A (2012) 109(9):3451–6. doi:10.1073/pnas.1201042109
79. Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF Jr, Tocker JE, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature (2010) 464(7293):1362–6. doi:10.1038/nature08901
80. Saenz SA, Siracusa MC, Monticelli LA, Ziegler CG, Kim BS, Brestoff JR, et al. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J Exp Med (2013) 210(9):1823–37. doi:10.1084/jem.20122332
81. Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med (2006) 203(4):1105–16. doi:10.1084/jem.20051615
82. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol (2012) 129(1):.e1–4. doi:10.1016/j.jaci.2011.09.041
83. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol (2012) 188(3):1503–13. doi:10.4049/jimmunol.1102832
84. Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol (2012) 129(1):.e1–6. doi:10.1016/j.jaci.2011.10.036
85. Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Köhler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature (1993) 362(6417):245–8. doi:10.1038/362245a0
86. Noben-Trauth N, Shultz LD, Brombacher F, Urban JF Jr, Gu H, Paul WE. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc Natl Acad Sci U S A (1997) 94(20):10838–43. doi:10.1073/pnas.94.20.10838
87. Voehringer D, Reese TA, Huang X, Shinkai K, Locksley RM. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J Exp Med (2006) 203(6):1435–46. doi:10.1084/jem.20052448
88. Perrigoue JG, Saenz SA, Siracusa MC, Allenspach EJ, Taylor BC, Giacomin PR, et al. MHC class II-dependent basophil-CD4+ T cell interactions promote T(H)2 cytokine-dependent immunity. Nat Immunol (2009) 10(7):697–705. doi:10.1038/ni.1740
89. Sokol CL, Chu NQ, Yu S, Nish SA, Laufer TM, Medzhitov R. Basophils function as antigen-presenting cells for an allergen-induced T helper type 2 response. Nat Immunol (2009) 10(7):713–20. doi:10.1038/ni.1738
90. Yoshimoto T, Yasuda K, Tanaka H, Nakahira M, Imai Y, Fujimori Y, et al. Basophils contribute to T(H)2-IgE responses in vivo via IL-4 production and presentation of peptide-MHC class II complexes to CD4+ T cells. Nat Immunol (2009) 10(7):706–12. doi:10.1038/ni.1737
91. Halim TY, Steer CA, Mathä L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity (2014) 40(3):425–35. doi:10.1016/j.immuni.2014.01.011
92. Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol (2014) 192(5):2442–8. doi:10.4049/jimmunol.1300974
93. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41(2):283–95. doi:10.1016/j.immuni.2014.06.016
94. Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy (2014) 69(10):1300–7. doi:10.1111/all.12446
95. Gold MJ, Antignano F, Halim TY, Hirota JA, Blanchet MR, Zaph C, et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol (2014) 133(4):1142–8. doi:10.1016/j.jaci.2014.02.033
96. Liu B, Lee JB, Chen CY, Hershey GK, Wang YH. Collaborative interactions between type 2 innate lymphoid cells and antigen-specific CD4+ Th2 cells exacerbate murine allergic airway diseases with prominent eosinophilia. J Immunol (2015) 194(8):3583–93. doi:10.4049/jimmunol.1400951
97. Bouchery T, Kyle R, Camberis M, Shepherd A, Filbey K, Smith A, et al. ILC2s and T cells cooperate to ensure maintenance of M2 macrophages for lung immunity against hookworms. Nat Commun (2015) 6:6970. doi:10.1038/ncomms7970
98. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513(7519):564–8. doi:10.1038/nature13577
99. Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Gröne A, Sibilia M, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity (2013) 38(2):275–84. doi:10.1016/j.immuni.2012.09.023
100. Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev (2009) 229(1):173–91. doi:10.1111/j.1600-065X.2009.00766.x
101. So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc Natl Acad Sci U S A (2006) 103(10):3740–5. doi:10.1073/pnas.0600205103
102. Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity (2015) 42(3):538–51. doi:10.1016/j.immuni.2015.02.007
103. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi:10.1038/16717
104. Wilhelm C, Stockinger B. Innate lymphoid cells and type 2 (th2) mediated immune responses – pathogenic or beneficial? Front Immunol (2011) 2:68. doi:10.3389/fimmu.2011.00068
105. Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science (2000) 288(5475):2369–73. doi:10.1126/science.288.5475.2369
106. Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol (2004) 5(1):64–73. doi:10.1038/ni1022
107. Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol (2009) 10(1):83–91. doi:10.1038/ni.1684
108. Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity (2008) 29(6):958–70. doi:10.1016/j.immuni.2008.11.001
109. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity (2011) 34(1):122–34. doi:10.1016/j.immuni.2010.12.009
110. Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med (2009) 206(1):35–41. doi:10.1084/jem.20072713
111. Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol (2009) 10(1):75–82. doi:10.1038/ni.1681
112. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol (2011) 29:71–109. doi:10.1146/annurev-immunol-031210-101312
113. Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity (1997) 7(4):493–504. doi:10.1016/S1074-7613(00)80371-4
114. Adachi S, Yoshida H, Kataoka H, Nishikawa S. Three distinctive steps in Peyer’s patch formation of murine embryo. Int Immunol (1997) 9(4):507–14. doi:10.1093/intimm/9.4.507
115. Finke D, Acha-Orbea H, Mattis A, Lipp M, Kraehenbuhl J. CD4+CD3- cells induce Peyer’s patch development: role of alpha4beta1 integrin activation by CXCR5. Immunity (2002) 17(3):363–73. doi:10.1016/S1074-7613(02)00395-3
116. Yoshida H, Honda K, Shinkura R, Adachi S, Nishikawa S, Maki K, et al. IL-7 receptor alpha+ CD3(-) cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int Immunol (1999) 11(5):643–55. doi:10.1093/intimm/11.5.643
117. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature (2009) 457(7230):722–5. doi:10.1038/nature07537
118. Schmutz S, Bosco N, Chappaz S, Boyman O, Acha-Orbea H, Ceredig R, et al. Cutting edge: IL-7 regulates the peripheral pool of adult ROR gamma+ lymphoid tissue inducer cells. J Immunol (2009) 183(4):2217–21. doi:10.4049/jimmunol.0802911
119. Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity (2008) 29(2):261–71. doi:10.1016/j.immuni.2008.05.014
120. Scandella E, Bolinger B, Lattmann E, Miller S, Favre S, Littman DR, et al. Restoration of lymphoid organ integrity through the interaction of lymphoid tissue-inducer cells with stroma of the T cell zone. Nat Immunol (2008) 9(6):667–75. doi:10.1038/ni.1605
121. Ota N, Wong K, Valdez PA, Zheng Y, Crellin NK, Diehl L, et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat Immunol (2011) 12(10):941–8. doi:10.1038/ni.2089
122. Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat Immunol (2014) 15(4):354–64. doi:10.1038/ni.2830
123. Kruglov AA, Grivennikov SI, Kuprash DV, Winsauer C, Prepens S, Seleznik GM, et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science (2013) 342(6163):1243–6. doi:10.1126/science.1243364
124. Taube C, Tertilt C, Gyülveszi G, Dehzad N, Kreymborg K, Schneeweiss K, et al. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PLoS One (2011) 6(7):e21799. doi:10.1371/journal.pone.0021799
125. Kim MY, Gaspal FM, Wiggett HE, McConnell FM, Gulbranson-Judge A, Raykundalia C, et al. CD4(+)CD3(-) accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where T cells collaborate with B cells. Immunity (2003) 18(5):643–54. doi:10.1016/S1074-7613(03)00110-9
126. Kim MY, McConnell FM, Gaspal FM, White A, Glanville SH, Bekiaris V, et al. Function of CD4+CD3- cells in relation to B- and T-zone stroma in spleen. Blood (2007) 109(4):1602–10. doi:10.1182/blood-2006-04-018465
127. Mackley EC, Houston S, Marriott CL, Halford EE, Lucas B, Cerovic V, et al. CCR7-dependent trafficking of RORgamma(+) ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat Commun (2015) 6:5862. doi:10.1038/ncomms6862
128. Gaspal FM, Kim MY, McConnell FM, Raykundalia C, Bekiaris V, Lane PJ. Mice deficient in OX40 and CD30 signals lack memory antibody responses because of deficient CD4 T cell memory. J Immunol (2005) 174(7):3891–6. doi:10.4049/jimmunol.174.7.3891
129. Withers DR, Gaspal FM, Mackley EC, Marriott CL, Ross EA, Desanti GE, et al. Cutting edge: lymphoid tissue inducer cells maintain memory CD4 T cells within secondary lymphoid tissue. J Immunol (2012) 189(5):2094–8. doi:10.4049/jimmunol.1201639
130. von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc Natl Acad Sci U S A (2014) 111(35):12835–40. doi:10.1073/pnas.1406908111
131. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature (2013) 498(7452):113–7. doi:10.1038/nature12240
132. Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity (2014) 40(4):594–607. doi:10.1016/j.immuni.2014.03.005
133. Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science (2015) 348(6238):1031–5. doi:10.1126/science.aaa4812
134. Radulovic K, Manta C, Rossini V, Holzmann K, Kestler HA, Wegenka UM, et al. CD69 regulates type I IFN-induced tolerogenic signals to mucosal CD4 T cells that attenuate their colitogenic potential. J Immunol (2012) 188(4):2001–13. doi:10.4049/jimmunol.1100765
135. Radulovic K, Rossini V, Manta C, Holzmann K, Kestler HA, Niess JH. The early activation marker CD69 regulates the expression of chemokines and CD4 T cell accumulation in intestine. PLoS One (2013) 8(6):e65413. doi:10.1371/journal.pone.0065413
136. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med (2012) 209(10):S1–19. doi:10.1084/jem.20120822
137. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med (2012) 209(10):1723–42,S1721. doi:10.1084/jem.20120914
138. Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature (2013) 501(7466):252–6. doi:10.1038/nature12428
139. Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A (2002) 99(1):351–8. doi:10.1073/pnas.231606698
140. Qiu J, Guo X, Chen ZM, He L, Sonnenberg GF, Artis D, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity (2013) 39(2):386–99. doi:10.1016/j.immuni.2013.08.002
141. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Bérard M, Kleinschek M, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol (2011) 12(4):320–6. doi:10.1038/ni.2002
142. Korn LL, Thomas HL, Hubbeling HG, Spencer SP, Sinha R, Simkins HM, et al. Conventional CD4+ T cells regulate IL-22-producing intestinal innate lymphoid cells. Mucosal Immunol (2014) 7(5):1045–57. doi:10.1038/mi.2013.121
143. Ivanov II, de Frutos RL, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe (2008) 4(4):337–49. doi:10.1016/j.chom.2008.09.009
144. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi:10.1016/j.cell.2006.07.035
145. Denning TL, Norris BA, Medina-Contreras O, Manicassamy S, Geem D, Madan R, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol (2011) 187(2):733–47. doi:10.4049/jimmunol.1002701
146. Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol (2012) 30:759–95. doi:10.1146/annurev-immunol-020711-074937
147. Ivanov II, Honda K. Intestinal commensal microbes as immune modulators. Cell Host Microbe (2012) 12(4):496–508. doi:10.1016/j.chom.2012.09.009
148. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science (2012) 336(6086):1321–5. doi:10.1126/science.1222551
149. Gaboriau-Routhiau V, Rakotobe S, Lécuyer E, Mulder I, Lan A, Bridonneau C, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity (2009) 31(4):677–89. doi:10.1016/j.immuni.2009.08.020
150. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139(3):485–98. doi:10.1016/j.cell.2009.09.033
151. Lécuyer E, Rakotobe S, Lengliné-Garnier H, Lebreton C, Picard M, Juste C, et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity (2014) 40(4):608–20. doi:10.1016/j.immuni.2014.03.009
152. Van Praet JT, Donovan E, Vanassche I, Drennan MB, Windels F, Dendooven A, et al. Commensal microbiota influence systemic autoimmune responses. EMBO J (2015) 34(4):466–74. doi:10.15252/embj.201489966
153. Geremia A, Arancibia-Cárcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi:10.1084/jem.20101712
154. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science (2014) 343(6178):1249288. doi:10.1126/science.1249288
155. Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature (2008) 454(7202):350–2. doi:10.1038/nature07021
156. Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol (2008) 9(6):650–7. doi:10.1038/ni.1613
157. Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol (2008) 9(6):641–9. doi:10.1038/ni.1610
158. Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity (2009) 31(1):15–23. doi:10.1016/j.immuni.2009.06.008
159. Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med (1998) 188(12):2375–80. doi:10.1084/jem.188.12.2375
160. Spits H, Lanier LL. Natural killer or dendritic: what’s in a name? Immunity (2007) 26(1):11–6. doi:10.1016/j.immuni.2007.01.004
161. Kashii Y, Giorda R, Herberman RB, Whiteside TL, Vujanovic NL. Constitutive expression and role of the TNF family ligands in apoptotic killing of tumor cells by human NK cells. J Immunol (1999) 163(10):5358–66.
Keywords: innate lymphoid cell, cytokine, T helper cell, immune response, antigen presentation
Citation: von Burg N, Turchinovich G and Finke D (2015) Maintenance of immune homeostasis through ILC/T cell interactions. Front. Immunol. 6:416. doi: 10.3389/fimmu.2015.00416
Received: 29 May 2015; Accepted: 29 July 2015;
Published: 13 August 2015
Edited by:
Stefan F. Martin, University of Freiburg, GermanyReviewed by:
Peter John Lane, Birmingham University, UKCopyright: © 2015 von Burg, Turchinovich and Finke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Finke, Department of Biomedicine, University of Basel Children’s Hospital, Mattenstrasse 28, Basel 4058, Switzerland,ZGFuaWVsYS5maW5rZUB1bmliYXMuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.