 Zachary Shriver
Zachary Shriver Jose M. Trevejo
Jose M. Trevejo Ram Sasisekharan
Ram Sasisekharan- 1Visterra Inc., Cambridge, MA, USA
- 2Department of Biological Engineering, Koch Institute of Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA
- 3Infectious Diseases Interdisciplinary Research Group, Singapore-MIT Alliance for Research and Technology, Singapore, Singapore
Passive immunization using antibodies is a promising alternative to other antiviral treatment options. The potential for seasonal protection arising from a single injection of antibodies is appealing and has been pursued for a number of infectious agents. However, until recently, antibody-based strategies to combat infectious agents have been hampered due to the fact that most antibodies have been found to be strain specific, with the virus evolving resistance in many cases. The discovery of broadly neutralizing antibodies (bNAbs) in influenza, dengue virus, and HIV, which bind to multiple, structurally diverse strains, has provided renewed interest in this area. This review will focus on new technologies that enable the discovery of bNAbs, the challenges and opportunities of immunotherapies as an important addition to existing antiviral therapy, and the role of antibody discovery in informing rational vaccine discovery – with agents targeting influenza specifically addressed. Multiple candidates have entered the clinic and raise the possibility that a single antibody or small combination of antibodies can effectively neutralize a wide variety of strains. However, challenges remain – including combating escape variants, pharmacodynamics of antibody distribution, and development of efficacy biomarkers beyond virologic endpoints.
Introduction
New alternative countermeasures for influenza are urgently needed. Vaccines to seasonal and pandemic influenza are foundational to provide widespread herd immunity to influenza. However, most inactivated and live-attenuated vaccine technologies are strain specific – requiring constant updating of the strains used in yearly multivalent vaccine preparations. In addition, severe influenza disease occurs each season in many high risk groups, to whom the vaccine provides limited or no protection, such as young children, the elderly, patients that are immunocompromised or who have pulmonary conditions, inflammatory conditions, or malignancies, as well as pregnant women (1, 2). In addition to vaccines, current treatment and prophylaxis of influenza is limited to the neuraminidase inhibitors oseltamivir (Tamiflu) and zanamivir (Relenza). Despite the availability of these treatments, 10–44% of hospitalized patients require intensive care and 25–50% of these patients die. In the United States, it is estimated that over 200,000 patients are hospitalized with influenza, with up to 48,000 deaths, per year (3). A comparison of annual mortality rates from infectious disease in the United States further demonstrates the lack of effective interventions against this deadly disease (Table S1 in Supplementary Material).
Furthermore, certain strains of influenza have resulted in infections with high mortality rates: the 1918 H1N1 Spanish Flu strain resulted in deaths of 1–3% of the world’s population, compared to the 1968 pandemic strain that resulted in the death of 0.03% of the world’s population (4). More recently, Avian H5 strains have had documented mortality rates up to 60% despite the use of currently licensed anti-viral treatments (5). Finally, the continued emergence of resistance to current anti-viral drugs increases the need for new therapeutics (6).
Passive Immunotherapy has a Long History
Prior to the advent of antibiotics, convalescent serum was the only antidote available for bacterial toxins, such as diphtheria and tetanus (7). Eventually, it was discovered that the protective properties within the serum were predominantly due to neutralizing antibodies. The therapeutic use of passive antibodies has been well established for several viral infections. There are licensed polyclonal antibody products for several viruses, including hepatitis B (HBIG), varicella (VZIG), cytomegalovirus (Cytogam), rabies (HRIG), and respiratory syncytial virus (RSV) (Respigam). More recently, monoclonal antibodies (mAbs) for viral infections have been developed, including licensure of Synagis for prevention of RSV infection. mAbs for the prevention and treatment of a number of other viral targets, including Hepatitis C, Rabies, and West Nile Virus, are in clinical development (8–11). Historically, these antibody products have primarily focused on the prevention of viral disease, although application to treatment of infection has been investigated for RSV (12). Of note, no monoclonal antibody-based solution has been approved for the treatment of active infection.
In the absence of development of a universal, broadly protective vaccine, passive immunization using antibodies potentially offers several benefits. First, passive immunity provides the opportunity to protect at-risk individuals from infection. At-risk segments of the population include first responders to a relatively novel strain as well as those who do not mount an immune response to vaccines including the immunocompromised, those in poor health, pregnant women, and critically ill patients. Indeed, recent modeling analyses completed by us and our collaborators (M. Boni, Oxford University Clinical Research Unit) indicate that for a sufficiently potent and long-lasting antibody (t1/2 = ~18 days, protective period = 3 half-lives), population coverage of only 4–6% would be required to significantly reduce hospitalization rates. Notably, given the current state-of-the-art in the production of antibodies, it is possible to readily generate enough monoclonal antibody to protect the population at the epicenter of an epidemic outbreak in a much shorter time scale than that of vaccine production (>6 months) (13). The potential for long-lasting protection, covering an entire exposure period (i.e., an entire season for influenza), arising from a single injection of antibodies is appealing and has been pursued for a number of infectious agents. For example, in the case of hepatitis A, prophylactic administration of immunoglobulins can protect against viral exposure. Additionally, post-exposure prophylaxis with immunoglobulin is >85% effective in preventing hepatitis A if administered within 2 weeks after viral exposure, and efficacy is even higher when administered early in the incubation period (14).
The use of antibody therapy to treat influenza has recently received more attention, with some clinical experience to support efficacy. A meta-analysis of studies conducted during the 1918 pandemic using blood products strongly supports a benefit for treated patients (15). Overall, the six studies documented a 21% reduced risk of mortality in treatment groups (16 vs. 37% mortality in controls, 95% of risk difference, CI: 15–27). Furthermore, a recent study evaluated the use of convalescent plasma in 93 patients with H1N1 2009 influenza in Hong Kong (16). In this prospective multi-center case-control study, patients with severe influenza, who were hospitalized and required intensive care unit support, were recruited and offered convalescent plasma containing influenza neutralizing antibody in addition to the standard of care with either oseltamivir or zanamivir. Mortality was significantly lower in the treatment groups who received convalescent plasma compared to the controls (20.0 vs. 54.8%, p = 0.01).
Given the overall promise (and current limitations) of passive immunization approaches, as well as the overarching goal of accounting for viral mutations, it is highly desirable to identify and/or engineer antibodies that bind with high affinity and that neutralize many or all strains that are capable of infecting humans. To this end, there is intense focus on antibodies that bind to influenza hemagglutinin (HA) since antibodies to HA are known to be protective in animal models and in humans. The challenge is that HA is a highly diverse protein; there are 18 subtypes categorized into two groups – 1 and 2. As such, the ability to identify an antibody or small collection of antibodies that can bind to and neutralize all clinically relevant strains is a substantial challenge. The ability to rapidly identify broadly neutralizing antibodies (bNAbs) has been enhanced by the development of several new high-throughput technologies that now promise to enable comprehensive tracking of all of the immunological cell subsets, extending even to the level of the individual clones of B cells that carry out adaptive responses (17, 18). Improvements in the toolkit for human immunological studies are continually evolving and are likely to increase our understanding/discovery of antibodies for therapeutic use.
Characterization of the overall B-cell response to infection or vaccination has provided potentially important insights into lasting immunity, including the heterogeneous nature of individualistic responses to vaccination/infection. However, with next-generation deep sequencing data, it has become clear that, in different individuals, expansion of B-cell clones in response to infection arising from similar or “convergent” antibody gene rearrangements can be detected. For example, tracking of B-cell clones following pandemic single antigen H1N1 vaccination revealed a strong clonal signature dominated by antibodies using the IGHV3-7V gene, and correspondence of highly similar CDR3 sequences in different humans. Convergent monoclonal antibody sequences display HA inhibition activity against H1N1 and other influenza strains (19). This raises the possibility of a so-called “universal” vaccine strategy-through selection of the appropriate immunogen(s) to elicit the most effective immune response. Other recent work using regions of or chimeric proteins derived from the stem region of HA seem to elicit a broadly protective response against multiple subtypes of influenza (20, 21). Furthermore, studies in the area of HIV have suggested that a broadly protective response may, in principle, be completed through eliciting specific B-cell responses (i.e., “training the immune system”) using temporally spaced immunization with different antigens (22).
In addition to the use of the above tools to study the adaptive immune response, there has recently been a concerted effort to identify, engineer, and characterize antibodies that bind to a number of influenza subtypes (so-called “broadly” neutralizing antibodies). Several of these antibodies are listed in Table 1. These broadly neutralizing mAbs are a new, promising modality for treatment of influenza, potentially across all strains of the virus. Such antibodies have been identified through panning the B-cell repertoire of vaccinated or infected individuals (23, 24) and are estimated to be ~0.001–0.01% of the total antibody response (23). Identification of such antibodies has generated interest for several reasons, including (i) mapping of the epitopes to which these antibodies bind provides insights into epitopes that can be targeted for vaccine development and (ii) the antibodies by themselves are useful products to provide passive immunization or therapeutic efficacy against a wide variety of influenza subtypes.
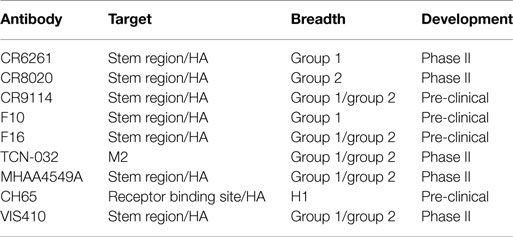
Table 1. Recent discoveries in broadly neutralizing antibodies to influenza.
Study of bNAbs has Led to Insight on Universal Vaccine Targets
In the context of vaccine efforts, identification of bNAbs to infectious agents provides a basis for the design of more protective vaccine strategies (25). Recent work on the evolution of bNAbs containing the Vh1-69 heavy chain demonstrates that somatic mutations to the germline, which result in recognition of a hydrophobic patch on the HA stem results in the antibodies becoming more hydrophobic and binding influenza HA with higher avidity (26). Another recent study characterized ~200 anti-stem antibodies and identified two key elements that are required for the initial development of most VH1-69 antibodies: a polymorphic germline-encoded phenylalanine at position 54 and a conserved tyrosine at position 98 in the third complementary determining region of the heavy chain (27). By tracing the development of such antibodies, these studies have demonstrated that it may be possible to develop an immunofocusing strategy to promote the production of bNAbs containing Vh1-69.
Properties of bNAbs
Antibodies to two major surface antigens, the M ion channel and HA, have been studied as potential passive immunotherapies (Table 1). Antibodies that target the highly conserved M2 protein possess the requisite breadth of binding across group 1 and group 2 (i.e., all subtypes of influenza A). However, such antibodies are non-neutralizing; the predominant mechanism of action for M2-specific antibody is indirect through ADCC-mediated killing of infected cells. This leads to incomplete protection. For example, in a lethal influenza mouse model; an M2-targeted antibody product required three injections with M2-specific antibodies at 24, 72, and 120 h post-infection and still provided only a partial (~60%) protective response (28).
In contrast, antibodies to HA can clearly neutralize influenza virus in vitro, provide complete protection after a single administration in vivo, and protect against multiple strains of influenza (24, 29, 30). Additionally, use of such antibodies likely also mitigates bacterial secondary infection, since rapid reduction in viral titers prevents bacterial adherence (31). These data are supported by the fact that the efficacy of current vaccine approaches to influenza (especially inactivated virus strategies) is measured by the HA neutralizing titer. However, through the processes of antigenic drift and shift, the HA of influenza virus can develop resistance to antibodies that target HA. Such has been the case with, for example, CR6261 (32), CR8020 (24), and F10 (33), where several mutations are known to lower the binding affinity of the antibody to HA and confer resistance. The fact that HA is subject to mutation and the virus can undergo reassortment questions whether an immunotherapy strategy can be adequately developed due to facile development of escape mutants.
Strategies to Design Effective Immunotherapy
There are at least two points that need to be considered with regard to an ideal immunotherapeutic strategy, particularly when considering a variable system like HA. The first is the epitope targeted by the antibody. Many of the bNAbs in Table 1 target the relatively conserved stem region of HA. While certain stem-binding-antibodies target epitopes that can mutate under selective pressure with apparently little or no fitness cost, other epitopes are less amenable to mutation and are more likely to engender a fitness cost (34). A structure-based network approach (35) can be used to provide insights into the tolerance to mutability of an amino acid in a protein system. This approach is based on analysis of sequences across different viral surface proteins that reveal amino acids that are highly networked (higher weighted contacts with neighboring amino acids), and therefore are more constrained in their ability to mutate (Figure 1A). By targeting these amino acids, it is possible to generate an antibody-based solution that is more refractory to resistance development while still maintaining binding to a potent and broadly neutralizing epitope. Additionally, in the context of therapy, it is likely that any antibody can be used in combination with a neuraminidase inhibitor, where there appears to be synergistic activities due to distinct mechanisms of action (36). Furthermore, as has become apparent in other viral diseases, such as HIV or hepatitis C, combination approaches are less likely to elicit resistance. Finally, recent studies have indicated that the activity of bNAbs is enhanced in the presence of the natural immune response (37).
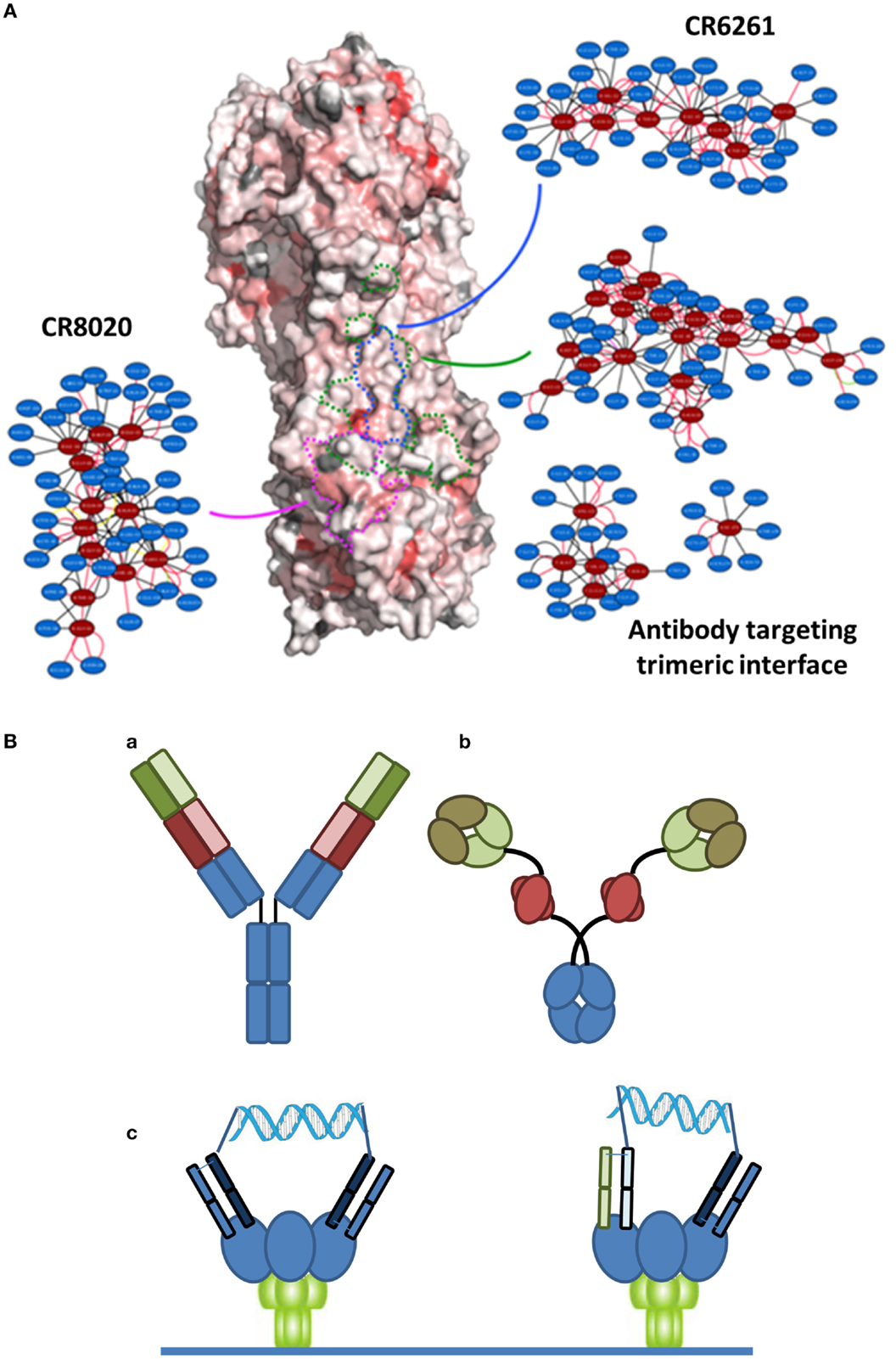
Figure 1. (A) Network-view of bNAb epitopes. HA trimer is represented in a solvent accessible surface format and colored based on normalized residue network scores. Coloring varies from white to red where white indicates poorly networked residues and red indicates highly networked residues. The three bNAb epitopes are highlighted by dotted borderlines (green: antibody targeting trimeric interface; blue: CR6261; pink: CR8020). The 2D network map of the epitope is also shown. A network is made up of nodes and edges. Nodes colored in red indicate functional epitope residues whereas nodes colored in blue indicate residues that are in the network environment of the epitope residues. (B) Different bispecific formats that have demonstrated activity against infectious disease targets. (a) A dual-variable domain immunoglobulin format containing two distinct Vh-Vl pairings (one in red and one in green) has demonstrated activity against hepatitis B. (b) A bispecific format where a single chain variable region against Psl (red) targets the antibody to the cell surface of Pseudomonas enables engagement of a traditional Vh-Vl paratope with the rarer PcrV target. (c) Crosslinking of binding domains of variable and constant regions (VH-CH1/VL-CL; Fabs), either homotypic (left) or heterotypic (right) with a defined DNA-based spacer enables more potent neutralization of HIV virus.
The second important point to an ideal strategy is that there are a number of engineering-based strategies outside of the variable or binding region that can be used to enhance the efficacy of the antibody-based solutions. Certainly, one of the relevant approaches is Fc engineering to enhance the recruitment of complement and/or innate immune cells. In the context of bNAbs against influenza, it is known that the various stem-binding antibodies are able to recruit complement and that Fc effector functions are critical to their protective effect (38). However, the efficiency of complement recruitment is based on the geometry of engagement, with some antibodies being able to better engage complement as compared to others (39). The effector functions can be further enhanced through engineering of Fc mutations and/or alteration of the glycosylation site to enhance ADCC. Finally, in other therapeutic areas, particularly oncology, there has been an emphasis on discovery of bispecific antibody formats (40) (Figure 1B). In the context of antibodies to infectious agents, initial examples have provided intriguing results. Alternative formats have been investigated including use of multiple antibody binding domains (VH-VL), or inclusion of antibody-like binding domains, such as scFv or Fab fragments. Recently, data have been reported for a bispecific antibody-like construct to Pseudomonas where one binding site binds to a high density ligand (Psl) and thus targets the antibody, and the other binding site targets a highly neutralizing epitope (PcrV) (41). Furthermore, with reference to viral diseases, a recent report of a bispecific antibody to hepatitis B reported synergistic activity compared to the activity of the parent antibodies alone (42). Finally, in HIV, where the density of the gp140 spike protein at the viral surface is highly limiting, bridging through the use of a bispecific antibody resulted in much higher activity (43). Such a dual-targeting strategy may also be useful for other viruses such as influenza.
Conclusion
If technologies can identify high affinity, bNAbs, passive immunization can likely provide an important adjunctive prophylactic and therapeutic option to supplement vaccination technologies. Antibody-based therapies are generally safe and well-tolerated, particularly when the antigen is an exogenous target. Even one of the more common effects of therapy, which is the development of anti-drug antibodies, at most serves to limit drug exposure rather than resulting in significant adverse effects. Recent maturation of several tools in antibody characterization, discovery, and engineering may enable a resurgence of passive immunotherapy strategies. With several antibody candidates that are currently in clinical development for influenza (Table 1) and potentially others, it is likely that we will determine in the near future whether an old idea becomes a new powerful tool to counteract the rapidly evolving threat of influenza and other virus infections.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded in part by National Institutes of Health Award (1R01AI111395), National Institutes of Health Merit Award (R37 GM057073-13), and National Research Foundation supported Interdisciplinary Research group in Infectious Diseases of SMART (Singapore MIT alliance for Research and Technology).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2015.00315
References
1. Treanor JJ, Hayden FG, Vrooman PS, Barbarash R, Bettis R, Riff D, et al. Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomized controlled trial. US Oral Neuraminidase Study Group. JAMA (2000) 283:1016–24. doi:10.1001/jama.283.8.1016
2. Schanzer DL, Langley JM, Tam TW. Co-morbidities associated with influenza-attributed mortality, 1994-2000, Canada. Vaccine (2008) 26:4697–703. doi:10.1016/j.vaccine.2008.06.087
3. Centers for Disease Control and Prevention. Estimates of deaths associated with seasonal influenza US, 1976-2007. MMWR (2012) 59:1057–62.
4. Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis (2012) 12:687–95. doi:10.1016/S1473-3099(12)70121-4
5. Writing Committee of the Second World Health Organization Consultation on Clinical Aspects of Human Infection with Avian InfluenzaAbdel-Ghafar AN, Chotpitayasunondh T, Gao Z, Hayden FG, Nguyen DH, et al. Update on avian influenza A (H5N1) virus infection in humans. N Engl J Med (2008) 358:261–73. doi:10.1056/NEJMra0707279
6. Hayden FG, de Jong MD. Emerging influenza antiviral resistance threats. J Infect Dis (2011) 203:6–10. doi:10.1093/infdis/jiq012
7. Saylor C, Dadachova E, Casadevall A. Monoclonal antibody-based therapies for microbial diseases. Vaccine (2009) 27(Suppl 6):G38–46. doi:10.1016/j.vaccine.2009.09.105
8. Beigel JH, Nordstrom JL, Pillemer SR, Roncal C, Goldwater DR, Li H, et al. Safety and pharmacokinetics of single intravenous dose of MGAWN1, a novel monoclonal antibody to West Nile virus. Antimicrob Agents Chemother (2010) 54:2431–6. doi:10.1128/AAC.01178-09
9. Babcock GJ, Iyer S, Smith HL, Wang Y, Rowley K, Ambrosino DM, et al. High-throughput sequencing analysis of post-liver transplantation HCV E2 glycoprotein evolution in the presence and absence of neutralizing monoclonal antibody. PLoS One (2014) 23:e100325. doi:10.1371/journal.pone.0100325
10. Leav B, Kulkarni P, Gogtay N, Ambrosino DA. Human monoclonal to rabies virus provides protective neutralizing activity: results of a Phase I study. In: ICAAC. Boston (2010).
11. Chung RT, Gordon FD, Curry MP, Schiano TD, Emre S, Corey K, et al. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. Am J Transplant (2013) 13:1047–54. doi:10.1111/ajt.12083
12. Malley R, DeVincenzo J, Ramilo O, Dennehy PH, Meissner HC, Gruber WC, et al. Reduction of respiratory syncytial virus (RSV) in tracheal aspirates in intubated infants by use of humanized monoclonal antibody to RSV F protein. J Infect Dis (1998) 178:1555–61. doi:10.1086/314523
13. Lambert LC, Fauci AS. Influenza vaccines for the future. N Engl J Med (2010) 363:2036–44. doi:10.1056/NEJMra1002842
14. Winokur PL, Stapleton JT. Immunoglobulin prophylaxis for hepatitis A. Clin Infect Dis (1992) 14:580–6. doi:10.1093/clinids/14.2.580
15. Luke TC, Kilbane EM, Jackson JL, Hoffman SL. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann Intern Med (2006) 145:599–609. doi:10.7326/0003-4819-145-8-200610170-00139
16. Hung IF, To KK, Lee CK, Lee KL, Chan K, Yan WW, et al. Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clin Infect Dis (2011) 52:447–56. doi:10.1093/cid/ciq106
17. DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD, et al. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat Med (2015) 21:86–91. doi:10.1038/nm.3743
18. Laserson U, Vigneault F, Gadala-Maria D, Yaari G, Uduman M, Vander Heiden JA, et al. High-resolution antibody dynamics of vaccine-induced immune responses. Proc Natl Acad Sci U S A (2014) 111:4928–33. doi:10.1073/pnas.1323862111
19. Jackson KJ, Liu Y, Roskin KM, Glanville J, Hoh RA, Seo K, et al. Human responses to influenza vaccination show seroconversion signatures and convergent antibody rearrangements. Cell Host Microbe (2014) 16:105–14. doi:10.1016/j.chom.2014.05.013
20. Mallajosyula VV, Citron M, Ferrara F, Lu X, Callahan C, Heidecker GJ, et al. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc Natl Acad Sci U S A (2014) 111:E2514–23. doi:10.1073/pnas.1402766111
21. Krammer F, Palese P, Steel J. Advances in universal influenza virus vaccine design and antibody mediated therapies based on conserved regions of the hemagglutinin. Curr Top Microbiol Immunol (2015) 386:301–21. doi:10.1007/82_2014_408
22. Wang S, Mata-Fink J, Kriegsman B, Hanson M, Irvine DJ, Eisen HN, et al. Manipulating the selection forces during affinity maturation to generate cross-reactive HIV antibodies. Cell (2015) 160:785–97. doi:10.1016/j.cell.2015.01.027
23. Hashem AM, Van Domselaar G, Li C, Wang J, She YM, Cyr TD, et al. Universal antibodies against the highly conserved influenza fusion peptide cross-neutralize several subtypes of influenza A virus. Biochem Biophys Res Commun (2010) 403:247–51. doi:10.1016/j.bbrc.2010.11.030
24. Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science (2011) 333:843–50. doi:10.1126/science.1204839
25. Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature (2011) 477:466–70. doi:10.1038/nature10373
26. Lingwood D, McTamney PM, Yassine HM, Whittle JR, Guo X, Boyington JC, et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature (2012) 489:566–70. doi:10.1038/nature11371
27. Pappas L, Foglierini M, Piccoli L, Kallewaard NL, Turrini F, Silacci C, et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature (2014) 516:418–22. doi:10.1038/nature13764
28. Grandea AG 3rd, Olsen OA, Cox TC, Renshaw M, Hammond PW, Chan-Hui PY, et al. Human antibodies reveal a protective epitope that is highly conserved among human and nonhuman influenza A viruses. Proc Natl Acad Sci U S A (2010) 107:12658–63. doi:10.1073/pnas.0911806107
29. Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science (2011) 333:850–6. doi:10.1126/science.1205669
30. Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, et al. Antibody recognition of a highly conserved influenza virus epitope. Science (2009) 324:246–51. doi:10.1126/science.1171491
31. Li N, Ren A, Wang X, Fan X, Zhao Y, Gao GF, et al. Influenza viral neuraminidase primes bacterial coinfection through TGF-beta-mediated expression of host cell receptors. Proc Natl Acad Sci U S A (2015) 112:238–43. doi:10.1073/pnas.1414422112
32. Throsby M, van den Brink E, Jongeneelen M, Poon LL, Alard P, Cornelissen L, et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One (2008) 3:e3942. doi:10.1371/journal.pone.0003942
33. Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol (2009) 16:265–73. doi:10.1038/nsmb.1566
34. Wu NC, Young AP, Al-Mawsawi LQ, Olson CA, Feng J, Qi H, et al. High-throughput profiling of influenza A virus hemagglutinin gene at single-nucleotide resolution. Sci Rep (2014) 4:4942. doi:10.1038/srep04942
35. Soundararajan V, Zheng S, Patel N, Warnock K, Raman R, Wilson IA, et al. Networks link antigenic and receptor-binding sites of influenza hemagglutinin: mechanistic insight into fitter strain propagation. Sci Rep (2011) 1:200. doi:10.1038/srep00200
36. Nakamura G, Chai N, Park S, Chiang N, Lin Z, Chiu H, et al. An in vivo human-plasmablast enrichment technique allows rapid identification of therapeutic influenza A antibodies. Cell Host Microbe (2013) 14:93–103. doi:10.1016/j.chom.2013.06.004
37. He W, Mullarkey CE, Duty JA, Moran TM, Palese P, Miller MS. Broadly-neutralizing anti-influenza virus antibodies: enhancement of neutralizing potency in polyclonal mixtures and IgA backbones. J Virol (2015) 89:3610–8. doi:10.1128/JVI.03099-14
38. DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat Med (2014) 20:143–51. doi:10.1038/nm.3443
39. Tharakaraman K, Subramanian V, Cain D, Sasisekharan V, Sasisekharan R. Broadly neutralizing influenza hemagglutinin stem-specific antibody CR8020 targets residues that are prone to escape due to host selection pressure. Cell Host Microbe (2014) 15:644–51. doi:10.1016/j.chom.2014.04.009
40. Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol (2015). doi:10.1016/j.molimm.2015.01.003
41. DiGiandomenico A, Keller AE, Gao C, Rainey GJ, Warrener P, Camara MM, et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med (2014) 6:262ra155. doi:10.1126/scitranslmed.3009655
42. Tan W. A bispecific antibody against two different epitopes on hepatitis B surface antigen has potent hepatitis B virus neutralizing activity. mAbs (2013) 5:946–55.
Keywords: influenza A virus, monoclonal antibodies, mutations, therapeutics, hemagglutinins, viral
Citation: Shriver Z, Trevejo JM and Sasisekharan R (2015) Antibody-based strategies to prevent and treat influenza. Front. Immunol. 6:315. doi: 10.3389/fimmu.2015.00315
Received: 14 April 2015; Accepted: 02 June 2015;
Published: 13 July 2015
Edited by:
Arun Kumar, GlaxoSmithKline Vaccines, ItalyReviewed by:
Graham Robert Leggatt, University of Queensland, AustraliaOwen Kavanagh, Queen’s University Belfast, UK
Copyright: © 2015 Shriver, Trevejo and Sasisekharan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ram Sasisekharan, Department of Biological Engineering, Koch Institute of Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, 76-461C, Cambridge, MA, USA,cmFtc0BtaXQuZWR1