 Hany Ibrahim Kenawy
Hany Ibrahim Kenawy Ismet Boral
Ismet Boral Alan Bevington
Alan Bevington- 1Department of Microbiology and Immunology, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt
- 2Department of Infection, Immunity and Inflammation, College of Medicine, Biological Sciences and Psychology, University of Leicester, Leicester, UK
The complement system is a major constituent of the innate immune system. It not only bridges innate and adaptive arms of the immune system but also links the immune system with the coagulation system. Current understanding of the role of complement has extended far beyond fighting of infections, and now encompasses maintenance of homeostasis, tissue regeneration, and pathophysiology of multiple diseases. It has been known for many years that complement activation is strongly pH sensitive, but only relatively recently has the physiological significance of this been appreciated. Most complement assays are carried out at the physiological pH 7.4. However, pH in some extracellular compartments, for example, renal tubular fluid in parts of the tubule, and extracellular fluid at inflammation loci, is sufficiently acidic to activate complement. The exact molecular mechanism of this activation is still unclear, but possible cross-talk between the contact system (intrinsic pathway) and complement may exist at low pH with subsequent complement activation. The current article reviews the published data on the effect of pH on the contact system and complement activity, the nature of the pH sensor molecules, and the clinical implications of these effects. Of particular interest is chronic kidney disease (CKD) accompanied by metabolic acidosis, in which therapeutic alkalinization of urine has been shown significantly to reduce tubular complement activation products, an effect, which may have important implications for slowing progression of CKD.
Introduction – The Physiological and Clinical Importance of pH Effects on Complement
It has been known for many years that complement is strongly activated by low pH, especially when pH falls below about 7.1 (1–7). Owing to the tight regulation of arterial blood pH close to the normal physiological value of 7.4 that occurs even under pathological conditions, complement and plasma proteins in major blood vessels are unlikely to be exposed to such low pH. However, at sites of infection or inflammation, a significant localized fall in pH can occur, reaching pH 6 or even lower (8–10). Furthermore, other fluid compartments in mammals, notably the fluid within the lumen of the renal tubule, routinely maintain pH values below 7.1 (Table 1).
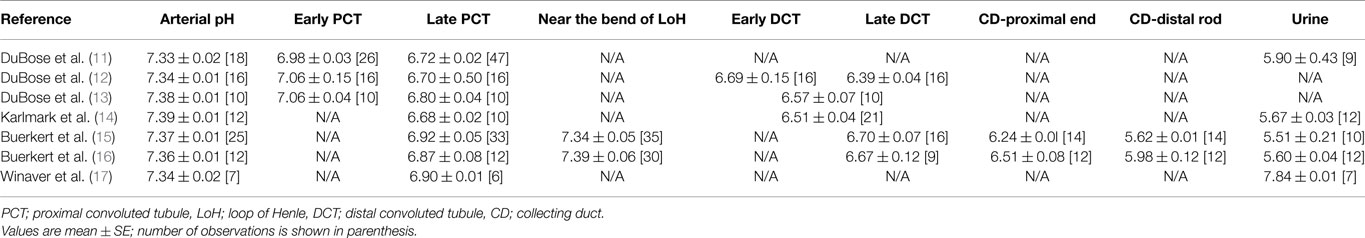
Table 1. Summary of intraluminal pH measurements obtained by in situ micro-puncture studies in renal tubules of healthy rats.
In healthy individuals, the lumen of the renal tubule is not routinely exposed to plasma proteins (including complement proteins). However, in chronic kidney disease (CKD), leakage of such proteins commonly occurs, resulting in proteinuria. There is now abundant evidence (18) that proteinuria is a major factor driving progression of CKD, and that the leakage of plasma proteins into the tubular lumen triggers an array of pathological changes in proximal tubular epithelial cells (PTEC) (18–20), including hyperplasia and epithelial–mesenchymal transition (EMT), which culminate in end-stage tubulointerstitial fibrosis. The complement system is widely recognized as a key mediator of renal injury (21) and there is mounting evidence that activation of plasma complement proteins leaking into the tubular lumen during proteinuria, followed by strong activation of locally synthesized complement (22) leads to progressive tubulointerstitial damage. Significant amounts of complement activation products are excreted in urine of patients with many forms of proteinuric nephropathy (23) and this excretion of activation products is blunted when metabolic acidosis in these patients is treated with sodium bicarbonate (NaHCO3) (23), even though bicarbonate has no long-term effect on proteinuria (23, 24). This implies that, in addition to the well-documented glomerular effects of complement (25), filtered complement, strongly augmented by endogenously expressed tubular complement (22), is activated by the low intratubular pH (Table 1). This may explain the important clinical observation (24) that progression of CKD is significantly slowed in response to therapy with oral alkali (sodium bicarbonate), much of which is excreted into the tubular lumen thus raising intraluminal pH.
While renal complement-activation during metabolic acidosis has traditionally been ascribed to covalent activation of complement C3 by ammoniagenesis (26), more recent direct measurements have failed to substantiate this (7), and direct activation of complement by physiological low pH (4–7) is a more likely explanation, possibly through activation of the alternative pathway (AP) (7, 27) and through pH-sensitive cross-talk between the coagulation (contact) and complement systems.
The current article reviews and compares the basic features of the complement and coagulation systems, cross-talk between these two systems, and the mechanisms whereby low pH may activate complement; in particular, the possibility that low pH is sensed initially by the contact system (intrinsic pathway) and that complement is then activated through contact system-complement cross-talk.
The Complement and the Coagulation Systems
The complement and the coagulation systems are two closely linked systems that serve a vital role in maintaining homeostasis. Their activities rely on a delicate balance between activator and inhibitor signals of sequential enzymatic reactions that include activation of zymogens and assembly of new proteolytic complexes. Complement is now thought to be involved in several activities besides its role in fighting infections: these include tissue regeneration (28), clearance of debris (29), and pathophysiology of multiple diseases (30, 31). Likewise, the coagulation system plays a role in fighting infections (32) and is implicated in pathophysiology of several diseases besides its role in the maintenance of hemostasis. Furthermore, complex cross-talk between complement and the coagulation system has been described that will be addressed in the current review, particularly with regard to mediating the activation of complement by physiologically attainable low pH.
The Complement System
Complement, as an integral part of the innate immune system has a major role in defense against invading pathogens. It achieves this through three main strategies; recruitment of immune cells to sites of infection, labeling of the invading pathogens via opsonization for uptake and destruction by phagocytes, and/or direct lysis of susceptible pathogens. Besides bridging innate and adaptive immunity, complement also bridges the immune and coagulation systems. More than 35 proteins, including circulating zymogens, and an array of fluid phase and membrane-bound regulators and cell-bound receptors, participate in complement activities. Three pathways have been recognized for complement activation; the classical pathway (CP), the AP, and the more recently discovered lectin pathway (LP) (33–35). The CP and LP are analogous, differing only in the initiator molecular complexes and the triggering signals (36). Complement C1q in association with two molecules of each of the serine proteases C1r and C1s makes the initiator complex of the CP (37). The C1qrs complex is activated on binding to antigen–antibody complex; however, it can also be activated in an antigen–antibody independent manner by binding to a number of molecules including C-reactive protein (CRP), lipopolysaccharide (LPS), polyanions, viral proteins, pneumolysin (38), and myelin. The LP-initiating complexes are made up of carbohydrate recognition molecules of either mannose binding lectin (MBL), ficolins or collectin-11 (Cl-11) associated with serine proteases, namely, MASP-1, MASP-2, and MASP-3 (39). LP is initiated upon recognition of certain sugar or acetylated sugar patterns decorating surfaces of invading microbes by the broad-spectrum carbohydrate recognition molecules of the LP. Unlike CP and LP, AP activation does not proceed via specific recognition molecules, but occurs instead through an imbalance between activating and inactivating signals acting on a steady state tick over process (40, 41). This kind of imbalance occurs on susceptible surfaces that lack complement regulators or do not support the binding of such regulators that normally occur on host cells. In addition, AP acts as a loop for amplification of signals from the other two pathways (42, 43).
Binding of recognition complexes from either CP or LP to their target structures leads to conformational changes within the molecules that result in activation of the attached serine proteases; C1r/C1s and MASP-1/MASP-2, respectively. The activated serine proteases C1s and MASP-2 cleave C4 into C4a and C4b. C4a is released into the fluid phase, whereas C4b attaches to the target surface. C2 binds to the attached C4b and is cleaved by C1s or MASP-2 releasing C2b into the fluid phase, whereas C2a remains attached to C4b. The resulting complex C4b2a represents the C3-convertase of the CP/LP that then activates C3. The slow and spontaneous hydrolysis of C3 into C3(H2O) is considered to provide the flux that maintains the AP activity. The resulting C3(H2O) is able to bind to factor B (FB), rendering it susceptible to cleavage by factor D (FD). This produces a limited amount of the fluid phase AP C3-convertase [C3(H2O)Bb] that is able to cleave C3 into C3a and C3b. In absence of surface regulators or surfaces that do not support factor H (FH) binding (the main negative regulator of AP), the C3b formed binds to target surfaces and assembles with FB (in presence of FD) the surface AP C3-convertase (C3bBb) (44) that is stabilized by properdin (45, 46).
All of the three complement activation pathways converge in the proteolytic cleavage of C3, where C3 is cleaved into the anaphylatoxin C3a, and the opsonin C3b that binds covalently to the target surface. Binding of several molecules of the C3b to C3-convertases (either from CP/LP or AP) results in the assembly of C5-convertase that cleaves C5 into the powerful anaphylatoxin C5a and the opsonin C5b. Opsonization of target cells with C5b allows, in certain cases, the assembly of terminal complement components C6, 7, 8, and 9 into the membrane attack complex (MAC) that inserts into target membranes forming channels that disrupt membrane function and lead to lysis of target cells (47, 48).
The Coagulation System
The intrinsic pathway of coagulation was first described by two independent laboratories that proposed the waterfall model (49) and the cascade model (50) of coagulation (Figure 1). Following the discovery of tissue factor (TF) (also called factor III or CD142), the waterfall/cascade models were refined to include the extrinsic pathway. However, these models could not fully explain in vivo hemostasis or the normal bleeding tendency in patients lacking some of the early components of the intrinsic pathway [the contact system components: FXII, prekallikrein (PK) and high molecular-weight kininogen (HMWK)]. In addition, it was unclear why patients with functional deficiency of the intrinsic pathway FVIII (hemophilia A) or FIX (hemophilia B) have severe bleeding dysfunction despite the presence of the extrinsic pathway activity. In 1990s, the emergence of the cell-based model of coagulation provided plausible answers to these anomalies (51, 52).
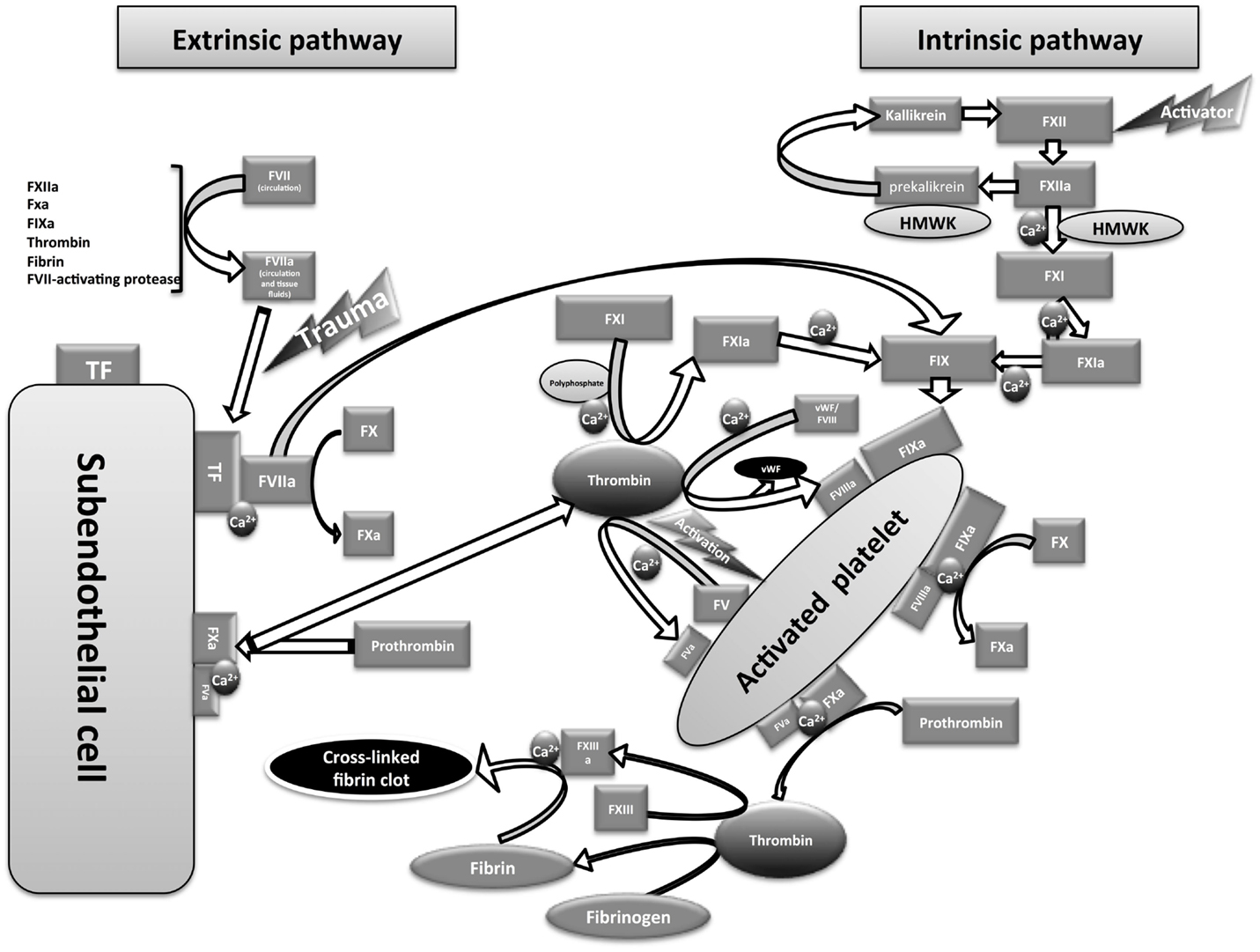
Figure 1. Diagram showing an overview of coagulation pathways.
The Extrinsic Pathway
Blood clotting through the extrinsic pathway of coagulation depends on the interaction between the circulating active form of FVII (FVIIa) and the membrane-bound TF to form a serine protease complex that activates FX into FXa (see Figure 1). FVII is the only coagulation factor found in circulation in active (1%) and inactive forms (53). The mechanism of FVII activation is still unconfirmed; however, autoactivation is suggested to provide the circulating FVIIa (54, 55). Other factors including FXIIa, FXa, FIXa, thrombin, plasmin, and FVII-activating protease also show the ability to activate FVII (56). Under physiological conditions, TF is not accessible to blood components, and only becomes accessible following injury to the endothelial cells lining the blood vessels. TF is expressed in a number of cells, including adventitial cells in the layers surrounding blood vessels. It has been reported that TF is also expressed in a number of activated cells (or cell-derived particles) in the blood, including monocytes, monocyte-derived microparticles, neutrophils, eosinophils, and platelets (57–62). However, intravascular TF exists in an encrypted form that cannot interact with FVIIa unless decrypted, possibly through the enzymatic activity of protein disulfide isomerase (63–65). Intravascular TF is thought to be involved in thrombosis (a pathological form of coagulation) rather than normal hemostasis (57, 59, 66). The extrinsic pathway is believed to be the only physiological trigger of coagulation in vivo that is activated immediately upon blood vessel injury (67).
The Intrinsic Pathway and the Contact System
The intrinsic pathway of coagulation is triggered upon the activation of the first component of the contact system “FXII” into FXIIa by an activator surface that is usually a negatively charged surface (68). The in vivo physiological activators of FXII are still unclear; however, platelet-derived polyphosphate (54, 69, 70) and mast cell heparin (71) are suggested to be contributory physiological activators of the contact system. Other known activators of FXII include extracellular RNA (72), DNA (73), collagen, Kaolin (74), dextran sulfate (75, 76), oversulfated chondroitin sulfate (77), glass, and plastic. FXIIa activates the second component of the contact system – PK into kallikrein. PK circulates with the cofactor HMWK (the third contact system component). The resulting kallikrein promotes the activation of additional FXII molecules via a positive feedback loop (Figure 1). These first two steps in contact system activation do not require the presence of calcium ions. The resulting FXIIa in the presence of calcium ions, phospholipids (phosphatidylserine provided by activated platelet surfaces), and the cofactor HMWK activates FXI into FXIa (78). FXIa then activates FIX into FIXa in the presence of calcium ions and phospholipids. FIXa forms (with the cofactor FVIIIa, in presence of calcium ions and phospholipids) a serine protease “Tenase” that activates FX into FXa. The resulting FXa either from the extrinsic or the intrinsic pathway forms, with the cofactor “FVa,” a serine protease called “prothrombinase” that activates prothrombin (FII) into thrombin (the common pathway). The resulting thrombin then acts on fibrinogen, releasing fibrin monomer that is cross-linked in the presence of FXIIIa to form a stable fibrin polymer clot. Besides the procoagulant activity, kallikrein can cleave HMWK to release the proinflammatory peptide “bradykinin” (kallikrein-kinin pathway) that acts on multiple target cells with the production of inflammatory mediators that include prostacyclin, prostaglandins, leukotrienes, endothelial-derived hyperpolarizing factor, and nitric oxide (79). By generation of bradykinin, the contact system plays an additional role, promoting other immune defense mechanisms during infection [for review, see Ref. (80)].
Growing evidence suggests that the contact system has surprisingly little impact on physiological hemostasis. Deficiency of FXII in humans and other animal species is not associated with deficient hemostasis. Moreover, non-mammalian vertebrates and cetaceans do not have FXII (81). On the other hand, it is strongly suspected that the contact system is associated with thrombosis (82). Deficiency of the contact system is protective against development of thrombosis and stroke (32, 83, 84). It is now believed that the mechanisms of physiological hemostasis are different from those of thrombosis (82) and thrombosis can be triggered via the activation of FXII by platelets and erythrocyte-derived microparticles (85). Current therapeutic strategies in thrombosis focus on targeting the contact system, hence affording protection from thrombosis or embolism without interfering with the hemostatic capacity. In view of the apparent links between the contact system and the activation of complement at low pH (discussed below), the contact system might also be a suitable therapeutic target in blocking the detrimental effects of complement activation under acidic conditions.
The Cell-Based Model of Coagulation
A more complete understanding of in vivo coagulation requires the cell-based model of coagulation, in which cell surfaces play a role distinct from providing the phospholipids required for assembly of protease complexes via platelet surfaces. The initiation step is believed to occur continuously in the extravascular tissue fluids and lymph, into which coagulation factors such as FVII, FIX, FX, prothrombin, and other low molecular-weight coagulation factors can diffuse from blood (51). In this step, FVIIa binds to TF on TF-bearing cells to form TF/FVIIa complex that activates FX to FXa. The resulting FXa activates prothrombin to thrombin before being rapidly inactivated by tissue factor pathway inhibitor (TFPI) and antithrombin III (86, 87). At the same time, TF/FVIIa complex activates FIX into IXa (88) that is required in further steps. Thus, this initiation step provides a continuous supply of trace amounts of extravascular thrombin that is required for further steps in the event of blood vessel injury. If such injury occurs, the following amplification and propagation steps will be triggered, involving blood coagulation factors and platelets that cannot normally diffuse from blood vessels into surrounding tissues. This occurs when these come into contact with the thrombin generated from the initiation phase (as described above), and the TF-bearing cells and subendothelial collagen.
Subsequently, platelets will be activated at the injury site by thrombin and collagen (89), releasing FV that becomes activated by thrombin on the platelet surface (see Figure 1). At the same time, thrombin will release FVIII from von Willebrand factor (vWF)-FVIII complex and activate it to FVIIIa. During this amplification step, the activated platelets are covered with the cofactors FVIIIa and FVa. The amount of thrombin generated at this stage is not sufficient to drive clot formation; however, it is very important for the amplification of the procoagulant signal.
In the subsequent propagation step, FIXa produced either from TF/FVIIa complex or from FXIa generated via thrombin action on FXI [in the presence of polyphosphate (90)] will bind to FVIIIa on platelets in the presence of calcium ions to form tenase that is 50 times more efficient than TF/FVIIa complex in FX activation (87), thus releasing large amounts of FXa. This FXa assembles with FVa and calcium to form prothrombinase on the activated platelet surface that will generate copious amounts of thrombin, which drive fibrin clot formation. Accordingly, TF/FVIIa appears to be the primary physiological trigger of in vivo coagulation (67) and contact system components (FXII, PK, and HMWK) do not seem to have a major role.
Cross-Talk between Complement and Coagulation Systems
The complement and coagulation systems share a number of common features. Activation of both systems leads to conversion of zymogens and assembly of proteolytic complexes that are mostly serine proteases of high-substrate specificity. Interactions between complement and coagulation systems have been described in a number of publications. For example, some complement regulators such as complement C1 inhibitor are involved in regulation of the contact system (91), and serine proteases from either of the two systems may act on substrates from the other system. For this reason, severe trauma and acute blood loss are not only associated with disseminated intravascular coagulopathy (DIC) but also with massive complement activation. This generates the potent anaphylatoxins C3a and C5a, and these may in their turn intensify coagulation (92–94). Activated platelets, which are critical participants in coagulation, can also activate both the CP (95) and the AP (96, 97); however, the physiological impact of this activation is still unknown, although complement activation products are known to activate platelets (97), which may lead to a positive feedback loop.
Thrombin generated from the coagulation system can activate complement C3 and C5 independent of the established complement activation pathways (98). Furthermore, C3 and C5 activation can proceed independent of each other (98). Similarly, Amara et al. (92, 99) reported the cleavage of C3 and C5, with the generation of C3a and C5a, respectively, by the coagulation factors FIXa, FXa, FXIa, and plasmin (100) independent of the known complement activation pathways. Thrombin and plasmin have been suggested to activate complement during liver regeneration in the absence of C4 and AP activity (28). In addition, FXIIa has been shown to activate the CP of complement via activation of C1qrs complex (101). Surprisingly, fluid phase activation of FXII by oversulfated chondroitin sulfate activated not only the contact (Kallikrein-Kinin) system but also C3 and C5 in the presence of EDTA (77). EDTA is a well-known inhibitor of complement via all of the known activation pathways by sequestering the divalent cations (Ca2+ and Mg2+) necessary for complement activation. Thus, C3 and C5 activation can proceed by a mechanism that is independent of the known C3- and C5-convertases. Depletion of FXII from plasma abolished this activation without affecting the normal complement activity; and reconstitution of depleted plasma with purified FXII restored complement activation (77).
However, not all such effects involve FXII: the use of aprotonin – a protease inhibitor of kallikrein and plasmin (but not of FXII) – inhibited C5 activation by oversulfated chondroitin sulfate (77). Furthermore, plasminogen-depleted plasma also failed to induce C5a production by oversulfated chondroitin sulfate (77). Thus, it seems that the contact system activity – not exclusively the individual activities of FXIIa or kallikrein – may drive complement C3 and C5 activation through generation of plasmin that has previously been reported to cleave C3 and C5 (77, 92, 100).
Wiggins et al. (102) showed that purified rabbit kallikrein was able to generate from rabbit C5 an activity that was chemotactic for rabbit neutrophils, suggesting that Kallikrein may cleave C5. Besides that, kallikrein was shown to play a role similar to factor D in cleaving C3bB, generating the AP C3-convertase C3bBb. However, this activity required the presence of divalent cations (103).
Conversely, complement components have been shown to influence coagulation activity in multiple ways. The key enzyme of LP activation (MASP-2) is able to generate thrombin through direct cleavage of prothrombin (104). Likewise, the terminal complement component complex C5b–9 has similar activity toward prothrombin even in absence of FV (105). Moreover, both the sublytic MAC and the cytolytically inactive terminal complement complex exhibit procoagulant activity mediated via the induction of TF expression by endothelial cells (106). The anaphylatoxin C5a promotes procoagulant activity by several actions on cells. C5a induces the upregulation of TF expression by endothelial cells (107) and by neutrophils (108). In addition, C5a induces the switch of mast cell and basophil activities from profibrinolytic to prothrombotic through the upregulation of plasminogen activator inhibitor-1 (PAI-1) (109). Interaction between the two membrane receptors TF and complement C5a receptor (C5a R) further suggests a cross-talk between the two systems (108). Accordingly, there is considerable evidence for two-way communication between complement and coagulation system components, influencing the activities of both systems.
Complement and Contact System Activities at Acidic pH
Several in vitro studies have demonstrated potent activation of complement activity, at acidic pH values, some of which are in the physiologically relevant range shown in Table 1 (1–7). However, care is needed in interpreting these effects for two reasons: first because in vivo the low pH may have multiple sites of action (as in the case of renal tubules or inflammatory foci); and second because not all such effects observed in vitro may be directly physiologically relevant because they require extremely low pH or non-physiological temperature. For example, Hammer et al. (1) reported that acidification of serum or C5 and C6 to pH 6.4 at 0°C followed by neutralization was associated with complement activation that led to lysis of non-sensitized red blood cells in presence of terminal complement components C7, C8, and C9. They attributed the observed activity to the formation of a complex between C5 and C6 similar in activity to that of C5b6 generated via the AP or the CP. This complex was thought to be formed as a result of C6-mediated cleavage of C5 α-chain aided by low pH, which changed the tertiary structure of either or both of C5 and C6. However, the physiological significance of this complex is uncertain, as it was unstable at physiological temperature (37°C).
Low pH may also exert multiple effects through the AP. AP hemolytic activity on erythrocytes from paroxysmal nocturnal haemglobinuria (PNH) patients (110, 111) and rabbit erythrocytes was enhanced at pH 6.4 compared to pH 7.4 (2), which forms the basis of Ham’s test used in diagnosis of PNH (110). They explained the enhancement of activity through the increased formation of the two C3-convertases; C3(H2O)Bb and C3bBb, in addition to the enhanced binding of FB and C5 to C3b deposited on erythrocytes at pH 6.4. At the same time, the inhibitory effect of CR1 and factor I (FI) was also diminished at this pH. Similarly, Peake et al. (7) suggested that maximal complement deposition on cultured PTEC occurred via the AP at acidic pH.
Complement activation under mildly acidic conditions has also been attributed to human CRP via the CP (3). CRP is known to trigger CP activation upon interaction with phosphocholine-containing or polycationic agents. However, even in absence of these agents, CRP has been reported to activate complement, with optimal activity at pH 6.3. Furthermore, Hammond et al. (112) showed that CRP was able to interact with FH, which may be a way of partially regulating the enhanced complement activity mediated by CRP at low pH. However, it should be noted that this interaction of CRP with FH required more acidic pH (5.2–4.6).
Contact System-Complement Cross-Talk as a Mediator of the Effect of Low pH on Complement
Apart from the evidence cited above, relatively little is known of direct effects of mildly acidic pH on complement proteins or complement regulatory proteins. An alternative explanation of the activation of complement by low pH is that pH is initially sensed by component(s) of the contact system and this then leads indirectly to complement activation through complement-coagulation cross-talk. In the study involving CRP cited above (3), complement activation occurred in glass tubes, but not in polypropylene tubes. Addition of Kaolin to polypropylene tubes restored complement activation, suggesting a requirement for the presence of negatively charged surfaces to support the pH-dependent CRP-mediated complement activation (3). These authors attributed these effects of low pH to the conformational changes that CRP underwent at this pH. However, a possible alternative explanation is that the contact system is involved in this CRP-mediated complement activation at mild acidic pH (3), as activation occurred only in the presence of negatively charged surfaces, conditions that activate the contact system as well.
Further evidence in support of a role for the contact system in mediating the activation of complement by low pH (through complement-coagulation cross-talk) comes from the observation that low pH results in accumulation of FXIIa as well as complement activation products. Generation of complement activation products C3a and C5a was observed by Emeis et al. (4) upon acidification of blood with either HCl or lactic acid. This complement activation was attributed to the effect of acidic pH itself, not to the lactate anion, as addition of lactate at control (non-acidic) pH had no such effect. Likewise, respiratory acidosis of blood was also associated with increase in C3a and C5a levels. Lactate acidosis could activate not only C3 and C5 but also more distal steps in the complement system, with the formation of the soluble terminal complement complex (sC5b–9) either in adults or neonates (113). In addition, Sonntag et al. (5, 6) showed that acidification of blood and plasma with lactic acid was associated with dose-dependent increase in complement C5a and contact system FXIIa generation, even in the absence of cellular components. Similarly, Renaux et al. (114) and Thomas et al. (115) reported increased contact system activity with increased kallikrein activity and bradykinin formation during hemodialysis upon lowering the pH of diluted plasma from pH 7.4 to 7.1.
In addition to the in vitro studies, parallel activation of complement and the contact system has also been observed in vivo in conditions associated with tissue acidosis, for example, in cases of myocardial infarction, shock, and perinatal asphyxia, where the anaphylatoxins C3a and C5a, and FXIIa levels were elevated (5, 6, 116, 117).
The molecular basis of this apparent strong relation between complement activation and contact system activation under mildly acidic pH conditions, where the contact system is probably the driving force for the complement activation, is still unclear. The pH sensor molecule(s) involved are unknown. A highly pH-sensitive plasma protein is the histidine proline-rich glycoprotein [HPRG – reviewed in Ref. (118)] in which much of the pH sensitivity is attributable to the “histidine rich” region of the molecule. Even though HPRG is known to associate with complement proteins (118, 119), no direct acid-dependent activation of complement by this molecule has ever been demonstrated. However, an interesting potential link with the contact system arises from the observation that HPRG and HMWK show about 50% sequence identity in their “histidine rich” region (118), indicating a possible direct pH-sensing role by HMWK.
In view of the apparent links between acid-dependent complement activation and the progression of CKD that were reviewed at the start of this article, the molecular basis of this effect of low pH, and the involvement of possible pH sensing in the contact system, merits further investigation; particularly as conventional therapy for correction of low pH by administering oral sodium bicarbonate to CKD patients may carry with it cardiovascular risks associated with sodium loading. Selective inhibition of the contact system (which may be possible without impairing hemostasis) might be a suitable alternative therapeutic target. However, before this possibility is pursued, two important points about the activation of complement at low pH remain to be clarified:
First, if the complement activation ultimately arises from complement-coagulation system cross-talk, with the cleavage of C3 and C5, by FIXa, FXa, FXIa, and plasmin (100), it needs to be confirmed that these proteases are still active at the relevant low pH. If this is important in the renal tubular lumen during proteinuria, it also needs to be shown that the failure of glomerular permselectivity during proteinuria is sufficient to allow the relevant contact system components (and not just complement) to leak into the acidic renal tubular lumen.
Second, it needs to be confirmed that the effect of elevated pH in vitro, or the therapeutic effect of alkalinizing the tubular lumen with bicarbonate therapy in vivo (24) arises from inhibiting the contact system, rather than through blocking the other proposed effects of low pH on complement, such as acid-induced amplification of spontaneous AP activity or enhanced binding of FB and C5 to C3b (2).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hammer CH, Hansch G, Gresham HD, Shin ML. Activation of the fifth and sixth components of the human complement system: C6-dependent cleavage of C5 in acid and the formation of a bimolecular lytic complex, C5b,6a. J Immunol (1983) 131(2):892–8.
2. Fishelson Z, Horstmann RD, Muller-Eberhard HJ. Regulation of the alternative pathway of complement by pH. J Immunol (1987) 138(10):3392–5.
3. Miyazawa K, Inoue K. Complement activation induced by human C-reactive protein in mildly acidic conditions. J Immunol (1990) 145(2):650–4.
4. Emeis M, Sonntag J, Willam C, Strauss E, Walka MM, Obladen M. Acidosis activates complement system in vitro. Mediators Inflamm (1998) 7(6):417–20.
5. Sonntag J, Emeis M, Strauss E, Obladen M. In vitro activation of complement and contact system by lactic acidosis. Mediators Inflamm (1998) 7(1):49–51.
6. Sonntag J, Wagner MH, Strauss E, Obladen M. Complement and contact activation in term neonates after fetal acidosis. Arch Dis Child Fetal Neonatal Ed (1998) 78(2):F125–8. doi:10.1136/fn.78.2.F125
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
7. Peake PW, Pussell BA, Mackinnon B, Charlesworth JA. The effect of pH and nucleophiles on complement activation by human proximal tubular epithelial cells. Nephrol Dial Transplant (2002) 17(5):745–52. doi:10.1093/ndt/17.5.745
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
8. Menkin V. The role of hydrogen ion concentration and the cytology of an exudate. In: Menkin V, editor. Biochemical Mechanisms in Inflammation. Springfield, IL: Charles C Thomas (1956). p. 66–103.
9. Rotstein OD, Nasmith PE, Grinstein S. The Bacteroides by-product succinic acid inhibits neutrophil respiratory burst by reducing intracellular pH. Infect Immun (1987) 55(4):864–70.
10. Grinstein S, Swallow CJ, Rotstein OD. Regulation of cytoplasmic pH in phagocytic cell function and dysfunction. Clin Biochem (1991) 24(3):241–7. doi:10.1016/0009-9120(91)80014-T
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
11. DuBose TD Jr, Pucacco LR, Lucci MS, Carter NW. Micropuncture determination of pH, PCO2, and total CO2 concentration in accessible structures of the rat renal cortex. J Clin Invest (1979) 64(2):476–82. doi:10.1172/JCI109485
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
12. DuBose TD Jr, Pucacco LR, Seldin DW, Carter NW, Kokko JP. Microelectrode determination of pH and PCO2 in rat proximal tubule after benzolamide: evidence for hydrogen ion secretion. Kidney Int (1979) 15(6):624–9. doi:10.1038/ki.1979.82
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
13. DuBose TD Jr, Pucacco LR, Carter NW. Determination of disequilibrium pH in the rat kidney in vivo: evidence of hydrogen secretion. Am J Physiol (1981) 240(2):F138–46.
14. Karlmark B, Jaeger P, Giebisch G. Luminal buffer transport in rat cortical tubule: relationship to potassium metabolism. Am J Physiol (1983) 245(5 Pt 1):F584–92.
15. Buerkert J, Martin D, Trigg D. Segmental analysis of the renal tubule in buffer production and net acid formation. Am J Physiol (1983) 244(4):F442–54.
16. Buerkert J, Martin D, Trigg D, Simon E. Effect of reduced renal mass on ammonium handling and net acid formation by the superficial and juxtamedullary nephron of the rat. Evidence for impaired reentrapment rather than decreased production of ammonium in the acidosis of uremia. J Clin Invest (1983) 71(6):1661–75. doi:10.1172/JCI110921
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
17. Winaver J, Walker KA, Kunau RT Jr. Effect of acute hypercapnia on renal and proximal tubular total carbon dioxide reabsorption in the acetazolamide-treated rat. J Clin Invest (1986) 77(2):465–73. doi:10.1172/JCI112325
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
18. Brunskill NJ. Albumin signals the coming of age of proteinuric nephropathy. J Am Soc Nephrol (2004) 15(2):504–5. doi:10.1097/01.ASN.0000112912.40303.81
19. Baines RJ, Brunskill NJ. The molecular interactions between filtered proteins and proximal tubular cells in proteinuria. Nephron Exp Nephrol (2008) 110(2):e67–71. doi:10.1159/000161982
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
20. Dixon R, Brunskill NJ. Activation of mitogenic pathways by albumin in kidney proximal tubule epithelial cells: implications for the pathophysiology of proteinuric states. J Am Soc Nephrol (1999) 10(7):1487–97.
21. Brown KM, Sacks SH, Sheerin NS. Mechanisms of disease: the complement system in renal injury – new ways of looking at an old foe. Nat Clin Pract Nephrol (2007) 3(5):277–86. doi:10.1038/ncpneph0465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
22. Sheerin NS, Risley P, Abe K, Tang Z, Wong W, Lin T, et al. Synthesis of complement protein C3 in the kidney is an important mediator of local tissue injury. FASEB J (2008) 22(4):1065–72. doi:10.1096/fj.07-8719com
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
23. Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S. Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol (2000) 11(4):700–7.
24. de Brito-Ashurst I, Varagunam M, FAU Raftery MJ, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol (2009) 20(9):2075–84. doi:10.1681/ASN.2008111205
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
25. Abbate M, Zoja C, Corna D, Rottoli D, Zanchi C, Azzollini N, et al. Complement-mediated dysfunction of glomerular filtration barrier accelerates progressive renal injury. J Am Soc Nephrol (2008) 19(6):1158–67. doi:10.1681/ASN.2007060686
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
26. Nath KA, Hostetter MK, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest (1985) 76(2):667–75. doi:10.1172/JCI112020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
27. Gaarkeuken H, Siezenga MA, Zuidwijk K, van Kooten C, Rabelink TJ, Daha MR, et al. Complement activation by tubular cells is mediated by properdin binding. Am J Physiol Renal Physiol (2008) 295(5):F1397–403. doi:10.1152/ajprenal.90313.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
28. Clark A, Weymann A, Hartman E, Turmelle Y, Carroll M, Thurman JM, et al. Evidence for non-traditional activation of complement factor C3 during murine liver regeneration. Mol Immunol (2008) 45(11):3125–32. doi:10.1016/j.molimm.2008.03.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
29. Pickering MC, Walport MJ. Links between complement abnormalities and systemic lupus erythematosus. Rheumatology (Oxford) (2000) 39(2):133–41. doi:10.1093/rheumatology/39.2.133
30. Alchi B, Jayne D. Membranoproliferative glomerulonephritis. Pediatr Nephrol (2010) 25(8):1409–18. doi:10.1007/s00467-009-1322-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
31. Sweigard JH, Yanai R, Gaissert P, Saint-Geniez M, Kataoka K, Thanos A, et al. The alternative complement pathway regulates pathological angiogenesis in the retina. FASEB J (2014) 28(7):3171–82. doi:10.1096/fj.14-251041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
32. Muller F, Renne T. Novel roles for factor XII-driven plasma contact activation system. Curr Opin Hematol (2008) 15(5):516–21. doi:10.1097/MOH.0b013e328309ec85
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
33. Fujita T. Evolution of the lectin-complement pathway and its role in innate immunity. Nat Rev Immunol (2002) 2(5):346–53. doi:10.1038/nri800
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
34. Schwaeble W, Dahl MR, Thiel S, Stover C, Jensenius JC. The mannan-binding lectin-associated serine proteases (MASPs) and MAp19: four components of the lectin pathway activation complex encoded by two genes. Immunobiology (2002) 205(4–5):455–66. doi:10.1078/0171-2985-00146
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
35. Matsushita M, Fujita T. The lectin pathway. Res Immunol (1996) 147(2):115–8. doi:10.1016/0923-2494(96)87185-9
36. Wallis R, Mitchell DA, Schmid R, Schwaeble WJ, Keeble AH. Paths reunited: initiation of the classical and lectin pathways of complement activation. Immunobiology (2010) 215(1):1–11. doi:10.1016/j.imbio.2009.08.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
37. Cooper NR. The classical complement pathway: activation and regulation of the first complement component. Adv Immunol (1985) 37:151–216.
38. Ali YM, Kenawy HI, Muhammad A, Sim RB, Andrew PW, Schwaeble WJ. Human L-ficolin, a recognition molecule of the lectin activation pathway of complement, activates complement by binding to pneumolysin, the major toxin of Streptococcus pneumoniae. PLoS One (2013) 8(12):e82583. doi:10.1371/journal.pone.0082583
39. Ali YM, Lynch NJ, Haleem KS, Fujita T, Endo Y, Hansen S, et al. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog (2012) 8(7):e1002793. doi:10.1371/journal.ppat.1002793
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
40. Nilsson B, Nilsson Ekdahl K. The tick-over theory revisited: is C3 a contact-activated protein? Immunobiology (2012) 217(11):1106–10. doi:10.1016/j.imbio.2012.07.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
41. Lachmann PJ. The amplification loop of the complement pathways. In:Alt FW, Advances in Immunology. Amsterdam: Elsevier Inc (2009). p. 115–49. doi:10.1016/S0065-2776(08)04004-2
42. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344(14):1058–66. doi:10.1056/NEJM200104053441406
43. Walport MJ. Complement. Second of two parts. N Engl J Med (2001) 344(15):1140–4. doi:10.1056/NEJM200104123441506
44. Xu Y, Narayana SV, Volanakis JE. Structural biology of the alternative pathway convertase. Immunol Rev (2001) 180:123–35. doi:10.1034/j.1600-065X.2001.1800111.x
45. Kemper C, Hourcade DE. Properdin: new roles in pattern recognition and target clearance. Mol Immunol (2008) 45(16):4048–56. doi:10.1016/j.molimm.2008.06.034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
46. Fearon DT, Austen KF. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med (1975) 142(4):856–63.
47. Wang Y, Bjes ES, Esser AF. Molecular aspects of complement-mediated bacterial killing. Periplasmic conversion of C9 from a protoxin to a toxin. J Biol Chem (2000) 275(7):4687–92. doi:10.1074/jbc.275.7.4687
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
48. Podack ER, Muller-Eberhard HJ, Horst H, Hoppe W. Membrane attach complex of complement (MAC): three-dimensional analysis of MAC-phospholipid vesicle recombinants. J Immunol (1982) 128(5):2353–7.
49. Davie E, Ratnoff O. Waterfall sequence for intrinsic blood clotting. Science (1964) 145(3638):1310–2. doi:10.1126/science.145.3638.1310
50. MacFarlane R. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature (1964) 202:498–9. doi:10.1038/202498a0
51. Mann KG, Krishnaswamy S, Lawson JH. Surface-dependent hemostasis. Semin Hematol (1992) 29(3):213–26.
52. Hoffman M. A cell-based model of coagulation and the role of factor VIIa. Blood Rev (2003) 17(Suppl 1):S1–5. doi:10.1016/S0268-960X(03)90000-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
53. Morrissey JH. Plasma factor VIIa: measurement and potential clinical significance. Haemostasis (1996) 26(Suppl 1):66–71.
54. Morrissey JH, Tajkhorshid E, Sligar SG, Rienstra CM. Tissue factor/factor VIIa complex: role of the membrane surface. Thromb Res (2012) 129(Suppl 2):S8–10. doi:10.1016/j.thromres.2012.02.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
55. Wildgoose P, Nemerson Y, Hansen LL, Nielsen FE, Glazer S, Hedner U. Measurement of basal levels of factor VIIa in hemophilia A and B patients. Blood (1992) 80(1):25–8.
56. McMichael M. New models of hemostasis. Top Companion Anim Med (2012) 27(2):40–5. doi:10.1053/j.tcam.2012.07.005
57. Giesen PL, Rauch U, Bohrmann B, Kling D, Roqué M, Fallon JT, et al. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci U S A (1999) 96(5):2311–5. doi:10.1073/pnas.96.5.2311
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
58. Zillmann A, Luther T, Müller I, Kotzsch M, Spannagl M, Kauke T, et al. Platelet-associated tissue factor contributes to the collagen-triggered activation of blood coagulation. Biochem Biophys Res Commun (2001) 281(2):603–9. doi:10.1006/bbrc.2001.4399
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
59. Müller I, Klocke A, Alex M, Kotzsch M, Luther T, Morgenstern E, et al. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J (2003) 17(3):476–8. doi:10.1096/fj.02-0574fje
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
60. Maugeri N, Brambilla M, Camera M, Carbone A, Tremoli E, Donati MB, et al. Human polymorphonuclear leukocytes produce and express functional tissue factor upon stimulation. J Thromb Haemost (2006) 4(6):1323–30. doi:10.1111/j.1538-7836.2006.01968.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
61. Moosbauer C, Morgenstern E, Cuvelier SL, Manukyan D, Bidzhekov K, Albrecht S, et al. Eosinophils are a major intravascular location for tissue factor storage and exposure. Blood (2007) 109(3):995–1002. doi:10.1182/blood-2006-02-004945
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
62. Darbousset R, Thomas GM, Mezouar S, Frère C, Bonier R, Mackman N, et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood (2012) 120(10):2133–43. doi:10.1182/blood-2012-06-437772
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
63. Reinhardt C, von Brühl ML, Manukyan D, Grahl L, Lorenz M, Altmann B, et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J Clin Invest (2008) 118(3):1110–22. doi:10.1172/JCI32376
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
64. Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest (2008) 118(3):1123–31. doi:10.1172/JCI34134
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
65. Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L, et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest (2012) 122(6):2104–13. doi:10.1172/JCI61228
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
66. Rivers RP, Hathaway WE, Weston WL. The endotoxin-induced coagulant activity of human monocytes. Br J Haematol (1975) 30(3):311–6. doi:10.1111/j.1365-2141.1975.tb00547.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
67. Smith SA. The cell-based model of coagulation. J Vet Emerg Crit Care (San Antonio) (2009) 19(1):3–10. doi:10.1111/j.1476-4431.2009.00389.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
68. Revak SD, Cochrane CG, Griffin JH. The binding and cleavage characteristics of human Hageman factor during contact activation. A comparison of normal plasma with plasmas deficient in factor XI, prekallikrein, or high molecular weight kininogen. J Clin Invest (1977) 59(6):1167–75. doi:10.1172/JCI108741
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
69. Smith SA, Choi SH, Davis-Harrison R, Huyck J, Boettcher J, Rienstra CM, et al. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood (2010) 116(20):4353–9. doi:10.1182/blood-2010-01-266791
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
70. Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci U S A (2006) 103(4):903–8. doi:10.1073/pnas.0507195103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
71. Renne T, Schmaier AH, Nickel KF, Blomback M, Maas C. In vivo roles of factor XII. Blood (2012) 120(22):4296–303. doi:10.1182/blood-2012-07-292094
72. Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A (2007) 104(15):6388–93. doi:10.1073/pnas.0608647104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
73. Oehmcke S, Morgelin M, Herwald H. Activation of the human contact system on neutrophil extracellular traps. J Innate Immun (2009) 1(3):225–30. doi:10.1159/000203700
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
74. Kunapuli SP, DeLa Cadena RA, Colman RW. Deletion mutagenesis of high molecular weight kininogen light chain. Identification of two anionic surface binding subdomains. J Biol Chem (1993) 268(4):2486–92.
75. Siebeck M, Cheronis JC, Fink E, Kohl J, Spies B, Spannagl M, et al. Dextran sulfate activates contact system and mediates arterial hypotension via B2 kinin receptors. J Appl Physiol (1985) (1994) 77(6):2675–80.
76. Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem (1991) 266(12):7353–8.
77. Kishimoto TK, Viswanathan K, Ganguly T, Elankumaran S, Smith S, Pelzer K, et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med (2008) 358(23):2457–67. doi:10.1056/NEJMoa0803200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
78. Scott CF, Colman RW. Fibrinogen blocks the autoactivation and thrombin-mediated activation of factor XI on dextran sulfate. Proc Natl Acad Sci U S A (1992) 89(23):11189–93. doi:10.1073/pnas.89.23.11189
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
79. Smith SA. Overview of hemostasis. In: Weiss D, Wardrop J, editors. Schlam’s Veterinary Hematology. Philadelphia, PA: Lippincott Williams and Wilkins (2011). p. 635–53.
80. Oehmcke S, Herwald H. Contact system activation in severe infectious diseases. J Mol Med (Berl) (2010) 88(2):121–6. doi:10.1007/s00109-009-0564-y
81. Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood (2010) 115(13):2569–77. doi:10.1182/blood-2009-09-199182
82. Renné T, Pozgajová M, Grüner S, Schuh K, Pauer HU, Burfeind P, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med (2005) 202(2):271–81. doi:10.1084/jem.20050664
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
83. Gailani D, Renne T. The intrinsic pathway of coagulation: a target for treating thromboembolic disease? J Thromb Haemost (2007) 5(6):1106–12. doi:10.1111/j.1538-7836.2007.02446.x
84. Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood (2008) 111(8):4113–7. doi:10.1182/blood-2007-10-120139
85. Van Der Meijden PE, Van Schilfgaarde M, Van Oerle R, Renne T, ten Cate H, Spronk HM. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J Thromb Haemost (2012) 10(7):1355–62. doi:10.1111/j.1538-7836.2012.04758.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
86. Lu G, Broze GJ Jr, Krishnaswamy S. Formation of factors IXa and Xa by the extrinsic pathway: differential regulation by tissue factor pathway inhibitor and antithrombin III. J Biol Chem (2004) 279(17):17241–9. doi:10.1074/jbc.M312827200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
87. Baugh RJ, Broze GJ Jr, Krishnaswamy S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J Biol Chem (1998) 273(8):4378–86. doi:10.1074/jbc.273.8.4378
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
88. Osterud B, Rapaport SI. Activation of factor IX by the reaction product of tissue factor and factor VII: additional pathway for initiating blood coagulation. Proc Natl Acad Sci U S A (1977) 74(12):5260–4. doi:10.1073/pnas.74.12.5260
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
89. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med (2008) 359(9):938–49. doi:10.1056/NEJMra0801082
90. Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood (2011) 118(26):6963–70. doi:10.1182/blood-2011-07-368811
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
91. Davis AE III. Biological effects of C1 inhibitor. Drug News Perspect (2004) 17(7):439–46. doi:10.1358/dnp.2004.17.7.863703
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
92. Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol (2008) 632:71–9.
93. Ganter MT, Brohi K, Cohen MJ, Shaffer LA, Walsh MC, Stahl GL, et al. Role of the alternative pathway in the early complement activation following major trauma. Shock (2007) 28(1):29–34. doi:10.1097/shk.0b013e3180342439
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
94. Hecke F, Schmidt U, Kola A, Bautsch W, Klos A, Kohl J. Circulating complement proteins in multiple trauma patients – correlation with injury severity, development of sepsis, and outcome. Crit Care Med (1997) 25(12):2015–24. doi:10.1097/00003246-199712000-00019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
95. Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost (2006) 4(9):2035–42. doi:10.1111/j.1538-7836.2006.02065.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
96. Saggu G, Cortes C, Emch HN, Ramirez G, Worth RG, Ferreira VP. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol (2013) 190(12):6457–67. doi:10.4049/jimmunol.1300610
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
97. Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med (2005) 201(6):871–9. doi:10.1084/jem.20041497
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
98. Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med (2006) 12(6):682–7. doi:10.1038/nm1419
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
99. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol (2010) 185(9):5628–36. doi:10.4049/jimmunol.0903678
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
100. Schaiff WT, Eisenberg PR. Direct induction of complement activation by pharmacologic activation of plasminogen. Coron Artery Dis (1997) 8(1):9–18. doi:10.1097/00019501-199701000-00002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
101. Ghebrehiwet B, Silverberg M, Kaplan AP. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med (1981) 153(3):665–76. doi:10.1084/jem.153.3.665
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
102. Wiggins RC, Giclas PC, Henson PM. Chemotactic activity generated from the fifth component of complement by plasma kallikrein of the rabbit. J Exp Med (1981) 153(6):1391–404. doi:10.1084/jem.153.6.1391
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
103. DiScipio RG. The activation of the alternative pathway C3 convertase by human plasma kallikrein. Immunology (1982) 45(3):587–95.
104. Krarup A, Wallis R, Presanis JS, Gal P, Sim RB. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One (2007) 2(7):e623. doi:10.1371/journal.pone.0000623
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
105. Wiedmer T, Esmon CT, Sims PJ. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood (1986) 68(4):875–80.
106. Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med (1997) 185(9):1619–27. doi:10.1084/jem.185.9.1619
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
107. Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, Niho Y. C5a induces tissue factor activity on endothelial cells. Thromb Haemost (1997) 77(2):394–8.
108. Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol (2006) 177(7):4794–802. doi:10.4049/jimmunol.177.7.4794
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
109. Wojta J, Kaun C, Zorn G, Ghannadan M, Hauswirth AW, Sperr WR, et al. C5a stimulates production of plasminogen activator inhibitor-1 in human mast cells and basophils. Blood (2002) 100(2):517–23. doi:10.1182/blood.V100.2.517
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
110. Ham TH. Chronic hemolytic anemia with paroxysmal nocturnal hemoglobinuria. A study of the mechanism of hemolysis in relation to acid-base equilibrium. N Engl J Med (1937) 217:215–7. doi:10.1056/NEJM193712022172307
111. Hinz CF Jr, Jordan WS Jr, Pıllemer L. The properdin system and immunity. IV. The hemolysis of erythrocytes from patients with paroxysmal nocturnal hemoglobinuria. J Clin Invest (1956) 35(5):453–7. doi:10.1172/JCI103296
112. Hammond DJ Jr, Singh SK, Thompson JA, Beeler BW, Rusiñol AE, Pangburn MK, et al. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J Biol Chem (2010) 285(46):36235–44. doi:10.1074/jbc.M110.142026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
113. Hecke F, Hoehn T, Strauss E, Obladen M, Sonntag J. In-vitro activation of complement system by lactic acidosis in newborn and adults. Mediators Inflamm (2001) 10(1):27–31. doi:10.1080/09629350123788
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
114. Renaux JL, Thomas M, Crost T, Loughraieb N, Vantard G. Activation of the kallikrein-kinin system in hemodialysis: role of membrane electronegativity, blood dilution, and pH. Kidney Int (1999) 55(3):1097–103. doi:10.1046/j.1523-1755.1999.0550031097.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
115. Thomas M, Valette P, Mausset AL, Dejardin P. High molecular weight kininogen adsorption on hemodialysis membranes: influence of pH and relationship with contact phase activation of blood plasma. Influence of pre-treatment with poly(ethyleneimine). Int J Artif Organs (2000) 23(1):20–6.
116. Buerke M, Murohara T, Lefer AM. Cardioprotective effects of a C1 esterase inhibitor in myocardial ischemia and reperfusion. Circulation (1995) 91(2):393–402. doi:10.1161/01.CIR.91.2.393
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
117. Heideman M, Hugli TE. Anaphylatoxin generation in multisystem organ failure. J Trauma (1984) 24(12):1038–43. doi:10.1097/00005373-198412000-00006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
118. Jones AL, Hulett MD, Parish CR. Histidine-rich glycoprotein: a novel adaptor protein in plasma that modulates the immune, vascular and coagulation systems. Immunol Cell Biol (2005) 83(2):106–18. doi:10.1111/j.1440-1711.2005.01320.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
119. Gorgani NN, Parish CR, Easterbrook Smith SB, Altin JG. Histidine-rich glycoprotein binds to human IgG and C1q and inhibits the formation of insoluble immune complexes. Biochemistry (1997) 36(22):6653–62. doi:10.1021/bi962573n
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: complement, coagulation, contact system, alternative pathway, lectin pathway, classical pathway, pH
Citation: Kenawy HI, Boral I and Bevington A (2015) Complement-coagulation cross-talk: a potential mediator of the physiological activation of complement by low pH. Front. Immunol. 6:215. doi: 10.3389/fimmu.2015.00215
Received: 25 January 2015; Paper pending published: 16 March 2015;
Accepted: 18 April 2015; Published: 06 May 2015
Edited by:
Timothy B. Niewold, Mayo Clinic, USAReviewed by:
Francesco Tedesco, University of Trieste, ItalyViviana P. Ferreira, University of Toledo, USA
Copyright: © 2015 Kenawy, Boral and Bevington. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hany Ibrahim Kenawy, Microbiology and Immunology Department, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt,aGFueS5rZW5hd3lAZ29vZ2xlbWFpbC5jb20=