 Caroline Blackwell
Caroline Blackwell Sophia Moscovis
Sophia Moscovis Sharron Hall
Sharron Hall Christine Burns1,2,3
Christine Burns1,2,3 Rodney J. Scott
Rodney J. Scott- 1Faculty of Health and Medicine, Hunter Medical Research Institute, School of Biomedical Sciences, University of Newcastle, Newcastle, NSW, Australia
- 2Information Based Medicine, Hunter Medical Research Institute, New Lambton, NSW, Australia
- 3Hunter Area Pathology Service Immunology, John Hunter Hospital, New Lambton, NSW, Australia
- 4Hunter Area Pathology Service Genetics, John Hunter Hospital, New Lambton, NSW, Australia
The risk factors for sudden infant death syndrome (SIDS) parallel those associated with susceptibility to or severity of infectious diseases. There is no evidence that a single infectious agent is associated with SIDS; the common thread appears to be induction of inflammatory responses to infections. In this review, interactions between genetic and environmental risk factors for SIDS are assessed in relation to the hypothesis that many infant deaths result from dysregulation of inflammatory responses to “minor” infections. Risk factors are assessed in relation to three important stages of infection: (1) bacterial colonization (frequency or density); (2) induction of temperature-dependent toxins; (3) induction or control of inflammatory responses. In this article, we review the interactions among risk factors for SIDS for their effects on induction or control of inflammatory responses. The risk factors studied are genetic factors (sex, cytokine gene polymorphisms among ethnic groups at high or low risk of SIDS); developmental stage (changes in cortisol and testosterone levels associated with 2- to 4-month age range); environmental factors (virus infection, exposure to cigarette smoke). These interactions help to explain differences in the incidences of SIDS observed between ethnic groups prior to public health campaigns to reduce these infant deaths.
Introduction
Sudden infant death syndrome (SIDS) is still the major cause of death between 1 month to 1 year of age among infants in industrialized countries. SIDS is a diagnosis of exclusion. The original definition was “… the sudden death of any infant or young child, which is unexpected by history, and in which a thorough post mortem examination fails to demonstrate an adequate cause of death” (1).The definition was revised in 1989 to “the sudden death of an infant under one year of age, which remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of the clinical history” (2) Comparison of epidemiological data from different countries found that infants of some ethnic groups had an increased risk of SIDS (Table 1).

Table 1. Variation in the incidence of SIDS among ethnic groups within countries before the “reduce the risks” campaigns.
The pathogenesis of SIDS has been examined by many disciplines. These have made significant contributions to the study of infant deaths and put forward hypotheses; however, many cannot explain the risk factors or the positive effects of the public health campaigns to reduce the risk (10). The idea that inflammation might be involved in these infant deaths is not new. In an article published in 1956, 126 non-traumatic sudden (“unexplained”) infant deaths were investigated; 106 (84%) revealed microscopic inflammatory changes in 1 or more sites of the respiratory tract, and there was histologic evidence of inflammatory disease in other organs in many cases (11). Table 2 summarizes some of the evidence for inflammatory responses in sudden infant deaths.
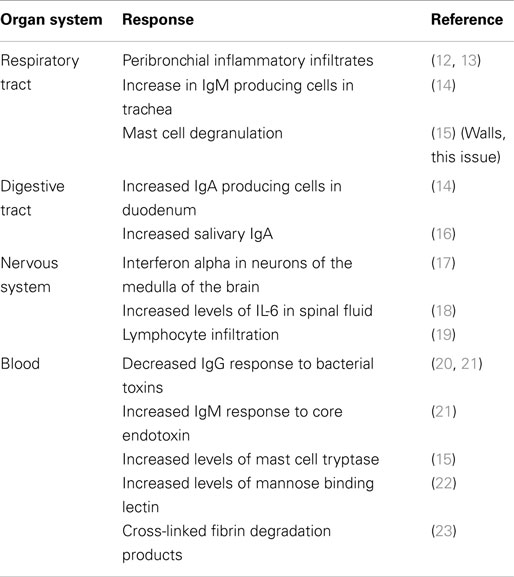
Table 2. Inflammatory or immune responses identified in SIDS infants.
Risk Factors for SIDS and Susceptibility to Infection
Epidemiological studies found significant associations between SIDS and a variety of genetic, developmental, and environmental factors (Table 3). When these factors are compared with those associated with increased susceptibility to bacterial infections, there are remarkable parallels. Each of these factors can affect one or more of the important stages in susceptibility to or severity of infections: frequency or density of bacterial colonization of mucosal surfaces; induction of temperature-dependent toxins; induction or control of inflammatory responses to infection. Each of these is described in detail below.
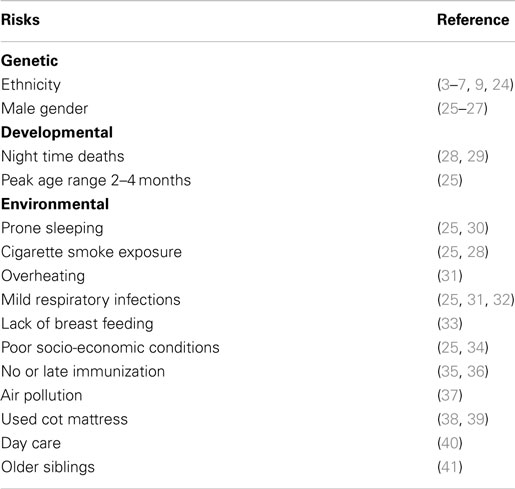
Table 3. Risk factors for SIDS that parallel risk factors for susceptibility of infants to infection.
Infectious agents or their products have been identified during autopsies of SIDS infants and more recently in sudden unexpected death of infancy (SUDI), which is defined as the sudden and unexpected death of an infant under 1 year of age; SUDI includes explained deaths and those that, after investigation of the death scene and meticulous post-mortem examination, remain unexplained (42). For many years, microbiological findings were dismissed as contamination, overgrowth, or normal flora. More recent studies have questioned these assertions and have provided evidence to support the likelihood that post-mortem bacteriology is a true reflection of infection in these infant deaths (42–44). The common bacterial toxin hypothesis (45–47) considered a number of the risk factors in a mathematical model. It has been supported by identification of potentially toxigenic bacteria or their toxins in SIDS and sudden unexpected deaths in infancy (SUDI) (Table 4). Many of the bacterial toxins or components of the bacteria implicated can act as super antigens, eliciting powerful cytokine storm responses such as those seen in toxic shock syndrome or bacterial sepsis.
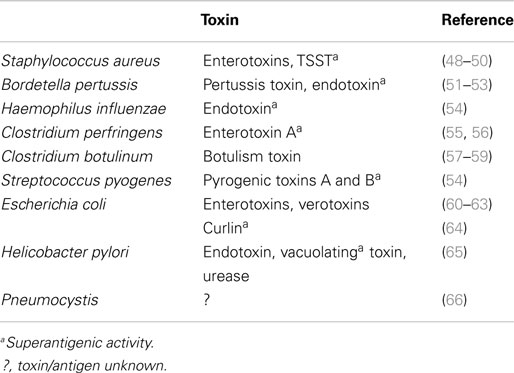
Table 4. Toxigenic bacteria and their toxins implicated in sudden death in infancy.
The infection hypothesis does not fit Koch’s postulates. That is, there is no single organism implicated and there is no widely accepted animal model that reflects the genetic, developmental, and environmental risk factors identified in epidemiological studies (see Blood-Siegfried, this issue). In addition, a variety of viruses have been identified in SIDS infants (67–69). Sterile site infections have been identified in SUDI, and a variety of toxigenic bacteria or their toxins have been reported in these infant deaths (Table 4). Our hypothesis is that there is not a particular organism or toxin, the factor in these deaths is the dysregulation of the inflammatory responses elicited by what appear to be mild or asymptomatic infections.
The objective of this review was to assess how the risk factors identified in epidemiological studies of SIDS affect susceptibility to infection and/or alter inflammatory responses to infections. It addresses the interactions between these identified risk factors and the three key stages of infection: (1) increased frequency or density of bacterial colonization; (2) induction of temperature-dependent toxins; (3) induction or control of inflammatory responses (Table 5).

Table 5. How do risk factors for SIDS affect susceptibility to infection.
Bacterial Colonization
Factors affecting colonization have been elucidated in previous studies. Virus infections, which often precede SIDS or SUDI, can enhance bacterial binding through induction of host receptors for bacteria or induction of new receptors (70, 71). The prone position can lead to pooling of respiratory secretions and increased numbers and varieties of bacteria, particularly in the presence of virus infection (72). Active smoking can predispose individuals to virus infections, and smokers are colonized more heavily and more frequently with potential pathogens (73). In addition to enhancing susceptibility to virus infections, material in cigarette smoke can passively coat epithelial cells and enhance “stickiness” for potential pathogens (74). Exposure of infants to new infectious agents can be enhanced by day care with other children or older siblings attending nursery or school outside the home environment.
Temperature-Dependent Toxins
The pyrogenic toxins of Staphylococcus aureus have been identified in over half of SIDS infants from five different countries (50). S. aureus is the most common isolate from the nasopharynx of healthy infants, and 64% of these have the capacity to produce these toxins (75). The toxins are induced only between 37 and 40°C, and the temperature of the nasopharynx is usually below this range (76). In the prone position, the temperature in the nasopharynx of children is increased and 15% had temperatures ≥37°C. While the toxigenic organisms are present in most infants, induction of the toxin is likely to be dependent on risk factors such as overheating, prone position, or virus infection with associated blocked nasal passages that result in reduced cooling effects of the passage of air.
Inflammation and SIDS
The common thread in these deaths is not a single organism or toxin; it is dysregulation of the innate inflammatory responses in the non-immune infant or young child who encounters a new potential pathogen. There is recent evidence that a balanced inflammatory response during infancy is important for survival. In a study of low birth weight infants, cytokine responses were assessed in vitro by stimulation of whole blood cultures with lipopolysaccharide (LPS), phytohemagglutinin (PHA), or purified protein derivative (PPD). Infants whose cytokine responses to LPS or PHA were very low or very high were at increased risk of death assessed by survival of the cohort tested. The interpretation of these findings was that a balanced response to new pathogens was more likely to result in survival (77). A variety of bacteria implicated in SIDS/SUDI possess structural antigens or exotoxins that can act as superantigens (e.g., LPS) that can activate large populations of inflammatory cells (Table 3).
Inflammation can be assessed from clinical evidence, histological findings, or molecular markers. Clinical evidence of inflammation has been identified in some of these infant deaths. Infants were very hot when found and remained hot for several hours after death (78). Histological signs of inflammation are considered the gold standard by the clinical pathologist and some of these are summarized in Table 2. It has been suggested that molecular markers of inflammation need to be measured; however, these are relatively expensive and are not usually included in standard autopsy protocols. To date, there have been few studies of inflammatory markers in sudden deaths (18, 79, 80). Because these components are not regularly examined during autopsy, co-operation with a research group with an interest in inflammation would help to obtain evidence for these markers.
Inflammation in Relation to Proposed Mechanisms of Death in SIDS
Cytokines produced in the inflammatory responses to infectious agents could have an impact on most of the mechanisms of death proposed: anaphylaxis; poor arousal; hypoxia and apnea; shock; cardiac arrythmias; hyperthermia; hypoglycemia (81). Anaphylaxis was the first mechanism proposed in an animal model in which guinea pigs were sensitized to cow’s milk (82, 83). Studies of SIDS infants have not found evidence to support the hypothesis that IgE-mediated anaphylaxis is involved in these infant deaths. There is evidence, however, for degranulation of mast cells in some infants (15) (Walls, this issue). In addition, in an in vitro study of first degree relatives of SIDS infants or infants who had suffered an acute life threatening episode (ALTE), there was evidence of increased mast cell hyper-releasability and degranulation (84). This could be mediated by some of the pyrogenic staphylococcal toxins through non-antibody activation of mast cells. The toxins, which are super antigens, can bind directly to the Vβ antigens on mast cells and trigger degranulation independent of antibody and complement. A mild virus infection might enhance colonization by S. aureus and increased temperature of the oropharynx due to blocked nasal passages. This could result in temperatures that are permissive for induction of the staphylococcal toxins. Increased production of interferon-γ (IFN-γ) and/or other mediators could enhance expression of Vβ antigens which are receptors for the toxins. Cross-linking of the Vβ antigens by the toxins could lead to non-IgE-mediated degranulation of mast cells (Figure 1). Evidence of mast cell tryptase and other products of mast cells have been published (15) (Walls, this volume).
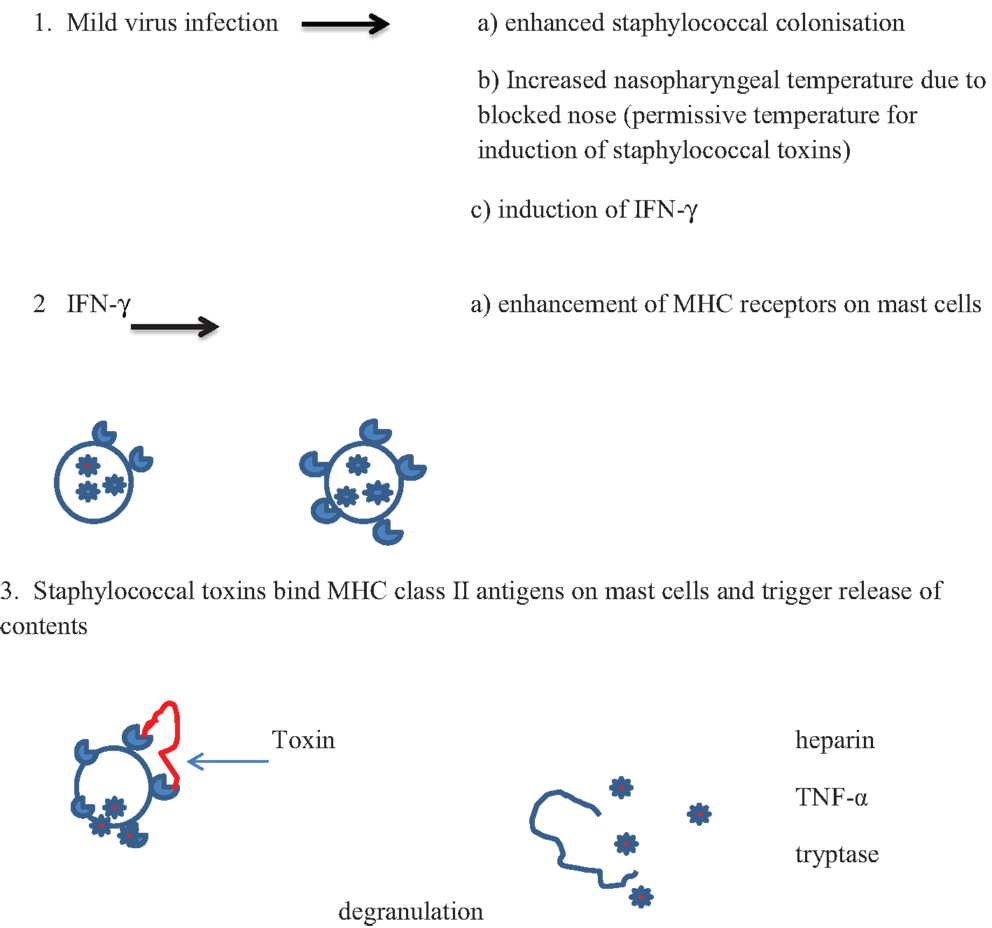
Figure 1. Proposed scheme for interactions between virus infections and pyrogenic staphylococcal toxins in non-antibody-mediated induction of mast cell degranulation (see text).
In vitro Studies of Inflammation in Human Material
The inflammatory responses can be affected by genetic, developmental, and environmental factors. In vitro studies need to consider these potential confounding factors. Taking blood from young infants is not practical for most studies, so cell lines or adult leukocytes have been used to assess the effects of risk factors for SIDS on inflammation. In addition to controlling for confounding factors, it is important to use biologically relevant concentrations of substances to be assessed in the model system (85).
While bacteria and their toxins have been implicated in SIDS, there is epidemiological and experimental evidence for virus infections acting as a cofactor. Many parents reported their child had a mild cold or sniffle prior to death. The presence of a risk factor such as prone sleeping, head covered, or parental smoking combined with infection was associated with a greater risk of SIDS than the individual risk factor alone (32).
In animal models, the lethal doses of both staphylococcal exotoxins (86) and endotoxin (87–89) were significantly reduced if the animals had mild/asymptomatic virus infection. Virus infections induce IFN-γ, which can significantly enhance production of pro-inflammatory cytokines (Moscovis et al., this volume). IL-6 has been identified in the CSF of SIDS infants (18). We demonstrated a dose-dependent enhancement by IFN-γ of IL-6 elicited by LPS in a human cell line and human peripheral blood monocytic cells (PBMC) (85). In addition, in a model system, IFN-γ has been demonstrated to reduce significantly the anti-inflammatory IL-10 responses elicited from human PBMC exposed to endotoxin (Moscovis et al., this volume).
In contrast, components of cigarette smoke, such as nicotine (or its liver metabolite cotinine), can suppress endotoxin-induced IFN-γ, IL-1β, TNF-α, and IL-8. There are, however, limitations in using single purified reagents for in vitro studies as the responses elicited might not reflect accurately the complex interactions that occur in vivo. Nicotine is only one of approximately 4,000 chemicals in cigarette smoke. A water soluble cigarette smoke extract (CSE) enhanced IL-8 responses but reduced other pro-inflammatory cytokines (85). In vitro studies found cells from smokers produced significantly lower anti-inflammatory IL-10 responses than cells of non-smokers (90). This could impair the ability to control pro-inflammatory responses to infection.
Developmental Stage and SIDS
The mathematical model used for the common bacterial toxin hypothesis predicted the 2- to 4-month age range as that during which the peak of SIDS occurred. This prediction fits the epidemiological data. There are a number of factors that could contribute to susceptibility to infection during his period: (1) loss of maternal antibody; (2) development of circadian rhythms and the associated changes in night time cortisol; (3) the testosterone surge in males between 1 and 5 months.
Antibody Levels and the Effects of Immunization
An important factor is the loss of maternal antibodies, which make the infants more reliant on their innate inflammatory responses to cope with new infections. This can include exposure to new infections brought home by older siblings or attendance at day care, both of which have been identified as risk factors for SIDS in epidemiological studies. Immunization has been demonstrated to be a protective factor in relation to SIDS (35). Most infants start their immunizations during the period of peak vulnerability, but they will not be fully immunized to common childhood illnesses implicated in some epidemiological studies (e.g., whooping cough) by 4 months of age. The protective effect of immunization was noted in early studies (35), and a shift in the age range of SIDS in infants in both Scotland and England was observed following changes in the immunization of infants in UK. That is, immunization began at 2 months instead of 3 months (91). It has also been demonstrated that some cross protection against staphylococcal exotoxins was elicited in animals by tetanus toxoid (91).
Development of Circadian Rhythm and Hormonal Changes
During the first months of life, significant physiological changes occur which could affect control of the inflammatory responses. Circadian rhythm develops between 4 and 16 weeks in Caucasian infants in Britain, but this does not occur until 12–20 weeks in Asian infants (92). The switch to circadian rhythm is measured by the night time drop in core body temperature. This physiological switch is accompanied by a dramatic drop in night time, but not daytime, cortisol levels (Figure 2).
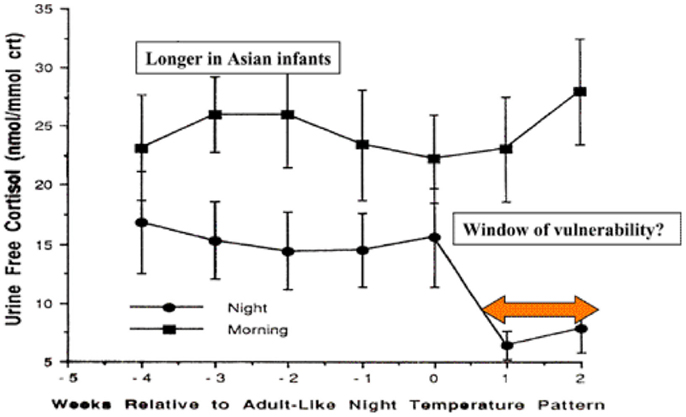
Figure 2. Day time and night time cortisol levels of infants in relation to development of circadian rhythm and potential for control of inflammatory responses to infection [adapted from Ref. (92)].
The levels of night time cortisol after the switch were not capable of reducing pro-inflammatory responses in a model system examining inflammatory responses of peripheral blood monocytes to staphylococcal toxins (93). Daytime levels and night time levels prior to the switch were able to reduce these responses. If these in vitro studies reflect the activity of cortisol in vivo in the infant, this 2- to 4-month period could be a window of vulnerability to new infections, particularly at night. The circadian switch occurs later among Asian infants (12–20 weeks), which allows time for natural exposure to infection or immunization to boost specific immunity. Prior to the public health campaigns to reduce the risks of SIDS, there was a lower incidence of SIDS among Asian infants in UK compared with infants of European origin (4).
The Effect of Testosterone
The male excess in SIDS noted in epidemiological studies and assessed in mathematical models (26) might reflect hormonal changes that occur during the period in which SIDS is most prevalent. There is a rise in testosterone production associated with the period during which SIDS is most prevalent. Between 1 and 5 months, testosterone levels range from 0.03 to 6.14 nMol L−1 for males and 0.03 to 0.17 nMol L−1 for females. In males, these levels decrease to 0.07–0.24 at 6–11 months (94). In a previous study (95), the ranges of testosterone in the adult females (<0.4–3.1 nMol L−1) tested were within the range for males in the 1- to 5-month age range (94). In the model system examined, there was a positive correlation between testosterone levels and pro-inflammatory responses to LPS when the cells were pre-treated with IFN-γ or IFN-γ and CSE. No correlations were observed for the higher levels (6.3–21 nMol L−1) found in adult males (95).
These findings indicate that in addition to low levels of night time cortisol present in infants over the age range of greatest risk of SIDS (93), dysregulation of the inflammatory responses to apparently “mild” infections might be amplified by the increase in testosterone in male infants at this time. As this does not occur in female infants, this could be an additional factor contributing to the higher proportion of males among SIDS infants. In vitro studies using the PBMC model might provide additional insights into the interactions between cortisol and testosterone levels noted in infants during this critical age range.
Interactions with Genetic Factors
Among the ethnic groups at increased risk of SIDS (e.g., indigenous populations in Australia and North America and Black Americans), there is a higher proportion of genotypes associated with strong pro-inflammatory responses (96–98). While these disparities have been ascribed primarily to socio-economic disadvantage, there is emerging evidence that genetic background and interactions between environmental factors such as cigarette smoke might contribute to susceptibility and severity of infections (85, 90). Of particular importance is the effect of cigarette smoke on the genotype of IL-10 (G-1082A) associated with lower levels of the anti-inflammatory response. The AA genotype is predominant among the groups at higher risk of SIDS, and in our in vitro studies, cells from smokers with this genotype had the lowest responses (90). Among the ethnic groups at increased risk of SIDS, there is also a higher incidence of maternal smoking. South Asians have genetic profiles similar to those for the higher risk groups; however, smoking is much less prevalent among south Asian women in UK.
Our experimental studies indicate that higher IFN-γ responses might elicit higher pro-inflammatory responses to bacterial toxins (Moscovis et al., this volume). Indigenous Australians tested had the highest proportion of individuals with the TT genotype of IFNG T + 874A (96), which is associated with high IFN-γ responses observed in vitro (99).
Conclusion
There is a wealth of knowledge about the risk factors for sudden death in infancy; it is important, however, to attempt to explain how these risk factors could result in death. There is a growing body of evidence that infection and inflammatory responses might trigger the events leading to sudden death in infancy. A recent review on infectious causes of SIDS concluded no specific organism was involved in SIDS (100). The evidence of Anderson et al. (77) indicates that a balanced inflammatory response is important for dealing with new infections. Our hypothesis is that the common thread in these deaths is dysregulation of the inflammatory responses to apparently mild infections. The risk factors identified in epidemiological studies can have significant effects on inflammatory responses that could affect the different physiological mechanisms proposed to explain these infant deaths (81). Many of the genetic, developmental, and environmental factors identified could affect this balance resulting in enhanced pro-inflammatory cytokine levels, which can affect glucose levels, heart rate, apnea, arousal, anaphylaxis, and shock. The components of the inflammatory responses involved might differ in individual children; however, it is the dysregulation of the responses that lead to the death of the child. We need to investigate further the interactions between genetic, developmental, and environmental risk factors on inflammatory responses to attempt to identify infants at increased risk and to attempt to introduce measures to prevent induction of these lethal responses.
Author Contributions
Each of the authors made substantial contributions to the conception, design, analyses, and interpretations of the work. They assisted in preparing the article, critically assessed the final version, and agree to be accountable for the accuracy and integrity of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was funded by the Foundation for the Study of Infant Death (FSID) (UK), Babes in Arms (UK), Hunter Medical Research Institute (HMRI), the University of Newcastle, and the John Hunter Children’s Research Foundation.
References
1. Beckwith J. Discussion of terminology and definition of the sudden infant death syndrome. In: Bergman AB, Beckwith J, Ray G, editors. Sudden Infant Death Syndrome; Proceedings of the Second International Conference on the Causes of Sudden Death in Infants. Seattle: University of Washington Press (1970). p. 14–22.
2. Rognum TO. Definition and pathologic features. In: Byard R, Krouse H, editors. Sudden Infant Death Syndrome: Problems, Progress and Possibilities. London: Arnold (2001). p. 4–30.
3. Alessandri LM, Read AW, Stanley FJ, Burton PR, Dawes VP. Sudden infant death syndrome in aboriginal and non-aboriginal infants. J Paediatr Child Health (1994) 30(3):234–41. doi: 10.1111/j.1440-1754.1994.tb00625.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Balarajan R, Soni Raleigh V, Botting B. Sudden infant death syndrome and postneonatal mortality in immigrants in England and Wales. BMJ (1989) 298(6675):716–20. doi:10.1136/bmj.298.6675.716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Bulterys M. High incidence of sudden infant death syndrome among Northern Indians and Alaska natives compared with Southwestern Indians: possible role of smoking. J Community Health (1990) 15(3):185–94. doi:10.1007/BF01350256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Adams MM. The descriptive epidemiology of sudden infant deaths among natives and whites in Alaska. Am J Epidemiol (1985) 122(4):637–43.
7. Spiers PS, Guntheroth WG. The black infant’s susceptibility to sudden infant death syndrome and respiratory infection in late infancy. Epidemiology (2001) 12(1):33–7. doi:10.1097/00001648-200101000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Mitchell EA, Scragg R. Observations on ethnic differences in SIDS mortality in New Zealand. Early Hum Dev (1994) 38(3):151–7. doi:10.1016/0378-3782(94)90206-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Mitchell EA, Stewart AW, Scragg R, Ford RP, Taylor BJ, Becroft DM, et al. Ethnic differences in mortality from sudden infant death syndrome in New Zealand. BMJ (1993) 306(6869):13–6. doi:10.1136/bmj.306.6869.13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Goldwater PN, Bettelheim KA. Pediatr Res Intern J [Internet]. (2013); 2013:[14 p.]. Available from: http://www.ibimapublishing.com/journals/PRIJ/2013/867520/a867520.html.
11. Adelson L, Kinney ER. Sudden and unexpected death in infancy and childhood. Pediatrics (1956) 17(5):663–99.
12. Baxendine JA, Moore IE. Pulmonary eosinophilia in sudden infant death syndrome. J Pathol (1995) 177(4):415–21. doi:10.1002/path.1711770413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Howat WJ, Moore IE, Judd M, Roche WR. Pulmonary immunopathology of sudden infant death syndrome. Lancet (1994) 343(8910):1390–2. doi:10.1016/S0140-6736(94)92523-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Stoltenberg I, Vege A, Opdal S, Saugstad O, Rognum TO. Does immunostimulation play a role in SIDS? In: Rognum T, editor. Sudden Infant Death Syndrome, New Trends in the Nineties. Oslo: Scandinavian University Press (1995). p. 179–81.
15. Holgate ST, Walters C, Walls AF, Lawrence S, Shell DJ, Variend S, et al. The anaphylaxis hypothesis of sudden infant death syndrome (SIDS): mast cell degranulation in cot death revealed by elevated concentrations of tryptase in serum. Clin Exp Allergy (1994) 24(12):1115–22. doi:10.1111/j.1365-2222.1994.tb03316.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Gleeson M, Clancy RL, Cripps AW. Mucosal immune response in a case of sudden infant death syndrome. Pediatr Res (1993) 33(6):554–6. doi:10.1203/00006450-199306000-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Howatson AG. Viral infection and alpha interferon in SIDS. J Clin Pathol (1992) 45(11 Suppl):25–8.
18. Vege Å, Rognum TO, Scott H, Aasen AO, Saugstad OD. S cases have increased levels of interleukin-6 in cerebrospinal fluid. Acta Paediatr (1995) 84(2):193–6. doi:10.1111/j.1651-2227.1995.tb13608.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Morris JA, Harrison LM, Telford DR. Postmortem cerebrospinal fluid pleocytosis: a marker of inflammation or postmortem artifact? Int J Pediatr (2012) 2012:964074. doi:10.1155/2012/964074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Siarakas S, Brown AJ, Murrell WG. Immunological evidence for a bacterial toxin aetiology in sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25(1–2):37–50. doi:10.1111/j.1574-695X.1999.tb01325.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Oppenheim BA, Barclay GR, Morris J, Knox F, Barson A, Drucker DB, et al. Antibodies to endotoxin core in sudden infant death syndrome. Arch Dis Child (1994) 70(2):95–8. doi:10.1136/adc.70.2.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Kilpatrick DC, James VS, Blackwell CC, Weir DM, Hallam NF, Busuttil A. Mannan binding lectin and the sudden infant death syndrome. Forensic Sci Int (1998) 97(2–3):135–8. doi:10.1016/S0379-0738(98)00149-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Goldwater PN, Williams V, Bourne AJ, Byard RW. Sudden infant death syndrome: a possible clue to causation. Med J Aust (1990) 153(1):59–60.
25. Fleming P, Blair P, Bacon C, Berry P. The CESDI SUDI Studies 1993-1996. London: The Stationery Office (2000).
26. Mage DT, Donner M. A unifying theory for SIDS. Int J Pediatr (2009) 368270(10):29. doi:10.1155/2009/368270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Brooke H, Gibson A, Tappin D, Brown H. Case-control study of sudden infant death syndrome in Scotland, 1992-5. BMJ (1997) 314(7093):1516–20. doi:10.1136/bmj.314.7093.1516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Daltveit AK, Irgens LM, Øyen N, Skjærven R, Markestad T, Wennergren G. Circadian variations in sudden infant death syndrome: associations with maternal smoking, sleeping position and infections. The Nordic epidemiological SIDS study. Acta Paediatr (2003) 92(9):1007–13. doi:10.1080/08035250310004360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Mitchell EA, Williams SM. Does circadian variation in risk factors for sudden infant death syndrome (SIDS) suggest there are two (or more) SIDS subtypes? Acta Paediatr (2003) 92(9):991–3. doi:10.1111/j.1651-2227.2003.tb02561.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Gilbert R, Salanti G, Harden M, See S. Infant sleeping position and the sudden infant death syndrome: systematic review of observational studies and historical review of recommendations from 1940 to 2002. Int J Epidemiol (2005) 34(4):874–87. doi:10.1093/ije/dyi088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Gilbert R, Rudd P, Berry PJ, Fleming PJ, Hall E, White DG, et al. Combined effect of infection and heavy wrapping on the risk of sudden unexpected infant death. Arch Dis Child (1992) 67(2):171–7. doi:10.1136/adc.67.2.171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Helweg-Larsen K, Lundemose JB, Ôyen N, Skjærven R, Alm B, Wennergren G, et al. Interactions of infectious symptoms and modifiable risk factors in sudden infant death syndrome. The Nordic epidemiological SIDS study. Acta Paediatr (1999) 88(5):521–7. doi:10.1111/j.1651-2227.1999.tb00168.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Alm B, Wennergren G, Norvenius SG, Skjaerven R, Lagercrantz H, Helweg-Larsen K, et al. Breast feeding and the sudden infant death syndrome in Scandinavia, 1992-95. Arch Dis Child (2002) 86(6):400–2. doi:10.1136/adc.86.6.400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Fleming PJ, Blair PS, Ward Platt M, Tripp J, Smith IJ, Group CSR. Sudden infant death syndrome and social deprivation: assessing epidemiological factors after post-matching for deprivation. Paediatr Perinat Epidemiol (2003) 17(3):272–80. doi:10.1046/j.1365-3016.2003.00465.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Hoffman HJ, Hunter JC, Damus K, Pakter J, Peterson DR, van Belle G, et al. Diphtheria-tetanus-pertussis immunization and sudden infant death: results of the national institute of child health and human development cooperative epidemiological study of sudden infant death syndrome risk factors. Pediatrics (1987) 79(4):598–611.
36. Fleming PJ, Blair PS, Platt MW, Tripp J, Smith IJ, Golding J. The UK accelerated immunisation programme and sudden unexpected death in infancy: case-control study. BMJ (2001) 322(7290):822. doi:10.1136/bmj.322.7290.822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Dales R, Burnett RT, Smith-Doiron M, Stieb DM, Brook JR. Air pollution and sudden infant death syndrome. Pediatrics (2004) 113(6):e628–31. doi:10.1542/peds.113.6.e628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Sherburn RE, Jenkins RO. Cot mattresses as reservoirs of potentially harmful bacteria and the sudden infant death syndrome. FEMS Immunol Med Microbiol (2004) 42(1):76–84. doi:10.1016/j.femsim.2004.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Tappin D, Brooke H, Ecob R, Gibson A. Used infant mattresses and sudden infant death syndrome in Scotland: case-control study. BMJ (2002) 325(7371):1007. doi:10.1136/bmj.325.7371.1007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Moon RY, Patel KM, Shaefer SJ. Sudden infant death syndrome in child care settings. Pediatrics (2000) 106(2 Pt 1):295–300. doi:10.1542/peds.106.2.295
41. Daltveit AK, Øyen N, Skjærven R, Irgens LM. The epidemic of SIDS in Norway 1967-93: changing effects of risk factors. Arch Dis Child (1997) 77(1):23–7. doi:10.1136/adc.77.1.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Weber MA, Klein NJ, Hartley JC, Lock PE, Malone M, Sebire NJ. Infection and sudden unexpected death in infancy: a systematic retrospective case review. Lancet (2008) 371(9627):1848–53. doi:10.1016/S0140-6736(08)60798-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Morris JA, Harrison LM, Partridge SM. Postmortem bacteriology: a re-evaluation. J Clin Pathol (2006) 59(1):1–9. doi:10.1136/jcp.2005.028183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Goldwater PN. Sterile site infection at autopsy in sudden unexpected deaths in infancy. Arch Dis Child (2009) 94(4):303–7. doi:10.1136/adc.2007.135939
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Morris JA, Haran D, Smith A. Hypothesis: common bacterial toxins are a possible cause of the sudden infant death syndrome. Med Hypotheses (1987) 22(2):211–22. doi:10.1016/0306-9877(87)90145-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Morris JA. The common bacterial toxins hypothesis of sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25(1–2):11–7. doi:10.1111/j.1574-695X.1999.tb01322.x
47. Morris JA. Common bacterial toxins and physiological vulnerability to sudden infant death: the role of deleterious genetic mutations. FEMS Immunol Med Microbiol (2004) 42(1):42–7. doi:10.1016/j.femsim.2004.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Malam JE, Carrick GF, Telford DR, Morris JA. Staphylococcal toxins and sudden infant death syndrome. J Clin Pathol (1992) 45(8):716–21. doi:10.1136/jcp.45.8.716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Zorgani A, Essery SD, Madani OA, Bentley AJ, James VS, MacKenzie DA, et al. Detection of pyrogenic toxins of Staphylococcus aureus in sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25(1–2):103–8. doi:10.1111/j.1574-695X.1999.tb01332.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Blackwell CC, Gordon AE, James VS, MacKenzie DA, Mogensen-Buchanan M, El Ahmer OR, et al. The role of bacterial toxins in sudden infant death syndrome (SIDS). Int J Med Microbiol (2002) 291(6–7):561–70. doi:10.1078/1438-4221-00168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lindgren C, Milerad J, Lagercrantz H. Sudden infant death and prevalence of whooping cough in the Swedish and Norwegian communities. Eur J Pediatr (1997) 156(5):405–9. doi:10.1007/s004310050626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Heininger U, Stehr K, Schmidt-Schläpfer G, Penning R, Vock R, Kleemann W, et al. Bordetella pertussis infections and sudden unexpected deaths in children. Eur J Pediatr (1996) 155(7):551–3. doi:10.1007/BF01957903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Heininger U, Kleemann WJ, Cherry JD, Group SIDSS. A controlled study of the relationship between Bordetella pertussis infections and sudden unexpected deaths among German infants. Pediatrics (2004) 114(1):e9–15. doi:10.1542/peds.114.1.e9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Telford DR, Morris JA, Hughes P, Conway AR, Lee S, Barson AJ, et al. The nasopharyngeal bacterial flora in the sudden infant death syndrome. J Infect (1989) 18(2):125–30.
55. Murrell WG, Stewart BJ, O’Neill C, Siarakas S, Kariks S. Enterotoxigenic bacteria in the sudden infant death syndrome. J Med Microbiol (1993) 39(2):114–27. doi:10.1099/00222615-39-2-114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Lindsay J, Mach A, Wilkinson M, Martin LM, Wallace FM, Keller A, et al. Clostridium perfringens type A cytotoxic-enterotoxin(s) as triggers for death in the sudden infant death syndrome: development of a toxico-infection hypothesis. Curr Microbiol (1993) 27(1):51–9. doi:10.1007/BF01576834
57. Arnon SS, Damus K, Chin J. Infant botulism: epidemiology and relation to sudden infant death syndrome. Epidemiol Rev (1981) 3(1):45–66.
58. Arnon S, Midura T, Damus K, Wood R, Chin J. Intestinal infection and toxin production by Clostirdium botulinum as one cause of sudden infant death syndrome. Lancet (1978) 311(8077):1273–7. doi:10.1016/S0140-6736(78)91264-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Sonnabend OR, Sonnabend WF, Krech U, Molz G, Sigrist T. Continuous microbiological and pathological study of 70 sudden and unexpected infant deaths: toxigenic intestinal Clostridium botulinum infection in 9 cases of sudden infant death syndrome. The Lancet (1985) 325(8423):237–41.
60. Bettelheim KA, Dwyer BW, Smith DL, Goldwater PN, Bourne AJ. Toxigenic Escherichia coli associated with sudden infant death syndrome. Med J Aust (1989) 151(9):538.
61. Bettelheim KA, Goldwater PN, Dwyer BW, Bourne AJ, Smith DL. Toxigenic Escherichia coli associated with sudden infant death syndrome. Scand J Infect Dis (1990) 22(4):467–76. doi:10.3109/00365549009027079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Pearce JL, Luke RKJ, Bettelheim KA. Extraintestinal Escherichia coli isolations from SIDS cases and other cases of sudden death in Victoria, Australia. FEMS Immunol Med Microbiol (1999) 25(1–2):137–44. doi:10.1111/j.1574-695X.1999.tb01337.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Pearce JL, Bettelheim KA, Luke RK, Goldwater PN. Serotypes of Escherichia coli in sudden infant death syndrome. J Appl Microbiol (2010) 108(2):731–5. doi:10.1111/j.1365-2672.2009.04473.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Goldwater PN, Bettelheim KA. Curliated Escherichia coli, soluble curlin and the sudden infant death syndrome (SIDS). J Med Microbiol (2002) 51(11):1009–12.
65. Stray-Pedersen A, Vege A, Rognum TO. Helicobacter pylori antigen in stool is associated with SIDS and sudden infant deaths due to infectious disease. Pediatr Res (2008) 64(4):405–10. doi:10.1203/PDR.0b013e31818095f7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Vargas SL, Ponce CA, Gallo M, Pérez F, Astorga J-F, Bustamante R, et al. Near-universal prevalence of Pneumocystis and associated increase in mucus in the lungs of infants with sudden unexpected death. Clin Infect Dis (2013) 56(2):171–9. doi:10.1093/cid/cis870
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. An SF, Gould S, Keeling JW, Fleming KA. Role of respiratory viral infection in sids: detection of viral nucleic acid by in situ hybridization. J Pathol (1993) 171(4):271–8. doi:10.1002/path.1711710407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Sedmak G, Nix WA, Jentzen J, Haupt TE, Davis JP, Bhattacharyya S, et al. Infant deaths associated with human parechovirus infection in Wisconsin. Clin Infect Dis (2010) 50(3):357–61. doi:10.1086/649863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Álvarez-Lafuente R, Aguilera B, Suárez-Mier MP, Morentin B, Vallejo G, Gómez J, et al. Detection of human herpesvirus-6, Epstein-Barr virus and cytomegalovirus in formalin-fixed tissues from sudden infant death: a study with quantitative real-time PCR. Forensic Sci Int (2008) 178(2):106–11. doi:10.1016/j.forsciint.2008.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Raza MW, El Ahmer OR, Ogilvie MM, Blackwell CC, Saadi AT, Elton RA, et al. Infection with respiratory syncytial virus enhances expression of native receptors for non-pilate Neisseria meningitidis on HEp-2 cells. FEMS Immunol Med Microbiol (1999) 23(2):115–24. doi:10.1111/j.1574-695X.1999.tb01230.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. El Ahmer OR, Raza MW, Ogilvie MM, Weir DM, Blackwell CC. Binding of bacteria to HEp-2 cells infected with influenza A virus. FEMS Immunol Med Microbiol (1999) 23(4):331–41. doi:10.1111/j.1574-695X.1999.tb01255.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Harrison LM, Morris JA, Telford DR, Brown SM, Jones K. The nasopharyngeal bacterial flora in infancy: effects of age, gender, season, viral upper respiratory tract infection and sleeping position. FEMS Immunol Med Microbiol (1999) 25(1–2):19–28. doi:10.1111/j.1574-695X.1999.tb01323.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Bagaitkar J, Demuth D, Scott D. Tobacco use increases susceptibility to bacterial infection. Tob Induc Dis (2008) 4(1):12. doi:10.1186/1617-9625-4-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. El Ahmer OR, Essery SD, Saadi AT, Raza MW, Ogilvie MM, Weir DM, et al. The effect of cigarette smoke on adherence of respiratory pathogens to buccal epithelial cells. FEMS Immunol Med Microbiol (1999) 23(1):27–36. doi:10.1016/S0928-8244(98)00114-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Blackwell C. Bacterial toxins and sudden unexpected death in infancy. Lancet (2008) 372(9640):714. doi:10.1016/S0140-6736(08)61296-9
76. Molony N, Blackwell CC, Busuttil A. The effect of prone posture on nasal temperature in children in relation to induction of staphylococcal toxins implicated in sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25(1–2):109–13. doi:10.1111/j.1574-695X.1999.tb01333.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Andersen A, Jensen KJ, Erikstrup C, Ravn H, Fisker AB, Lisse IM, et al. Both very low- and very high in vitro cytokine responses were associated with infant death in low-birth-weight children from Guinea Bissau. PLoS One (2014) 9(4):e93562. doi:10.1371/journal.pone.0093562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Sunderland R, Emery JL. Febrile convulsions and cot death. Lancet (1981) 318(8239):176–8. doi:10.1016/S0140-6736(81)90359-7
79. Kadhim H, Kahn A, Sebire G. Distinct cytokine profile in SIDS brain: a common denominator in a multifactorial syndrome? Neurology (2003) 61(9):1256–9. doi:10.1212/01.WNL.0000092014.14997.47
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Vennemann MM, Loddenkotter B, Fracasso T, Mitchell EA, Debertin AS, Larsch KP, et al. Cytokines and sudden infant death. Int J Legal Med (2012) 126(2):279–84. doi:10.1007/s00414-011-0638-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Raza MW, Blackwell CC. Sudden infant death syndrome, virus infections and cytokines. FEMS Immunol Med Microbiol (1999) 25(1–2):85–96. doi:10.1111/j.1574-695X.1999.tb01330.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Parish WE, Barrett AM, Coombs RRA, Gunther M, Camps F. Hypersensitivity to milk and sudden death in infancy. Lancet (1960) 276(7160):1106–10. doi:10.1016/S0140-6736(60)92187-5
83. Devey ME, Anderson KJ, Coombs RR, Henschel MJ, Coates ME. The modified anaphylaxis hypothesis for cot death. Anaphylactic sensitization in guinea-pigs fed cow’s milk. Clin Exp Immunol (1976) 26(3):542–8.
84. Gold Y, Goldberg A, Sivan Y. Hyper-releasibility of mast cells among family members of babies expired of sudden infant death syndrome and among babies post apparent life threatening events and their families. Pediatr Res (1999) 45(S5–2):28A–A. doi:10.1203/00006450-199905020-00110
85. Moscovis S, Hall S, Burns C, Scott R, Blackwell C. Development of an experimental model for assessing the effects of cigarette smoke and virus infections on inflammatory responses to bacterial antigens. Innate Immun (2014) 20(6):647–58. doi:10.1177/1753425913503893
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Sarawar SR, Blackman MA, Doherty PC. Superantigen shock in mice with an inapparent viral infection. J Infect Dis (1994) 170(5):1189–94. doi:10.1093/infdis/170.5.1189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Lundemose JB, Smith H, Sweet C. Cytokine release from human peripheral blood leucocytes incubated with endotoxin with and without prior infection with influenza virus: relevance to the sudden infant death syndrome. Int J Exp Pathol (1993) 74(3):291–7.
88. Blood-Siegfried J, Nyska A, Lieder H, Joe M, Vega L, Patterson R, et al. Synergistic effect of influenza A virus on endotoxin-induced mortality in rat pups: a potential model for sudden infant death syndrome. Pediatr Res (2002) 52(4):481–90. doi:10.1203/00006450-200210000-00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Blood-Siegfried J, Shelton B. Animal models of sudden unexplained death. FEMS Immunol Med Microbiol (2004) 42(1):34–41. doi:10.1016/j.femsim.2004.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Moscovis SM, Gordon AE, Al Madani OM, Gleeson M, Scott RJ, Roberts-Thomson J, et al. Interluekin-10 and sudden infant death syndrome. FEMS Immunol Med Microbiol (2004) 42(1):130–8. doi:10.1016/j.femsim.2004.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Essery SD, Raza MW, Zorgani A, MacKenzie DA, James VS, Weir DM, et al. The protective effect of immunisation against diphtheria, pertussis and tetanus (DPT) in relation to sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25(1–2):183–92. doi:10.1111/j.1574-695X.1999.tb01342.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Petersen SA, Wailoo MP. Interactions between infant care practices and physiological development in Asian infants. Early Hum Dev (1994) 38(3):181–6. doi:10.1016/0378-3782(94)90210-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Gordon AE, Al Madani O, Weir DM, Busuttil A, Blackwell C. Cortisol levels and control of inflammatory responses to toxic shock syndrome toxin-1 (TSST-1): the prevalence of night-time deaths in sudden infant death syndrome (SIDS). FEMS Immunol Med Microbiol (1999) 25(1–2):199–206. doi:10.1111/j.1574-695X.1999.tb01344.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Soldin S, Brugnara C, Wong E. Pediatric Reference Ranges. 4 ed. Washington, DC: AACC Press (2003).
95. Moscovis SM, Hall ST, Burns CJ, Scott RJ, Blackwell CC. The male excess in sudden infant deaths. Innate Immun (2014) 20(1):24–9. doi:10.1177/1753425913481071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Cox AJ, Moscovis SM, Blackwell CC, Scott RJ. Cytokine gene polymorphism among indigenous Australians. Innate Immun (2014) 20(4):431–9. doi:10.1177/1753425913498911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Larcombe L, Rempel JD, Dembinski I, Tinckam K, Rigatto C, Nickerson P. Differential cytokine genotype frequencies among Canadian aboriginal and Caucasian populations. Genes Immun (2004) 6(2):140–4. doi:10.1038/sj.gene.6364157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Ness RB, Haggerty CL, Harger G, Ferrell R. Differential distribution of allelic variants in cytokine genes among African Americans and white Americans. Am J Epidemiol (2004) 160(11):1033–8. doi:10.1093/aje/kwh325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Anuradha B, Rakh SS, Ishaq M, Murthy KJR, Valluri VL. Interferon-γ low producer genotype +874 overrepresented in bacillus Calmette-Guerin nonresponding children. Pediatr Infect Dis J (2008) 27(4):325–9. doi:10.1097/INF.0b013e31816099e6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Alfelali M, Khandaker G. Infectious causes of sudden infant death syndrome. Paediatr Respir Rev (2014) 15(4):307–11. doi:10.1016/j.prrv.2014.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: sudden infant death syndrome, inflammation, infection, cigarette smoke, ethnicity
Citation: Blackwell C, Moscovis S, Hall S, Burns C and Scott RJ (2015) Exploring the risk factors for sudden infant deaths and their role in inflammatory responses to infection. Front. Immunol. 6:44. doi: 10.3389/fimmu.2015.00044
Received: 05 December 2014; Paper pending published: 10 January 2015;
Accepted: 23 January 2015; Published online: 05 March 2015.
Edited by:
Masaaki Murakami, Hokkaido University, JapanReviewed by:
Yasunobu Arima, Osaka University, JapanFrédéric Velard, Université de Reims Champagne-Ardenne, France
Copyright: © 2015 Blackwell, Moscovis, Hall, Burns and Scott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Blackwell, Information Based Medicine, Hunter Medical Research Institute, New Lambton, NSW 2305, Australia e-mail:Y2Fyb2xpbmUuYmxhY2t3ZWxsQG5ld2Nhc3RsZS5lZHUuYXU=