 Stephanie Gurka
Stephanie Gurka Evelyn Hartung
Evelyn Hartung Martina Becker
Martina Becker Richard A. Kroczek
Richard A. Kroczek
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 04 February 2015
Sec. Antigen Presenting Cell Biology
Volume 6 - 2015 | https://doi.org/10.3389/fimmu.2015.00035
Since the identification of mouse dendritic cells (DC) in the early 70s, all attempts to consistently classify the identified functional DC subpopulations according to their surface molecule expression failed. In the absence of DC lineage markers, a great variety of non-congruent surface molecules were used instead. Recent advances in the understanding of the involvement of transcription factors in the differentiation of DC subpopulations, together with the identification of a lineage marker for cross-presenting DC, have now allowed to establish a consistent and unified DC classification in the mouse. We demonstrate in the present article that all conventional DC in the mouse can be universally subdivided into either XCR1+ (“cross-presenting”) DC or SIRPα+ DC, irrespective of their activation status. This advancement will greatly facilitate future work on the biology of mouse DC. We discuss this new classification in view of current DC classification systems in the mouse and the human.
Dendritic cells (DC) were discovered by Steinman et al. already in the early 70s (1). Nevertheless, it was until recently difficult to unequivocally distinguish them from other related cell types such as monocytes or macrophages. As a result, a combination of several markers had to be used to define DC in flow cytometry and histology. For practical purposes, mouse conventional DC today are identified in flow cytometry as cells, which express the integrin CD11c and high levels of MHC II, but lack expression of T-, B-, and plasmacytoid DC lineage markers, and also molecules characteristic for monocytes and macrophages. In the absence of (sub-) lineage markers, also DC subpopulations could not easily be defined using surface molecules. This has led over time to the use of a great variety of surface markers distinguishing supposedly functionally distinct DC subpopulations and made it difficult to directly compare results between laboratories. Further complexity was brought about by the observation that DC with an apparently similar function had different phenotypes in lymphoid tissues versus peripheral organs. As a consequence, the division of DC into subpopulations remained somewhat arbitrary, making the experimental results, including gene expression studies, less informative. Being central to the understanding of DC biology, the question how mouse conventional DC should be divided into subsets remained a matter of intensive scientific debate to this very day.
Recently, we identified the first molecule restricted in its expression to mouse DC (2). Based on a variety of experimental systems, we then could demonstrate that the chemokine receptor XCR1 is a lineage marker for cross-presenting DC (3), a DC subpopulation playing an important role in the induction of CD8+ T cell cytotoxicity (see below). The use of an antibody directed to XCR1 allowed for the first time the unequivocal identification and thus a precise phenotyping of cross-presenting DC in various body compartments of the mouse (3–6).
With the ability to define the cross-presenting DC population, it became possible to ask the question whether the remaining DC could also be defined by their surface characteristics. The use of an extended panel of antibodies directed to DC surface molecules indicated that all XCR1− DC were characterized by expression of SIRPα/CD172a in the intestine (4). In view of these results, together with the data presented here, we now propose a new classification based on the expression XCR1 and SIRPα, which can be used to define DC subpopulations in all lymphoid and non-lymphoid compartments of the mouse. We hope that this classification will greatly facilitate the study of DC biology in the future.
This article describes the various steps, which have led to the establishment of this new DC classification system, and discusses the implications for the understanding of human DC subpopulations. Gene expression profiles and functional aspects of DC subsets [recently reviewed by Ref. (7–10)] are taken into account only as far as they contribute to the classification of mouse DC. Monocyte-derived inflammatory DC and plasmacytoid DC are not considered here, since they represent different cell lineages.
Work of numerous groups has established that at least two major conventional DC populations exist in the mouse. In the late 90s, a subset of DC was identified in lymphoid tissues, which expressed the CD8α homodimer on the cell surface (and lacked CD8β and CD11b), and these DC were hence termed “CD8+ DC” (11). A major step forward in the understanding of DC biology was the demonstration that CD8+ DC excel in antigen “cross-presentation”, a process in which antigen is shunted into the MHC I pathway and presented to CD8+ T cells (12–14). Further studies demonstrated that mouse CD8+ DC are specialized in the uptake and proteolytic processing of stressed cells and the subsequent presentation of the derived peptides to CD8+ T cells (15–17). In general terms, antigen cross-presentation allows an efficient induction of CD8 T cell cytotoxicity to antigens originating from cell-invading pathogens or mutated (cancer) cells.
After identification of CD8α as a relevant subset marker on around 20% of DC in the spleen, the remaining splenic DC were classified as CD4+ DC (60%) and double-negative DC (DN DC, 20%) in C57BL/6 mice (18). When Edwards et al. (19) performed microarray gene expression profiling of splenic DC populations in the mouse, they confirmed the differing nature of CD8+ DC, but noticed only relatively minor differences in the gene expression profiles of CD4+ and DN DC, and thus concluded that these two populations are phylogenetically related. Later work based on targeting of antigen directly to splenic DC subsets has confirmed the superior capacity of CD8+ (DEC-205+/CD205+) DC for antigen cross-presentation and at the same time demonstrated a higher efficiency of CD8− (33D1+/DCIR2+) DC in the presentation of antigen to CD4+ T cells (20).
While the phenotypical and functional classification of splenic DC made substantial progress, the understanding of DC subpopulations in peripheral lymphoid tissues and organs lagged behind. CD8α was detectable on resident DC in all lymphoid organs, but absent on DC in certain peripheral tissues and on DC migrating from the periphery to lymph nodes (LN) (21, 22). Since CD8α was less useful in the periphery, a different classification system was established by subdividing DC into CD103+ and CD103− populations (22–25). Antigen cross-presentation was shown to be restricted to CD103+ DC residing in the lung, the intestine, and skin-draining LN (22, 24, 25), suggesting a functional relationship to the CD8+ splenic DC. However, it soon was recognized that the CD103+ DC population is not homogenous and therefore the exact relationship between peripheral DC and lymphoid-resident DC remained unresolved.
A major step forward was brought about by work on the involvement of transcription factors (TF) in the differentiation of DC. A series of studies demonstrated that development of CD8+ splenic DC and their peripheral counterparts critically depend on the TF IRF-8 (also designated ICSBP), Id2, and Batf3 (26–31). The most informative turned out to be the Batf3-KO mouse, where only the splenic CD8+ DC, the lung and dermal CD103+ DC, and the intestinal CD103+ CD11b− DC were absent and thus could be identified as developmentally related (only later it became apparent that other small DC populations with a differing phenotype were also Batf3-dependent, see below). At the same time, this animal model showed clear deficits in antigen cross-presentation (29, 31). Together, this work strongly indicated that the Batf3-dependent DC were the cross-presenting DC lineage in the mouse.
When searching for the function of XCL1, a chemokine secreted by activated CD8+ T cells and NK cells (32, 33), we found that the corresponding receptor XCR1 is exclusively expressed by a subset of DC. This observation represented the first instance of a surface molecule being restricted to conventional DC in the mouse. Analyzing a XCR1-lacZ-reporter mouse (a system which provides limited signal intensity and suffers from high background in some extra-splenic tissues when using fluorescein-di-β-d-galactopyranoside as substrate in flow cytometry), we found XCR1 to be expressed by 70–90% of CD8+ DC and by up to 8% of DN DC in the spleen. Histological analyses further indicated that other lymphoid tissues and peripheral organs contained XCR1+ DC (2). Using the same XCR1-reporter mice and extending the flow cytometry studies to DC in LN and several organs, Crozat et al. (34) found XCR1 signals essentially limited to CD103+ CD11b− DC, allowing them to postulate that expression of XCR1 defines mouse lymphoid-tissue resident and migratory DC of the “CD8α-type.”
Further understanding of XCR1-expressing DC became possible with the development of a mAb specific for murine XCR1, which offered high-resolution flow cytometry and also allowed sorting of XCR1+ DC for functional assays. These studies (3) confirmed earlier findings with the lacZ-reporter system in the spleen (2) that expression of XCR1 and CD8 overlap, but are not congruent. In the lung, the intestine, in skin-draining, and mesenteric LN, CD103+ CD11b− DC were found to be essentially XCR1+. However, additional XCR1+ DC populations could also be identified there and these were negative for CD103 or positive for CD11b (3, 4). Thus, the expression pattern of XCR1 differed from the “CD8+” and “CD103+ CD11b−” DC phenotypes associated with antigen cross-presentation in the past.
In experiments directly aimed to define the correlation between XCR1 expression and Batf3-dependence of DC, it became apparent that all XCR1+ DC (irrespective of their CD8, CD103, or CD11b expression status), were absent in Batf3-KO mice (3, 4). Congruent with this observation, the 20% of splenic CD8+ DC, which are negative for XCR1, were preserved in Batf3-KO animals (3); these particular CD8+ XCR1− DC apparently represent a distinct DC population with a very different gene expression profile (35). Together, the studies demonstrated for DC in all tissues an excellent and unique correlation between XCR1 surface expression and dependence on Batf3 [for data and discussion on the correlation between XCR1 and surface molecules preferentially expressed on Batf3-dependent DC such as CD8, CD205, Clec9A/DNGR-1, Itga8, and CADM1 see in Ref. (5)].
These correlation studies were very striking, but did not deliver direct information on the functional role of XCR1+ versus XCR1− DC. Only when functional assays using soluble and cell-associated antigen were performed with splenic (3) and intestinal DC (4, 6), it became apparent that antigen cross-presentation is the domain of XCR1+ DC, in particular with cell-associated antigen. Conversely, the CD8+ DC negative for XCR1 were found to be incapable of antigen cross-presentation (3).
The perfect correlation between expression of XCR1, Batf3-dependence, and the ability to cross-present (cell-associated) antigen in various organ systems (3, 4, 6) allow to conclude that XCR1 expression generally demarcates the Batf3-dependent cross-presenting DC, as postulated (3). Thus, XCR1 can be regarded as the lineage marker for cross-presenting DC in the mouse.
Is there a surface molecule which would define the remaining, the XCR1− DC population? To examine this question, we re-analyzed all of our correlation studies, which were based on the use of a panel of antibodies directed to DC surface molecules (among others CD11b, CD171a/SIRPα, DCIR2), and the use of two reporter mice (CD207, CX3CR1). In all of our analyses, the only molecule, which showed a consistent and full anti-correlation with XCR1 was CD172a/SIRPα, indicating that this surface molecule could be used to positively demarcate XCR1− DC (3, 4). Based on these studies, we have proposed that XCR1 and SIRPα can be used to classify intestinal DC and possibly all DC in the mouse (4, 36).
In order to test the general applicability of this new classification system and to make the XCR1 expression studies directly comparable, we have now isolated DC from a greater variety of lymphoid and non-lymphoid organs and analyzed them in parallel. As can be seen in Figure 1, XCR1 and SIRPα were found to be clearly anti-correlated in all organs tested. At the same time, all DC present in these organs could be assigned to either population. Thus, the anti-correlation between XCR1 and SIRPα can now be demonstrated in a great variety of tissues.
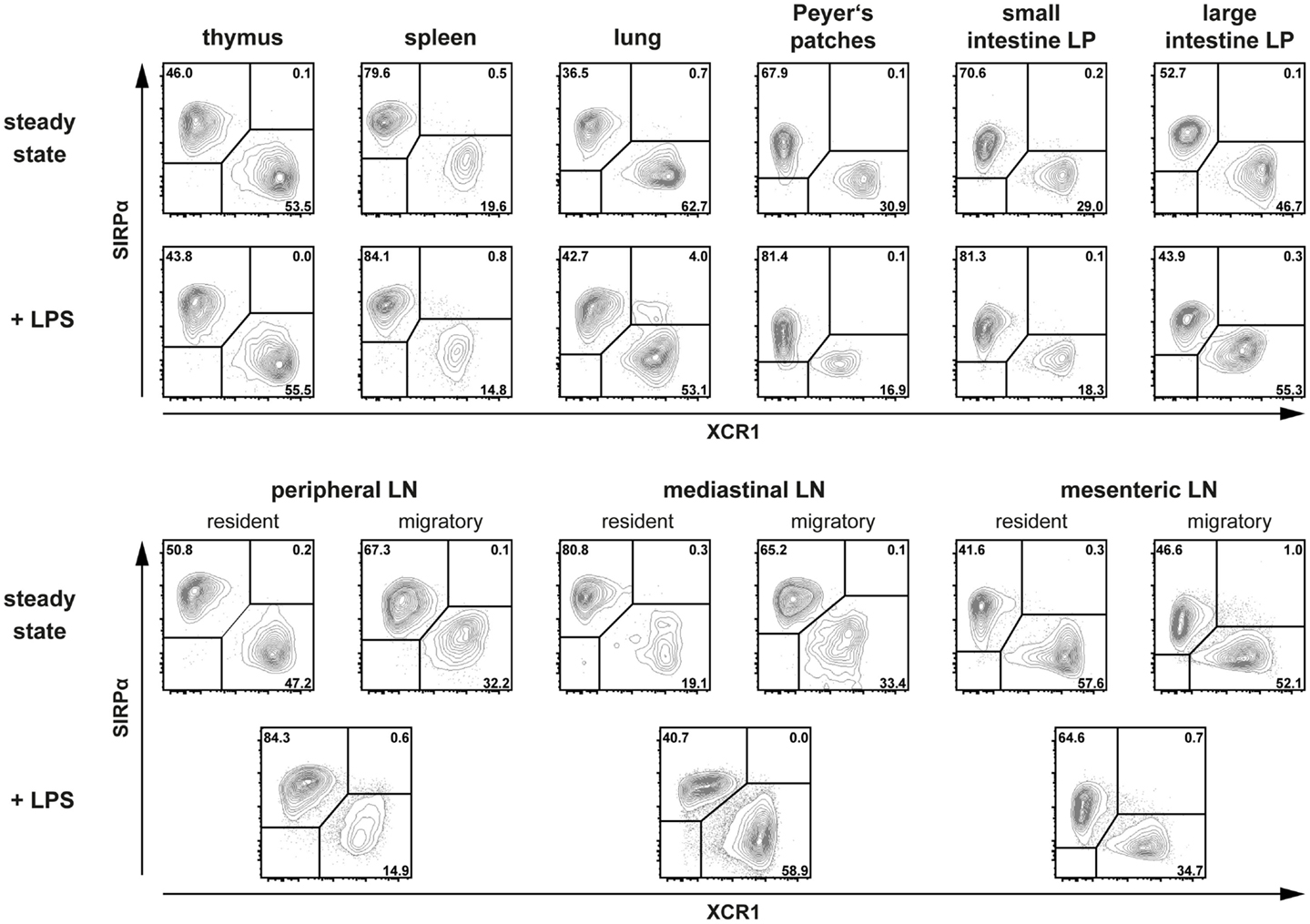
Figure 1. Expression of XCR1 and SIRPα on DC in various tissues at steady state and in inflammation. Cells from different tissues of C57BL/6 mice (either untreated or 14 h after injection of 10 μg LPS i.v.) were isolated after enzymatic digestion (except for the spleen), and DC from lamina propria (LP), Peyer’s patches, and mesenteric LN were additionally enriched by density gradient centrifugation as described before (3, 4). Cells from brachial, axillary, and inguinal LN were pooled for the study of peripheral LN DC. For flow cytometric analysis of XCR1 and SIRPα expression on DC, gates were set on live CD90− CD19− CD317− CD11c+ MHC II+ cells for thymus, spleen, peripheral and mediastinal LN, and on live CD45+ CD3− B220− F4/80− CD11c+ MHC II+ cells for lung, Peyer’s patches, LP, and mesenteric LN. DC from steady-state LN were separated based on their MHC II expression levels into resident (MHC IIint) and migratory (MHC IIhigh) populations.
All published data on the anti-correlation of XCR1 and SIRPα have been obtained in steady-state animals only. It was therefore important to test DC also under inflammatory conditions, when many DC surface molecules are up or down regulated. To this end, animals were injected with 10 μg LPS i.v. and DC examined 14 h later. Under these conditions, SIRPα expression remained rather stable, while XCR1 was slightly down regulated in some organs, however without compromising the discrimination of XCR1+ DC from SIRPα+ DC (Figure 1). Thus, the subdivision of conventional DC based on the expression of XCR1 and SIRPα appears to be universally applicable. With the commercial availability of an antibody directed to mouse (and rat) XCR1 (clone ZET) this classification can now be generally tested under all possible conditions.
SIRPα, also abundantly expressed on macrophages, neutrophils and some non-lymphoid tissues, has been implicated in the control of cell phagocytosis (37, 38). Cells expressing CD47, the ligand for SIRPα, appear to be protected from engulfment by phagocytic cells (37). It is thus intriguing to note that both CD205 and Clec9A/DNGR-1 on XCR1+ DC may regulate cell phagocytosis (and antigen processing) in a positive way (39, 40), and SIRPα on XCR1− DC in a negative fashion. This functional feature may possibly contribute to the division of labor between the XCR1+ and SIRPα+ DC populations.
All available data (gene expression profile, toll-like receptor expression pattern in particular), indicate that XCR1+ DC are a homogenous DC lineage. Does this mean that all XCR1+ DC function in the same way? This may not be the case. It is of interest in this context that only CD8+ (i.e., XCR1+) splenic DC expressing CD103 were capable to take up cells and to cross-present their antigen to CD8+ T cells in a previous study (41). Only approximately 50–60% of splenic CD8+/XCR1+DC express CD103 under non-inflammatory conditions (3, 41), and the situation appears similar in resident mesenteric LN (4, 42). It is therefore quite possible that XCR1+ DC are functionally heterogenous in terms of cross-presentation, depending on their activation state, their anatomical positioning, and the upregulation of other molecules in reaction to environmental cues.
This question is largely unresolved at present. Edwards et al. (19) showed that CD4+ DC and DN DC, which now would be grouped together as SIRPα+ DC, have a highly similar gene expression profile, suggesting one uniform population. Nevertheless, the authors found some genes to be quite specifically expressed in either population. Other reports also point to a possible ontogenic or functional subdivision of SIRPα+ DC. For example, only a small fraction of SIRPα+ DC in the spleen express CD8 and differ in their expression profile from the remaining DC populations (3, 35). Mice with a CD11c-cre driven deletion of the TF Notch2 showed a rather specific ablation of CD103+ CD11b+ DC in the intestinal system (representing an ablation of approximately 50% of SIRPα+ DC), and these DC were identified as an obligate source of IL-23 in the defense to C. rodentium (43). In another study, depletion of only CD301b+ DC in the CD103− (and thus most likely SIRPα+) fraction of dermal DC resulted in a severe impairment of skin Th2 immunity (44). More studies will be required to make the results comparable and to answer the question on the heterogeneity of SIRPα+ DC.
In the human, in vivo experiments on the function of DC subsets are not possible, access to DC in various compartments is limited, and the frequency of DC in the blood is very low. As a result, data on human DC are rather scarce. At the gene expression level, it was established that mouse CD8+ DC correspond to human CD141+ (BDCA3+) DC, and mouse CD11b+ DC to human CD1c+ (BDCA1+) DC (45), the two identified human DC populations (46). Support for this correlation came from functional studies, which demonstrated a superior capacity of CD141+ DC for cross-presentation (47–49). Gene expression studies rely on previous sorting of DC populations and thus depend on the use of “correct” surface markers. Regarding these surface markers, however, human DC somewhat differ from mouse DC. XCR1 is exclusively expressed on CD141+ DC, but not on all of them. Analyses of DC obtained from peripheral blood, thymus, and spleen demonstrated that an average of 80% of CD141+ DC express XCR1 (own unpublished data). At the same time, all of the CD141+ DC express Clec9A/DNGR-1, which in the human appears to be restricted to conventional DC, as it is not found on plasmacytoid DC (40, 50, 51). Thus, in the human, expression of XCR1 and Clec9A/DNGR-1 is not fully correlated (own unpublished data), as is the case with conventional DC in the mouse. CADM1, another surface molecule tightly associated with Batf3-dependent cross-presenting DC in the mouse, gives a bright signal in the human and is perfectly correlated with CD141 [(52), and own unpublished data]. Thus, it is possible that cross-presenting DC in the human can be demarcated by the co-expression of CD141, Clec9A, and CADM1. This assumption is further supported by the observation that SIRPα on DC in various human organs is correlated with CD1c and fully anti-correlated with CD141 (own unpublished data). Thus, on a broad scale, it is quite clear that the human CD141+ DC contain the cross-presenting DC population. Whether all of CD141+ DC can cross-present or whether this function is restricted to the XCR1+ CD141+ DC remains to be determined. Further detailed insight into this question will require gene expression and functional studies comparing the majority of CD141+ DC expressing XCR1 and the 20% fraction of CD141+ DC negative for XCR1.
In summary, recent developments in the field allowed major progress in the classification of mouse and human DC. Particularly in the mouse, where the subdivision of DC was notoriously difficult, the use of a general DC classification scheme based on the expression of XCR1 and SIRPα will make experimental results more precise and also more comparable. Without any doubt, the use of additional surface molecules will continue to be necessary in order to better understand the functional states of DC of a given lineage. Thus the old “markers” such as CD4, CD8, CD11b, or CD103 will not become obsolete, but they will obtain a new role in the functional analyses of XCR1+ versus SIRPα+ mouse DC subpopulations.
The Associate Editor, Dr. Christian Kurts, declares that despite having collaborated with author Richard A. Kroczek in the past 2 years, there has been no conflict of interest during the handling of this manuscript. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work was supported by grants of the Wilhelm Sander Foundation, the Fritz Thyssen Foundation, the VIP program of the Bundesministerium für Bildung und Forschung, and in part by the Deutsche Forschungsgemeinschaft (Kr 827/16-1 and Kr 827/18-1).
1. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I morphology, quantitation, tissue distribution. J Exp Med (1973) 137:1142–62. doi:10.1084/jem.137.5.1142
2. Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Güttler S, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity (2009) 31:823–33. doi:10.1016/j.immuni.2009.08.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Bachem A, Hartung E, Güttler S, Mora A, Zhou X, Hegemann A, et al. Expression of XCR1 characterizes the Batf3-dependent lineage of dendritic cells capable of antigen cross-presentation. Front Immunol (2012) 3:214. doi:10.3389/fimmu.2012.00214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Becker M, Güttler S, Bachem A, Hartung E, Mora A, Jäkel A, et al. Ontogenic, phenotypic, and functional characterization of XCR1+ dendritic cells leads to a consistent classification of intestinal dendritic cells based on the expression of XCR1 and SIRPα. Front Immunol (2014) 5:326. doi:10.3389/fimmu.2014.00326
5. Gurka S, Hartung E, Becker M, Kroczek RA. Mouse conventional dendritic cells can be universally classified based on the mutually exclusive expression of XCR1 and SIRPα. bioRxiv (2014). doi:10.1101/012567
6. Cerovic V, Houston SA, Westlund J, Utriainen L, Davison ES, Scott CL, et al. Lymph-borne CD8α+ dendritic cells are uniquely able to cross-prime CD8+ T cells with antigen acquired from intestinal epithelial cells. Mucosal Immunol (2015) 8:38–48. doi:10.1038/mi.2014.40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Hashimoto D, Miller J, Merad M. Dendritic cell and macrophage heterogeneity in vivo. Immunity (2011) 35:323–35. doi:10.1016/j.immuni.2011.09.007
8. Haniffa M, Collin M, Ginhoux F. Ontogeny and functional specialization of dendritic cells in human and mouse. Adv Immunol (2013) 120:1–49. doi:10.1016/B978-0-12-417028-5.00001-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi:10.1146/annurev-immunol-020711-074950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity (2014) 40:642–56. doi:10.1016/j.immuni.2014.04.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev (2010) 234:18–31. doi:10.1111/j.0105-2896.2009.00870.x
12. Kurts C, Heath WR, Carbone FR, Allison J, Miller JFAP, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med (1996) 184:923–30. doi:10.1084/jem.184.3.923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. den Haan JM, Lehar SM, Bevan MJ. CD8+ but not CD8- dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med (2000) 192:1685–96. doi:10.1084/jem.192.12.1685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Pooley JL, Heath WR, Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J Immunol (2001) 166:5327–30. doi:10.4049/jimmunol.166.9.5327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y, et al. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med (2002) 195:1289–302. doi:10.1084/jem.20020161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Schulz O, Reis E, Sousa C. Cross-presentation of cell-associated antigens by CD8α+ dendritic cells is attributable to their ability to internalize dead cells. Immunology (2002) 107:183–9. doi:10.1046/j.1365-2567.2002.01513.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci USA (2006) 103:10729–34. doi:10.1073/pnas.0601956103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Vremec D, Pooley J, Hochrein H, Wu L, Shortman K. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol (2000) 164:2978–86. doi:10.4049/jimmunol.164.6.2978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Edwards AD, Chaussabel D, Tomlinson S, Schulz O, Sher A, Reis E, et al. Relationships among murine CD11chigh dendritic cell subsets as revealed by baseline gene expression patterns. J Immunol (2003) 171:47–60. doi:10.4049/jimmunol.171.1.47
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (2007) 315:107–11. doi:10.1126/science.1136080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Sung SS, Fu SM, Rose CE Jr, Gaskin F, Ju ST, Beaty SR. A major lung CD103 (αE)-β7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol (2006) 176:2161–72. doi:10.4049/jimmunol.176.9.5683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. del Rio ML, Rodriguez-Barbosa JI, Kremmer E, Förster R. CD103- and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J Immunol (2007) 178:6861–6. doi:10.4049/jimmunol.178.11.6861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J Exp Med (2005) 202:1051–61. doi:10.1084/jem.20040662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med (2008) 205:2139–49. doi:10.1084/jem.20080414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol (2009) 10:488–95. doi:10.1038/ni.1724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC III, et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8α+ dendritic cells. J Exp Med (2002) 196:1415–25. doi:10.1084/jem.20021263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reis E, Sousa C, et al. Essential role for ICSBP in the in vivo development of murine CD8α+ dendritic cells. Blood (2003) 101:305–10. doi:10.1182/blood-2002-04-1088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol (2003) 4:380–6. doi:10.1038/ni903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi:10.1126/science.1164206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med (2009) 206:3115–30. doi:10.1084/jem.20091756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J Exp Med (2010) 207:823–36. doi:10.1084/jem.20091627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Müller S, Dorner B, Korthäuer U, Mages HW, D’apuzzo M, Senger G, et al. Cloning of ATAC, an activation-induced, chemokine-related molecule exclusively expressed in CD8+ T lymphocytes. Eur J Immunol (1995) 25:1744–8. doi:10.1002/eji.1830250638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Dorner BG, Scheffold A, Rolph MS, Hüser MB, Kaufmann SH, Radbruch A, et al. MIP-1α, MIP-1β, RANTES, and ATAC/lymphotactin function together with IFN-γ as type 1 cytokines. Proc Natl Acad Sci USA (2002) 99:6181–6. doi:10.1073/pnas.092141999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Crozat K, Tamoutounour S, Vu Manh TP, Fossum E, Luche H, Ardouin L, et al. Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8α+ type. J Immunol (2011) 187:4411–5. doi:10.4049/jimmunol.1101717
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Bar-On L, Birnberg T, Lewis KL, Edelson BT, Bruder D, Hildner K, et al. CX3CR1+ CD8α+ dendritic cells are a steady-state population related to plasmacytoid dendritic cells. Proc Natl Acad Sci USA (2010) 107:14745–50. doi:10.1073/pnas.1001562107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Becker M, Güttler S, Bachem A, Hartung E, Mora A, Jäkel A, et al. Ontogenic, phenotypic, and functional characterization of XCR1+ dendritic cells leads to a consistent classification of intestinal dendritic cells based on the expression of XCR1 and SIRPα. bioRxiv (2014). doi:10.1101/004648
37. Matozaki T, Murata Y, Okazawa H, Ohnishi H. Functions and molecular mechanisms of the CD47-SIRPα signalling pathway. Trends Cell Biol (2009) 19:72–80. doi:10.1016/j.tcb.2008.12.001
38. Nuvolone M, Kana V, Hutter G, Sakata D, Mortin-Toth SM, Russo G, et al. SIRPα polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. J Exp Med (2013) 210:2539–52. doi:10.1084/jem.20131274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Shrimpton RE, Butler M, Morel AS, Eren E, Hue SS, Ritter MA. CD205 (DEC-205): a recognition receptor for apoptotic and necrotic self. Mol Immunol (2009) 46:1229–39. doi:10.1016/j.molimm.2008.11.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Sancho D, Mourao-Sa D, Joffre OP, Schulz O, Rogers NC, Pennington DJ, et al. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest (2008) 118:2098–110. doi:10.1172/JCI34584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Qiu CH, Miyake Y, Kaise H, Kitamura H, Ohara O, Tanaka M. Novel subset of CD8α+ dendritic cells localized in the marginal zone is responsible for tolerance to cell-associated antigens. J Immunol (2009) 182:4127–36. doi:10.4049/jimmunol.0803364
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Pribila JT, Itano AA, Mueller KL, Shimizu Y. The α1β1 and αEβ7 integrins define a subset of dendritic cells in peripheral lymph nodes with unique adhesive and antigen uptake properties. J Immunol (2004) 172:282–91. doi:10.4049/jimmunol.172.1.282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol (2013) 14:937–48. doi:10.1038/ni.2679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A. CD301b+ dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity (2013) 39:733–43. doi:10.1016/j.immuni.2013.08.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Robbins SH, Walzer T, Dembele D, Thibault C, Defays A, Bessou G, et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol (2008) 9:R17. doi:10.1186/gb-2008-9-1-r17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Ju X, Clark G, Hart DN. Review of human DC subtypes. Methods Mol Biol (2010) 595:3–20. doi:10.1007/978-1-60761-421-0_1
47. Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med (2010) 207:1273–81. doi:10.1084/jem.20100348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Crozat K, Guiton R, Guilliams M, Henri S, Baranek T, Schwartz-Cornil I, et al. Comparative genomics as a tool to reveal functional equivalences between human and mouse dendritic cell subsets. Immunol Rev (2010) 234:177–98. doi:10.1111/j.0105-2896.2009.00868.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Jongbloed SL, Kassianos AJ, Mcdonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med (2010) 207:1247–60. doi:10.1084/jem.20092140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Caminschi I, Proietto AI, Ahmet F, Kitsoulis S, Shin TJ, Lo JC, et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood (2008) 112:3264–73. doi:10.1182/blood-2008-05-155176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Huysamen C, Willment JA, Dennehy KM, Brown GD. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J Biol Chem (2008) 283:16693–701. doi:10.1074/jbc.M709923200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Galibert L, Diemer GS, Liu Z, Johnson RS, Smith JL, Walzer T, et al. Nectin-like protein 2 defines a subset of T-cell zone dendritic cells and is a ligand for class-I-restricted T-cell-associated molecule. J Biol Chem (2005) 280:21955–64. doi:10.1074/jbc.M502095200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: dendritic cells, mouse, classification, XCR1, SIRPα, cross-presentation
Citation: Gurka S, Hartung E, Becker M and Kroczek RA (2015) Mouse conventional dendritic cells can be universally classified based on the mutually exclusive expression of XCR1 and SIRPα. Front. Immunol. 6:35. doi: 10.3389/fimmu.2015.00035
Received: 05 December 2014; Accepted: 19 January 2015;
Published online: 04 February 2015.
Edited by:
Christian Kurts, Friedrich Wilhelms-Universität Bonn, GermanyReviewed by:
Diana Dudziak, University Hospital of Erlangen, GermanyCopyright: © 2015 Gurka, Hartung, Becker and Kroczek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard A. Kroczek, Molecular Immunology, Robert Koch-Institute, Nordufer 20, Berlin 13353, Germany e-mail:a3JvY3pla0Bya2kuZGU=
†Stephanie Gurka, Evelyn Hartung and Martina Becker have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.