 Stephen M. Laidlaw
Stephen M. Laidlaw Lynn B. Dustin
Lynn B. Dustin- Kennedy Institute of Rheumatology, Nuffield Department of Orthopaedics, Rheumatology, and Musculoskeletal Sciences, University of Oxford, Oxford, UK
Innate immunity is key to the fight against the daily onslaught from viruses that our bodies are subjected to. Essential to this response are the interferons (IFNs) that prime our cells to block viral pathogens. Recent evidence suggests that the Type III (λ) IFNs are intimately associated with the immune response to hepatitis C virus (HCV) infection. Genome-wide association studies have identified polymorphisms within the IFN-λ gene locus that correlate with response to IFNα-based antiviral therapy and with spontaneous clearance of HCV infection. The mechanisms for these correlations are incompletely understood. Restricted expression of the IFN-λ receptor, and the ability of IFN-λ to induce IFN-stimulated genes in HCV-infected cells, suggest potential roles for IFN-λ in HCV therapy even in this era of directly acting antivirals. This review summarizes our current understanding of the IFN-λ family and the role of λ IFNs in the natural history of HCV infection.
Hepatitis C Virus
Hepatitis C virus (HCV) is a positive sense, single-stranded RNA virus in the family Flaviviridae. It is estimated that as many as 160 million people are chronically infected worldwide with 3–4 million new infections every year (1, 2). With a global prevalence of 2.35%, estimated to range between 0.14% on the island of Reunion and 14% in Egypt (1), there is a large economic cost and burden to society.
Transmission of HCV usually occurs following contact with infected blood through the percutaneous route, e.g., unsafe needle sharing, but may also occur through sexual transmission, iatrogenic, or vertical transmission routes. Following acute infection spontaneous resolution can occur while infected individuals that fail to clear the virus develop a chronic infection leading to liver disease, including fibrosis, cirrhosis, and hepatocellular carcinoma. Estimates of spontaneous clearance rates range from 14 to 45% depending on the population studied (3). One of the major risk factors for chronicity is HCV genotype. Currently, seven genotypes and multiple sub-types of HCV have been identified (4). Most patients are infected with only one genotype rather than multiple genotypes and within an individual the virus will mutate to form multiple genetic variants, called a quasispecies (5).
HCV Cell Culture
In vivo and vitro, HCV primarily infects human hepatocytes. It has been possible to reconstitute the replication cycle of HCV in human non-hepatic cells such as 293T cells in vitro (6), and even in mice (7). Both of these approaches required the addition of extra host factors; both 293T cells and mice required the HCV entry factors claudin-1 (CLDN1) and CD81, in addition, 293T cells required occludin (OCLN), scavenger receptor class B type I (SR-BI), and the human micro RNA miR-122, while effective HCV RNA replication in mice also required the knockdown of murine innate immune mediators (7–9). Key to the replication in non-human cells seems to be the expression of the human micro RNA miR-122 (9). miR-122 has been shown to bind to the 5’ UTR of HCV and to enhance translation and replication of HCV RNA (10–14), thus, enhancing HCV propagation (15).
Experimental investigations with HCV are mainly carried out using cell-culture models. Historically, HCV was first propagated in permissive human hepatoma cell lines derived from Huh-7. Initially, only low-level replication was possible, but selection using interferon alpha (IFN-α) allowed for the isolation of cell lines that were highly permissive for replication, producing high-viral titers, including Huh-7.5 (16, 17). Key to the progression of HCV research was the development of HCV replicon systems (18). These consist of a minimal HCV genome (NS3 to NS5B non-structural genes flanked by the 5′ and 3′ UTRs) combined with a selection marker and/or reporter gene that is incapable of producing infectious virus but is capable of RNA replication in vitro. Another great leap occurred in 2005 when a number of groups (19–21) advanced the replicon system to show that HCV could undertake a full cycle in cell culture to produce infectious virus. This followed the identification of an HCV isolate that was replication competent without requiring tissue culture-adapting mutations (22).
Due to the limited host range of HCV, it has not been possible until recently to use small animal models of infection, and serum clinical isolates of HCV replicate poorly in tissue culture. The use of adult primary human hepatocytes (PHH) is the closest in vitro model of HCV infection that is currently available. Unfortunately, the availability of these is limited, resulting only from organ donation or patient biopsy. Infection efficiency of PHH is low and outcome of infection is highly variable. Even so, a number of studies are now being published using these cells (23–26).
In contrast to adult PHH, only a handful of studies have used primary fetal human hepatocytes (FHH) (27–31). These cells are long-lived and support sustained low levels of HCV replication.
Interferons
As a positive sense single-stranded RNA virus, HCV replication necessitates the generation of double-stranded RNA intermediates (32). The infected cell identifies this as a major pathogen associated molecular pattern (PAMP), recognized by pattern recognition receptors (PRRs). These immune sensing molecules can be classified into groups, i.e., the RIG-I like receptors (RLRs), the toll-like receptors (TLRs), and the viral DNA sensors (33). Once activated these sensing molecules trigger a number of signaling pathways, resulting in the generation of Type I and III interferons (IFNs) and proinflammatory cytokines.
Identified more than 50 years ago by Alick Isaacs and Jean Lindenmann (34, 35), IFNs are the mainstay for fighting viral infections. In human beings, there are three classes of IFNs, Types I, II, and III, mainly classified on their binding to specific IFN receptors. There are multiple type I IFNs, including multiple sub-types of IFN-α, -β, -ε, -κ, and -ω, and all signal through the IFNAR complex (36). In contrast, Type II IFN comprises only one molecule, IFN-γ, which signals through the IFN-γR complex (37). Receptor binding stimulates a cascade of signal transduction events, discussed in detail below, and triggers an expression of IFN-stimulated genes (ISGs) that mediate a host of antiviral effects [reviewed by Schoggins and Rice (38)].
There are four known Type III IFNs, namely, IFNλ1–4 (gene names: IFNL1–IFNL4). These four genes are all located in a small region on the long arm of chromosome 19 (39–41) and are thought to have arisen as a result of gene duplication (42), see Figure 1. Upon the discovery of IFNL4, the HUGO Nomenclature Committee renamed IL29, IL28A, and IL28B to IFNL1, -L2, and -L3 respectively (39–41). These cytokines signal through a heterodimeric complex consisting of the ligand-binding chain, IFN-λR1 (IL-28Rα) and the accessory chain IL-10R2 (39, 40, 43, 44).
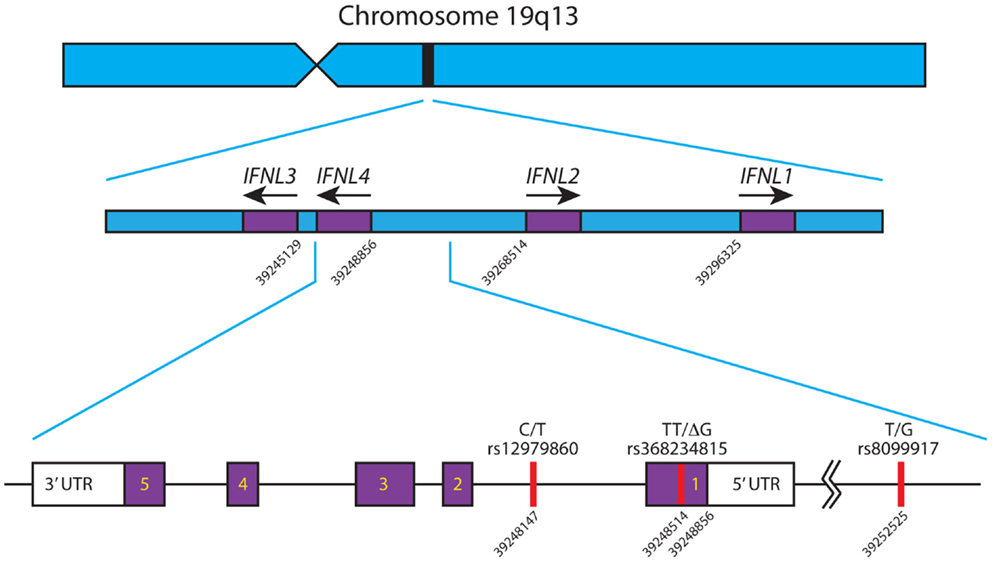
Figure 1. Schematic of the IFNL gene locus. The IFNL1-4 genes are shown in their correct orientation and position in the region of chromosome 19 q13. Based on the human genome sequence, the nucleotide positions of the first exon of each gene are indicated. The primary SNPs, rs12979860, rs368234815, and rs8099917, affecting IFNL3 and IFNL4 are depicted with their corresponding nucleotide position. IFNL4 is located on the reverse strand of chromosome 19, has 5 exons and 3′/5′ untranslated regions. All nucleotide positions are based on Ensembl genome assembly GRCh38 (December 2013), updated August 2014, database version 76 (45). Adapted from Ref. (46).
Restricted Expression of the IFN-λ Receptor
Whereas the Type I IFN receptor IFNAR and the IL-10R2 subunit of the IFN-λ receptor are present on virtually all human cell types, the second IFN-λ receptor subunit IFN-λR1 is expressed primarily on cells of epithelial cell origin (47) so only organs with high-epithelial cell numbers express detectable levels of IFN-λ (e.g., skin, intestine, and lungs). As expected, in vitro response to IFN-λ depends on the expression of IFN-λR1. In cells that lack the receptor, overexpression of IFN-λR1 can restore IFN-λ responses (48).
The greater induction of ISGs following stimulation by IFN-α compared to IFN-λ may be due in part to the number of receptors expressed by individual cells.
Human hepatic cells express both subunits of the IFN-λ receptor (IFNλ-R1 and IL10R2) (49). Addition of IFN-λ to hepatic cell lines (such as Huh-7, HepG2), Huh-7 HCV replicon expressing cell lines (49) and human primary hepatocytes (50) causes STAT1 phosphorylation and induction of ISGs, while induction was not seen following the addition of IFN-λ to primary human monocytes and lymphocytes (50) or mouse hepatocytes (51). As leukocytes and peripheral blood mononuclear cells (PBMCs) from human donors express only 6% of the level of IFNλ-R1 compared to Huh-7, this may explain their lack of response (49).
Signal Transduction in Response to Type III IFNs
Signal transduction in response to Type III IFNs is similar to that seen with Type I IFN [reviewed by Au-Yeung et al. (52)], and is summarized in Figure 2. When Type III interferon binds to IFN-λR1, a conformational change allows the binding of IL-10R2 activating the receptor-associated tyrosine kinases Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2) to cross-tyrosine-phosphorylate the IFN-λR1/IL-10R2 receptor complex allowing the recruitment of signal transducer and activator of transcription (STAT) 2 via its Src Homology 2 (SH2) domain. Further JAK tyrosine-phosphorylation of STAT2 allows the binding of the SH2 domain from cytoplasmic STAT1, resulting in the formation of a STAT1/STAT2 heterodimer. STAT2 is normally bound to IFN regulatory factor 9 (IRF9) in the cytosol, where it shuttles between the nucleus and cytosol. The addition of IRF9 to the complex forms a heterotrimeric transcription complex called IFN-stimulated gene factor 3 (ISGF3). IFN-λ may also activate STATs 3–5, although the significance of this activation has not been studied (53). Once assembled, ISGF3 relocates from the cytosol to the nucleus where it binds to interferon stimulated response elements (ISREs) upstream of ISGs [reviewed by Reich (54)], encoding proteins such as myxovirus (influenza virus) resistance 1 (MX1), melanoma differentiation-associated protein 5 (MDA5), and 2′-5′ oligoadenylate synthetase (OAS). The IFN-α and IFN-λ induction of these ISGs is reduced in STAT1 deficient hepatocytes, consistent with the requirement of STAT1 for formation of the ISGF3 complex (50). In HepG2 cells, induction of IFN-α and IFN-β is not seen when cells are stimulated with IFN-α or IFN-λ but these same cytokines do induce IFN-λ (55). Therefore, in contrast to the Type I IFNs, Type III IFNs are themselves ISGs.
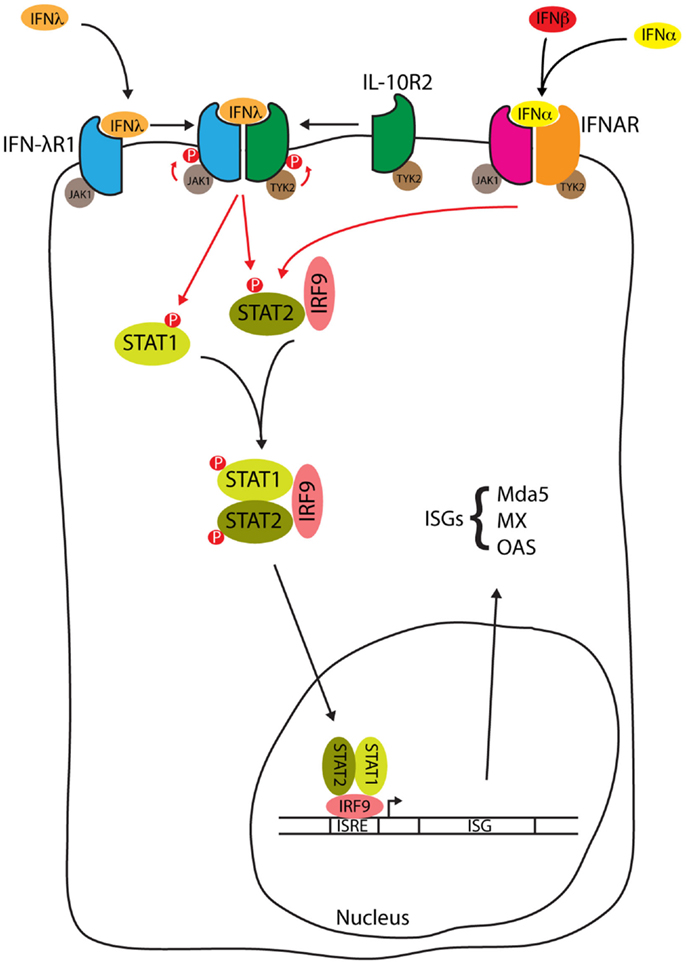
Figure 2. IFN-λ signal transduction. Type III interferon binding to the IFN-λ receptor and type I interferon binding to the IFNAR both activate the JAK1/STAT pathway to form the ISGF3 complex. ISGF3 relocates from the cytosol to the nucleus whereupon binding to ISREs induces the expression of ISGs such as myxovirus (influenza virus) resistance 1 (MX1), melanoma differentiation-associated protein 5 (MDA5), and 2′-5′ oligoadenylate synthetase (OAS), see text for details.
Other pathways may also be activated by IFN-λ, including the AKT signaling pathway (56). In common with Type I IFN, stimulation of cells with IFN-λ leads to phosphorylation, and hence activation of the mitogen-activated protein kinase (MAPK) c-Jun N-terminal kinase (JNK), although evidence for the activation of MAPK p38 or extracellular-signal-regulated kinases (ERK) 1/2 is conflicting (48, 57) and may depend on cell type.
As Type I and Type III IFN share similar signal transduction mechanisms, the repertoire of ISGs induced upon cytokine stimulation is predicted to be similar. Most microarray analysis and RT-PCR studies have shown IFN-λ induces essentially the same genes as IFN-α in HepG2 cells, Huh-7.5 cells containing a full-length HCV replicon (FL-neo), Huh-7.5 cells, and primary mouse and human hepatocytes (50, 58–60). Of note, a study by Bauhofer et al. (61) suggests that the differentiation state of these cells may alter the repertoire of gene induction. They showed that a wider range of ISGs were induced in IFN-α or -λ treated dimethyl sulfoxide differentiated Huh-7.5 (Huh-7.5dif) and PHH cells, as compared to IFN-α or -λ treated wild-type Huh-7.5 cells (61).
Although most of the above-mentioned studies show similar gene expression repertoires after stimulation with IFN-α or IFN-λ, the kinetics, and magnitude of induction are different. In a study comparing global transcriptional profiles over time, Bolen et al. identified a difference in the scale of ISG induction between different cytokines in both Huh-7 cells and primary hepatocytes (62) with IFN-β > IFN-α > IFN-λ3 > IFN-λ1 > IFN-λ2. In this experiment, the same set of genes were induced with all the cytokines but whereas IFN-α stimulated gene expression wanes after approximately 6 h, IFN-β and IFN-λ1–3 stimulated gene expression continues at a high level for >24 h. The greater specific activity (ISG induction) of IFN-λ3 over other Type III IFNs as shown by Bolen et al. has also been replicated by Dellgren et al. (63) who showed that IFN-λ3 is 16-fold more active than IFN-λ2 and 2-fold more active than IFN-λ1. A recent comparison between IFN-λ3 and IFN-λ4 has shown comparable specific activity (44). One possible explanation for the differences in the scale of ISG induction between IFN-α and IFN-λ may be due to transcription factor binding differences. Chromatin immunoprecipitation assays show that less efficient remodeling at promoter sites occurs following stimulation by IFN-λ compared to IFN-α (48). Blocking signaling through the Type I receptor, IFNAR, does not abrogate the activity of IFN-λ (50). IFN-λ signaling was shown to be enhanced by HCV infection of Huh-7.5 cells, an observation, which has been attributed to up-regulation of IFN-λR1 and prolongation of JAK-STAT signaling within those cells (60).
Expression of IFN-λ
Up-regulation of IFN-λ is seen following infection of different cells by diverse viruses such as encephalomyocarditis virus (EMCV), murine cytomegalovirus, reovirus, Sendai virus, dengue virus, and measles virus (39, 40, 64, 65), probably following RLR signaling from the peroxisome (65). Although expression of both IFN Type I and III are triggered in similar ways, differential expression can clearly be seen in organs such as the brain and central nervous system where IFN Type III expression is minimal compared to Type I IFN expression, following viral infection (47). Dendritic cells (DCs) can express IFN-λ on stimulation with double-stranded RNA and TLR3 ligand (66).
Within the liver there is an expression of both IFN-α and -λ but identification of the cell types that express IFN-λ within the infected liver is difficult to determine. It is known that freshly isolated primary FHH express IFN-λ when infected with HCV (30, 31). Until recently, it was not possible to distinguish a particular cytokine expressing cell within a population. However, in an elegant set of experiments carried out by Sheahan et al. (31), the expression profiles of individual fetal hepatocytes were determined following laser capture micro-dissection. Type III interferon induction was observed only in HCV-infected FHH cells and not by surrounding bystander cells. Furthermore IFNL1 expression was found to be dependant on HCV replication as blocking viral replication using 2′CMA (a viral replicase inhibitor) abrogated IFN-λ1 protein secretion (31).
Although there are no studies addressing the issue of IFN-λ secretion by DCs within the HCV-infected liver, it is known that DCs can secrete IFN-λ following in vitro stimulation (55, 66–68). As it is unlikely that DCs support significant HCV replication, exposure to the viral PAMP is thought to occur via interaction with infected hepatocytes, endocytosis of HCV virions, or exposure to viral RNA following immune cell induced death of HCV-infected cells.
There is conflicting evidence for the up-regulation of IFN-λ in HCV chronically infected liver. Some studies have shown upregulated serum levels of IFN-λ in chronic HCV patients when compared to serum levels of patients with either non-viral diseased livers or control non-diseased livers (49, 69), while others have shown lower IFN-λ serum levels in chronic HCV livers compared to non-diseased livers (70). During analysis of IFN-λ transcripts in liver biopsies, Mihm et al. showed no difference between HCV diseased liver and non-viral diseased liver although an increase in IFN-λ transcripts can be seen when HCV-infected livers were compared to healthy livers (71, 72). In vitro studies show that HCV infection causes IFN-λ transcription in PHH, FHH, and the human hepatocyte cell line PH5CH8 (30, 72).
Sequence Homology of IFN-λ1–4
IFN-λ1–3 were discovered by two independent research groups following sequence analysis of the human genome (39, 40), while IFNλ-4 was discovered when genome-wide association study (GWAS) data were compared to RNA-seq analysis of PHHs treated with polyinosinic:polycytidylic acid (polyI:C), a synthetic mimic of double-stranded RNA (41).
IFN-λ1–4 are similar in amino-acid sequence (41–97% amino-acid conservation), especially within the first and last alpha helices, which are the primary regions of contact between Type III IFNs and their receptor, IFN-λR1 (see Figure 3; Table 1). Outside of these regions IFN-λ4 is less similar to IFN-λ1–3 having only 30% amino-acid identity. One notable region where amino-acid conservation is poor is α-helix 3, a region predicted to interact with the IFN-λ receptor subunit IL10R2 (44, 73). IFN-λ2 and IFN-λ3 are the most similar, with approximately 96% amino-acid identity (seven amino-acid differences) within their coding sequences and virtually identical within their non-coding upstream and downstream flanking sequences (41). Despite this similarity between IFN-λ2 and -3, it is remarkable that there is a 16-fold difference in specific activity (63). The reasons for this are as yet unclear, but it is interesting to note that these differences are also located in the region that is predicted to interact with the IFN-λ receptor subunit IL10R2.
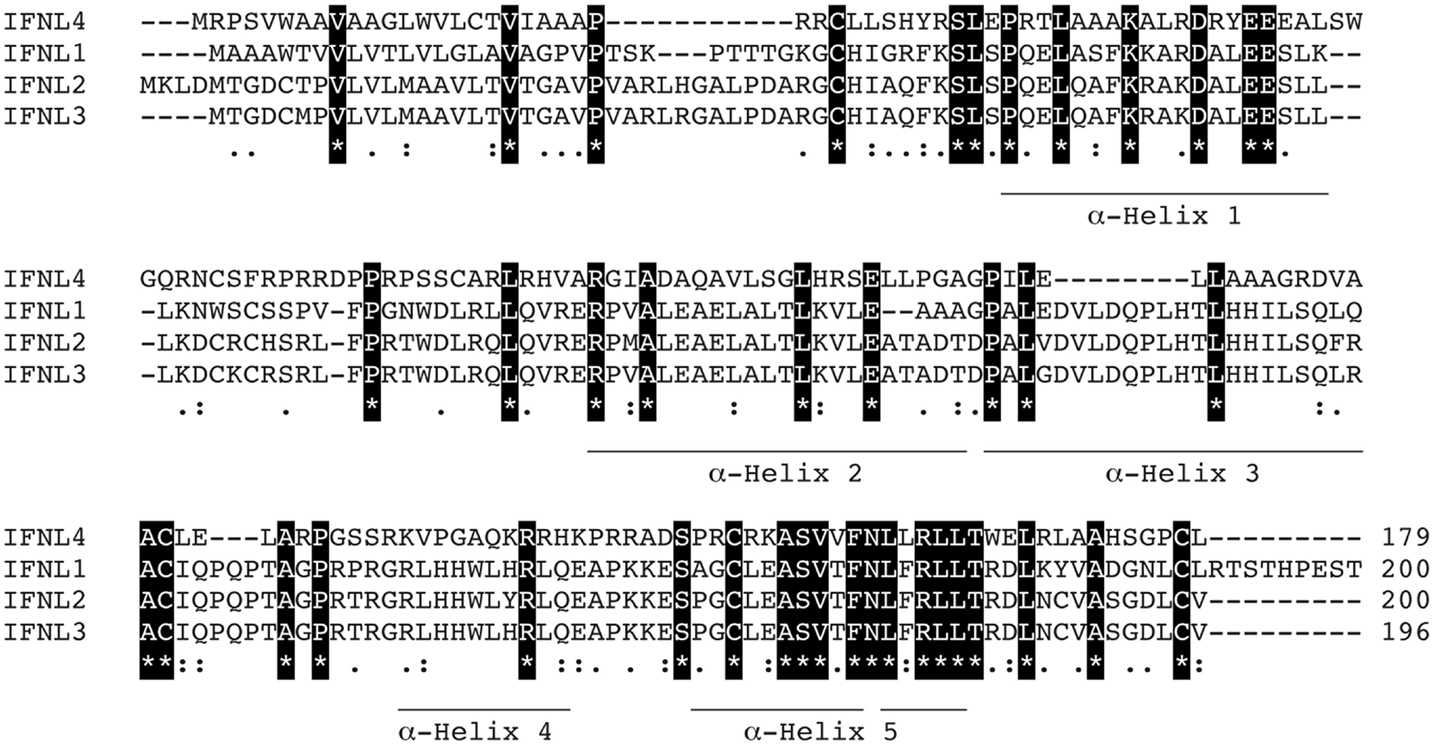
Figure 3. Protein sequence analysis. Clustal multiple sequence alignment by MUSCLE (3.8) for IFN-λs, IFNL1–4. Protein IDs are Q8IU54, Q8IZJ0, Q8IZI9, and K9M1U5-1, respectively. Identical amino-acids are shaded in black, conserved amino-acids are represented by a colon (:) and semi-conserved amino-acids by a period (.). Alpha helices are indicated and are based on the crystal structure of IFNL3 (3HHC chain A) (73–75).
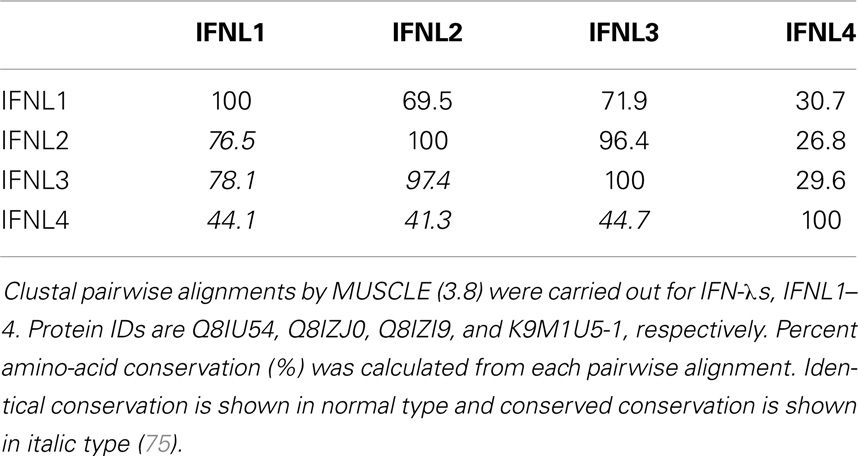
Table 1. Amino-acid conservation table.
Currently, two protein structures have been determined for IFN-λ; IFNλ3 (3HHC) (76) and IFNλ1 in complex with its receptor IFN-λR1 (3OG6) (77). These show that the IFN-λs exist as monomers and bind to the IFN-λR1 receptor as a 1:1 complex. The structure of IFN-λ3 shows that it is most closely related to IL-22, a member of the IL-10 family (76).
The transcription factors that regulate expression of the IFN-λ genes are currently unclear. Conflicting studies have shown requirements for IRF7 alone (IFN-α like), IRF7 plus IRF3 (IFN-β like), or IRF7, IRF3, and NF-κB (72, 78, 79). This uncertainty may be due to the wide range of cell lines used, induction protocols, and the species of IFN-λ used. Knockdown of the p65 subunit of NF-κB decreased IFNL1 transcription in response to HCV and PolyI:C, whereas NF-κB was dispensable for the expression of IFNL2 and IFNL3 (72). Lee et al. suggested that this differential requirement for NF-κB could be the basis of the differential expression of the IFN-λ gene family. Recently, another transcription factor of IFNLs, Med23, was discovered to interact directly with IRF7, leading to the up-regulation of IFN-λ (80).
IFN-λ Inhibits HCV
Studies of HepG2 cells expressing miR-122 and CD81, primary human hepatocytes, and chimpanzee and human in vivo studies have all shown that HCV induces primarily an IFN-λ response rather than IFN-α or -β (30, 31, 72, 81–83). IFN-λ can then act in an autocrine fashion, stimulating the expression of ISGs (30, 82). Exogenous addition of IFN-λ to hepatocytes inhibits replication of both HCV replicons (49, 58, 59, 84, 85) and cell culture-derived HCV (30, 31, 60, 70, 72, 82). In HCV replicon cell lines, recombinant IFNL1 had a greater effect on HCV replication than IFNL2 and IFNL3 (85). IFNL1 also induces STAT1 phosphorylation more readily than IFNL2/3 (49, 85), while no differences are seen between IFNL3 and IFNL4 (44). This is not the case when comparing IFN-λ activity against EMCV in HepG2 cells. Dellgren et al. (63) showed that against EMCV, IFNL3 was the most active of IFNL1–3. Concomitant with a reduction in HCV replication, IFN-λ also suppresses the microRNA miR-122 (72). The addition of both IFN-λ and the miR-122 inhibitor (miRIDIAN) increased the suppression of HCV replication (72).
IFN-λ as a Therapy
Until recently, the standard therapy for chronic HCV was the use of pegylated interferon-α (Peg-IFN) in combination with ribavirin (RBV). Unfortunately, these regimens were poorly tolerated and often ineffective, with treatment efficacy varying among different HCV genotypes and among patients of different ethnic backgrounds or different comorbid conditions (86). Treatment with Peg-IFN and RBV results in sustained virologic response (SVR) in approximately 40–50% of genotype 1-infected people and 80% in those infected with genotypes 2 or 3 (2). HCV treatment is evolving rapidly, and current regimens for genotype 1-infected patients include either telaprevir or boceprevir, first-generation direct-acting antivirals (DAAs). These protease inhibitors (PIs) need to be administered in combination with Peg-IFN and RBV in order to reduce the likelihood of viral drug resistance. The addition of PIs leads to an increase in the likelihood of SVR to between 63 and 92% (2).
Following the discovery that the receptor for IFN-λ is cell-type restricted compared to the IFNAR receptor, it was thought that administration of IFN-λ would have fewer off-target effects and therefore patients would suffer fewer adverse side effects. As reports from in vitro studies suggested that HCV was sensitive to IFN-λ, there was hope that IFN-λs could be used therapeutically to combat HCV. Those hopes have largely been realized following Phase 1 trials looking at the use of pegylated IFN-λ in the treatment of genotype 1 chronic HCV infection (87, 88). The Muir et al. study compared treatment regimens consisting of Peg-IFN-λ or Peg-IFN-λ + RBV in IFN-α treatment-relapsed patients (patients who had relapsed following at least 12 weeks treatment with Peg-IFN-α + RBV) and Peg-IFN-λ + RBV in treatment-naïve patients. Although SVR was not evaluated in this study (being only 4 weeks long), the majority of patients displayed antiviral activity with a >2-log10 decrease in HCV RNA; in the case of treatment-naïve patients, 2 out of 7 achieved transient undetectable HCV RNA. Encouragingly, the administration of Peg-IFN-λ was well tolerated in both of the phase 1 trials with few adverse events. A possible reason for this effect with Peg-IFN-λ is that unlike IFN-α, which can produce a long-lasting refractoriness in JAK-STAT signaling (89), the use of Peg-IFN-λ induces signaling even following multiple or prolonged stimulations (90) and may also be able to overcome pre-existing refractoriness due to previous treatment with IFN-α.
Although the treatment for chronic HCV is heading toward IFN-free regimens with the development of DAAs that target either the HCV RNA polymerase (e.g., sofosbuvir) or non-structural protein NS5A (e.g., daclatasvir or ledipasvir), these treatments are costly and it is not yet known whether these treatments will be applicable to all genotypes. It may be some time until the use of IFN is discontinued, be it α or λ.
Genome-Wide Association Studies
A number of independent groups have identified single nucleotide polymorphisms (SNPs) located near the IFN-λ3 locus that correlate with HCV treatment response and spontaneous clearance of HCV infection. These are summarized in Table 2. The polymorphisms include rs12979860 (C/T) (91–93) and rs8099917 (T/G) (94–97). The rs12979860 SNP is located approximately 3 kb upstream of the IFNL3 gene, within an intron of IFNL4, while the rs8099917 SNP is located within an intergenic region between the IFNL2 and IFNL3 genes.
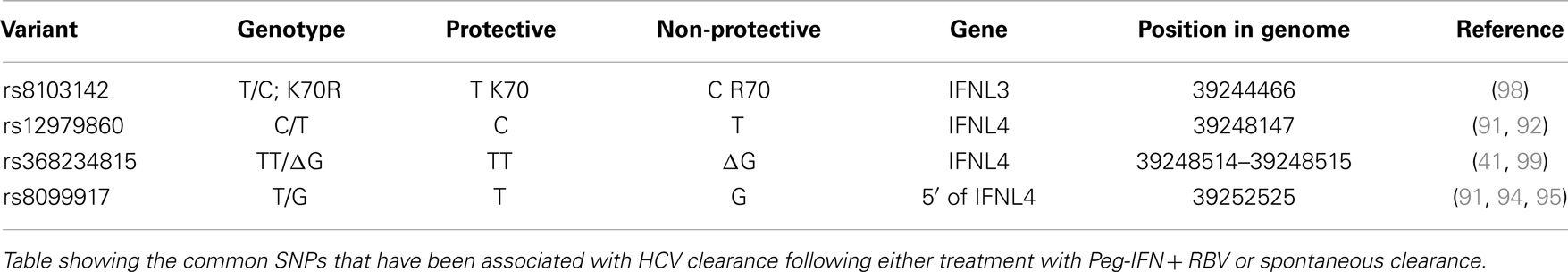
Table 2. SNPs at the IFNL locus.
These SNPs have been associated with SVR following treatment of chronic HCV with Peg-IFN and RBV (91, 94, 95, 97) and also with spontaneous clearance of HCV infection (92, 96, 100) [reviewed by O’Brien et al. (46)]. It has been shown that individuals with the non-beneficial rs12979860-T allele (C/T or T/T) do not respond as well to standard therapy as those individuals with two copies of the beneficial rs12979860-C (C/C) allele. The prevalence of the non-beneficial rs12979860-T allele is higher in people of African ancestry than in those of European or Asian ancestry. In a similar manner, individuals with two copies of the beneficial rs8099917-T (T/T) allele respond better than those individuals with one or no copies of the non-beneficial T allele (T/G or G/G) (94). As the rs12979860 and rs8099917 SNPs are in linkage disequilibrium (LD) with each other, favorable rs8099917 alleles are also more prevalent in Europeans/Asians compared to Africans. Although Melis et al. did not look at patient data they have developed a method to analyze both rs12979860 and rs8099917 (101), which may provide greater predictability in treatment outcomes.
Recently, two groups identified a possible causal SNP upstream of the IFNL3 gene, rs368234815 (TT or ΔG) (originally designated ss469415590) (41, 46, 99). This SNP results in a frameshift mutation leading to the polyI:C mediated transient expression of a new IFN-λ, now termed IFNL4 (41). The IFNL4 protein is made in IFNL4-ΔG carriers but not IFNL4-TT homozygotes. In addition, due to its location within a CpG island upstream of IFNL3, the TT/ΔG polymorphism is responsible for the methylation of a cytosine residue, which may influence gene expression (99). IFNL4-ΔG is strongly associated with impaired spontaneous clearance. In African-Americans, the IFNL4-ΔG SNP has been shown to be a better marker for predicting response following Peg-IFN and RBV than rs12979860 (41, 102). This is in contrast to European- and Asian-Americans where the two SNPs are equally informative (41). This discrepancy is due to the differences in the degree of LD between these SNPs within these populations (99). Recently, another possible causal SNP was detected, rs8103142 (98). This polymorphism is present within the second exon of IFNL3 and changes the amino-acid lysine at position 70 to arginine (K70R). Interestingly, this amino-acid is one of only seven that are different between IFNL2 and IFNL3 (73). Given that IFNL3 has a 16-fold greater specific activity than IFNL2, a number of groups have studied the functional consequences of this substitution in vitro (85, 103, 104); however, they were unable to show any difference in anti-HCV replicon activity or in the stimulation of its activity.
The molecular mechanism for the association between treatment outcome and IFN-λ SNPs is not known. The positions of these SNPs, in the vicinity of IFNL3 and IFNL4, suggest that they may be involved in transcriptional regulation although the evidence for this is conflicting. Ge et al. (91) were unable to detect a relationship between IFNL3 mRNA levels and rs12979860 polymorphism in PBMCs from non-HCV-infected patients; Urban et al. showed that there was no significant difference in IFNL2/3 mRNA expression in HCV-infected liver tissue (103). In contrast, the presence of the non-beneficial G allele for SNP rs8099917 was associated with lower levels of IFN-λ expression in PBMC taken from HCV-infected patients (95) and also whole blood from healthy individuals (94), but there was no significant difference in IFNL3 mRNA expression between rs8099917 genotypes in a study carried out by Honda et al. (105). Conversely, carriers of the protective rs12979860 C allele were shown to have higher serum protein levels of IFN-λ (70), although this may be due to LD with the IFNL4 associated SNP, rs368234815. In a publication by Bibert et al., PBMCs from individuals carrying different allelic combinations of rs12979860 and rs368234815 were stimulated with polyI:C and the expression of IFNL3 mRNA was measured. Their study showed lower expression of IFNL3 mRNA in PBMCs from individuals carrying one or two copies of the mutant ΔG allele but not by rs12979860 (99). This study also showed that the plasma level of the ISG, IFN-γ-inducible protein 10 (IP-10), was reduced in individuals carrying the rs368234815 non-beneficial allele but not in those carrying the rs12979860 non-beneficial allele. Another mechanism may be the alteration of IFNL3 transcript stability via the action of HCV induced miRNA targeting of the polymorphic region of the IFNL3 3′ UTR (106).
As previously mentioned, miR-122 may play an important role in HCV replication (10). No correlation between expression of miR-122 and SNP rs12979860 genotype was seen in human liver biopsies by Urban et al. (103) although a correlation was seen between SVR and miR-122 expression with greater expression of miR-122 in responders compared to non-responders (NR) (103, 107). In contrast, Estrabaud et al. showed that there was a significant increase of miR-122 at baseline in rs12979860 CC genotype patients regardless of their response status (108). The authors argue that the larger numbers of patients in their study may explain this discrepancy (108). As miR-122 stimulates HCV replication in vitro, and antagonism of miR-122 by the oligonucleotide SPC3649 results in a decrease in serum HCV RNA in vivo (109), the finding that a beneficial allele may increase miR-122 is surprising. In the case of rs12979860, genotype C/C patients have a lower baseline ISG expression (103, 105, 110), and hence a stronger ISG activation upon infection. This innate activation may swamp any effect on HCV replication brought about by an increase in miR-122 (108).
In the case of IFNL4, the frameshift produced by the unfavorable, non-protective ΔG allele causes the production of IFN-λ4 protein. This protein was postulated to have a weak signal peptide (SP) and subsequent poor secretion (41) but subsequent experiments have shown that the SP functions correctly and that poor secretion is due to the lack of N-linked glycosylation (44). IFN-λ4 is active without glycosylation as Escherichia coli expressed protein is able to induce ISGs in HepG2 cells to a similar level as IFN-λ3, and addition of recombinant IFN-λ4 to Huh-7-Lunet and HepG2 cells inhibited HCV replicon replication (44). The presence of an IFN-λ4 protein that is able to induce ISG expression, is active against HCV, yet is associated with poorer HCV clearance rates is counter-intuitive. Various theories have been put forward for this effect including intracellular accumulation of non-glycosylated IFN-λ4 could be cytotoxic and result in cell death, or that IFN-λ4 could impede signaling by other IFN-λs by blocking the IFN-λ receptor and causing exhaustion of the IFN response pathways (46). IFNL4 transcripts were exclusively detected in chronic HCV liver biopsies and not in control uninfected, HBV infected, and inflammatory liver diseased liver biopsies (111), suggesting that the IFNL4 gene is specifically activated in HCV infection. In addition, Amanzada et al. were able to show a positive correlation between HCV viral loads and the amount of IFNL4 transcripts. As expected, IFNL4 transcripts were detected in all IFNL4 SNP rs368234815 genotypes but IFN-λ4 protein can only be produced in those patients with the ΔG genotype.
It needs to be borne in mind that although GWAS studies have allowed us to narrow down the area in which a causal variant may reside, neither the identified SNPs nor the expression of the novel IFNL4 gene may be the true causal variant, and these associations may be due to some other as yet unidentified mechanism.
The IFN-λ gene region seems to have undergone genetic selection pressure, especially in the case of IFN-λ4. The negative selection of the ΔG allele and its replacement with the TT variant in non-African populations suggests that infection with another infectious agent may have driven this change. This geographical selective force probably occurred after the colonization of the New World (92). Candidates such as hepatitis B and HIV have been discounted as they do not show any association with the known IFN-λ SNPs (112–114). Also, any such disease would need to predate the movement of modern human beings out of Africa and therefore pathogens such as HIV are unlikely candidates. It remains to be seen if this polymorphism affects the outcome of other infections.
IFN-λ and the Adaptive Immune Response
As well as its role in innate immunity, IFN-λ may have an immunomodulatory function (115). There is currently sparse literature describing the action of IFN-λ and its effect on adaptive immunity directed toward HCV, perhaps because leukocytes in general do not express the IFN-λR1 receptor (116). Though some authors have demonstrated both IFN-λR1 mRNA and cell-surface expression in T-cells, others have not been able to replicate these findings (117). It has been reported that DCs can acquire IFN-λ responsiveness following up-regulation of IFN-λR1 during their maturation from monocytes (115). If this is the case, then even though IFN-λR1 may be absent from T-cells their function may be altered by the interaction with IFN-λ stimulated DCs.
Conclusion
Recent advances in the treatment of HCV with newly developed DAAs will revolutionize the field of HCV research but the cost of these treatments and the side effects they may engender mean that it is too soon to abandon HCV basic research. Many questions still remain to be answered, especially in HCV innate immunity and the role of IFN-λ. The identification of genetic variation at the IFNL gene locus has allowed us to begin to understand the basis of HCV resolution, but further research is required to appreciate how the IFN-λs are involved in the progression to the chronic state following acute infection. Future research should concentrate on understanding IFN-λ gene regulation and the relevance of its differential expression.
As therapy moves toward IFN-free DAA-based regimens, will the patient genotype have an effect on SVR, and if so will this SVR be affected by the same SNPs seen with IFN-α therapy or will this new therapy throw up its own set of SNPs? The development of a more relevant culture system would be advantageous for our improved understanding of HCV pathogenesis and treatment. Although the techniques of micro-patterning and 3D cell culture are advancing, these models are not yet “user friendly.” In the absence of these, greater use of physiologically relevant cells such as primary hepatocytes will allow us to tease apart the intricate pathways and interactions that need to be discovered if we are to fully appreciate the complexities of the host:HCV interaction. Researchers should not be afraid of the inherent donor liver variation; as the maxim says variety is the spice of life.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the U.S. National Institutes of Health (grant R01AI089957).
References
1. Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect (2011) 17:107–15. doi: 10.1111/j.1469-0691.2010.03432.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Mutimer D, Aghemo A, Diepolder H, Negro F, Robaeys G, Ryder S, et al. EASL clinical practice guidelines: management of hepatitis C virus infection. J Hepatol (2014) 60:392–420. doi:10.1016/j.jhep.2013.11.003
3. Seeff LB. The history of the “natural history” of hepatitis C (1968-2009). Liver Int (2009) 29(Suppl 1):89–99. doi:10.1111/j.1478-3231.2008.01927.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology (2014) 59:318–27. doi:10.1002/hep.26744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Bukh J, Miller RH, Purcell RH. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin Liver Dis (1995) 15:41–63. doi:10.1055/s-2007-1007262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Da Costa D, Turek M, Felmlee DJ, Girardi E, Pfeffer S, Long G, et al. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J Virol (2012) 86:11919–25. doi:10.1128/JVI.01066-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature (2013) 501:237–41. doi:10.1038/nature12427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Vogt A, Scull MA, Friling T, Horwitz JA, Donovan BM, Dorner M, et al. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology (2013) 444:1–11. doi:10.1016/j.virol.2013.05.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Frentzen A, Anggakusuma, Gurlevik E, Hueging K, Knocke S, Ginkel C, et al. Cell entry, efficient RNA replication, and production of infectious hepatitis C virus progeny in mouse liver-derived cells. Hepatology (2014) 59:78–88. doi:10.1002/hep.26626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science (2005) 309:1577–81. doi:10.1126/science.1113329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, et al. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J (2008) 27:3300–10. doi:10.1038/emboj.2008.244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Jopling CL. Regulation of hepatitis C virus by microRNA-122. Biochem Soc Trans (2008) 36:1220–3. doi:10.1042/BST0361220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Lin LT, Noyce RS, Pham TN, Wilson JA, Sisson GR, Michalak TI, et al. Replication of subgenomic hepatitis C virus replicons in mouse fibroblasts is facilitated by deletion of interferon regulatory factor 3 and expression of liver-specific microRNA 122. J Virol (2010) 84:9170–80. doi:10.1128/JVI.00559-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Machlin ES, Sarnow P, Sagan SM. Masking the 5’ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci U S A (2011) 108:3193–8. doi:10.1073/pnas.1012464108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Jangra RK, Yi M, Lemon SM. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol (2010) 84:6615–25. doi:10.1128/JVI.00417-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol (2002) 76:13001–14. doi:10.1128/JVI.76.24.13001-13014.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Murray EM, Grobler JA, Markel EJ, Pagnoni MF, Paonessa G, Simon AJ, et al. Persistent replication of hepatitis C virus replicons expressing the beta-lactamase reporter in subpopulations of highly permissive Huh7 cells. J Virol (2003) 77:2928–35. doi:10.1128/JVI.77.5.2928-2935.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science (1999) 285:110–3. doi:10.1126/science.285.5424.110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, et al. Complete replication of hepatitis C virus in cell culture. Science (2005) 309:623–6. doi:10.1126/science.1114016
20. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med (2005) 11:791–6. doi:10.1038/nm0805-905b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A (2005) 102:9294–9. doi:10.1073/pnas.0503596102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Kato T, Furusaka A, Miyamoto M, Date T, Yasui K, Hiramoto J, et al. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J Med Virol (2001) 64:334–9. doi:10.1002/jmv.1055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Ploss A, Khetani SR, Jones CT, Syder AJ, Trehan K, Gaysinskaya VA, et al. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc Natl Acad Sci U S A (2010) 107:3141–5. doi:10.1073/pnas.0915130107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Podevin P, Carpentier A, Pene V, Aoudjehane L, Carriere M, Zaidi S, et al. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology (2010) 139:1355–64. doi:10.1053/j.gastro.2010.06.058
25. Gondeau C, Briolotti P, Razafy F, Duret C, Rubbo PA, Helle F, et al. In vitro infection of primary human hepatocytes by HCV-positive sera: insights on a highly relevant model. Gut (2013) 63:1490–500. doi:10.1136/gutjnl-2013-304623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Helle F, Brochot E, Fournier C, Descamps V, Izquierdo L, Hoffmann TW, et al. Permissivity of primary human hepatocytes and different hepatoma cell lines to cell culture adapted hepatitis C virus. PLoS One (2013) 8:e70809. doi:10.1371/journal.pone.0070809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Iacovacci S, Manzin A, Barca S, Sargiacomo M, Serafino A, Valli MB, et al. Molecular characterization and dynamics of hepatitis C virus replication in human fetal hepatocytes infected in vitro. Hepatology (1997) 26:1328–37. doi:10.1053/jhep.1997.v26.pm0009362380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Lazaro CA, Chang M, Tang W, Campbell J, Sullivan DG, Gretch DR, et al. Hepatitis C virus replication in transfected and serum-infected cultured human fetal hepatocytes. Am J Pathol (2007) 170:478–89. doi:10.2353/ajpath.2007.060789
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Andrus L, Marukian S, Jones CT, Catanese MT, Sheahan TP, Schoggins JW, et al. Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology (2011) 54:1901–12. doi:10.1002/hep.24557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, et al. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology (2011) 54:1913–23. doi:10.1002/hep.24580
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, Ploss A, et al. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe (2014) 15:190–202. doi:10.1016/j.chom.2014.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Targett-Adams P, Boulant S, McLauchlan J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J Virol (2008) 82:2182–95. doi:10.1128/JVI.01565-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev (2011) 243:61–73. doi:10.1111/j.1600-065X.2011.01048.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci (1957) 147:258–67. doi:10.1098/rspb.1957.0049
35. Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. Some properties of interferon. Proc R Soc Lond B Biol Sci (1957) 147:268–73. doi:10.1098/rspb.1957.0049
36. de Weerd NA, Samarajiwa SA, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem (2007) 282:20053–7. doi:10.1074/jbc.R700006200
37. Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol (1997) 15:563–91. doi:10.1146/annurev.immunol.15.1.563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol (2011) 1:519–25. doi:10.1016/j.coviro.2011.10.008
39. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4:69–77. doi:10.1038/ni875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol (2003) 4:63–8. doi:10.1038/ni873
41. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet (2013) 45:164–71. doi:10.1038/ng.2521
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res (2010) 30:555–64. doi:10.1089/jir.2010.0078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Dumoutier L, Lejeune D, Hor S, Fickenscher H, Renauld JC. Cloning of a new type II cytokine receptor activating signal transducer and activator of transcription (STAT)1, STAT2 and STAT3. Biochem J (2003) 370:391–6. doi:10.1042/BJ20021935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Hamming OJ, Terczynska-Dyla E, Vieyres G, Dijkman R, Jorgensen SE, Akhtar H, et al. Interferon lambda 4 signals via the IFNlambda receptor to regulate antiviral activity against HCV and coronaviruses. EMBO J (2013) 32:3055–65. doi:10.1038/emboj.2013.232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2014. Nucleic Acids Res (2014) 42:D749–55. doi:10.1093/nar/gkt1196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. O’Brien TR, Prokunina-Olsson L, Donnelly RP. IFN-lambda4: the paradoxical new member of the interferon lambda family. J Interferon Cytokine Res (2014). doi:10.1089/jir.2013.0136
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog (2008) 4:e1000017. doi:10.1371/journal.ppat.1000017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the JAK-STAT pathway and the mitogen-activated protein kinases. J Virol (2007) 81:7749–58. doi:10.1128/JVI.02438-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Diegelmann J, Beigel F, Zitzmann K, Kaul A, Goke B, Auernhammer CJ, et al. Comparative analysis of the lambda-interferons IL-28A and IL-29 regarding their transcriptome and their antiviral properties against hepatitis C virus. PLoS One (2010) 5:e15200. doi:10.1371/journal.pone.0015200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Dickensheets H, Sheikh F, Park O, Gao B, Donnelly RP. Interferon-lambda (IFN-lambda) induces signal transduction and gene expression in human hepatocytes, but not in lymphocytes or monocytes. J Leukoc Biol (2013) 93:377–85. doi:10.1189/jlb.0812395
51. Hermant P, Demarez C, Mahlakoiv T, Staeheli P, Meuleman P, Michiels T. Human but not mouse hepatocytes respond to interferon-lambda in vivo. PLoS One (2014) 9:e87906. doi:10.1371/journal.pone.0087906
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Au-Yeung N, Mandhana R, Horvath CM. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAKSTAT (2013) 2:e23931. doi:10.4161/jkst.23931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Dumoutier L, Tounsi A, Michiels T, Sommereyns C, Kotenko SV, Renauld JC. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J Biol Chem (2004) 279:32269–74. doi:10.1074/jbc.M404789200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Reich NC. STATs get their move on. JAKSTAT (2013) 2:e27080. doi:10.4161/jkst.27080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol (2006) 80:4501–9. doi:10.1128/JVI.80.9.4501-4509.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Ank N, West H, Paludan SR. IFN-lambda: novel antiviral cytokines. J Interferon Cytokine Res (2006) 26:373–9. doi:10.1089/jir.2006.26.373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, et al. IL-28A and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am J Physiol Gastrointest Liver Physiol (2005) 289:G960–8. doi:10.1152/ajpgi.00126.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology (2006) 44:896–906. doi:10.1002/hep.21312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, Macdonald MR, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology (2006) 131:1887–98. doi:10.1053/j.gastro.2006.09.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kohli A, Zhang X, Yang J, Russell RS, Donnelly RP, Sheikh F, et al. Distinct and overlapping genomic profiles and antiviral effects of Interferon-lambda and -alpha on HCV-infected and noninfected hepatoma cells. J Viral Hepat (2012) 19:843–53. doi:10.1111/j.1365-2893.2012.01610.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Bauhofer O, Ruggieri A, Schmid B, Schirmacher P, Bartenschlager R. Persistence of HCV in quiescent hepatic cells under conditions of an interferon-induced antiviral response. Gastroenterology (2012) 143(429–438):e428. doi:10.1053/j.gastro.2012.04.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Bolen CR, Ding S, Robek MD, Kleinstein SH. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology (2014) 59:1262–72. doi:10.1002/hep.26657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Dellgren C, Gad HH, Hamming OJ, Melchjorsen J, Hartmann R. Human interferon-lambda3 is a potent member of the type III interferon family. Genes Immun (2009) 10:125–31. doi:10.1038/gene.2008.87
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine (2005) 31:109–18. doi:10.1016/j.cyto.2005.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol (2014) 15:717–26. doi:10.1038/ni.2915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Lauterbach H, Bathke B, Gilles S, Traidl-Hoffmann C, Luber CA, Fejer G, et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med (2010) 207:2703–17. doi:10.1084/jem.20092720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Yin Z, Dai J, Deng J, Sheikh F, Natalia M, Shih T, et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol (2012) 189:2735–45. doi:10.4049/jimmunol.1102038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Stone AE, Giugliano S, Schnell G, Cheng L, Leahy KF, Golden-Mason L, et al. Hepatitis C virus pathogen associated molecular pattern (PAMP) triggers production of lambda-interferons by human plasmacytoid dendritic cells. PLoS Pathog (2013) 9:e1003316. doi:10.1371/journal.ppat.1003316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Dolganiuc A, Kodys K, Marshall C, Saha B, Zhang S, Bala S, et al. Type III interferons, IL-28 and IL-29, are increased in chronic HCV infection and induce myeloid dendritic cell-mediated FoxP3+ regulatory T cells. PLoS One (2012) 7:e44915. doi:10.1371/journal.pone.0044915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Langhans B, Kupfer B, Braunschweiger I, Arndt S, Schulte W, Nischalke HD, et al. Interferon-lambda serum levels in hepatitis C. J Hepatol (2011) 54:859–65. doi:10.1016/j.jhep.2010.08.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Mihm S, Frese M, Meier V, Wietzke-Braun P, Scharf JG, Bartenschlager R, et al. Interferon type I gene expression in chronic hepatitis C. Lab Invest (2004) 84:1148–59. doi:10.1038/labinvest.3700135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Lee HC, Narayanan S, Park SJ, Seong SY, Hahn YS. Transcriptional regulation of IFN-lambda genes in hepatitis C virus-infected hepatocytes via IRF-3.IRF-7.NF-kappaB complex. J Biol Chem (2014) 289:5310–9. doi:10.1074/jbc.M113.536102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Gad HH, Hamming OJ, Hartmann R. The structure of human interferon lambda and what it has taught us. J Interferon Cytokine Res (2010) 30:565–71. doi:10.1089/jir.2010.0062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res (2000) 28:235–42. doi:10.1093/nar/28.1.235
75. Edgar RC. Muscle: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics (2004) 5:113. doi:10.1186/1471-2105-5-113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Gad HH, Dellgren C, Hamming OJ, Vends S, Paludan SR, Hartmann R. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem (2009) 284:20869–75. doi:10.1074/jbc.M109.002923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Miknis ZJ, Magracheva E, Li W, Zdanov A, Kotenko SV, Wlodawer A. Crystal structure of human interferon-lambda1 in complex with its high-affinity receptor interferon-lambdaR1. J Mol Biol (2010) 404:650–64. doi:10.1016/j.jmb.2010.09.068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Osterlund PI, Pietila TE, Veckman V, Kotenko SV, Julkunen I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol (2007) 179:3434–42. doi:10.4049/jimmunol.179.6.3434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Thomson SJ, Goh FG, Banks H, Krausgruber T, Kotenko SV, Foxwell BM, et al. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc Natl Acad Sci U S A (2009) 106:11564–9. doi:10.1073/pnas.0904477106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Griffiths SJ, Koegl M, Boutell C, Zenner HL, Crump CM, Pica F, et al. A systematic analysis of host factors reveals a Med23-interferon-lambda regulatory axis against herpes simplex virus type 1 replication. PLoS Pathog (2013) 9:e1003514. doi:10.1371/journal.ppat.1003514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Park H, Serti E, Eke O, Muchmore B, Prokunina-Olsson L, Capone S, et al. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology (2012) 56:2060–70. doi:10.1002/hep.25897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, et al. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology (2012) 142:978–88. doi:10.1053/j.gastro.2011.12.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Israelow B, Narbus CM, Sourisseau M, Evans MJ. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology (2014) 60(4):1170–9. doi:10.1002/hep.27227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits hepatitis B and C virus replication. J Virol (2005) 79:3851–4. doi:10.1128/JVI.79.6.3851-3854.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Friborg J, Levine S, Chen C, Sheaffer AK, Chaniewski S, Voss S, et al. Combinations of lambda interferon with direct-acting antiviral agents are highly efficient in suppressing hepatitis C virus replication. Antimicrob Agents Chemother (2013) 57:1312–22. doi:10.1128/AAC.02239-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Itoh Y, Nishimura T, Hashimoto H, Yamaguchi K, Niimi T, Yokomizo C, et al. Simple formula to predict response to peginterferon alpha2b and ribavirin combination therapy in genotype 1 chronic hepatitis C patients with high viral loads. Hepatol Res (2011) 41:126–32. doi:10.1111/j.1872-034X.2010.00750.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Muir AJ, Shiffman ML, Zaman A, Yoffe B, De La Torre A, Flamm S, et al. Phase 1b study of pegylated interferon lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology (2010) 52:822–32. doi:10.1002/hep.23743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Ramos EL. Preclinical and clinical development of pegylated interferon-lambda 1 in chronic hepatitis C. J Interferon Cytokine Res (2010) 30:591–5. doi:10.1089/jir.2010.0066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Sarasin-Filipowicz M, Wang X, Yan M, Duong FH, Poli V, Hilton DJ, et al. Alpha interferon induces long-lasting refractoriness of JAK-STAT signaling in the mouse liver through induction of USP18/UBP43. Mol Cell Biol (2009) 29:4841–51. doi:10.1128/MCB.00224-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Makowska Z, Duong FH, Trincucci G, Tough DF, Heim MH. Interferon-beta and interferon-lambda signaling is not affected by interferon-induced refractoriness to interferon-alpha in vivo. Hepatology (2011) 53:1154–63. doi:10.1002/hep.24189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature (2009) 461:399–401. doi:10.1038/nature08309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature (2009) 461:798–801. doi:10.1038/nature08463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Thompson AJ, Muir AJ, Sulkowski MS, Ge D, Fellay J, Shianna KV, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology (2010) 139(120–129):e118. doi:10.1053/j.gastro.2010.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet (2009) 41:1100–4. doi:10.1038/ng.447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet (2009) 41:1105–9. doi:10.1038/ng.449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology (2010) 138(1345):1338–45. doi:10.1053/j.gastro.2009.12.056
97. Ochi H, Maekawa T, Abe H, Hayashida Y, Nakano R, Imamura M, et al. IL-28B predicts response to chronic hepatitis C therapy – fine-mapping and replication study in Asian populations. J Gen Virol (2011) 92:1071–81. doi:10.1099/vir.0.029124-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. di Iulio J, Ciuffi A, Fitzmaurice K, Kelleher D, Rotger M, Fellay J, et al. Estimating the net contribution of interleukin-28B variation to spontaneous hepatitis C virus clearance. Hepatology (2011) 53:1446–54. doi:10.1002/hep.24263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Bibert S, Roger T, Calandra T, Bochud M, Cerny A, Semmo N, et al. IL28B expression depends on a novel TT/-G polymorphism which improves HCV clearance prediction. J Exp Med (2013) 210:1109–16. doi:10.1084/jem.20130012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Duggal P, Thio CL, Wojcik GL, Goedert JJ, Mangia A, Latanich R, et al. Genome-wide association study of spontaneous resolution of hepatitis C virus infection: data from multiple cohorts. Ann Intern Med (2013) 158:235–45. doi:10.7326/0003-4819-158-4-201302190-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Melis R, Fauron C, McMillin G, Lyon E, Shirts B, Hubley LM, et al. Simultaneous genotyping of rs12979860 and rs8099917 variants near the IL28B locus associated with HCV clearance and treatment response. J Mol Diagn (2011) 13:446–51. doi:10.1016/j.jmoldx.2011.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Aka PV, Kuniholm MH, Pfeiffer RM, Wang AS, Tang W, Chen S, et al. Association of the IFNL4-DeltaG allele with impaired spontaneous clearance of hepatitis C virus. J Infect Dis (2014) 209:350–4. doi:10.1093/infdis/jit433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Urban TJ, Thompson AJ, Bradrick SS, Fellay J, Schuppan D, Cronin KD, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology (2010) 52:1888–96. doi:10.1002/hep.23912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Sugiyama M, Tanaka Y, Wakita T, Nakanishi M, Mizokami M. Genetic variation of the IL-28B promoter affecting gene expression. PLoS One (2011) 6:e26620. doi:10.1371/journal.pone.0026620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Honda M, Sakai A, Yamashita T, Nakamoto Y, Mizukoshi E, Sakai Y, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology (2010) 139:499–509. doi:10.1053/j.gastro.2010.04.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. McFarland AP, Horner SM, Jarret A, Joslyn RC, Bindewald E, Shapiro BA, et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol (2014) 15:72–9. doi:10.1038/ni.2758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Sarasin-Filipowicz M, Krol J, Markiewicz I, Heim MH, Filipowicz W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat Med (2009) 15:31–3. doi:10.1038/nm.1902
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Estrabaud E, Lapalus M, Broet P, Appourchaux K, De Muynck S, Lada O, et al. Reduction of microRNA 122 expression in IFNL3 CT/TT carriers and during progression of fibrosis in patients with chronic hepatitis C. J Virol (2014) 88:6394–402. doi:10.1128/JVI.00016-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science (2010) 327:198–201. doi:10.1126/science.1178178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Dill MT, Duong FH, Vogt JE, Bibert S, Bochud PY, Terracciano L, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology (2011) 140:1021–31. doi:10.1053/j.gastro.2010.11.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Amanzada A, Kopp W, Spengler U, Ramadori G, Mihm S. Interferon-lambda4 (IFNL4) transcript expression in human liver tissue samples. PLoS One (2013) 8:e84026. doi:10.1371/journal.pone.0084026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, et al. A whole-genome association study of major determinants for host control of HIV-1. Science (2007) 317:944–7. doi:10.1126/science.1143767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet (2009) 41:591–5. doi:10.1038/ng.348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Martin MP, Qi Y, Goedert JJ, Hussain SK, Kirk GD, Hoots WK, et al. IL28B polymorphism does not determine outcomes of hepatitis B virus or HIV infection. J Infect Dis (2010) 202:1749–53. doi:10.1086/657146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Mennechet FJ, Uze G. Interferon-lambda-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood (2006) 107:4417–23. doi:10.1182/blood-2005-10-4129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Donnelly RP, Sheikh F, Kotenko SV, Dickensheets H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J Leukoc Biol (2004) 76:314–21. doi:10.1189/jlb.0204117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: innate immunity, interferon lambda, hepatitis C virus, hepatocyte, chronic infection
Citation: Laidlaw SM and Dustin LB (2014) Interferon lambda: opportunities, risks, and uncertainties in the fight against HCV. Front. Immunol. 5:545. doi: 10.3389/fimmu.2014.00545
Received: 01 September 2014; Accepted: 13 October 2014;
Published online: 31 October 2014.
Edited by:
Rene DeWaal Malefyt, Merck Research Laboratories, USAReviewed by:
Christian Schönbach, Nazarbayev University, KazakhstanDavid Escors, University College London, UK
Copyright: © 2014 Laidlaw and Dustin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lynn B. Dustin, Kennedy Institute of Rheumatology, Nuffield Department of Orthopaedics, Rheumatology, and Musculoskeletal Sciences, University of Oxford, South Parks Road, Oxford OX1 3SY, UK e-mail:bHlubi5kdXN0aW5Aa2VubmVkeS5veC5hYy51aw==