 Alisha Holtzhausen
Alisha Holtzhausen Fei Zhao
Fei Zhao Kathy S. Evans
Kathy S. Evans Brent A. Hanks
Brent A. Hanks- 1Department of Pharmacology and Cell Biology, Duke University Medical Center, Durham, NC, USA
- 2Division of Medical Oncology, Department of Medicine, Duke University Medical Center, Durham, NC, USA
Although prolonged genetic pressure has been conjectured to be necessary for the eventual development of tumor immune evasion mechanisms, recent work is demonstrating that early genetic mutations are capable of moonlighting as both intrinsic and extrinsic modulators of the tumor immune microenvironment. The indoleamine 2,3-dioxygenase-1 (IDO) immunoregulatory enzyme is emerging as a key player in tumor-mediated immune tolerance. While loss of the tumor suppressor, BIN-1, and the over-expression of cyclooxygenase-2 have been implicated in intrinsic regulation of IDO, recent findings have demonstrated the loss of TβRIII and the upregulation of Wnt5a by developing cancers to play a role in the extrinsic control of IDO activity by local dendritic cell populations residing within tumor and tumor-draining lymph node tissues. Together, these genetic changes are capable of modulating paracrine signaling pathways in the early stages of carcinogenesis to establish a site of immune privilege by promoting the differentiation and activation of local regulatory T cells. Additional investigation of these immune evasion pathways promises to provide opportunities for the development of novel strategies to synergistically enhance the efficacy of the evolving class of T cell-targeted “checkpoint” inhibitors.
Introduction
Indoleamine 2,3-dioxygenase-1 (IDO) is a heme-containing enzyme known to catalyze the rate limiting step in the degradation of the essential amino acid tryptophan to its metabolic byproducts known collectively as the kynurenines (1). Although initially felt to play primarily an anti-microbial role, pioneering work eventually showed this biochemical pathway to impact the immune system by inhibiting T cell proliferation and driving the differentiation and activation of regulatory T cells (Tregs) (2–6). While IDO has been broadly implicated in the progression of several cancers by aiding tumors to evade the host immune system, the mechanisms utilized by cancers to regulate IDO activity have remained poorly characterized (7, 8). Recent work in pre-clinical models has revealed novel mechanisms utilized by cancers to manipulate IDO activity within the tumor microenvironment. Interestingly, these mechanisms have been found to be regulated by previously defined tumor suppressors and oncogenes, which undergo genetic alteration relatively early during tumorigenesis. In contrast to the cancer immunoediting hypothesis, which proposes that immune-mediated selective pressure by the adaptive immune system is necessary before active immune tolerization mechanisms develop (9), this observation suggests that subversion of the immune system is necessary at relatively early stages of tumor development and, in fact, occur concurrently with the process of malignant transformation (Figure 1). This review discusses our understanding of IDO regulation, highlights mechanisms utilized by cancers to control IDO activity in the tumor immune microenvironment, and outlines pharmacological strategies for reversing these processes to ultimately augment our immunotherapeutic strategies for managing cancer patients.
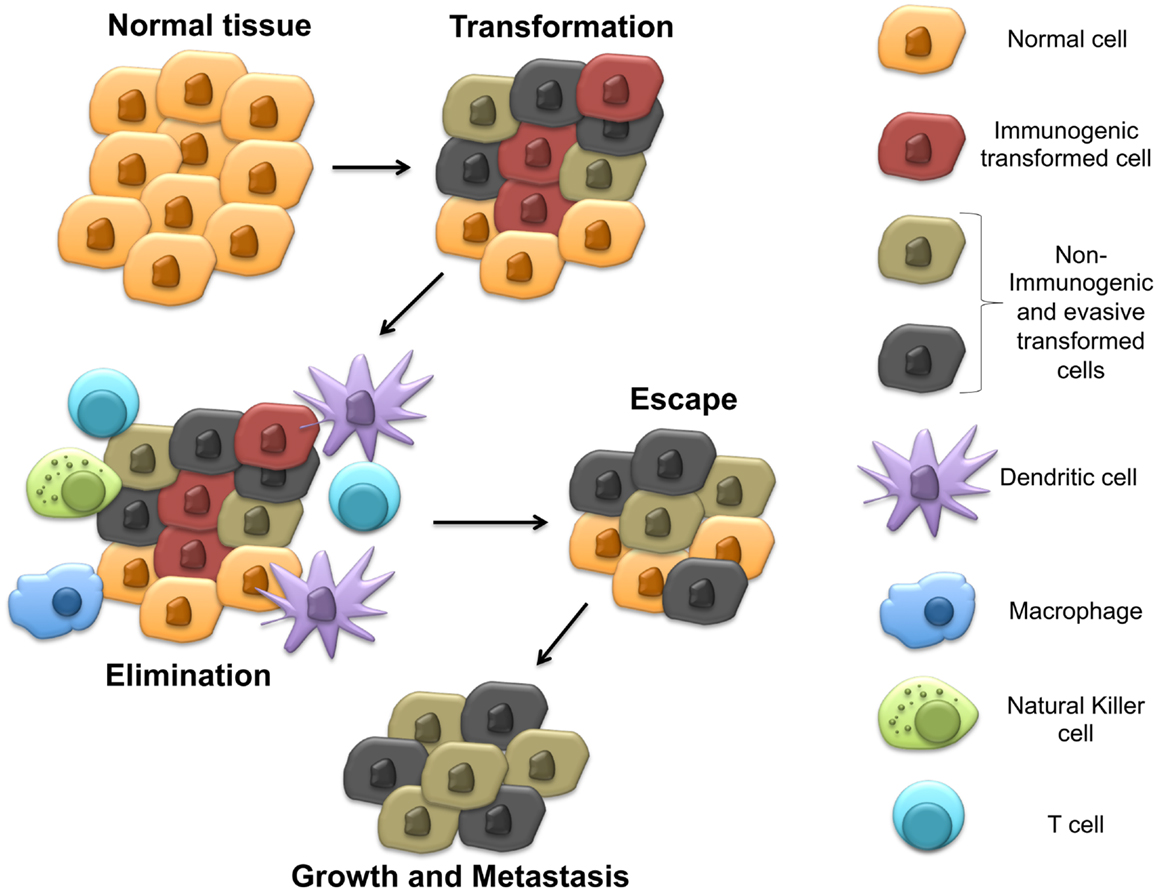
Figure 1. Early steps in carcinogenesis include immune evasion by upregulating IDO expression in the tumor microenvironment. This model proposes the development of immune evasion mechanisms early during transformation, which stimulate local IDO activity prior to the development of adaptive anti-tumor immunity and generation of the selective pressure responsible for cancer immunoediting.
Regulation of IDO Expression and Enzymatic Activity
Several factors have been shown to regulate the expression of IDO in a cell type-specific manner in monocytes, macrophages, endothelial cells, fibroblasts, some tumor cells, and various populations of dendritic cells (DCs). Initial studies focused on the process of inflammation, showing interferon-γ (IFN-γ) to be a potent inducer of IDO expression in many cell types and demonstrating this pathway of IDO induction to provide protection from intracellular pathogens by depleting local tryptophan levels (10, 11). Other inflammatory factors that have been shown to regulate IDO expression include IL-1, tumor necrosis factor-α (TNF-α), and lipopolysaccharide (LPS) (12, 13). Interestingly, prostaglandin E2 has been observed to induce the transcription of IDO in human monocyte-derived DCs; however, additional activation with TNF-α or LPS was noted to be necessary to achieve full enzymatic activity (14). This is consistent with observations by other investigators who have found that two-signals are often necessary to induce maximal IDO expression by specific DC subsets (5). Additional T cell-derived signals have also been demonstrated to play a role in regulating IDO expression including reverse signaling via B7 co-stimulatory molecule (CD80/CD86) cross-linking on the surface of antigen-presenting cells (APCs) (15–18). Using this mechanism, Tregs constitutively expressing CTLA-4 condition DCs by stimulating IDO expression and, in turn, suppressing local T cell proliferation, thereby establishing a state of immune tolerance. Later work revealed this B7 reverse signaling mechanism to be dependent upon the activation of the non-canonical NFκB signaling pathway, a mechanism also responsible for the induction of IDO following stimulation by other cell surface receptors including CD40 and the glucocorticoid-induced TNF receptor (GITR) (19).
It has been generally proposed that IDO activation signals have evolved to provide a negative feedback mechanism to dampen local inflammatory processes and prevent immune-mediated pathology. However, more recent studies have shown the immunosuppressive cytokine, TGF-β, to induce novel IDO functionality by specific subtypes of DCs including the murine CD8+ splenic DC subset as well as the plasmacytoid DC (pDC) population (20, 21). These studies revealed TGF-β to stimulate the expression and Fyn-dependent phosphorylation of IDO, enabling this protein to also serve as a scaffolding molecule for downstream signaling ultimately leading to the expression of both TGF-β and IDO itself. As opposed to the rapid and short-lived induction of IDO expression by IFN-γ, the stimulation of IDO expression by TGF-β is thought to be durable and to serve as a mechanism for generating long-term immune tolerance. This post-translational modification of IDO by TGF-β also has additional implications in terms of its regulation. In inflammatory conditions, exposure to IL-6 promotes the degradation of IDO by upregulating suppressor of cytokine signaling 3 (SOCS3), which binds to a phosphorylated immunoreceptor tyrosine-based inhibitory motif (ITIM) in IDO and promotes its proteosomal degradation (22).
Additional post-translational regulatory mechanisms have been elucidated, which are also capable of contributing to the regulation of IDO activity. This includes the nitration of various IDO tyrosine residues by peroxynitrite, a byproduct of nitric oxide (NO) and superoxide, which has also been shown to dampen IDO enzyme activity in macrophages (23). This is consistent with other findings showing NO to directly inhibit IDO activity by binding to its active site heme moiety (24). Indeed, previous work indicates that the reduction–oxidation status of the cell is capable of modulating activity of the IDO holoenzyme by interfering with the heme biosynthetic pathway (25).
It is clear that there are multiple mechanisms, which may regulate IDO on both the transcriptional and post-translational levels. However, the biological contexts in which these regulatory mechanisms affect IDO activity remain unclear. This is particularly true for the process of carcinogenesis, which occurs in a biochemically altered environment. Several studies have supported an important role for IDO in the generation of an immunotolerant tumor microenvironment that facilitates tumor progression (26–28). These findings indicate that the mechanisms utilized by cancers to modulate IDO expression and activity may be central to understanding the highly complex process of carcinogenesis. Here, we discuss recent studies investigating the mechanisms that cancers utilize to manipulate local IDO activity within the immune microenvironment in order to facilitate their metastatic progression.
Tumor-Mediated Regulation of Intrinsic IDO1 Expression
The expression of IDO by many cancer types has been correlated with inferior progression-free and overall survival (29). However, the regulation of IDO expression by malignant tissues has been poorly understood. In 2005, the first mechanism by which several solid tumors can regulate the intrinsic expression of IDO was described. This work focused on the BAR adapter-encoding gene BIN-1, a tumor suppressor previously found to be downregulated in several transformed cell lines and demonstrated to suppress the transformational activity of MYC by interacting with its N-terminus (30). Subsequent studies revealed BIN-1 to interfere with malignant transformation utilizing several mechanisms beyond the suppression of MYC activity and additional work showed BIN-1 to play the role of a tumor suppressor in several cancer types including melanoma, breast cancer, colon cancer, and prostate cancer (31–33). After noting that BIN-1 seemed to suppress the development of a transformed epithelial tumor model via an immune-dependent mechanism, Muller and colleagues noted that the deletion of Bin-1 significantly enhanced the IFN-γ-mediated upregulation of IDO expression by tumor cells (34). Indeed, the observed enhancement in tumor growth following Bin-1 deletion was reversed in the presence of the 1-methyltryptophan (1-MT) IDO inhibitor only in the setting of an intact immune system. These authors concluded that BIN-1 was capable of modulating IDO expression by regulating the STAT1 and NFκB signaling pathways that have been previously implicated in promoting the transcription of Indo, the IDO encoding gene (Figure 2A). This represented the initial study linking IDO regulation to an intrinsic tumor suppressor pathway by showing that the loss of Bin1 tumor expression contributes to tumorigenesis by driving cellular proliferation while simultaneously concealing itself from detection and destruction by the host immune system. This work prompted us to conjecture that early phases of tumor initiation and growth will often require the evolution of multifunctional genes, which regulate both cell division and/or survival, as well as elements of the local immune microenvironment.
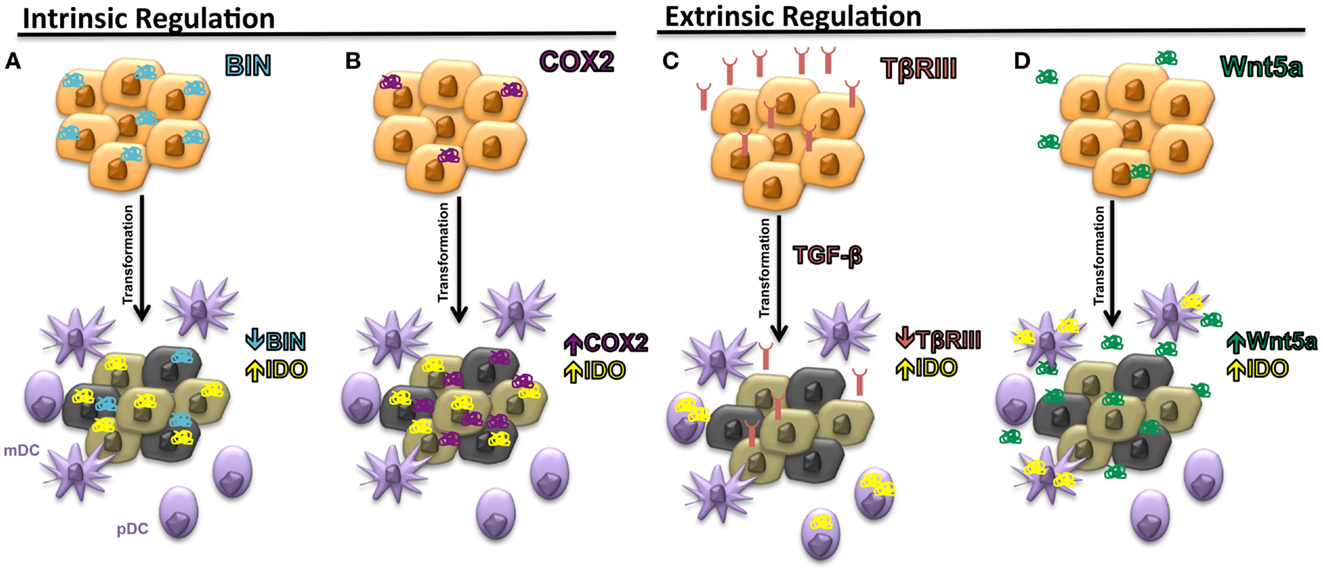
Figure 2. Intrinsic and extrinsic mechanisms of IDO regulation in the tumor microenvironment. (A) Downregulation of Bin1 expression leads to enhanced expression of IDO by tumor cells. (B) Upregulation of cyclooxygenase-2 (COX-2) expression by tumor cells stimulates intrinsic tumor expression of IDO. (C) Loss of TβRIII, the gene encoding the type IIII TGF-β receptor (TβRIII), allows for increased TGF-β paracrine signaling in the tumor microenvironment and the upregulation of IDO by local plasmacytoid DCs (pDCs). (D) Increased soluble Wnt5a secretion upregulates IDO expression by local myeloid DCs (mDCs).
Cyclooxygenase-2 (COX2), another modulator of IDO expression and activity, has also been implicated in the process of tumorigenesis and may be consistent with this hypothesis (35). COX2 represents the inducible isoform of the cyclooxygenases and plays a critical role in eicosanoid biosynthesis, including the prostaglandins and leukotrienes. The initial data linking COX2 biology with carcinogenesis was provided by genetic studies showing that when APCΔ716 mice, which model the colon cancer syndrome, familial adenomatous polyposis, are crossed with mice carrying an inactivating mutation in the Pgst2 gene encoding COX2, offspring develop exhibiting a diminished number of intestinal polyps (36). Since this study, several pre-malignant and malignant tissues have been shown to express COX2 at relatively early time points of tumorigenesis and several pro-tumorigenic functions have been ascribed to COX2 including the promotion of tumor-mediated angiogenesis, anti-apoptosis, and the generation of the epidermal growth factor receptor ligand, amphiregulin (37). One of the downstream products of COX2 activity, prostaglandin E2 (PGE2), has been previously demonstrated to interfere with T cell and DC function (38). Additional studies have shown COX2 and IDO expression to correlate in both human breast cancer cell lines and human breast cancer primary tissues while other investigators have found PGE2 to directly stimulate IDO expression (14, 39). Interestingly, COX2 inhibitors enhance the anti-tumor effects of DC-based vaccines and promote tumor-specific T cell responses in the MMTV-neu autochthonous murine mammary carcinoma model further suggesting an immunologic role for COX2 in cancer (35). Further studies have also shown COX2 inhibitors to augment a MUC1-based vaccine in a transgenic pancreatic cancer model in a manner that depended on suppressed IDO activity within tumor tissues (40). Similar roles for COX2 in promoting Tregs in non-small cell lung cancer and in elevating IDO expression in acute myeloid leukemia have also been described (41, 42). Together, these studies suggest that COX2 represents an important regulator of IDO function within malignant tissues (Figure 2B). While these studies focused on investigating the relationship between COX2 expression and the intrinsic regulation of IDO expression by tumor cells, a more recent study has shown a COX2-expressing MCF-7 breast cancer cell line to induce IDO expression by co-cultured fibroblasts, suggesting that paracrine IDO regulatory networks may also be relevant during the process of carcinogenesis (43).
Characterizing Tumor-Mediated Regulation of Extrinsic IDO1 Expression
Although it is unclear if the cell type expressing the IDO enzyme may affect its ultimate immunologic impact in the setting of cancer, previous investigators have shown a relationship between local DC expression of IDO and poor clinical prognosis in patients with melanoma (28, 44). In light of these data, we reasoned that tumor-derived soluble factors may have evolved to manipulate local DC expression of this critical immune regulatory mechanism. Further, in light of the dual role of the BIN-1 tumor suppressor described above, we searched for soluble factors already described to have a pro-tumorigenic role in the literature. These criteria led us to the type III TGF-β receptor (TβRIII) that functions as a co-receptor for the TGF-β signaling pathway by binding and presenting all three TGF-β isoforms to the type I and II TGF-β receptor signaling complex (45). TβRIII has been independently implicated in suppressing cellular migration in several experimental systems through a β-arrestin2-mediated mechanism (46, 47). Consistent with the criteria discussed above, earlier studies also revealed TβRIII to be shed at the cell surface and for its soluble form, sTβRIII, to bind and suppress downstream TGF-β-mediated signaling, effectively functioning as a molecular sink for the TGF-β cytokine (48, 49). Additional work has demonstrated human breast cancers to downregulate TβRIII expression by loss of heterozygosity and for TβRIII to impede metastatic progression of the 4T1 murine breast cancer model (50). This loss of TβRIII expression has since been shown to occur during the progression of several additional cancers including pancreatic, lung, and prostate cancers (51–53). Notably, the downregulation of TβRIII has also been demonstrated to occur at a relatively early stage of tumorigenesis, exemplified by the loss of TβRIII in tissue specimens histologically characterized as ductal carcinoma in situ, an early precursor to invasive ductal carcinoma of the breast. Additional studies showing the loss of TβRIII to promote the epithelial-to-mesenchymal transition (EMT) in pancreatic cancer further suggests that this genetic alteration occurs at a relatively early time point during carcinogenesis (53).
TGF-β has been described as a pleiotropic regulator of the immune system capable of modulating both IDO activity and several additional immunosuppressive pathways (54). By inhibiting both T cell and NK cell proliferation and activation, as well as promoting the differentiation of Treg populations, the TGF-β cytokine plays an important role in maintaining peripheral immune tolerance. We, therefore, hypothesized that the loss of TβRIII expression by developing malignancies would allow for enhanced TGF-β-dependent signaling in the tumor microenvironment, thereby inhibiting local immune surveillance mechanisms and ultimately promoting tumorigenesis (55). Using the 4T1 murine breast cancer model, we initially determined that TβRIII expression suppressed metastatic progression only in immunocompetent hosts. Consistent with this observation, further work revealed the loss of TβRIII to be associated with the development of an immunotolerant microenvironment characterized by a decrease in the number of CD8+ T cells and a corresponding increase in the CD4+FoxP3+ Treg population in both breast cancer and melanoma model systems.
These findings led to the identification of an association between TβRIII expression and suppressed levels of IDO in both the tumor bed and within the tumor-draining lymph nodes (TDLNs). We determined that the loss of TβRIII expression correlated with the upregulation of IDO by pDC populations within TDLN tissues, the same cell population that was previously noted to be important for the expression of IDO within TDLN tissues (Figure 2C) (21, 27). These results also corresponded with diminished pDC and whole TDLN tissue IDO enzyme activity when recovered from mice bearing TβRIII-expressing tumors. Since sTβRIII is an effective inhibitor of downstream TGF-β signaling, we hypothesized that TGF-β was a major mediator of pDC IDO activity in the tumor microenvironment and confirmed the findings of Pallotta and colleagues by showing TGF-β treatment of purified pDCs to enhance IDO expression and enzymatic activity (21). We then demonstrated this paracrine signaling mechanism to be functionally relevant by demonstrating purified pDCs derived from mice harboring TβRIII-downregulated tumors, to suppress in vitro T cell proliferation in an IDO-dependent manner. Further, using a doxycycline-inducible system, we showed that the earlier the alteration in TβRIII expression by a developing tumor, the more profound the effect on the local tumor microenvironment. These results suggest that modulation of the immune microenvironment at earlier stages of tumorigenesis is associated with a greater impact on tumor progression.
The work described above illustrates the potential for genetic alterations within the tumor to impact local DC function and to subvert immunosurveillance. Given the critical role that these APCs play in orchestrating the anti-tumor immune response, it follows that the evolution of various mechanisms to convert local DCs into a tolerogenic state would be quite advantageous for a developing tumor. The pathways for driving DC tolerogenesis in the setting of cancer remain poorly characterized; however, the β-catenin signaling pathway has emerged as a potentially important component of this process. This is supported by data showing that activation of the β-catenin pathway in myeloid DCs (mDCs) conditioned these APCs to promote the generation of IL-10-expressing CD4+ T cells capable of suppressing the autoimmune phenotype of a mouse model of multiple sclerosis (56). These findings were further substantiated by in vivo experiments using the CD11c-cre × β-catlox/lox transgenic model demonstrating the DC-specific β-catenin pathway as an important regulator of Treg differentiation in gastrointestinal tissues (57). These studies raised the possibility that tumors may promote DC tolerization in the tumor microenvironment via the expression of soluble Wnt ligands. Interestingly, several members of the Wnt ligand family have been noted to play a role in carcinogenesis. Indeed, mechanistic studies have revealed Wnt5a to promote melanoma cell migration and invasion, ultimately leading to disease metastases (58). In addition, increased Wnt5a expression levels in melanoma tissues as well as diminished levels of the soluble Wnt antagonist, Dkk-1, have been associated with an inferior clinical outcome in patients with advanced melanoma (59–63).
Together, the above reports led us to screen the conditioned media of several human melanoma cell lines for their ability to stimulate downstream β-catenin signaling activity. This work consistently showed that melanoma-derived conditioned media was capable of inducing this signaling pathway in both reporter cell lines and primary DCs in vitro (64). Further work using the Tyr:CreER;BrafCA;Ptenlox/lox inducible transgenic melanoma model, showed that TDLN DCs and tumor-infiltrating DCs associated with developing melanomas expressed elevated levels of known β-catenin target genes including Axin2, Ccnd1, C-myc, and Tcf-7 relative to DCs derived from more distant lymph node tissues. This local paracrine β-catenin signaling effect was then confirmed in situ within the melanoma stroma and within TDLN tissues by confocal microscopy using the Tg(TCF/Lef1-HIST1H2BB/EGFP)61Hadj/J transgenic reporter strain, which encodes an EGFP reporter downstream of a β-catenin-responsive promoter containing tandem TCF/LEF1 transcription factor binding elements (65).
After verifying that this signaling pathway could be induced within mDCs in an autochthonous model of melanoma, we demonstrated that β-catenin was promoting tolerogenic DC development by regulating the downstream expression of IDO. Although recent studies had shown Wnt-mediated signaling to stimulate expression of the immunosuppressive factor IL-10 by DCs, the effects on IDO expression were unknown (61). Using a variety of methods including pharmacological inhibitors, promoter–reporter systems, immunofluorescence microscopy, and chromatin immunoprecipitation, we were able to demonstrate that Wnt ligands robustly induced the durable upregulation of both IDO expression and enzymatic activity by bone marrow-derived DCs in a β-catenin-dependent manner. By performing blocking experiments, we further noted that the soluble Wnt5a ligand was the dominant factor in melanoma-conditioned media in the induction of IDO by DCs and that Wnt5a was capable of promoting IDO expression in an IFN-γ-independent manner (Figure 2D). Importantly, as opposed to several other stimuli including Wnt3a, additional work revealed Wnt5a conditioned DCs to significantly promote the differentiation and expansion of naïve CD4+ T cells into Tregs in an IDO-dependent manner. Indeed, further in silico gene expression analysis has revealed a significant association between Wnt5a and Foxp3 gene expression levels in human melanomas. Similar to BIN-1, COX2, and TβRIII above, this work implicates Wnt5a as a factor with dual roles in carcinogenesis including tumor invasion and metastasis, as well as the suppression of local immune surveillance. Similar to TβRIII, Wnt5a has also been implicated in the promotion of EMT in both pancreatic cancer and gastric cancer (66, 67). This finding implies that Wnt5a is upregulated and is capable of modulating the immune microenvironment at an early time point during the process of carcinogenesis suggesting that Wnt5a likely has an impact on the establishment of immune privilege during the earliest stages of transformation.
Therapeutic Implications of IDO Regulatory Pathways in Cancer-Mediated Immune Evasion
Although recent advances in immunotherapy have made substantial strides in improving clinical responses in patients with advanced cancers, a significant fraction of these patients continue to fail therapy. In light of the diverse array of immune evasion mechanisms that individual cancers are able to employ to escape detection and destruction by the host immune system, it seems that combinatorial therapies, which target different aspects of immune suppression will be necessary to fully realize the promise of immunotherapy. While the majority of immunotherapy development has targeted T cell-expressed negative regulators such as CTLA-4 and PD-1 (68, 69), few agents are currently under investigation, which are capable of modulating tolerogenic DCs in the tumor microenvironment. An exception to this includes the IDO inhibitor, 1-methyltryptophan, which was shown to enhance chemotherapeutic effects in both the murine orthotopic 4T1 and the autochthonous MMTV-neu breast cancer models (70). Given these encouraging findings, high throughput screening has been employed to identify improved compounds for further clinical trial development (71–73). Although single agent efficacy has been modest (74), recent reports are showing encouraging clinical responses when combined with anti-CTLA-4 monoclonal antibody therapy while several other combination studies are ongoing (75–79) (Table 1). These results suggest that immunotherapeutic regimens employing a combinatorial approach including T cell-targeted immune checkpoint inhibitors and agents capable of reversing tumor-mediated immune evasion mechanisms have great promise.
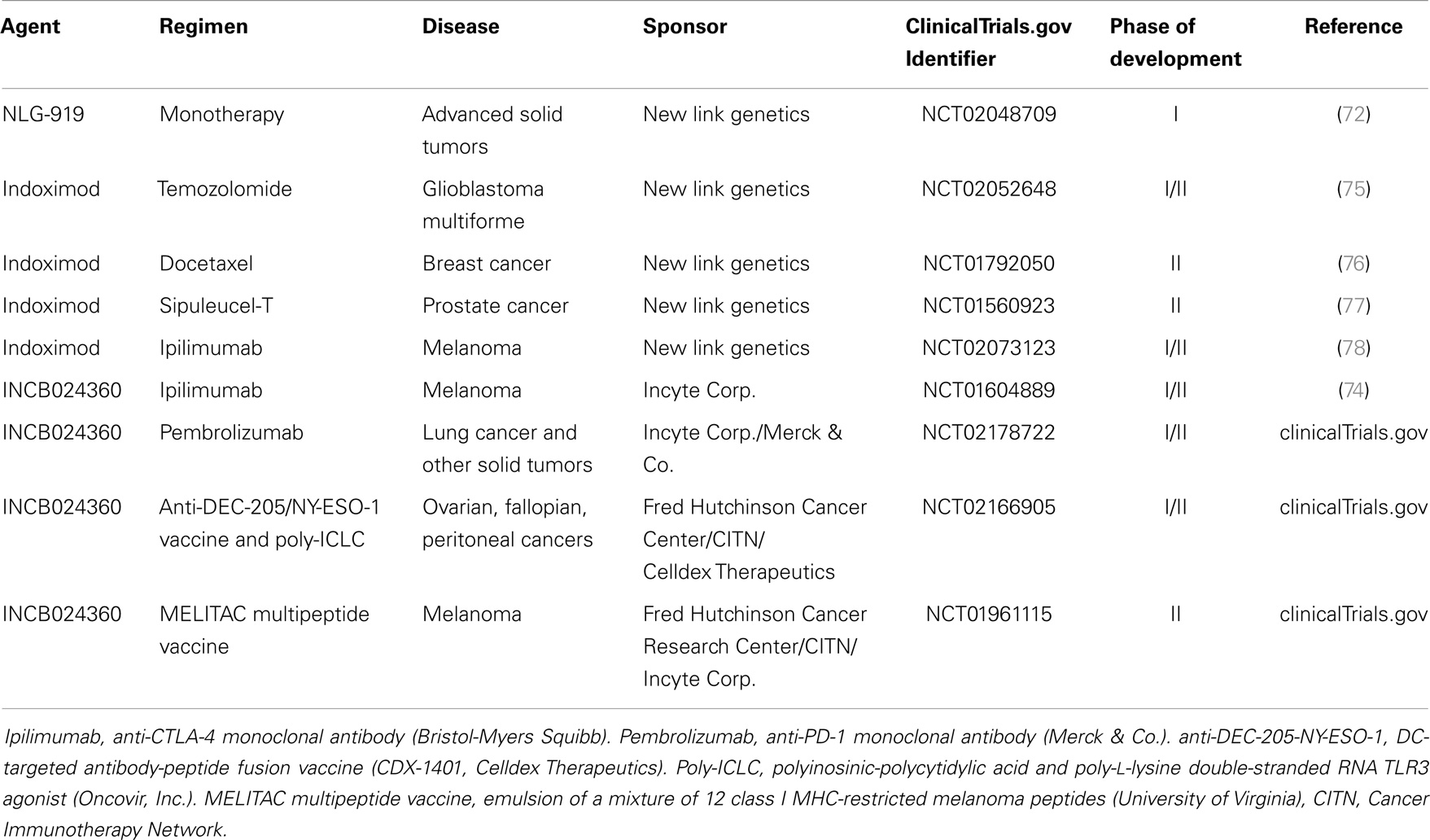
Table 1. Active clinical trials investigating the activity of IDO inhibitors in advanced cancer.
In addition to targeting the IDO immunoregulatory enzyme itself, pharmacological strategies designed to interfere with the previously discussed regulatory pathways of IDO have theoretical advantages (Figure 3). First, evidence showing that TGF-β-induced IDO in specific DC subsets is capable of maintaining immune tolerance via a signaling mechanism that is independent of its enzymatic activity suggests that inhibitors targeting the active site of IDO may have limited clinical benefit (21). Second, IDO is likely to only be a component of the tolerogenic DC program induced by specific tumor-derived mediators, implying that inhibition of the upstream signals that activate this program are more likely to have greater clinical efficacy. This is exemplified by TGF-β, which is known to induce other immunosuppressive pathways involving a variety of cellular targets (54).
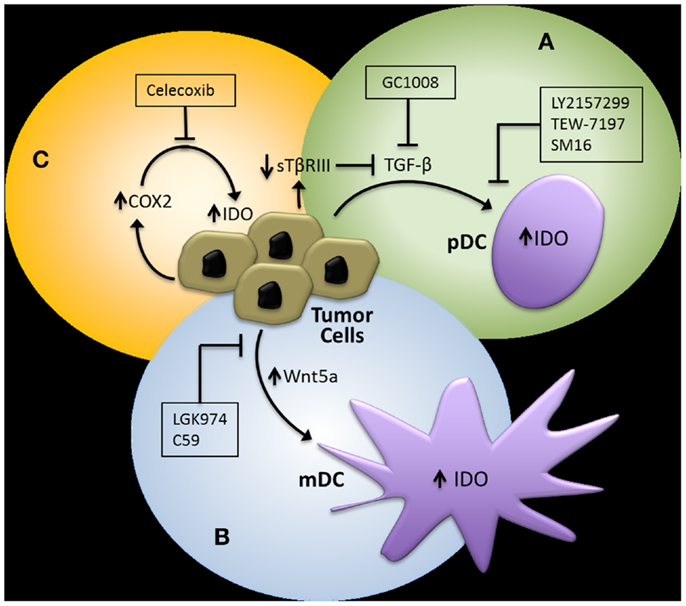
Figure 3. Potential pharmacological strategies for suppressing IDO expression in the tumor microenvironment. (A) TGF-β inhibitors. GC1008, pan-TGF-β isoform monoclonal antibody (Genzyme), LY2157299, type I TGF-β receptor serine/threonine kinase small molecule inhibitor (Eli Lilly), TEW-7197, type I TGF-β receptor serine/threonine kinase small molecule inhibitor (MedPacto, Inc.). (B) Wnt inhibitors. LGK974, Porcn acyl transferase small molecule inhibitor (Novartis). (C) Cyclooxygenase-2 (COX2) Inhibitors. Celecoxib (Pfizer, Inc.). pDC, plasmacytoid DC; mDC, myeloid DC.
Since our studies have indicated that tumor-derived TGF-β is capable of inducing IDO expression by pDCs and that this effect is enhanced upon loss of TβRIII in a murine breast cancer model, we investigated the ability of an oral type I TGF-β receptor serine/threonine kinase inhibitor (SM16) to augment the immunologic response of a Her2/neu vaccine. This work confirmed that a type I TGF-β receptor serine/threonine kinase inhibitor could synergistically enhance the CD8+ T cell host immune response to Her2/neu and effectively suppress the progression of a Her2/neu-expressing 4T1 breast cancer model (55). Given these findings as well as the supporting data suggesting that the loss of TβRIII in murine melanoma models also promotes the development of an immune suppressive microenvironment, we investigated the combination of another oral type I TGF-β receptor kinase inhibitor currently in clinical trial development, LY2157299 monohydrate (80, 81), with anti-CTLA-4 monoclonal antibody therapy in the Tyr:CreER;BrafCA;Ptenlox/lox transgenic melanoma mouse model (82). Consistent with our previous findings, this combinatorial treatment approach also generated a synergistic anti-tumor response, effectively enhancing the tumor-infiltrating CD8+ T cell/Treg ratio and suppressing both primary melanoma development and the establishment of distant metastasis.
As described previously, emerging data suggest that the tumor-mediated expression of Wnt5a contributes to the generation of an immunotolerant microenvironment. We, therefore, hypothesized that the inhibition of Wnt5a-mediated signaling would also augment immunotherapy efficacy in melanoma. Several strategies to inhibit the Wnt-β-catenin pathway have been investigated; however, one of the more promising approaches is targeting the membrane-bound O-acyltransferase, Porcn, which catalyzes the palmitoylation of all vertebrate Wnt ligands, a step necessary for effective secretion of the Wnt soluble protein family (83, 84). This work has led to the introduction of LGK974, a small molecule Porcn acyltransferase inhibitor, into early phase clinical trials (85). To determine if the inhibition of Wnt secretion by targeting Porcn would be an effective approach for reversing melanoma-mediated immune suppression, we genetically silenced Porcn expression by the B16 murine melanoma model and performed several in vivo tumor assays. This new B16-PORCNKD cell line was found to exhibit impaired Wnt secretion, suppressed tumor growth in vivo, and for this to be associated with both enhanced levels of infiltrating CD8+ T cells and suppressed levels of PD-1-expressing tumor-infiltrating lymphocytes. With this data, we then evaluated the ability of a commercially available pharmacological inhibitor of Porcn, C59, to reverse B16-mediated immune suppression (86). Given as monotherapy to mice bearing B16 melanomas, this agent did not seem to exhibit a significant anti-tumor effect, however, when combined with anti-CTLA-4 monoclonal antibody therapy, a synergistic enhancement in activated 41BB+ TRP2-specific CD8+ tumor-infiltrating lymphocytes were observed along with B16 tumor growth suppression. Together, these data support the use of combinatorial immunotherapy strategies that involve agents capable of interfering with tumor immune evasion pathways including the upregulation of local IDO expression.
Conclusion
Studies focused on understanding the interplay between tumor development and the host immune system are now revealing an intimate relationship between the processes of tumor invasion and metastasis and the active induction of immune tolerance. Rather than developing as a response to immune-mediated selective pressure, we hypothesize that some immune evasion mechanisms are capable of developing at a very early stage in carcinogenesis and simultaneously promoting tumor invasion while also interfering with tumor detection by the host immune system. The pathways that we have found to meet these criteria are induced by intrinsic genetic alterations, resulting in the downregulation of both the BIN-1 and TβRIII tumor suppressors and the upregulation of the pro-tumorigenic factors, COX2 and Wnt5a. Interestingly, this body of work also highlights important differences in cell-specific IDO expression kinetics. While IFN-γ is a rapid inducer of IDO expression in many cell types, studies are now demonstrating that the loss of TβRIII in a TGF-βhi tumor microenvironment promotes durable IDO expression by pDCs while the upregulation of the Wnt5a oncogene results in durable IDO expression by mDC populations (Figures 2C,D). We expect for several other tumor-mediated soluble factors or perhaps exosome-derived factors to also regulate IDO expression via similar paracrine signaling mechanisms. Their identification will be important for therapeutic development as well as for the establishment of predictive biomarkers to determine when these novel therapeutic strategies would be most effectively employed. Importantly, pre-clinical experimental investigation, to date, suggests that the use of a pharmacologic agent to inhibit these tumor-mediated evasion pathways which target IDO activity effectively synergize with immune checkpoint blockade. These data strongly support the physiologic relevance of these novel immune evasion pathways, which target IDO activity within the tumor microenvironment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding: Duke Cancer Institute start-up funding, Duke-Melanoma Research Alliance Young Investigator Award in Honor of Frank Courtney.
References
1. Shimizu T, Nomiyama S, Hirata F, Hayaishi O. Indoleamine 2,3-dioxygenase. Purification and some properties. J Biol Chem (1978) 253(13):4700–6.
2. Carlin JM, Borden EC, Sondel PM, Byrne GI. Interferon-induced indoleamine 2,3-dioxygenase activity in human mononuclear phagocytes. J Leukoc Biol (1989) 45(1):29–34.
3. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
4. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189(9):1363–72. doi:10.1084/jem.189.9.1363
5. Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol (2000) 164(7):3596–9. doi:10.4049/jimmunol.164.7.3596
6. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor {zeta}-chain and induce a regulatory phenotype in naive T cells. J Immunol (2006) 176:6752–61. doi:10.4049/jimmunol.176.11.6752
7. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117(5):1147–54. doi:10.1172/JCI31178
8. Katz JB, Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev (2008) 222:206–21. doi:10.1111/j.1600-065X.2008.00610.x
9. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331(6024):1565–70. doi:10.1126/science.1203486
10. Takikawa O, Kuroiwa T, Yamazaki F, Kido R. Mechanism of interferon-gamma action. Characterization of indoleamine 2,3-dioxygenase in cultured human cells induced by interferon-gamma and evaluation of the enzyme-mediated tryptophan degradation in its anticellular activity. J Biol Chem (1988) 263(4):2041–8.
11. Dai W, Gupta SL. Regulation of indoleamine 2,3-dioxygenase gene expression in human fibroblasts by interferon-gamma. Upstream control region discriminates between interferon-gamma and interferon-alpha. J Biol Chem (1990) 265(32):19871–7.
12. Babcock TA, Carlin JM. Transcriptional activation of indoleamine dioxygenase by interleukin 1 and tumor necrosis factor alpha in interferon-treated epithelial cells. Cytokine (2000) 12(6):588–94. doi:10.1006/cyto.1999.0661
13. Hissong BD, Byrne GI, Padilla ML, Carlin JM. Upregulation of interferon-induced indoleamine 2,3-dioxygenase in human macrophage cultures by lipopolysaccharide, muramyl tripeptide, and interleukin-1. Cell Immunol (1995) 160(2):264–9. doi:10.1016/0008-8749(95)80037-J
14. Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood (2005) 106(7):2375–81. doi:10.1182/blood-2005-03-0979
15. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4(10):762–74. doi:10.1038/nri1457
16. Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol (2002) 3(11):1097–101. doi:10.1038/ni846
17. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4(12):1206–12. doi:10.1038/ni1003
18. Munn DH, Sharma MD, Mellor AL. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J Immunol (2004) 172(7):4100–10. doi:10.4049/jimmunol.172.7.4100
19. Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med (2007) 13(5):579–86. doi:10.1038/nm1563
20. Belladonna M, Volpi C, Bianchi R, Vacca C, Orabona C, Pallotta M, et al. Cutting edge: autocrine TGFbeta sustains default tolerogenesis by IDO-competent dendritic cells. J Immunol (2008) 181(8):5194–8. doi:10.4049/jimmunol.181.8.5194
21. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol (2011) 12(9):870–8. doi:10.1038/ni.2077
22. Orabona C, Pallotta MT, Volpi C, Fallarino F, Vacca C, Bianchi R, et al. SOCS3 drives proteasomal degradation of indoleamine 2,3-dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis. Proc Natl Acad Sci U S A (2008) 105(52):20828–33. doi:10.1073/pnas.0810278105
23. Fujigaki H, Saito K, Lin F, Fujigaki S, Takahashi K, Martin BM, et al. Nitration and inactivation of IDO by peroxynitrite. J Immunol (2006) 176(1):372–9. doi:10.4049/jimmunol.176.1.372
24. Thomas SR, Terentis AC, Cai H, Takikawa O, Levina A, Lay PA, et al. Post-translational regulation of human indoleamine 2,3-dioxygenase activity by nitric oxide. J Biol Chem (2007) 282(33):23778–87. doi:10.1074/jbc.M700669200
25. Thomas SR, Salahifar H, Mashima R, Hunt NH, Richardson DR, Stocker R. Antioxidants inhibit indoleamine 2,3-dioxygenase in IFN-gamma-activated human macrophages: posttranslational regulation by pyrrolidine dithiocarbamate. J Immunol (2001) 166(10):6332–40. doi:10.4049/jimmunol.166.10.6332
26. Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer (2002) 101(2):151–5. doi:10.1002/ijc.10645
27. Sharma MD, Baban B, Chandler PR, Hou D-Y, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine-2,3-dioxygenase. J Clin Invest (2007) 117(9):2570–82. doi:10.1172/JCI31911
28. Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest (2004) 114(2):280–90. doi:10.1172/JCI21583
29. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res (2011) 17(22):6985–91. doi:10.1158/1078-0432.CCR-11-1331
30. Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat Genet (1996) 14(1):69–77. doi:10.1038/ng0996-69
31. Elliott K, Sakamuro D, Basu A, Du W, Wunner W, Staller P, et al. Bin1 functionally interacts with Myc and inhibits cell proliferation via multiple mechanisms. Oncogene (1999) 18(24):3564–73. doi:10.1038/sj.onc.1202670
32. Ge K, DuHadaway J, Du W, Herlyn M, Rodeck U, Prendergast GC. Mechanism for elimination of a tumor suppressor: aberrant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc Natl Acad Sci U S A (1999) 96(17):9689–94. doi:10.1073/pnas.96.17.9689
33. Ge K, Minhas F, Duhadaway J, Mao NC, Wilson D, Buccafusca R, et al. Loss of heterozygosity and tumor suppressor activity of Bin1 in prostate carcinoma. Int J Cancer (2000) 86(2):155–61. doi:10.1002/(SICI)1097-0215(20000415)86:2<155::AID-IJC2>3.0.CO;2-M
34. Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast G. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene, Bin1, potentiates cancer chemotherapy. Nat Med (2005) 11(3):312–9. doi:10.1038/nm1196
35. Basu GD, Tinder TL, Bradley JM, Tu T, Hattrup CL, Pockaj BA, et al. Cyclooxygenase-2 inhibitor enhances the efficacy of a breast cancer vaccine: role of IDO. J Immunol (2006) 177(4):2391–402. doi:10.4049/jimmunol.177.4.2391
36. Zha S, Yegnasubramanian V, Nelson WG, Isaacs WB, De Marzo AM. Cyclooxygenases in cancer: progress and perspective. Cancer Lett (2004) 215(1):1–20. doi:10.1016/j.canlet.2004.06.014
37. Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell (2003) 4(6):431–6. doi:10.1016/S1535-6108(03)00310-6
38. Pockaj BA, Basu GD, Pathangey LB, Gray RJ, Hernandez JL, Gendler SJ, et al. Reduced T-cell and dendritic cell function is related to cyclooxygenase-2 overexpression and prostaglandin E2 secretion in patients with breast cancer. Ann Surg Oncol (2004) 11(3):328–39. doi:10.1245/ASO.2004.05.027
39. Soliman H, Rawal B, Fulp J, Lee JH, Lopez A, Bui MM, et al. Analysis of indoleamine 2-3 dioxygenase (IDO1) expression in breast cancer tissue by immunohistochemistry. Cancer Immunol Immunother (2013) 62(5):829–37. doi:10.1007/s00262-013-1393-y
40. Mukherjee P, Basu GD, Tinder TL, Subramani DB, Bradley JM, Arefayene M, et al. Progression of pancreatic adenocarcinoma is significantly impeded with a combination of vaccine and COX-2 inhibition. J Immunol (2009) 182(1):216–24. doi:10.4049/jimmunol.182.1.216
41. Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res (2005) 65(12):5211–20. doi:10.1158/0008-5472.CAN-05-0141
42. Iachininoto MG, Nuzzolo ER, Bonanno G, Mariotti A, Procoli A, Locatelli F, et al. Cyclooxygenase-2 (COX-2) inhibition constrains indoleamine 2,3-dioxygenase 1 (IDO1) activity in acute myeloid leukaemia cells. Molecules (2013) 18(9):10132–45. doi:10.3390/molecules180910132
43. Chen JY, Li CF, Kuo CC, Tsai KK, Hou MF, Hung WC. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res (2014) 16(4):410. doi:10.1186/s13058-014-0410-1
44. Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science (2002) 297(5588):1867–70. doi:10.1126/science.1073514
45. Gatza CE, Oh SY, Blobe GC. Roles for the type III TGF-beta receptor in human disease. Cell Signal (2010) 22(8):1163–74. doi:10.1016/j.cellsig.2010.01.016
46. Blobe GC, Schiemann WP, Pepin MC, Beauchemin M, Moustakas A, Lodish HF, et al. Functional roles for the cytoplasmic domain of the type III transforming growth factor beta receptor in regulating transforming growth factor beta signaling. J Biol Chem (2001) 276(27):24627–37. doi:10.1074/jbc.M100188200
47. Mythreye K, Blobe G. The type III TGF-beta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42. Proc Natl Acad Sci U S A (2009) 106(20):8221–6. doi:10.1073/pnas.0812879106
48. Andres JL, Stanley K, Cheifetz S, Massague J. Membrane-anchored and soluble forms of betaglycan, a polymorphic proteoglycan that binds transforming growth factor-beta. J Cell Biol (1989) 109(6):3137–45. doi:10.1083/jcb.109.6.3137
49. Lopez-Casillas F, Payne H, Andres J, Massague J. Betaglycan can act as a dual modulator of TGF-beta access to signaling receptors: mapping of ligand binding and GAG attachment sites. J Cell Biol (1994) 124:557–68. doi:10.1083/jcb.124.4.557
50. Dong M, How T, Kirkbride K, Gordon K, Lee J, Hempel N, et al. The type III TGFbeta receptor suppresses breast cancer progression. J Clin Invest (2007) 117(1):206–17. doi:10.1172/JCI29293
51. Finger E, Turley R, Dong M, How T, Fields T, Blobe G. TbetaRIII suppresses non-small cell lung cancer invasiveness and tumorigenicity. Carcinogenesis (2008) 29(3):528–35. doi:10.1093/carcin/bgm289
52. Turley R, Finger E, Hempel N, How T, Fields T, Blobe G. The type III transforming growth factor-beta receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res (2007) 67(3):1090–8. doi:10.1158/0008-5472.CAN-06-3117
53. Gordon K, Dong M, Chislock E, Fields T, Blobe G. Loss of type III transforming growth factor beta receptor expression increases motility and invasiveness associated with epithelial to mesenchymal transition during pancreatic cancer progression. Carcinogenesis (2008) 29(2):252–62. doi:10.1093/carcin/bgm249
54. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol (2010) 10:554–67. doi:10.1038/nri2808
55. Hanks BA, Holtzhausen A, Jamieson R, Gimpel P, Campbell O, Sun L, et al. Type III TGF-β receptor downregulation generates an immunotolerant tumor microenvironment. J Clin Invest (2013) 123(9):3925–40. doi:10.1172/JCI65745
56. Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, et al. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity (2007) 27(4):610–24. doi:10.1016/j.immuni.2007.08.015
57. Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science (2010) 329(5993):849–53. doi:10.1126/science.1188510
58. Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, et al. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell (2002) 1(3):279–88. doi:10.1016/S1535-6108(02)00045-4
59. O’Connell MP, Weeraratna AT. Hear the Wnt Ror: how melanoma cells adjust to changes in Wnt. Pigment Cell Melanoma Res (2009) 22(6):724–39. doi:10.1111/j.1755-148X.2009.00627.x
60. Feldmann R, Schierl M, Fink AM, Sator PG, Maiweg J, Steiner A. Serum levels of glycoprotein Dickkopf-1 in patients with cutaneous malignant melanoma: a prospective pilot study. Dermatology (2011) 222(2):171–5. doi:10.1159/000324516
61. Valencia J, Hernandez-Lopez C, Martinez VG, Hidalgo L, Zapata AG, Vicente A, et al. Wnt5a skews dendritic cell differentiation to an unconventional phenotype with tolerogenic features. J Immunol (2011) 187(8):4129–39. doi:10.4049/jimmunol.1101243
62. Oderup C, LaJevic M, Butcher EC. Canonical and noncanonical Wnt proteins program dendritic cell responses for tolerance. J Immunol (2013) 190(12):6126–34. doi:10.4049/jimmunol.1203002
63. Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature (2000) 406(6795):536–40. doi:10.1038/35020115
64. Holtzhausen A, Evans K, Hanks BA. Role of the Wnt-beta-catenin signaling pathway in melanoma-mediated dendritic cell tolerization. J Immunother Cancer (2013) 1(Suppl 1):153. doi:10.1186/2051-1426-1-S1-P153
65. Ferrer-Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, Hadjantonakis AK. A sensitive and bright single-cell resolution live imaging reporter of Wnt/ss-catenin signaling in the mouse. BMC Dev Biol (2010) 10:121. doi:10.1186/1471-213X-10-121
66. Bo H, Zhang S, Gao L, Chen Y, Zhang J, Chang X, et al. Upregulation of Wnt5a promotes epithelial-to-mesenchymal transition and metastasis of pancreatic cancer cells. BMC Cancer (2013) 13:496. doi:10.1186/1471-2407-13-496
67. Kanzawa M, Semba S, Hara S, Itoh T, Yokozaki H. WNT5A is a key regulator of the epithelial-mesenchymal transition and cancer stem cell properties in human gastric carcinoma cells. Pathobiology (2013) 80(5):235–44. doi:10.1159/000346843
68. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
69. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369(2):134–44. doi:10.1056/NEJMoa1305133
70. Hou D, Muller AJ, Sharma MD, DuHadaway JB, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res (2007) 67(2):792–801. doi:10.1158/0008-5472.CAN-06-2925
71. Yue EW, Douty B, Wayland B, Bower M, Liu X, Leffet L, et al. Discovery of potent competitive inhibitors of indoleamine 2,3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model. J Med Chem (2009) 52(23):7364–7. doi:10.1021/jm900518f
72. Khleif S, Munn DH, Nyak-Kapoor A, Mario MR, Kennedy E, Vahanian NN, et al. First-in-human phase I study of the novel indoleamine-2,3-dioxygenase (IDO) inhibitor NLG-919. J Clin Oncol (2014) 32(15 Suppl):abstrTPS3121.
73. Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood (2010) 115(17):3520–30. doi:10.1182/blood-2009-09-246124
74. Beatty GL, O’Dwyer PJ, Clark J, Shih JG, Newton RC, Schaub R, et al. Phase I study of the safety, pharmacokinetics, and pharmacodynamics of the oral inhibitor of indoleamine 2,3-dioxygenase (IDO1) INCB024360 in patients with advanced malignancies. J Clin Oncol (2013) 31(15 Suppl):abstr3025.
75. Gibney GT, Hamid O, Gangadhar TC, Lutzky J, Olszanski AJ, Gajewski T, et al. Preliminary results from a phase 1/2 study of INCB024360 combined with ipilimumab in patients with melanoma. J Clin Oncol (2014) 32(15 Suppl):abstr3010.
76. Zakharia Y, Johnson TS, Colman H, Vahanian NN, Link CJ, Kennedy E, et al. A phase I/II study of the combination of indoximod and temozolomide for adult patients with temozolomide-refractory primary malignant brain tumors. J Clin Oncol (2014) 32(15 Suppl):abstrTPS2017.
77. Soliman HH, Minton SE, Ismail-Khan R, Han HS, Vahanian NN, Ramsey WJ, et al. A phase 2 study of docetaxel in combination with indoximod in metastatic breast cancer. J Clin Oncol (2014) 32(15 Suppl):abstrTPS3124.
78. Kennedy E, Rossi GR, Vahanian NN, Link CJ. Phase 1/2 trial of the indoleamine 2,3-dioxygenase pathway (IDO) inhibitor indoximod plus ipilimumab for the treatment of unresectable stage 3 or 4 melanoma. J Clin Oncol (2014) 32(15 Suppl):abstrTPS9117.
79. Jha GG, Miller JS. A randomized, double-blind phase 2 study of sipuleucel-T followed by indoximod or placebo in the treatment of patients with asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer. J Clin Oncol (2014) 32(15 Suppl):abstrTPS5111.
80. Suarez C, Rodon J, Desjardins A, Forsyth P, Gueorguieva I, Cleverly A, et al. Phase 1b study evaluating safety and pharmacokinetics of the oral transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate when combined with chemoradiotherapy in newly diagnosed malignant gliomas. J Clin Oncol (2013) 31(15 Suppl):abstr2039.
81. Faivre SJ, Santoro A, Kelley R, Merle P, Gane E, Douillard J, et al. A phase 2 study of a novel transforming growth factor-beta (TGF-beta1) receptor I kinase inhibitor, LY215299 monohydrate, in patients with advanced hepatocellular carcinoma. J Clin Oncol (2014) 32(3 Suppl):abstrLBA173.
82. Holtzhausen A, Evans K, Siska P, Rathmell J, Hanks BA. Combinatorial TGF-beta signaling blockade and anti-CTLA-4 antibody immunotherapy in a murine BRAF(V600E)-PTEN-/- transgenic model of melanoma. J Clin Oncol (2014) 32(15 Suppl):abstr3011.
83. Proffitt KD, Virshup DM. Precise regulation of porcupine activity is required for physiological Wnt signaling. J Biol Chem (2012) 287(41):34167–78. doi:10.1074/jbc.M112.381970
84. Lum L, Clevers H. Cell biology. The unusual case of porcupine. Science (2012) 337(6097):922–3. doi:10.1126/science.1228179
85. Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc Natl Acad Sci U S A (2013) 110(50):20224–9. doi:10.1073/pnas.1314239110
Keywords: indoleamine 2,3-dioxygenase, dendritic cells, tumor immune evasion, β-catenin, tumor immunotherapy, Wnt5a, type III TGF-β receptor, COX-2
Citation: Holtzhausen A, Zhao F, Evans KS and Hanks BA (2014) Early carcinogenesis involves the establishment of immune privilege via intrinsic and extrinsic regulation of indoleamine 2,3-dioxygenase-1: translational implications in cancer immunotherapy. Front. Immunol. 5:438. doi: 10.3389/fimmu.2014.00438
Received: 30 July 2014; Paper pending published: 12 August 2014;
Accepted: 29 August 2014; Published online: 06 October 2014.
Edited by:
Ursula Grohmann, University of Perugia, ItalyReviewed by:
Vincenzo Bronte, University of Verona, ItalyDavid Munn, Georgia Regents University, USA
Copyright: © 2014 Holtzhausen, Zhao, Evans and Hanks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brent A. Hanks, Department of Medicine, Duke University Medical Center, 203 Research Drive, MSRB1, Box 2639, Durham, NC 27710, USA e-mail:YnJlbnQuaGFua3NAZHVrZS5lZHU=