 Isabelle Debock
Isabelle Debock Véronique Flamand
Véronique Flamand
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 27 August 2014
Sec. Vaccines and Molecular Therapeutics
Volume 5 - 2014 | https://doi.org/10.3389/fimmu.2014.00393
This article is part of the Research TopicUnderstanding the Ontogeny of the Immune System to Promote Immune-Mediated Health for LifeView all 20 articles
In comparison to adults, newborns display a heightened susceptibility to pathogens and a propensity to develop allergic diseases. Particular properties of the neonatal immune system can account for this sensitivity. Indeed, a defect in developing protective Th1-type responses and a skewing toward Th2 immunity characterize today the neonatal T-cell immunity. Recently, new findings concerning Th17, regulatory helper T-cell, and follicular helper T-cell subsets in newborns have emerged. In some circumstances, development of effector inflammatory Th17-type responses can be induced in neonates, while differentiation in regulatory T-cells appears to be a default program of neonatal CD4+ T-cells. Poor antibody production, affinity maturation, and germinal center reaction in vaccinated neonates are correlated with a limiting expansion of TFH lymphocytes. We review herein the factors accounting for and the implications of the unbalanced neonatal helper T-cell immunity.
Pioneer experiments by Medawar, Billingham, and Brent showed that mice exposed to allogeneic cells at birth did not reject a graft expressing the same alloantigens (1). This transplantation tolerance or neonatal tolerance was associated with intrathymic clonal deletion of donor-specific cytotoxic CD8+ T-cells (2, 3). Further studies actually demonstrated that allospecific CD4+ T-cells differentiated into IL-4-producing Th2 cells in mice tolerating the allogeneic cells and tissues, while IL-2 and IFN-γ production by Th1 cells was defective (4–6). These discoveries indicated that neonates were not immunodeficient as first thought, since they were able to mount effective, while biased, immune responses. The neonatal stage was therefore considered as a transitory period characterized by an absence of memory cells and a slow adaptive immune response with tolerance features. Furthermore, the immune deviation to the Th2 subset and the defect in developing protective Th1-type responses could account for the higher susceptibility to infectious agents presented by neonates, as well as their propensity to develop allergic diseases in developed countries (7). This particular immunological status also led to consider the neonatal period as a beneficial time window to develop immune tolerance to specific antigens, like autoantigens.
The precise mechanisms underlying the polarization of the neonatal CD4+ T-cell compartment were delineated quite recently. When appropriately stimulated, neonates may achieve an immune response comparable to adult counterparts. Several works in mice first established that Th1-type responses could be induced in neonates with Th1-promoting inflammatory treatments, and brought some clues to explain the unbalanced Th1/Th2 neonatal immunity. This induction of Th1-cell development and, consequently, the modulation of the Th2-type response could be achieved by administration of IFN-γ itself (8), antigen-presenting cells (APCs)-derived IL-12 (9), dendritic cells (DCs) (10), live virus (11, 12), DNA vaccines (13), CpG nucleotides (14), or by CD40 triggering (15). These studies all suggested that particular properties of neonatal CD4+ T-cells and APCs could be involved in the skewed neonatal adaptive immune system.
Zaghouani and colleagues actually identified a mechanism contributing to the Th2 immune deviation in neonates at the molecular level (16). They demonstrated that mouse neonates could mount antigen-specific Th1 and Th2-type primary responses following antigenic stimulation. However, upon recall with antigen, neonatally stimulated Th1 cells died by apoptosis, while neonatally stimulated Th2 cells were not affected and mounted a secondary response. The apoptosis of primary Th1 cells was driven by the Th2 prototypic cytokine IL-4. To mediate this effect, IL-4 signaled through its alternative heteroreceptor composed of the IL-4Rα and IL-13Rα1 chains that is only expressed on neonatal IFN-γ-producing Th1 cells when IL-12p70-producing DCs are not present, thus favoring the development of neonatal Th2 cells. In line with this, studies have demonstrated the importance of age at the time of initial respiratory syncytial virus (RSV) infection in determining the pathophysiological responses. In human infants, a Th2-skewed immune response is particularly dominant in the lung in response to RSV and leads to severe bronchiolitis and subsequent asthma development with persisting respiratory dysfunction into adulthood. A significant up-regulation of IL-4Rα expression was recently detected on human cord blood CD4+ T-cells, in contrast to adult peripheral blood CD4+ T-cells, following in vitro RSV stimulation (17). Furthermore, the IL-4Rα-mediated reinforced Th2 response was confirmed in vivo upon neonatal infection in mice. Indeed, an increased expression of IL-4Rα on CD4+ T-cells (more pronounced on Th2 than on Th1 cells) from mice neonatally infected with RSV and reinfected with RSV as adults was observed. Thus, the neonatal RSV pathogenesis was associated with the expression of IL-4Rα on CD4+ T-cells that become functional upon RSV reinfection and positively control the Th2 response, the airway hyper-activity, and the inflammation. Although, here, the mechanism explaining the skewed Th2-dominant response is a selective higher induction of Th2 proliferation as a selective Th1 apoptosis triggered through the IL-4Rα/IL-13Rα1 heteroreceptor during reinfection in neonatally infected mice was not monitored (17).
Moreover, neonatal CD4+ T-cells display intrinsic epigenetic conformation within the Th2 genomic locus promoting the Th2 cytokines genes expression and the subsequent development of Th2 cells (18). In mice, the enhancer and regulatory region conserved non-coding sequence-1 (CNS-1) of the Th2 locus is indeed hypomethylated in thymic and peripheral neonatal CD4+ T-cells, allowing a rapid and high production of IL-4 and IL-13. In addition, as in adults, the transcription factors regulating Th1 and Th2 differentiations, T-bet, and STAT6, respectively, are already functional in early life (19). The neonatal helper T-cell compartment thus presents unique properties contributing to the unbalanced Th1/Th2 response in early life. This intrinsic bias was recently confirmed in human neonatal CD4+ T-cells. Indeed, a novel subset of naive CD31+CD4+ T-cells that express and store intracellularly an unglycosylated isoform of IL-4 was identified in infant adenoids (20). These IL-4 isoform-expressing cells are not present in adults and are suspected to be early-age restricted Th2 precursors’ cells that would spontaneously differentiate in mature IL-4 secreting Th2 cells. At the level of APCs, in mice, the absence of IL-12p70 (composed of IL-12p35 and IL-12p40 chains) producing neonatal DCs in the first days of life favors the expression of the alternative receptor of IL-4 on neonatal Th1 cells, rending them sensitive to IL-4-induced apoptosis, and promotes Th2-cell development (21). By day 6 after birth, a particular subset of CD11c+ CD8α+ IL-12-producing DCs that would allow a shift to Th1 immunity arises in newborn mice (21). These data confirmed the previously reported adult level of IL-12p70 secretion by 7-day-old splenic CD11c+ sorted DCs stimulated in vitro with TLR9 ligand (22). A defect in IL-12p70 secretion by neonatal monocyte derived DCs is observed in humans, a repressive chromatin state hampering IL-12p35 gene transcription (23). Furthermore, the promoter of the IFN-γ gene is hypermethylated in cord blood CD4+ T-cells, while neonatal CD8+ T-cells do not present this epigenetic modification. The Ifng promoter hypermethylation is associated with impaired production of this Th1 cytokine (24). Conversely, the IL-13 locus is maintained in an accessible chromatin architecture with the appearance of DNase I hypersensitivity sites and extensive DNA demethylation (25) that could account for the elevated IL-13 expression by human neonatal CD4+ T-cells upon TCR triggering (26). An unbalanced Th1/Th2 adaptive immunity might therefore be also at play in human neonates.
With this demonstration of the Th2-skewed immunity and the possible induction of Th1 immunity under appropriate conditions in newborns, the question about the capacity of neonates to develop other helper T-cell responses has been raised. In particular, some attention has been paid to the development of inflammatory Th17-type responses that could compensate for the defective neonatal Th1 immunity. Th17 cells were initially identified in mouse models of autoimmunity (27, 28) and established as an independent CD4+ helper T-cell subset soon after their identification (29, 30). Murine studies elucidate their differentiation process as initiated in the presence of two cytokines, TGF-β and IL-6 (31–33) or TGF-β and IL-21 (34–36), although the need of TGF-β has been challenged (37). TGF-β produced by murine Th17 cells is also necessary for stabilizing in an autocrine manner the commitment of the Th17 cell lineage (38). The Th17 cell development is sustained in the presence of IL-23, a member of the IL-12 family composed of the IL-23p19 chain and the IL-12p40 chain that is shared with IL-12. Th17 cells typically produce IL-17A, IL-17F, and IL-22, which can recruit neutrophils and confer host protection against extracellular bacteria and fungi at epithelial barriers (39). Study of the Th17 cell development in neonates is of particular interest as preterm infants are at higher risk of developing bacterial and fungal infections. As for every helper T-cell subset (40), the Th17 cell development is negatively regulated by the key cytokines of the other subsets. Indeed, the presence of IFN-γ or IL-4 in the naive CD4+ T-cells environment inhibits their differentiation in Th17 lymphocytes (41).
In the context of the neonatal Th2-biased immune response, the inhibitory effect of IL-4 on the development of inflammatory Th17-type responses could represent a major regulation mechanism. A first study on experimental autoimmune encephalomyelitis showed that neonates could mount mixed Th1 and Th17-type autoantigen-specific immune responses (42). However, there were fewer IL-17-producing cells in neonates compared to adult mice, which is in agreement with another report (43). The question of the direct inhibition of neonatal Th17-type responses by IL-4 was addressed in the neonatal tolerance context (44). Mouse neonates immunized with allogeneic cells were also submitted to IL-4 deprivation that inhibited the development of the allospecific Th2-type response (45) and allowed the mRNA up-regulation of Th17-type cytokines and the master regulator of the Th17 pathway RORγt (46), thereby favoring the development of alloreactive IL-17A-producing Th17 cells (44). Likewise, neutralization of IL-25, a Th2-prone cytokine, following murine neonatal infection with pneumonia virus, which belongs to the same family as RSV, reduces some key features of asthma and promotes a Th17-type response. In the infected lung, IL-25 is expressed locally by the airway epithelial cells and can initiate the Th2 differentiation in an IL-4-dependent manner (47). Recently, other works in mice suggested that neonates could mount IL-17A-associated immunity upon infection with the enteropathogen Yersinia enterolitica (48) or anti-mycobacterial vaccination with particular adjuvant (49, 50). In humans, stimulation of cord blood naive T-cells, particularly CD8+ T-cells, with IL-23 induces the expression of IL-23 receptor and the production of IFN-γ and IL-17 (51). In addition, a strong production of IL-23 by neonatal DCs, as well as monocytes, has been reported (52, 53), suggesting that there is no defect in the expression of the IL-12p40 chain, and that human neonates could develop effective Th17 immunity. Accordingly, IL-17-producing cells originated exclusively from cord blood CD161+CD4+ T-cell progenitors were detected in response to IL-1β and IL-23 (54). In agreement with this notion, an in vitro study suggested that naive CD4+ T-cells from preterm and term infants exhibited higher expression of IL-23R, RORC, and STAT3 mRNA than adult counterparts and were accordingly capable of differentiating in Th17 cells under polarizing conditions (with added IL-1, IL-6, IL-23, and TGF-β) and stimulation with anti-CD3 and anti-CD28 antibodies (55). However, in another culture system with anti-CD3 antibodies and cord blood monocytes as viable APCs, naive neonatal CD4+ T-cells were defective in Th17 development and addition of IL-1β, IL-6, and IL-23 only partially restored the IL-17 production. This defect was correlated with a clear reduced RORC2 mRNA levels in neonatal CD4+ T-cells. Human neonatal T-cells could therefore be considered as having intrinsic mechanism that prevents Th17 differentiation through the regulation of RORγT expression (56). Interestingly, a recently described cord blood population of CD4+ T-cell precursors with an effector memory phenotype was shown to display features of IFN-γ producing Th1-like cells, IL-4 producing Th2-like cells but IL-17-deficient Th17-like cells unless they were stimulated with IL-1β and IL-23 (57).
We may therefore consider that under strong TCR/CD28 and cytokine polarizing signaling with no dominance of Th2-type immunity, development of effective Th17 cells can be induced in neonates.
Since the Th2 bias impairs Th1 and Th17-type immune responses in early life, neonatal immunity is typically considered to exhibit an anti-inflammatory profile. In this context, one may suspect that regulatory T-cells (Tregs) play an important role in early life to maintain this anti-inflammatory status. Indeed, it was shown that the lack of cytotoxic activity by CD8+ T-cells reactive to allogeneic cells is controlled by CD4+ CD25+ Tregs (58). Additionally, Tregs and Th17 cells have a reciprocal development pathway. In the presence of TGF-β, their transcriptional master regulators, i.e., Foxp3 for Tregs and RORγT for Th17 cells, respectively, are upregulated. In the absence of proinflammatory cytokines like IL-6 (in mice), or IL-6, IL-1β, and IL-23 (in humans), Foxp3 dominates RORγT function and prevents Th17 development. Such mechanism could impair the differentiation of Th17 cells in early life.
More recently, mechanism about the contribution of Tregs in neonatal immunity was proposed. Murine neonatal CD4+ thymocytes and T-cells actually seem to be prone to intrinsically differentiate into suppressive Foxp3+ Treg cells (59), even though newborn mice display low frequency of Tregs (60). Strikingly, TCR triggering alone was shown to be sufficient to induce enhanced Tregs differentiation in neonates. Exogenous addition of IL-2 or TGF-β, both required to differentiate adult Tregs, was indeed unnecessary (59). In humans, activation of naive CD45RO− CD25− CD4+ T-cells in the presence of APCs showed increased numbers of functional Foxp3+ cells presenting an enhanced expression of PD-1 molecules in cord blood than in adult peripheral blood (61). These studies suggest that Tregs are functional in early life. While neonatal Tregs are thought to be essential for the early life induction of immune tolerance to self antigens, their presence can conversely impair neonatal effector and protective responses to vaccines and pathogens, as proposed by a report on neonatal HSV infection in mice (62) and during Plasmodium falciparum infection in humans. Indeed, CD4+ CD25+ Foxp3+ T-cells downregulated IFN-γ production of cord blood monocytes in response to the parasite through production of IL-10 (63).
Furthermore, development of Tregs seems to occur very early in life. During pregnancy and weaning, the developing immune system actually faces allogeneic stimulation. The fetus and neonate indeed encounter maternal allogeneic cells that present non-inherited maternal alloantigens (NIMA) and are transferred across the placenta and through ingestion of breast milk (64, 65), leading to the establishment of maternal microchimerism. This exposure to maternal alloantigens is associated with the induction of Foxp3+ Tregs specific of NIMA in newborn mice (66, 67). Development of tolerogenic Tregs upon NIMA stimulation is also described in human fetuses (68). However, exposure to NIMA can also lead to priming of cytotoxic and Th1 T-cell responses when the dose of NIMA encountered is low (69), high doses being associated with the development of Tregs (66, 70). Maternal microchimerism thus influences adaptive immunity in early life. Furthermore, it was proposed in a model of juvenile diabetes that maternal microchimeric cells could initiate inflammation to autoantigens and shape the fetal immune response (71). Besides this, the allogeneic stimulation of fetal and neonatal immune system by NIMA-expressing cells is likely to be the first immunological event of life.
Another important question in neonatal T-cell immunity concerns the status of follicular helper T-cells (TFH). This CD4+ helper T-cell subset was first described in human tonsils (72, 73) and is defined as the specialized provider of help to B-cells (74). Indeed, TFH cells play a key role in germinal center (GC) reaction, isotype class switching and antibody affinity maturation through their interactions with cognate B-cells. The development of effective TFH cells requests the presence of various molecules, including Bcl6, CXCR5, PD-1, IL-6, and IL-21 (74). Among these markers, Bcl6 is the transcriptional master regulator of TFH cells essential for the expression of the TFH cell genetic program (75–77). Notably, Bcl6 induces the expression of the chemokine receptor CXCR5 (78), which is crucial for the positioning of TFH cells in B-cell follicles and GCs in secondary lymphoid organs. TFH cells produce IL-21, a cytokine acting on B-cells and on TFH cells themselves via an autocrine loop (79–81). With regards to the relationship between TFH cells and other CD4+ T-cell subsets, a negative regulation exists as Bcl6 represses the expression or function of the master regulators of other Th-cell lineages, i.e., T-bet (Th1), GATA3 (Th2), and RORγt (Th17) (75–77). However, this effect appears to be partial, as TFH cells can produce IFN-γ (82), IL-4 (83, 84), or IL-17 (85), hallmarks of Th1, Th2, and Th17 cells.
Two recent studies in mice sought to determine if newborns can mount effective TFH cell responses (86, 87). Both works showed that CD4+ CXCR5+ PD-1+ TFH cells can be induced in mice neonates subjected to antigenic immunization, but that their full development is impaired. Indeed, neonatal TFH cells expressed lower levels of Bcl6 and IL-21 and were poorly located in GCs, while they were highly present in interfollicular regions (86). This localization outside of GCs correlates with the limited expansion and differentiation in GC TFH cells of newborn TFH cells (87). Corresponding to this limited TFH cell response, the humoral immune response of immunized neonates was defective, as revealed by a reduced antibody production and maturation and a decreased and delayed formation of GCs (86).
Both studies also indicated IL-4 expression by neonatal TFH cells. The role of IL-4 in the emergence of TFH cells in newborns was addressed by Debock et al. (86). It was shown that this cytokine promotes the development of TFH cells in early life. Indeed, the proportion of TFH cells in immunized IL-4-deficient newborns was greatly reduced. Moreover, in the absence of IL-4, neonatal TFH cells highly expressed RORγt and IL-17, features of Th17 cells. This particular phenotype was associated with an enhanced production of IgG2a in immunized neonates, suggesting that IL-4 impacts the genetic profile and the B-cell help specificity of neonatal TFH cells.
The finding that TFH cells are present and functional in early life, although in a limited extent compared to adults, is of interest to improve neonatal and pediatric vaccines. Indeed, in the context of defective follicular DCs and B-cells, the other partners of humoral immunity, in newborns (88), early priming of neonatal TFH cells could promote the induction of high-affinity antibody responses when the other cellular actors become functional. Interestingly, administration of a CpG-adjuvanted vaccine in mouse neonates circumvented the limited expansion and development in GC TFH cells of neonatal TFH cells (87), suggesting that this could represent a beneficial strategy for efficient vaccination of newborns.
Our understanding of neonatal helper T-cell immunity was greatly improved in the last years. Precise mechanisms underlying the profound defect of Th1 immunity and the subsequent Th2-biased polarization in early life were indeed identified at the molecular and cellular levels. This imbalance in neonatal T-cell responses may imprint development of adult T-cell responses as illustrated in a study showing the higher susceptibility to allergic inflammation of mice neonatally immunized with a pro-Th2 vaccine and a protection when pro-Th1 vaccine was used (10). In addition to this, neonates also appear capable of mounting inflammatory Th17-type responses when properly stimulated. This could be a potential target to ensure neonatal protection to pathogens and to prevent development of allergic diseases, as neonatal Th17 immunity could compensate for the lack of Th1-type immune responses and oppose the Th2 pathway. However, caution should be taken as induction of strong Th17-type responses could possibly break the induction of immune tolerance to self antigens and favor the development of autoimmunity. The default generation of Tregs in neonates could furthermore promote the Th17 pathway by producing one of its differentiation cytokine, TGF-β (31–33), which could lead to an uncontrolled inflammatory response. Notwithstanding this, Treg cells appear to play a critical role in the developing immune system, contributing to the neonatal general anti-inflammatory status and limiting protective immune responses developed by newborns. The presence and functionality of TFH cells in neonates were also recently delineated. These providers of B-cell help are induced upon vaccination in early life, but their development is limited compared to adults. This correlates well with the defective humoral immune responses observed in newborns. Targeting this particular T-cell population with appropriate adjuvants could enhance vaccine responses in newborns and therefore reduce the need of several booster doses in early life to reach effective IgG responses while increasing their resistance to vaccine-preventable life-threatening infections like tuberculosis, malaria, and HIV (Figure 1).
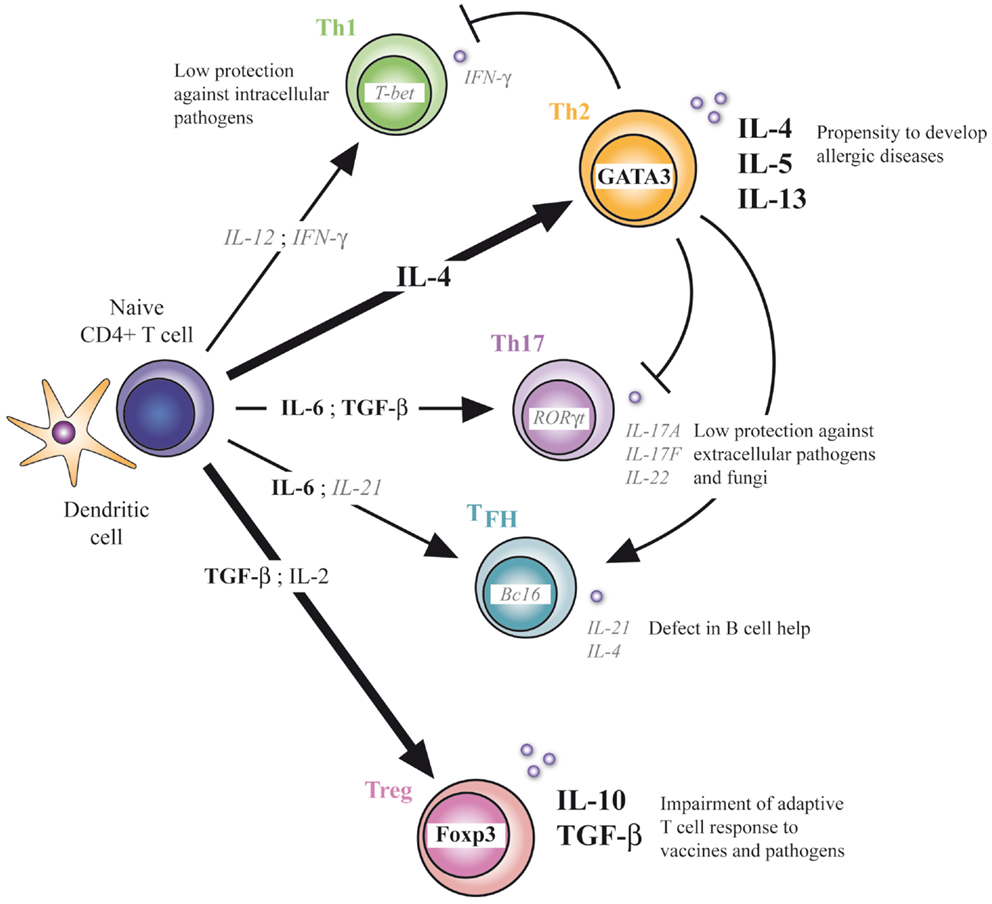
Figure 1. CD4+ T-cell differentiation in early life. Defect in Th1, Th17, and TFH differentiation and enhanced Th2 and Treg differentiation characterize the neonatal T-cell immunity. At the molecular level, respective transcriptional factors and cytokines are positively (bold) or negatively (italic) regulated. Implications of this unbalanced adaptive immunity are depicted.
Regarding the aspects that still need to be addressed in neonatal T-cell immunity, the phenotype and the differentiation pathway of neonatal Th17 cells require further study. TGF-β-stimulated Th17 cells can indeed produce IL-17 and IL-10, thus displaying a more regulatory phenotype, while Th17 cells differentiated in the presence of IL-23 seem to be more pathogenic, these inflammatory properties being due in part to their production of IFN-γ (37). Since IL-23 is highly produced in human neonates, the inflammatory Th17 cell subpopulation could be more represented in newborns. However, considering the neonatal regulatory context, regulatory Th17 cells producing IL-10 and limiting inflammatory response could arise preferentially. In line with this hypothesis, neonatal B-cells secreting IL-10 have been shown to control DCs response upon TLR triggering (89, 90).
More clarification is also needed for the development and function of TFH cells in early life, notably with regards to their potential link with the other Th-cell subsets. While there are now some insights in the relationship between the newborn TFH cells and the Th2 and Th17 populations (86), the potential link of neonatal TFH cells with Tregs is also of interest since follicular Tregs expressing CXCR5, PD-1, and CTLA-4 or GITR have been described (91). Also, the role in early life of recently described TFH cell regulators like the transcription factor achaete-scute homolog 2 (92) or the Notch signaling (93), as well as of molecules involved in the TFH cell/B-cell interactions such as the SAP/SLAM family of molecules (74) or BLyS (94), requests further attention. In addition, better phenotypical characterization of neonatal TFH cells could lead to the identification of potential new targets for vaccination.
Another intriguing question is the potential development of IL-9-producing Th9 cells in neonates, since they are induced in the presence of IL-4 and TGF-β (95). Considering the heightened production of IL-4 and the default generation of Tregs displayed by neonates, Th9 cells could represent an important Th-cell subset in newborns.
Finally, epigenetic modifications hampering the Th1 pathway and favoring the Th2 response have been described in neonates. It would be interesting to determine, which transcription factors and histones and DNA modifying enzymes are responsible for the Il12p35 and Ifng genes repressive chromatin state, and if a particular chromatin conformation is also present in the Il17a, Il10, and Foxp3 genes. This could provide therapeutic targets to induce protective immunity in early life.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Support for this work was generously provided by grants from the government of the Walloon Region, GlaxoSmithKline Biologicals, the Fonds National de la Recherche Scientifique (FNRS, Belgium), an Interuniversity Attraction Pole of the Belgian Federal Science Policy and by a European grant from Feder-Convergence.
1. Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature (1953) 172(4379):603–6. doi: 10.1038/172603a0
2. Feng HM, Glasebrook AL, Engers HD, Louis JA. Clonal analysis of T cell unresponsiveness to alloantigens induced by neonatal injection of F1 spleen cells into parental mice. J Immunol (1983) 131(5):2165–9.
3. Nossal GJ, Pike BL. Functional clonal deletion in immunological tolerance to major histocompatibility complex antigens. Proc Natl Acad Sci U S A (1981) 78(6):3844–7. doi:10.1073/pnas.78.6.3844
4. Abramowicz D, Vandervorst P, Bruyns C, Doutrelepont JM, Vandenabeele P, Goldman M. Persistence of anti-donor allohelper T cells after neonatal induction of allotolerance in mice. Eur J Immunol (1990) 20(8):1647–53. doi:10.1002/eji.1830200805
5. Powell TJ Jr, Streilein JW. Neonatal tolerance induction by class II alloantigens activates IL-4-secreting, tolerogen-responsive T cells. J Immunol (1990) 144(3):854–9.
6. Schurmans S, Heusser CH, Qin HY, Merino J, Brighouse G, Lambert PH. In vivo effects of anti-IL-4 monoclonal antibody on neonatal induction of tolerance and on an associated autoimmune syndrome. J Immunol (1990) 145(8):2465–73.
7. Holt PG. The role of genetic and environmental factors in the development of T-cell mediated allergic disease in early life. Paediatr Respir Rev (2004) 5(Suppl A):S27–30. doi:10.1016/S1526-0542(04)90006-1
8. Donckier V, Abramowicz D, Bruyns C, Florquin S, Vanderhaeghen ML, Amraoui Z, et al. IFN-gamma prevents Th2 cell-mediated pathology after neonatal injection of semiallogenic spleen cells in mice. J Immunol (1994) 153(6):2361–8.
9. Donckier V, Flamand V, Desalle F, Vanderhaeghen ML, de Veerman M, Thielemans K, et al. IL-12 prevents neonatal induction of transplantation tolerance in mice. Eur J Immunol (1998) 28(4):1426–30. doi:10.1002/(SICI)1521-4141(199804)28:04<1426::AID-IMMU1426>3.0.CO;2-P
10. Dubois A, Deruytter N, Adams B, Kanda A, Delbauve S, Fleury S, et al. Regulation of Th2 responses and allergic inflammation through bystander activation of CD8+ T lymphocytes in early life. J Immunol (2010) 185(2):884–91. doi:10.4049/jimmunol.0903287
11. Fadel SA, Ozaki DA, Sarzotti M. Enhanced type 1 immunity after secondary viral challenge in mice primed as neonates. J Immunol (2002) 169(6):3293–300. doi:10.4049/jimmunol.169.6.3293
12. Sarzotti M, Robbins DS, Hoffman PM. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science (1996) 271(5256):1726–8. doi:10.1126/science.271.5256.1726
13. Martinez X, Brandt C, Saddallah F, Tougne C, Barrios C, Wild F, et al. DNA immunization circumvents deficient induction of T helper type 1 and cytotoxic T lymphocyte responses in neonates and during early life. Proc Natl Acad Sci U S A (1997) 94(16):8726–31. doi:10.1073/pnas.94.16.8726
14. Brazolot Millan CL, Weeratna R, Krieg AM, Siegrist CA, Davis HL. CpG DNA can induce strong Th1 humoral and cell-mediated immune responses against hepatitis B surface antigen in young mice. Proc Natl Acad Sci U S A (1998) 95(26):15553–8. doi:10.1073/pnas.95.26.15553
15. Flamand V, Donckier V, Demoor FX, Le Moine A, Matthys P, Vanderhaeghen ML, et al. CD40 ligation prevents neonatal induction of transplantation tolerance. J Immunol (1998) 160(10):4666–9.
16. Li L, Lee HH, Bell JJ, Gregg RK, Ellis JS, Gessner A, et al. IL-4 utilizes an alternative receptor to drive apoptosis of Th1 cells and skews neonatal immunity toward Th2. Immunity (2004) 20(4):429–40. doi:10.1016/S1074-7613(04)00072-X
17. You D, Marr N, Saravia J, Shrestha B, Lee G, Turvey SE. IL-4Ra on CD4+ T cells plays a pathogenic role in respiratory syncytial virus reinfection in mice infected initially as neonates. J Leukoc Biol (2013) 93(6):933–42. doi:10.1189/jlb.1012498
18. Rose S, Lichtenheld M, Foote MR, Adkins B. Murine neonatal CD4+ cells are poised for rapid Th2 effector-like function. J Immunol (2007) 178(5):2667–78. doi:10.4049/jimmunol.178.5.2667
19. Rose S, Guevara P, Farach S, Adkins B. The key regulators of adult T helper cell responses, STAT6 and T-bet, are established in early life in mice. Eur J Immunol (2006) 36(5):1241–53. doi:10.1002/eji.200535563
20. Hebel K, Weinert S, Kuropka B, Knolle J, Kosak B, Jorch G, et al. CD4+ T cells from human neonates and infants are poised spontaneously to run a nonclassical IL-4 program. J Immunol (2014) 192:5160–70. doi:10.4049/jimmunol.1302539
21. Lee HH, Hoeman CM, Hardaway JC, Guloglu FB, Ellis JS, Jain R, et al. Delayed maturation of an IL-12-producing dendritic cell subset explains the early Th2 bias in neonatal immunity. J Exp Med (2008) 205(10):2269–80. doi:10.1084/jem.20071371
22. Sun CM, Fiette L, Tanguy M, Leclerc C, Lo-Man R. Ontogeny and innate properties of neonatal dendritic cells. Blood (2003) 102(2):585–91. doi:10.1182/blood-2002-09-2966
23. Goriely S, Van Lint C, Dadkhah R, Libin M, De Wit D, Demonte D, et al. A defect in nucleosome remodeling prevents IL-12(p35) gene transcription in neonatal dendritic cells. J Exp Med (2004) 199(7):1011–6. doi:10.1084/jem.20031272
24. White GP, Watt PM, Holt BJ, Holt PG. Differential patterns of methylation of the IFN-gamma promoter at CpG and non-CpG sites underlie differences in IFN-gamma gene expression between human neonatal and adult CD45RO- T cells. J Immunol (2002) 168(6):2820–7. doi:10.4049/jimmunol.168.6.2820
25. Webster RB, Rodriguez Y, Klimecki WT, Vercelli D. The human IL-13 locus in neonatal CD4+ T cells is refractory to the acquisition of a repressive chromatin architecture. J Biol Chem (2007) 282(1):700–9. doi:10.1074/jbc.M609501200
26. Ribeiro-do-Couto LM, Boeije LC, Kroon JS, Hooibrink B, Breur-Vriesendorp BS, Aarden LA, et al. High IL-13 production by human neonatal T cells: neonate immune system regulator? Eur J Immunol (2001) 31(11):3394–402. doi:10.1002/1521-4141(200111)31:11<3394::AID-IMMU3394>3.0.CO;2-B
27. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421(6924):744–8. doi:10.1038/nature01355
28. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201(2):233–40. doi:10.1084/jem.20041257
29. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6(11):1123–32. doi:10.1038/ni1254
30. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi:10.1038/ni1261
31. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi:10.1038/nature04753
32. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441(7090):231–4. doi:10.1038/nature04754
33. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24(2):179–89. doi:10.1016/j.immuni.2006.01.001
34. Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature (2007) 448(7152):484–7. doi:10.1038/nature05970
35. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448(7152):480–3. doi:10.1038/nature05969
36. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol (2007) 8(9):967–74. doi:10.1038/ni1488
37. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature (2010) 467(7318):967–71. doi:10.1038/nature09447
38. Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-b1 promotes in vivo Th17 cell differentiation. Immunity (2011) 34:396–408. doi:10.1016/j.immuni.2011.03.005
39. Korn T, Bettelli E, Oukka M, Kuchroo VK. . IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
40. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
41. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol (2007) 25:821–52. doi:10.1146/annurev.immunol.25.022106.141557
42. Hofstetter HH, Kovalovsky A, Shive CL, Lehmann PV, Forsthuber TG. Neonatal induction of myelin-specific Th1/Th17 immunity does not result in experimental autoimmune encephalomyelitis and can protect against the disease in adulthood. J Neuroimmunol (2007) 187(1–2):20–30. doi:10.1016/j.jneuroim.2007.04.001
43. Bogaert D, Weinberger D, Thompson C, Lipsitch M, Malley R. Impaired innate and adaptive immunity to Streptococcus pneumoniae and its effect on colonization in an infant mouse model. Infect Immun (2009) 77(4):1613–22. doi:10.1128/IAI.00871-08
44. Debock I, Delbauve S, Dubois A, Petein M, Leo O, Goldman M, et al. Th17 alloimmunity prevents neonatal establishment of lymphoid chimerism in IL-4-deprived mice. Am J Transplant (2012) 12(1):81–9. doi:10.1111/j.1600-6143.2011.03778.x
45. Donckier V, Wissing M, Bruyns C, Abramowicz D, Lybin M, Vanderhaeghen ML, et al. Critical role of interleukin 4 in the induction of neonatal transplantation tolerance. Transplantation (1995) 59(11):1571–6. doi:10.1097/00007890-199506000-00012
46. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi:10.1016/j.cell.2006.07.035
47. Siegle JS, Hansbro N, Dong C, Angkasekwinai P, Foster PS, Kumar RK. Blocking induction of T helper type 2 responses prevents development of disease in a model of childhood asthma. Clin Exp Immunol (2011) 165:19–28. doi:10.1111/j.1365-2249.2011.04392.x
48. Echeverry A, Saijo S, Schesser K, Adkins B. Yersinia enterocolitica promotes robust mucosal inflammatory T-cell immunity in murine neonates. Infect Immun (2010) 78(8):3595–608. doi:10.1128/IAI.01272-09
49. Kamath AT, Rochat AF, Christensen D, Agger EM, Andersen P, Lambert PH, et al. A liposome-based mycobacterial vaccine induces potent adult and neonatal multifunctional T cells through the exquisite targeting of dendritic cells. PLoS One (2009) 4(6):e5771. doi:10.1371/journal.pone.0005771
50. Kamath AT, Rochat AF, Valenti MP, Agger EM, Lingnau K, Andersen P, et al. Adult-like anti-mycobacterial T cell and in vivo dendritic cell responses following neonatal immunization with Ag85B-ESAT-6 in the IC31 adjuvant. PLoS One (2008) 3(11):e3683. doi:10.1371/journal.pone.0003683
51. Vanden Eijnden S, Goriely S, De Wit D, Willems F, Goldman M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T cells in human. Eur J Immunol (2005) 35(2):469–75. doi:10.1002/eji.200425677
52. Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol (2009) 183(11):7150–60. doi:10.4049/jimmunol.0901481
53. Vanden Eijnden S, Goriely S, De Wit D, Goldman M, Willems F. Preferential production of the IL-12(p40)/IL-23(p19) heterodimer by dendritic cells from human newborns. Eur J Immunol (2006) 36(1):21–6. doi:10.1002/eji.200535467
54. Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med (2008) 205:1903–16. doi:10.1084/jem.20080397
55. Black A, Bhaumik S, Kirkman RL, Weaver CT, Randolph DA. Developmental regulation of Th17-cell capacity in human neonates. Eur J Immunol (2012) 42(2):311–9. doi:10.1002/eji.201141847
56. de Roock S, Stoppelenburg AJ, Scholman R, Hoeks SB, Meerding J, Prakken B, et al. Defective Th17 development in human neonatal T cells involves reduced RORC2 mRNA content. J Allergy Clin Immunol (2013) 132(3):754–6. doi:10.1016/j.jaci.2013.04.014
57. Zhang X, Mozeleski B, Lemoine S, Dériaud E, Lim A, Zhivaki D, et al. CD4 T cells with effector memory phenotype and function develop in the sterile environment of the fetus. Sci Transl Med (2014) 6(238):238ra72. doi:10.1126/scitranslmed.3008748
58. Gao Q, Rouse TM, Kazmerzak K, Field EH. CD4+CD25+ cells regulate CD8 cell anergy in neonatal tolerant mice. Transplantation (1999) 68(12):1891–7. doi:10.1097/00007890-199912270-00013
59. Wang G, Miyahara Y, Guo Z, Khattar M, Stepkowski SM, Chen W. “Default” generation of neonatal regulatory T cells. J Immunol (2010) 185(1):71–8. doi:10.4049/jimmunol.0903806
60. Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental regulation of Foxp3 expression during ontogeny. J Exp Med (2005) 202(7):901–6. doi:10.1084/jem.20050784
61. de Roock S, Hoeks SB, Meurs L, Steur A, Hoekstra MO, Prakken BJ, et al. Critical role for programmed death 1 signaling and protein kinase B in augmented regulatory T-cell induction in cord blood. J Allergy Clin Immunol (2011) 128(6):754–6. doi:10.1016/j.jaci.2011.08.006
62. Fernandez MA, Puttur FK, Wang YM, Howden W, Alexander SI, Jones CA. T regulatory cells contribute to the attenuated primary CD8+ and CD4+ T cell responses to herpes simplex virus type 2 in neonatal mice. J Immunol (2008) 180(3):1556–64. doi:10.4049/jimmunol.180.3.1556
63. Bisseye C, van der Sande M, Morgan WD, Holder AA, Pinder M, Ismaili J. Plasmodium falciparum infection in the placenta impacts on the T helper type 1 (Th1)/Th2 balance of neonatal T cells through CD4+CD25+ forkhead box P3+ regulatory T cells and interleukin-10. Clin Exp Immunol (2009) 158:287–93. doi:10.1111/j.1365-2249.2009.04014.x
64. Shimamura M, Ohta S, Suzuki R, Yamazaki K. Transmission of maternal blood cells to the fetus during pregnancy: detection in mouse neofnatal spleen by immunofluorescence flow cytometry and polymerase chain reaction. Blood (1994) 83(4):926–30.
65. Zhou L, Yoshimura Y, Huang Y, Suzuki R, Yokoyama M, Okabe M, et al. Two independent pathways of maternal cell transmission to offspring: through placenta during pregnancy and by breast-feeding after birth. Immunology (2000) 101(4):570–80. doi:10.1046/j.1365-2567.2000.00144.x
66. Dutta P, Molitor-Dart M, Bobadilla JL, Roenneburg DA, Yan Z, Torrealba JR, et al. Microchimerism is strongly correlated with tolerance to noninherited maternal antigens in mice. Blood (2009) 114(17):3578–87. doi:10.1182/blood-2009-03-213561
67. Molitor-Dart ML, Andrassy J, Kwun J, Kayaoglu HA, Roenneburg DA, Haynes LD, et al. Developmental exposure to noninherited maternal antigens induces CD4+ T regulatory cells: relevance to mechanism of heart allograft tolerance. J Immunol (2007) 179(10):6749–61. doi:10.4049/jimmunol.179.10.6749
68. Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science (2008) 322(5907):1562–5. doi:10.1126/science.1164511
69. Opiela SJ, Levy RB, Adkins B. Murine neonates develop vigorous in vivo cytotoxic and Th1/Th2 responses upon exposure to low doses of NIMA-like alloantigens. Blood (2008) 112(4):1530–8. doi:10.1182/blood-2007-08-106500
70. Andrassy J, Kusaka S, Jankowska-Gan E, Torrealba JR, Haynes LD, Marthaler BR, et al. Tolerance to noninherited maternal MHC antigens in mice. J Immunol (2003) 171(10):5554–61. doi:10.4049/jimmunol.171.10.5554
71. Roy E, Leduc M, Guegan S, Rachdi L, Kluger N, Scharfmann R, et al. Specific maternal microchimeric T cells targeting fetal antigens in beta cells predispose to auto-immune diabetes in the child. J Autoimmun (2010) 36(3–4):253–62. doi:10.1016/j.jaut.2011.02.003
72. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192(11):1545–52. doi:10.1084/jem.192.11.1545
73. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med (2000) 192(11):1553–62. doi:10.1084/jem.192.11.1553
74. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
75. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi:10.1126/science.1175870
76. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325(5943):1001–5. doi:10.1126/science.1176676
77. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31(3):457–68. doi:10.1016/j.immuni.2009.07.002
78. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34(6):932–46. doi:10.1016/j.immuni.2011.03.023
79. Eddahri F, Denanglaire S, Bureau F, Spolski R, Leonard WJ, Leo O, et al. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood (2009) 113(11):2426–33. doi:10.1182/blood-2008-04-154682
80. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity (2008) 29(1):138–49. doi:10.1016/j.immuni.2008.05.009
81. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity (2008) 29(1):127–37. doi:10.1016/j.immuni.2008.06.001
82. Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity (2011) 35(6):919–31. doi:10.1016/j.immuni.2011.11.012
83. Harada Y, Tanaka S, Motomura Y, Harada Y, Ohno S, Ohno S. The 3’ enhancer CNS2 is a critical regulator of interleukin-4-mediated humoral immunity in follicular helper T cells. Immunity (2012) 36(2):188–200. doi:10.1016/j.immuni.2012.02.002
84. Vijayanand P, Seumois G, Simpson LJ, Abdul-Wajid S, Baumjohann D, Panduro M, et al. Interleukin-4 production by follicular helper T cells requires the conserved Il4 enhancer hypersensitivity site V. Immunity (2012) 36(2):175–87. doi:10.1016/j.immuni.2011.12.014
85. Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho IC, Sharpe AH, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol (2009) 10(2):167–75. doi:10.1038/ni.1690
86. Debock I, Jaworski K, Chadlaoui H, Delbauve S, Passon N, Twyffels L, et al. Neonatal follicular Th cell responses are impaired and modulated by IL-4. J Immunol (2013) 191(3):1231–9. doi:10.4049/jimmunol.1203288
87. Mastelic B, Kamath AT, Fontannaz P, Tougne C, Rochat AF, Belnoue E. Environmental and T cell-intrinsic factors limit the expansion of neonatal follicular T helper cells but may be circumvented by specific adjuvants. J Immunol (2012) 189(12):5764–72. doi:10.4049/jimmunol.1201143
88. Siegrist CA, Aspinall R. B-cell responses to vaccination at the extremes of age. Nat Rev Immunol (2009) 9(3):185–94. doi:10.1038/nri2508
89. Walker WE, Goldstein DR. Neonatal B cells suppress innate toll-like receptor immune responses and modulate alloimmunity. J Immunol (2007) 179(3):1700–10. doi:10.4049/jimmunol.179.3.1700
90. Zhang X, Deriaud E, Jiao X, Braun D, Leclerc C, Lo-Man R. Type I interferons protect neonates from acute inflammation through interleukin 10-producing B cells. J Exp Med (2007) 204(5):1107–18. doi:10.1084/jem.20062013
91. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
92. Liu X, Chen X, Zhong B, Wang A, Wang X, Chu F, et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature (2014) 507(7493):513–8. doi:10.1038/nature12910
93. Auderset F, Schuster S, Fasnacht N, Coutaz M, Charmoy M, Koch U, et al. Notch signaling regulates follicular helper T cell differentiation. J Immunol (2013) 191(5):2344–50. doi:10.4049/jimmunol.1300643
94. Goenka R, Matthews AH, Zhang B, O’Neill PJ, Scholz JL, Migone TS, et al. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J Exp Med (2014) 211(1):45–56. doi:10.1084/jem.20130505
Keywords: vaccine, T helper subsets, Th2-biased response, defective Th1 response, follicular helper T-cells, Th17, Tregs
Citation: Debock I and Flamand V (2014) Unbalanced neonatal CD4+ T-cell immunity. Front. Immunol. 5:393. doi: 10.3389/fimmu.2014.00393
Received: 30 May 2014; Accepted: 31 July 2014;
Published online: 27 August 2014.
Edited by:
Arnaud Marchant, Université Libre de Bruxelles, BelgiumReviewed by:
Yasmin Thanavala, Roswell Park Cancer Institute, USACopyright: © 2014 Debock and Flamand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Véronique Flamand, Institut d’Immunologie Médicale, Université Libre de Bruxelles, Rue Adrienne Bolland 8, Gosselies 6041, Belgium e-mail:dmZsYW1hbmRAdWxiLmFjLmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.