 Dhanusha Yesudhas
Dhanusha Yesudhas Vijayakumar Gosu
Vijayakumar Gosu Muhammad Ayaz Anwar
Muhammad Ayaz Anwar Sangdun Choi
Sangdun Choi- Department of Molecular Science and Technology, Ajou University, Suwon, South Korea
Toll-like receptor (TLR) signaling has been implicated in the inflammatory responses in intestinal epithelial cells (IECs). Such inflammatory signals mediate complex interactions between commensal bacteria and TLRs and are required for IEC proliferation, immune response, repair, and homeostasis. The upregulation of certain TLRs in colorectal cancer (CRC) tissues suggests that TLRs may play an essential role in the prognosis of chronic and inflammatory diseases that ultimately culminate in CRC. Here, we provide a comprehensive review of the literature on the involvement of the TLR pathway in the initiation, progression, and metastasis of CRC, as well as inherited genetic variation and epigenetic regulation. The differential expression of TLRs in epithelial cells has also been discussed. In particular, we emphasize the physiological role of TLR4 in CRC development and pathogenesis, and propose novel and promising approaches for CRC therapeutics with the aid of TLR ligands.
Introduction
The innate immune system possesses a robust mechanism in the form of evolutionarily conserved toll-like receptors (TLRs) that can detect the signature pattern of invading microorganisms for the protection of the host. TLRs are a class of type I trans-membrane glycoproteins. Human and mouse cells comprises of 13 types of TLRs that can detect different kinds of bacterial and viral-associated patterns (1–3). TLR1–9 are highly conserved in both species; while the mouse TLR10 is non-functional due to retroviral insertion, TLR11–13 are undetected in the human genome. Examples of TLR-specific ligands are: lipopeptides for TLR1/2 and 2/6 (4–6), dsRNA for TLR3 (7), lipopolysaccharide (LPS) for TLR4 (8), flagellin for TLR5 (9), ssRNA for TLR7/8 (10, 11), and CpG DNA for TLR9 (12–14). TLRs not only detect invading microbes but also recognize intracellular anomalies and mount an immune response, thereby playing a cardinal role in the homeostasis of the human immune system (15, 16). The abnormal activation of TLRs can jeopardize normal physiological processes and cause several inflammatory diseases, cancers, and autoimmune diseases (17, 18).
Toll-like receptors are ubiquitously expressed, although their expression level may vary according to the circumstances and the tissues. In addition, induced expression of TLRs has been observed when ligands bind to their cognate TLRs (19). Research in the last decade has focused on elucidating various functions, intermediate molecules, and ligands associated with TLRs. There is a well-established link between TLR-induced inflammation and the development and progression of cancer (20, 21). Similarly, TLRs are also known to play a vital role in colorectal cancer (CRC) that affects the large intestine and the rectum. This region is heavily populated by intestinal microbes, highlighting the crucial role of TLRs in CRC pathogenesis (17, 22).
Colorectal cancer is one of the most complex diseases and causes death in many cases in the United States (23). Globally, more than one million new cases of CRC are reported annually (24, 25). The complexity of CRC is primarily attributed to environmental factors, while genetic factors play a minor role. The known risk factors for CRC are food-borne mutagens, pollution, certain commensal bacteria, and chronic intestinal inflammation (25). Commonly, CRC occurs in the right ascending colon with the most common symptom being blood in the stool or rectal bleeding. Genetically, inherited colon polyps also contribute to the development of CRC (26). Since CRC can damage the host immune system during their proliferation period, stimulating it against CRC promises to be an attractive approach for drug discovery (27).
In this review, we discuss the role of TLRs in the maintenance of homeostasis and the development of CRC in intestinal epithelial cells (IECs). Improved techniques to detect dysfunctional TLR signaling in carcinogenesis may stimulate the development of novel therapies to prevent or treat CRC. Recent studies have improved the understanding of TLR-targeted applications such as identifying their differential expression, their role in tumor progression, potential use as immune modulating agents, and development of novel TLR ligands in anti-cancer therapies.
TLR Signaling: An Overview
The localization of TLRs is heterogeneous and varies from the cell surface (TLR1, 2, 4, 5, 6, 10, and mouse TLR11, 12) to the endosomes (TLR3, 7, 8, and 9) (28), depending on the localization of pathogen-associated molecular patterns (PAMPs). TLRs comprises of the following three domains: ectodomain [contains leucine rich repeats (LRR)] that recognizes PAMPs, a trans-membrane region, and a cytosolic toll/interleukin-1 (IL-1) receptor (TIR) domain that interacts with adaptor molecules (such as MyD88/MAL and TRIF/TRAM) to propagate downstream signaling. Ligand binding triggers the dimerization of TLRs, facilitating the binding of adaptor molecules, which subsequently activate the IL-1 receptor-associated kinase (IRAK) family (29). Upon IRAK recruitment, IRAK4 phosphorylates IRAK1 at key serine and threonine residues, and enables IRAK1 to eventually activate tumor necrosis factor receptor-associated factor 6 (TRAF6) (30) that subsequently activates transforming growth factor-β-activated protein kinase 1 (TAK1), a member of the mitogen-activated protein (MAP) kinase kinase kinase (MAP3K) family. TAK1 forms a complex with TGF-β-activated kinase 1/MAP3K7 binding protein 1 (TAB1), TAB2, and TAB3 and then activates nuclear factor (NF)-κB by phosphorylating IKK that in turn phosphorylates IκB for proteasomal degradation. Following the degradation of IκB, NF-κB translocates into the nucleus and induces inflammatory mediators. Moreover, TAK1 activates members of the MAP kinase kinase 3 (MKK3) and MKK6 to activate an alternative closely related pathway that phosphorylates c-Jun N-terminal Kinase (JNK) and p38. TLR signaling can also activate extracellular signal-regulated kinase (ERK) via the activation of MEK1/2. In response to various TLR ligands, reduced activity of NF-κB, JNK, and p38 was observed in B cells and embryonic fibroblasts derived from TAK1-deficient mice (31). In the TRIF-dependent pathway triggered by TLR3 and TLR4, TRIF recruits TRAF3, TAB1, and IKK and activates the type I IFN. The TRIF-dependent pathway also activates TRAF6 and TAB1, which regulate the delayed activation of NF-κB and MAP kinases (32) (Figure 1).
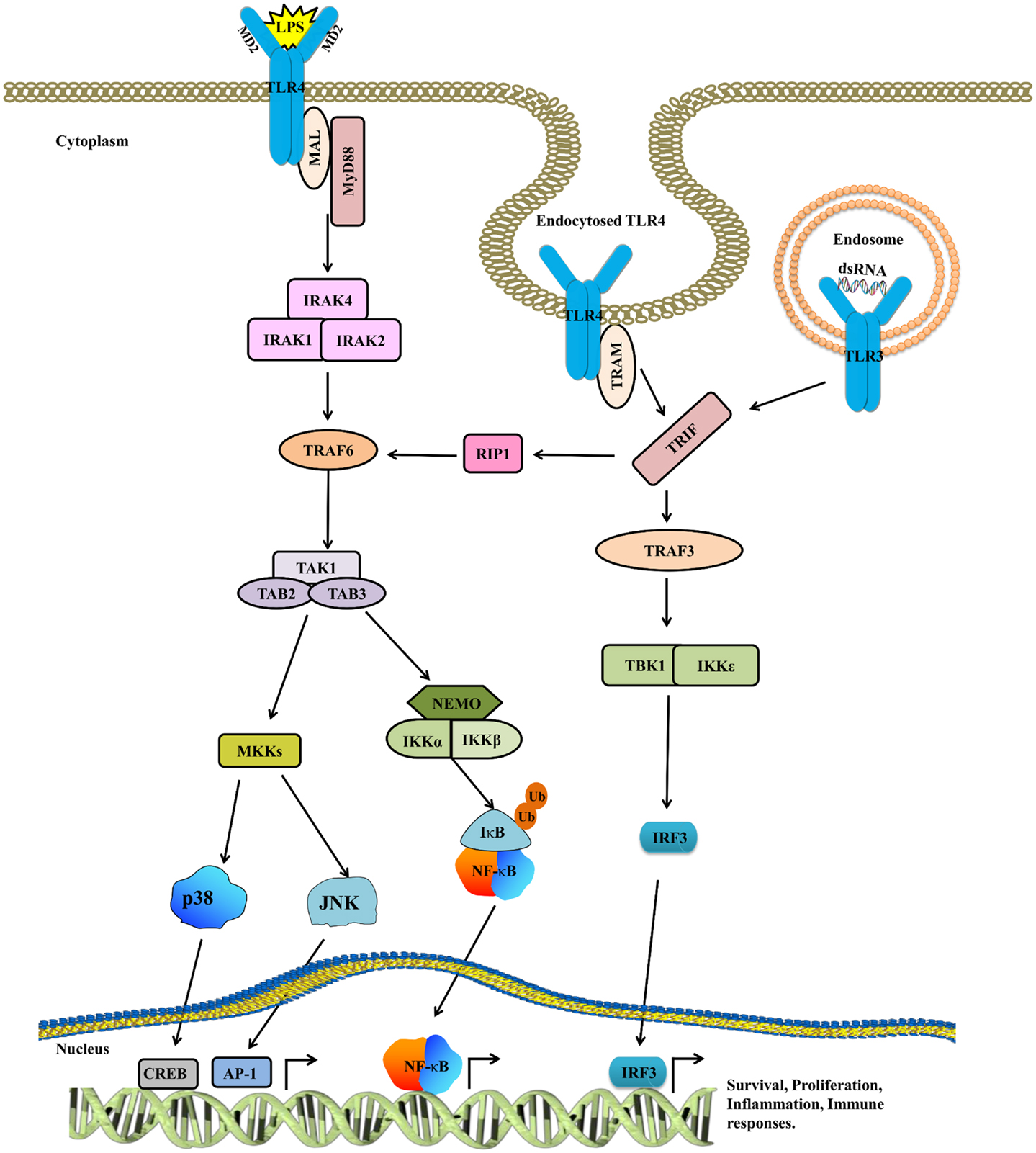
Figure 1. The TLR4 signaling pathway. TLR4 is activated by LPS, whereas CD14 and MD2 act as accessory proteins for LPS/TLR4 binding. Upon ligand binding, TLR4 dimerizes, and recruits downstream adaptor molecules such as MyD88/MAL and TRIF/TRAM to mount an inflammatory response. The activated MyD88/MAL then activates IRAK4, TRAF6, TAK1, and IKK complexes, while TRIF/TRAM signals through RIP1 to TRAF6/TAK1 and IKK. After this, both these pathways converge at NF-κB. The cytoplasmic NF-κB complex is maintained in the inactive state by IκB, which is in turn degraded by proteasomes, resulting in the translocation of NF-κB into the nucleus. Besides activating NF-κB, TAK1 also phosphorylates MAPKs to further reinforce the inflammatory response. The TRIF/TRAM pathway not only activates NF-κB but also triggers IRF3 to mount an antiviral response. Cumulatively, all these signaling pathways assist in eradicating infection as well as play an important role in sustaining the normal physiological functions in IECs.
TLRs and Their Expression Patterns in IECs
The human intestinal tract plays a crucial role in maintaining the complex ecosystem of commensal bacteria and also physically isolates the countless resident bacteria from the lamina propria (33). It was originally believed that IECs prevent bacteria from invading the body. However, IECs have a complicated and common beneficial link with the microorganisms in the intestinal gut flora. The commensal bacteria metabolize carbohydrates and the IECs break down the short-chain fatty acids produced as a result of bacterial fermentation of undigested carbohydrates and use them as an energy source (34). IEC membranes express TLRs that detect the commensal PAMPs and mediate signaling to maintain epithelial cell integrity and tight junctions, cell proliferation, immunoglobulin A (IgA) production, and antimicrobial peptide expression (34). In addition, they can also induce a pro-inflammatory response by interacting with the immune cells in the lamina propria (35, 36). Therefore, tight regulation of TLRs is imperative to prevent adverse effects since anomalous or dysregulated TLR signaling can mediate cancer induction and propagation.
Colorectal cancer pathogenesis is governed by TLR expression that is difficult to detect due to the heterogeneous nature of IECs (33). To elucidate the expression profile of TLR2–5 in epithelial cells, small intestinal, and colonic biopsy specimens from patients with inflammatory bowel disease (IBD) were assessed by immunofluorescence histochemistry using polyclonal antibodies against TLR2, 3, 4, and 5. This study showed that TLR3 and TLR5 are ubiquitously expressed while TLR2 and TLR4 are expressed at a very low level in normal cells (37). Conversely, the diseased tissue specimens demonstrated significant overexpression of TLR4 and a decline in TLR3 expression. The expression pattern of TLR2 and TLR5 remained unaltered between the normal and diseased specimens. Furthermore, in normal human IECs, TLR2, and TLR4 were marginally expressed, while TLR3 expression was relatively high. While TLR2 was expressed in the colonic tissue from the epithelium and lamina propria, TLR3 was expressed in the mature epithelial cells of the crypts. Furthermore, TLR5 was moderately overexpressed in a basolateral fashion in the epithelial cells of normal human tissues (38). Tissues from CRC patients demonstrated increased expression of TLR7, 8, 9, and 10 (39); this study also showed that TLR8 expression is an independent marker for CRC.
TLRs and Intestinal Homeostasis
Toll-like receptor activation is responsible for fighting against microbial infections, while leaving the host cell intact. This is usually accomplished by producing antimicrobial peptides, inflammatory mediators, adenomatous polyposis coli (APCs) maturation, and triggering of cell survival and tissue repairing pathways (40). TLRs are marginally expressed on IECs and are primarily localized on the basolateral surface or in the endosomal vesicles (41). Moreover, regulatory mechanisms such as the expression of TLR inhibitors like single immunoglobulin IL-1-related receptor (SIGIRR), toll-interacting protein (TOLLIP), A20, and IRAK3 are involved in the regulation of TLR signaling (42); these inhibitory molecules prevent TLRs from mounting an immune response even during continuous interaction (34, 43) and nurturing the anti-inflammatory phenotype of homing leukocytes (44). SIGIRR-deficient mice demonstrate defective intestinal homeostasis, and these defects are associated with the microbiota and hyper-expression of inflammatory mediators. Notably, these defects also render the azoxymethane (AOM)-dextran sodium sulfate (DSS)-treated SIGIRR−/− mice prone to colitis and colitis-associated CRC. Interestingly, the rescue of SIGIRR expression in the IECs of SIGIRR−/− mice restored the immune tolerance and abolished the risk of tumor development in these mice (45).
Although elevated TLR activity disrupts the recognition of intestinal microbes by TLR2 and TLR4, TLR signaling is necessary for maintaining homeostasis and regulation of tissue repair in IECs. MyD88-deficient mice, which hamper signaling through IL-1 family members including TLRs, possess profound abnormalities in the mucosa with higher proliferation rates in the crypts (46). Cumulatively, this leads to defects in repair of the intestinal barrier following injury, and increased risk of colitis and CRC (46, 47). Moreover, mice in which normal flora is disrupted by antibiotics display a similar phenotype to mice lacking MyD88, as well as decreased expression of factors [i.e., tumor necrosis factor (TNF), CXC-chemokine ligand 1, IL-6, and heat shock proteins] required for normal intestinal homeostasis (48).
Relationship between Inflammation and CRC
The direct link between intestinal inflammation and CRC prognosis is well-established and is also supported by numerous genetic, pharmacological, and epidemiological studies conducted during the last decade (49). Recent reports demonstrate the complex interplay between distinct immune cells, and also show that pro-inflammatory mediators influence almost all the steps of CRC progression. However, the mechanisms by which inflammation stimulates the development of cancer remain elusive and are expected to vary from colitis-associated CRC to other forms of CRC (25, 50). The relationship between inflammatory responses caused by multiple factors such as the microbiota, IBD, and CRC has been demonstrated by comparative experiments conducted in wild type and Il10−/− mice. When treated with AOM, Il10-/- mice were found to show an increased risk of colon tumor development, spontaneous colitis, and CRC, while AOM-WT mice were devoid of colitis and rarely progressed to adenomas. In addition, mice with Bacteroides vulgatus or dual knockout mice (Il10- and MyD88-deficient mice) treated with AOM showed reduced transcription of Il12p40 and TNF-α and remained tumor-free (51).
TLR-induced inflammation is a well-established phenomenon and is perpetuated by several cytokines, ILs, and TNF-α, all of which are known to substantially regulate immune cells and inflammatory responses against cancer (48, 52). Among these, TNF-α is of particular importance and is now recognized as a pro- as well as anti-tumorigenic protein (53). The activation of the TLR4 signaling pathway induces TNF-α and NF-κB, leading to the promotion of CRC (17, 54–56); TNF-α knockout mice treated with AOM/DSS show significantly less tumor formation, representing the pro-tumorigenic role of TNF-α (57). Immunohistochemistry analyses of mononuclear cells in the lamina propria and colons of patients with advanced stage CRC demonstrate the expression of TNF-α (57). TNF-α also promotes the activation of NF-κB, which reinforces inflammation by inducing cyclooxygenase-2 (COX-2), IL-6, IL-8, and TNF-α to favor tumorigenesis (55, 58, 59). However, inflammation alone is not sufficient for colon cancer and the contribution of other risk factors is equally essential to the pathogenesis of this complex disease.
Contribution of TLR4 to CRC Development
Although IECs are in close proximity to LPS, they do not mount an immune response on the commensal bacteria under normal circumstances. However, in the diseased state, disruption of the coexistence between IECs, and bacteria leads to an inflammatory response. This raises an important question: when and how much inflammation should have to be raised in order to equilibrate the bacterial threat (Figure 2). Numerous studies have been conducted to address this dilemma (60–64). For instance, IFN-α and IFN-γ are known to increase the LPS response in IECs, which is directly linked to the expression of TLR4 and MD2 (63, 65). Moreover, continuous LPS stimulation culminates in reduced TLR4 expression and increased expression of inhibitory proteins (62). However, a conflicting report demonstrated that long-term LPS exposure does not alter TLR4 expression (66). Moreover, hypoxia and numerous endotoxins are known to be prevalent in the inflamed intestinal lining, possibly causing induced TLR4 expression (60). Hung et al. observed an increase in the TLR4 expression from the mucosa of CRC patients of different ages and sexes as well as from a variety of CRC cell lines (HT29, SW480, and KM20) (67). In addition, Maria and colleagues showed that TLR4 expression is required for dysplasia and polyp formation. This finding is consistent with results of experiments performed in TLR4 gene knockout mice (56, 68). Collectively, these data present a clear association between TLR4 and CRC development.
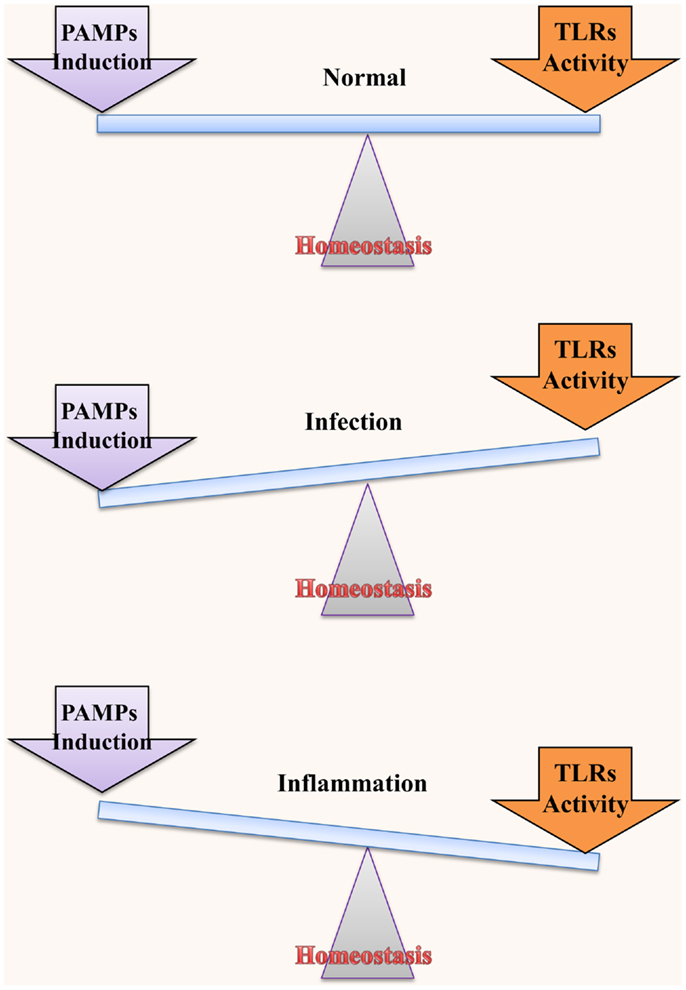
Figure 2. Homeostatic interaction between microbiota and TLRs. TLRs play an important role in maintaining normal functions of IECs; however, regulation of the activation and induction of TLRs through various mechanisms is necessary for this role. TLRs in the intestine exist in close proximity to and may be stimulated by commensal bacteria. Therefore, it is extremely necessary to regulate their functions. Under normal conditions, homeostasis between bacterial induction and TLR activation is maintained to ensure a disease-free status. On the other hand, if TLRs are inappropriately activated or if they mount an exaggerated immune response to a low level stimulus, they may culminate in bacterial infection and inflammatory disease/cancer, respectively.
In CRC, elevated TLR4 expression is observed in all tumor components such as the epithelial, endothelial, and stromal layers (69). However, the level of this expression varies depending on the type of cancer. Although all TLRs are expressed at the minimal basal mRNA level, IECs can upregulate the TLR expression, based on the inflammatory signals or other stimuli (70). An alternate study demonstrated a low level of TLR4/MD2 expression in normal human colonic epithelial cells and the lamina propria, which is consistent with the level of TLR4/MD2 expression detected in various epithelial cell lines (71). These studies establish the fact that any alteration in the prevalent inflammatory conditions or the population of luminal bacteria may influence the strength and nature of TLR signaling, paving the way for initiation of inflammatory responses in IECs. Besides this negative role, studies in TLR4- and TLR9-deficient mice have shown that TLR signaling in IECs is essential for protecting the host from inflammation-related damage and for homeostasis (46, 72).
TLR4 Crosstalk in CRC Progression
TLR4 is overexpressed in the liver metastasis of CRC (73). In response to LPS binding, over-stimulation of the TLR4/MD2 complex enhances the phosphorylation of protein kinase B (also known as AKT), which in turn activates the function of β1 integrin. This complex interplay between multiple pathways promotes the adhesiveness and metastatic behavior of CRC (74). The enhanced AKT phosphorylation can be blocked by eritoran (a TLR4 antagonist), PI 103 [a phosphatidylinositide 3-kinases (PI3K) inhibitor], or anti-β1 integrin antibodies that are known to ameliorate CRC and its metastatic behavior (75–77), indicating that the PI3K/AKT signaling pathway is induced by TLR4 in response to LPS binding and plays a central role in the growth and progression of CRC. Furthermore, LPS is known to induce the expression of the urokinase plasminogen activator (uPA) system through TLR4 and NF-κB in human colorectal cell lines. During tumor progression, vital extracellular matrix (ECM) interactions occur, in which uPA and the expression and activity of its receptor facilitate the growth and metastasis of CRC (78). Conversely, inhibition of TLR4, NF-κB, or the uPA system can attenuate CRC progression. Although NF-κB is known to impair apoptosis in tumor cells (55, 79, 80), NF-κB activation through TNFR signaling also protects cells from apoptosis. Studies performed in the Saos-2 cell line reveal that p53-induced cell death is dependent on NF-κB, and the ablation of NF-κB leads to the abrogation of p53-dependent cell death (81). Thus, the TNF-α/NF-κB interaction plays a vital role in CRC and IBD-related diseases and manipulation of this interaction may improve the treatment of CRC.
TLR4 is overexpressed during inflammation-associated colorectal neoplasia in humans and mice. Similarly, mice lacking TLR4 are largely protected from colon carcinogenesis (56). A dissection of this mechanism reveals that TLR4 triggers elevated production of prostaglandin E2, increases Cox-2 induction, and influences epidermal growth factor receptor signaling (EGFR) in chronic colitis. TLR4 can thus manipulate numerous pathways and cause further deterioration of the neoplastic situation. A recent comparative immunohistochemistry analysis between normal mucosa and adenomas showed that TLR4 and MD2 are overexpressed in 20 and 23% of the adenomas, respectively (82), further substantiating the involvement of the TLR4 pathway in CRC. Furthermore, mutations in the APC gene cause pre-disposition to CRC. A correlation between the TLR/MyD88 signaling pathway and APC mutations was recently proposed (82, 83) since MyD88 signaling was found to facilitate the growth of intestinal polyps while the ablation of MyD88 restricted polyp growth in Apcmin/+/Myd88−/− mice, but not in Apcmin/+ mice (83, 84). In addition, MyD88 induces ERK to block the degradation of the oncoprotein c-Myc, and such cells with continued activation of c-Myc are prone to neoplastic transformation (85). Similarly, c-Myc is also important for APC-mediated tumorigenesis (86), since knocking out c-Myc in IECs of Apcmin/+ mice impedes tumor growth (84). Furthermore, reduced expression of c-Myc has been reported in Apcmin/+/Myd88-/- of both normal and tumor mice (84, 87). Treatment of Apcmin/+ mice with PD03259012, an inhibitor of MEK1/2, which is the kinase directly upstream of ERK, also inhibits tumor growth. These data indicate that a complex interplay of protein signaling brings about tumor proliferation in the IECs of various transgenic mouse models. Moreover, heritable changes in the APC gene frequently lead to familial adenomatous polyposis (FAP). FAP is the most dominant inherited syndrome of CRC (88, 89) and Apcmin/+ mice show increased propensity for the development of adenomatous polyps after the loss of the wild type APC allele (88). Up to 80% of sporadic CRCs are known to be initiated by DNA damage of the genes involved in the APC signaling pathway (87).
Correlation between CRC Development and Inherited Genetic Variations of TLR4
The human TLR4 gene is located on the long (q) arm of chromosome 9 at position 33.1, and contain four exons. The dominant expression of TLR4 has been observed in lymphocytes, monocytes, leukocytes, and splenocytes (90). Besides CRC, many human pathologies and carcinomas are associated with the polymorphisms of TLR4 (91–93). The TLR4 gene contains two single-nucleotide polymorphisms (SNPs), namely, Asp299Gly and Thr399Ile that are significantly important in tumor development (94, 95). Both these SNPs are located in the coding sequence for the TLR4 ectodomain and mediate an amino acid substitution. These Asp299Gly and Thr399Ile SNPs in TLR4 are known to attenuate cytokine expression, leading to an increased propensity for the development of gastric cancer and CRC (94, 96–99). The detection of these two SNPs was carried out using allele-specific polymerase chain reaction and the primer extension method (SNaPshot) for gastric cancer and CRC, respectively. For gastric cancer, only Thr399Ile showed a significant correlation, while both the SNPs were significantly correlated to CRC (94, 100, 101). In addition, the association of the TLR3 (rs3775291) polymorphism and IL-10 promoter variation (rs1800872) to CRC pathogenesis was evaluated in a large cohort of German CRC patients. This study found that the IL-10 promoter variant is significantly associated with an increased risk of lymph node metastasis (for carriers of the TT genotype). Interestingly, a TLR3 gene polymorphism was found to correlate with patient survival, and the TT genotype was responsible for increased mortality. This TLR3 variation was limited only to stage II patients who were devoid of adjuvant therapy (102, 103).
The LPS-sensing complex is comprises TLR4, MD2, LPS binding protein, and CD14. A positive link between CD14–260 polymorphisms and the occurrence of CRC in the Chinese Han population was demonstrated (104, 105), in which the CD14 polymorphism C/C, but not C/T, was significantly correlated to CRC; no correlation between TLR4 Asp299Gly and CRC was found. However, it is possible that the polymorphism in TLR4 was associated with the population under study (106). A multi-racial study (22 Malays, 20 Chinese, and 18 Indians) conducted in Malaysia showed that there is no correlation between TLR4 polymorphisms (Asp299Gly; Thr399Ile) and the risk of CRC (107). However, a study on Russian population revealed that IL1B_1473G/C and TLR4_896A/G SNPs are involved in rectal cancer development (108). A conflicting report validated the potential link between TLR4 polymorphisms (Asp299Gly and Thr399Ile) and the digestive tract cancer and CRC (101). This study retrieved and analyzed extensive data from various databases and concluded that Asp299Gly is significantly correlated with an increased risk of gastric cancer, while there was no correlation between this polymorphism and digestive tract cancer and CRC. Moreover, it was also observed that the T allele of Thr399Ile does not influence digestive tract, gastric, or CRC. It is evident that additional studies are necessary to support these findings.
Epigenetic Regulation of TLR4 in CRC
Intestinal epithelial cells are stimulated by the commensal bacteria in the intestinal lumen with the help of TLRs for the maintenance of homeostasis. This stimulation from the commensal bacteria is finite, should not trigger an excessive inflammatory response, and is known to influence epigenetic modification in the host cells (109). These epigenetic modifications involve DNA methylation and histone deacetylation that suppress and promote the transcription process, respectively, and in turn regulate gene expression (110).
The TLR4 gene is methylated in the 5′ region; also, the degree of methylation in epithelial cells is higher than that in the splenic cells, caused by the interaction of the commensal bacterial with IECs. Takahashi and colleagues showed that commensal bacteria modulate the epigenetic regulation in IECs by DNA methylation of TLR4 (111). In their study, the authors compared the methylation levels in the IECs from the small and large intestine obtained from conventional (CV) mice with commensal bacteria and germ-free (GF) mice without commensal bacteria. The methylation level of CpG motifs in the 5′ region of TLR4 from the large intestine was lower in the GF mice compared with CV mice, while in the small intestine, the methylation levels remained unchanged between the GF and CV mice. The frequency of methylation is also found to depend on the MyD88 adaptor molecule. Results from in vivo experiments show that the frequency of CpG methylation is less in the GF mice (MyD88 knockout mice) compared to CV mice (111).
Environmental factors also play a crucial role in regulating epigenetic modifications. In the presence of factors such as myriad food habits and increasing pollution, intestinal commensal bacteria produce short-chain fatty acids known as butyrates that inhibit histone deacetylation (112, 113). Besides TLR4, MD2 can also be downregulated to attenuate the LPS response. IECs are known to poorly express MD2, which directly correlates to DNA hypermethylation (114). In IBD, IECs exhibit elevated expression of MD2 and TLR4 mRNA, while in normal cells; TLR4/MD2 transcription is reduced due to DNA methylation. The deacetylation and blocking of methylation enables cells to express higher amounts of TLR4 and MD2 mRNA. This study demonstrates how epigenetic regulation of TLR4 and MD2 prevents dysregulation of inflammation in IECs and thus provides a novel approach to target CRC.
Therapeutic Targeting of TLR4
Synthetic TLR4 ligands are potential targets for therapeutic applications for cancer, allergies, and viral infections (115). By virtue of their cell surface location, quick induction, and the ability to mount a wide array of inflammatory responses, TLRs are one of the most promising targets for therapeutics (91). The clinical trials of various TLR4 ligands are enlisted in Table 1.
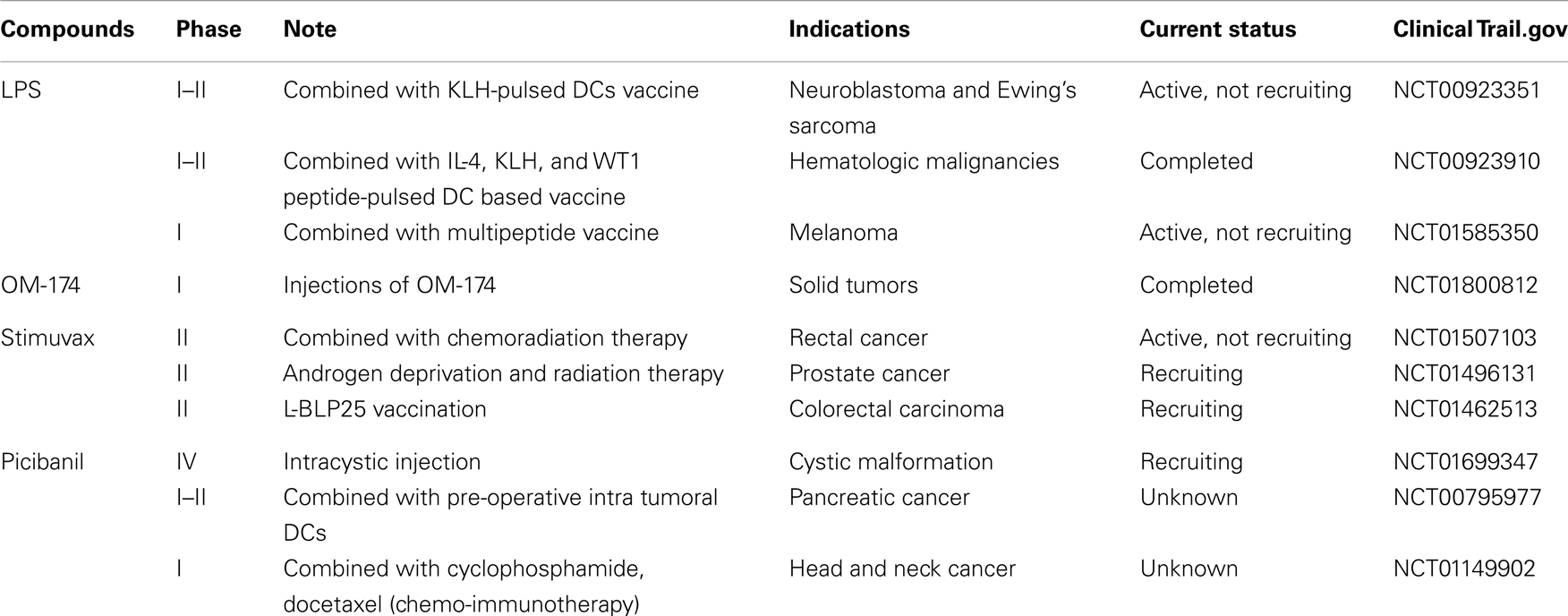
Table 1. TLR4 agonists in clinical trials.
TLR4 agonists have immune regulatory applications as adjuvants in vaccines and in the treatment of chronic viral infection and cancer therapy. LPS was the first microbial product identified as a potential TLR4 agonist and implemented for therapeutic applications (116). LPS is very toxic since it induces excessive inflammatory cytokines. However, low-dose LPS combined with non-steroidal anti-inflammatory drug ibuprofene was proved to be safe, with higher levels of TNF-α and IL-1 in all patients (117, 118). Marginal to encouraging results were observed when ibuprofene combined with Salmonella abortus equi LPS for non-small cell lung carcinoma (NSCLC) and CRC patients, respectively (119). Currently, a few clinical trials are being conducted for oncological indications involving cell-based vaccination to treat Ewing sarcoma, neuroblastoma, and rhabdomyosarcoma patients (NCT00923351). Besides, peptide-pulsed dendritic cells (DCs) were combined with LPS to treat hematological malignancies (NCT00923910), and to treat melanoma patients (NCT01585350), LPS along with oil-based adjuvant and a peptide vaccine are being investigated (119). A less toxic TLR4 agonist, monophosphoryl lipid A (MPLA), is an immunity modulating agent that activates MyD88-independent pathway in TLR4 signaling, triggers the induction of IFN-γ, and regulation of CD80/86, which forms the crucial aspect of adjuvancy (27, 120). MPLA adjuvant plays a dual role in defending the host from pathogens by stimulating the innate immune system, and induces the long-term adaptive immune system (115, 121, 122). Food and Drug Administration has approved MPLA to use as a vaccine against HPV associated cervical cancer (Cervarix). It also enhances the inflammatory behavior of immune cells, which may be useful in a variety of cancers to overcome the cancer-induced immune suppression. However, this may not be helpful in case of CRC, where TLR should emphasize the tolerance of immune system, not the over-activation. Furthermore, it is established that TLRs can act as double-edge sword that may be exploited in pathologies-dependent circumstances to avoid the undesirable consequences (123). OM-174 is a triacyl lipid A analog that activates TLR4 and culminates in tumor growth regression by increasing the IFN-γ production (124, 125) This is well-tolerated at biological concentrations with strong antitumor effects (NCT01800812) (126). A new anti-cancer vaccine, BLP25 liposome vaccine (Stimuvax), can identify and destroy the cancer antigen MUC1, thereby inducing an immune response against cancer cells (127, 128). However, this could not significantly improve the NSCLC (128). Now, stimuvax is being investigated for the treatment of rectal and prostate cancers (NCT01507103 and NCT01496131). Group A Streptococcus pyogenes [in lyophilized form OK-432 (Picibanil)] is shown to stimulate TLR4, which is used to treat gastric, cervical, and oral cancers (119). This compound is currently being examined to treat pancreatic cancer patients in pre-operative settings using intra tumoral injection of DCs (NCT00795977), combined with chemotherapy (cyclophosphamide + docetaxel) for head and neck squamous cell carcinoma (HNSCC) patients (NCT01149902), and via intracystic injection at cystic malformation (NCT01699347). Currently, most of the TLR4 antagonists are being evaluated against cancer-unrelated symptoms.
Conclusion and Future Perspectives
In this review, we highlight the correlation between CRC and TLRs, in particular, TLR4. We also propose that a beneficial link exists between commensal bacteria and TLRs in order to maintain intestinal homeostasis. In IECs, TLRs are involved in epithelial cell proliferation, IgA production, regulating the permeability of the intestinal barrier, antimicrobial peptide expression, and defense against invading pathogens. Over-stimulation of TLRs in response to minor signals (due to dysregulation) may result in colitis and CRC. Several studies suggested the relationship of TLR4 signaling with CRC, therefore therapeutic benefit can be achieved by targeting TLR4. However, the development of CRC is highly complex. Experimental studies supported that the gut microbiota contributed to CRC. The studies involving human subjects and considering their microbiota composition revealed the vivid differences in microbial density and population. Therefore, modulating the microbial population, usage of probiotics to favor the growth of certain bacteria, and delineating the interaction of microbiota with the epithelial cells can potentially be used to limit the CRC development.
Furthermore, inflammation is central to the development of cancer, and there are few clinical trials being conducted for anti-inflammatory drugs, but by combining molecular approaches with CV therapies, i.e., chemo- or radiotherapy, anti-inflammatory drugs would increase the efficacy to treat CRC. Additionally, targeting the downstream molecules in TLR4 pathway involved in CRC is also expected to have a tremendous impact on CRC therapeutics. Moreover, differential expression of TLRs leads to tumor development, in which the contribution of TLR4 is considerably higher than in the other TLRs. We hope that extensive studies involving the TLR4 pathway will eventually provide therapeutic targets to treat CRC. Recently developed techniques may also prove helpful in the analyses of differential expression levels of TLRs, their mutations, and epigenetic modifications. These analyses would further aid in the design and development of novel therapeutic approaches for CRC treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Mid-Career Researcher Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (2012R1A2A2A02016803), and partly supported by a grant from the Priority Research Centers Program (NRF 2012-0006687).
References
1. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4(7):499–511. doi: 10.1038/nri1391
2. Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science (2012) 337(6098):1111–5. doi:10.1126/science.1220363
3. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol (2010) 11(5):373–84. doi:10.1038/ni.1863
4. Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, et al. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J Immunol (2000) 164(2):554–7. doi:10.4049/jimmunol.164.2.554
5. Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol (2002) 169(1):10–4. doi:10.4049/jimmunol.169.1.10
6. Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, et al. Discrimination of bacterial lipoproteins by toll-like receptor 6. Int Immunol (2001) 13(7):933–40. doi:10.1093/intimm/13.7.933
7. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature (2001) 413(6857):732–8. doi:10.1038/35099560
8. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282(5396):2085–8. doi:10.1126/science.282.5396.2085
9. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by toll-like receptor 5. Nature (2001) 410(6832):1099–103. doi:10.1038/35074106
10. Diebold SS, Kaisho T, Hemmi H, Akira S, Reis E, Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (2004) 303(5663):1529–31. doi:10.1126/science.1093616
11. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (2004) 303(5663):1526–9. doi:10.1126/science.1093620
12. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature (2000) 408(6813):740–5. doi:10.1038/35047123
13. Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity (2004) 21(1):107–19. doi:10.1016/j.immuni.2004.06.007
14. Gosu V, Basith S, Kwon OP, Choi S. Therapeutic applications of nucleic acids and their analogues in toll-like receptor signaling. Molecules (2012) 17(11):13503–29. doi:10.3390/molecules171113503
15. Lavelle EC, Murphy C, O’Neill LA, Creagh EM. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol (2010) 3(1):17–28. doi:10.1038/mi.2009.124
16. Rakoff-Nahoum S, Medzhitov R. Role of toll-like receptors in tissue repair and tumorigenesis. Biochemistry (Mosc) (2008) 73(5):555–61. doi:10.1134/S0006297908050088
17. So EY, Ouchi T. The application of toll like receptors for cancer therapy. Int J Biol Sci (2010) 6(7):675–81. doi:10.7150/ijbs.6.675
18. Keogh B, Parker AE. Toll-like receptors as targets for immune disorders. Trends Pharmacol Sci (2011) 32(7):435–42. doi:10.1016/j.tips.2011.03.008
19. Shi Z, Cai Z, Sanchez A, Zhang T, Wen S, Wang J, et al. A novel toll-like receptor that recognizes vesicular stomatitis virus. J Biol Chem (2011) 286(6):4517–24. doi:10.1074/jbc.M110.159590
20. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature (2006) 441(7092):431–6. doi:10.1038/nature04870
21. Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol (2005) 5(10):749–59. doi:10.1038/nri1703
22. Frolova L, Drastich P, Rossmann P, Klimesova K, Tlaskalova-Hogenova H. Expression of toll-like receptor 2 (TLR2), TLR4, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. J Histochem Cytochem (2008) 56(3):267–74. doi:10.1369/jhc.7A7303.2007
23. Smith RA, Brooks D, Cokkinides V, Saslow D, Brawley OW. Cancer screening in the United States, 2013. CA Cancer J Clin (2013) 63(2):87–105. doi:10.3322/caac.21174
24. Tenesa A, Dunlop MG. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat Rev Genet (2009) 10(6):353–8. doi:10.1038/nrg2574
25. Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology (2010) 138(6): 2101–14.e5. doi:10.1053/j.gastro.2010.01.058
26. Adelstein BA, Macaskill P, Chan SF, Katelaris PH, Irwig L. Most bowel cancer symptoms do not indicate colorectal cancer and polyps: a systematic review. BMC Gastroenterol (2011) 11:65. doi:10.1186/1471-230X-11-65
27. Evans JT, Cluff CW, Johnson DA, Lacy MJ, Persing DH, Baldridge JR. Enhancement of antigen-specific immunity via the TLR4 ligands MPL adjuvant and Ribi.529. Expert Rev Vaccines (2003) 2(2):219–29. doi:10.1586/14760584.2.2.219
28. Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity (2010) 32(3):305–15. doi:10.1016/j.immuni.2010.03.012
29. Gosu V, Basith S, Durai P, Choi S. Molecular evolution and structural features of IRAK family members. PLoS One (2012) 7(11):e49771. doi:10.1371/journal.pone.0049771
30. Chen ZJ. Ubiquitin signalling in the NF-κB pathway. Nat Cell Biol (2005) 7(8):758–65. doi:10.1038/ncb0805-758
31. Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol (2005) 6(11):1087–95. doi:10.1038/ni1255
32. Takeda K, Akira S. TLR signaling pathways. Semin Immunol (2004) 16(1):3–9. doi:10.1016/j.smim.2003.10.003
33. Moossavi S, Rezaei N. Toll-like receptor signalling and their therapeutic targeting in colorectal cancer. Int Immunopharmacol (2013) 16(2):199–209. doi:10.1016/j.intimp.2013.03.017
34. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol (2010) 10(2):131–44. doi:10.1038/nri2707
35. Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, et al. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun (2006) 74(5):2522–36. doi:10.1128/IAI.74.5.2522-2536.2006
36. Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J Immunol (2013) 179:566–77. doi:10.4049/jimmunol.179.1.566.
37. Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun (2000) 68(12):7010–7. doi:10.1128/IAI.68.12.7010-7017.2000
38. Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol (2001) 167(4):1882–5. doi:10.4049/jimmunol.167.4.1882
39. Grimm M, Kim M, Rosenwald A, Heemann U, Germer C-T, Waaga-Gasser AM, et al. Toll-like receptor (TLR) 7 and TLR8 expression on CD133 + cells in colorectal cancer points to a specific role for inflammation-induced TLRs in tumourigenesis and tumour progression. Eur J Cancer (2010) 46(15):2849–57. doi:10.1016/j.ejca.2010.07.017
40. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454(7203):428–35. doi:10.1038/nature07201
41. Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol (2005) 5(6):446–58. doi:10.1038/nri1630
42. Anwar MA, Basith S, Choi S. Negative regulatory approaches to the attenuation of toll-like receptor signaling. Exp Mol Med (2013) 45:e11. doi:10.1038/emm.2013.28
43. Lebeer S, Vanderleyden J, De Keersmaecker SC. Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat Rev Microbiol (2010) 8(3):171–84. doi:10.1038/nrmicro2297
44. Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol (2010) 10(3):159–69. doi:10.1038/nri2710
45. Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, et al. The toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity (2007) 26(4):461–75. doi:10.1016/j.immuni.2007.02.012
46. Rakoff NS, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell (2004) 118(2):229–41. doi:10.1016/j.cell.2004.07.002
47. Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med (2010) 207(8):1625–36. doi:10.1084/jem.20100199
48. Clevers H. At the crossroads of inflammation and cancer. Cell (2004) 118(6):671–4. doi:10.1016/j.cell.2004.09.005
49. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer (2013) 13(11):759–71. doi:10.1038/nrc3611
50. Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology (2011) 140(6):1807–16. doi:10.1053/j.gastro.2011.01.057
51. Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One (2009) 4(6):e6026. doi:10.1371/journal.pone.0006026
52. Swann JB, Vesely MD, Silva A, Sharkey J, Akira S, Schreiber RD, et al. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A (2008) 105(2):652–6. doi:10.1073/pnas.0708594105
53. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer (2009) 9(5):361–71. doi:10.1038/nrc2628
54. Govind S. Control of development and immunity by rel transcription factors in Drosophila. Oncogene (1999) 18(49):6875–87. doi:10.1038/sj.onc.1203223
55. Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol (2004) 14(2):64–9. doi:10.1016/j.tcb.2003.12.004
56. Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology (2007) 133(6):1869–81. doi:10.1053/j.gastro.2007.09.008
57. Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest (2008) 118(2):560–70. doi:10.1172/JCI32453
58. Oshima H, Oshima M. The inflammatory network in the gastrointestinal tumor microenvironment: lessons from mouse models. J Gastroenterol (2012) 47(2):97–106. doi:10.1007/s00535-011-0523-6
59. Ogino S, Kirkner GJ, Nosho K, Irahara N, Kure S, Shima K, et al. Cyclooxygenase-2 expression is an independent predictor of poor prognosis in colon cancer. Clin Cancer Res (2008) 14(24):8221–7. doi:10.1158/1078-0432.CCR-08-1841
60. Leaphart CL, Cavallo J, Gribar SC, Cetin S, Li J, Branca MF, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol (2007) 179(7):4808–20. doi:10.4049/jimmunol.179.7.4808
61. Jilling T, Simon D, Lu J, Meng FJ, Li D, Schy R, et al. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol (2006) 177(5):3273–82. doi:10.4049/jimmunol.177.5.3273
62. Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology (2004) 126(4):1054–70. doi:10.1053/j.gastro.2004.01.007
63. Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol (2001) 167(3):1609–16. doi:10.4049/jimmunol.167.3.1609
64. Suzuki M, Hisamatsu T, Podolsky DK. Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular toll-like receptor 4-MD-2 complex. Infect Immun (2003) 71(6):3503–11. doi:10.1128/IAI.71.6.3503-3511.2003
65. Abreu MT, Arnold ET, Thomas LS, Gonsky R, Zhou Y, Hu B, et al. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J Biol Chem (2002) 277(23):20431–7. doi:10.1074/jbc.M110333200
66. Hornef MW, Frisan T, Vandewalle A, Normark S, Richter-Dahlfors A. Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med (2002) 195(5):559–70. doi:10.1084/jem.20011788
67. Doan HQ, Bowen KA, Jackson LA, Evers BM. Toll-like receptor 4 activation increases Akt phosphorylation in colon cancer cells. Anticancer Res (2009) 29(7):2473–8.
68. Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, et al. Cox-2 is regulated by toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology (2006) 131(3):862–77. doi:10.1053/j.gastro.2006.06.017
69. Cammarota R, Bertolini V, Pennesi G, Bucci EO, Gottardi O, Garlanda C, et al. The tumor microenvironment of colorectal cancer: stromal TLR-4 expression as a potential prognostic marker. J Transl Med (2010) 8(1):112. doi:10.1186/1479-5876-8-112
70. Fukata M, Abreu MT. TLR4 signalling in the intestine in health and disease. Biochem Soc Trans (2007) 35(Pt 6):1473–8. doi:10.1042/BST0351473
71. Abreu MT, Thomas LS, Arnold ET, Lukasek K, Michelsen KS, Arditi M. TLR signaling at the intestinal epithelial interface. J Endotoxin Res (2003) 9(5):322–30. doi:10.1179/096805103225002593
72. Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9 – induced type I IFN protects mice from experimental colitis. J Clin Invest (2005) 115(3):695–702. doi:10.1172/JCI200522996C1
73. Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, et al. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J Leukoc Biol (2009) 85(6):966–77. doi:10.1189/jlb.1208763
74. Sheng H, Shao J, Townsend CM Jr, Evers BM. Phosphatidylinositol 3-kinase mediates proliferative signals in intestinal epithelial cells. Gut (2003) 52(10):1472–8. doi:10.1136/gut.52.10.1472
75. Somanath PR, Kandel ES, Hay N, Byzova TV. Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. J Biol Chem (2007) 282(31):22964–76. doi:10.1074/jbc.M700241200
76. Hsu RY, Chan CH, Spicer JD, Rousseau MC, Giannias B, Rousseau S, et al. LPS-induced TLR4 signaling in human colorectal cancer cells increases beta1 integrin-mediated cell adhesion and liver metastasis. Cancer Res (2011) 71(5):1989–98. doi:10.1158/0008-5472.CAN-10-2833
77. Earl TM, Nicoud IB, Pierce JM, Wright JP, Majoras NE, Rubin JE, et al. Silencing of TLR4 decreases liver tumor burden in a murine model of colorectal metastasis and hepatic steatosis. Ann Surg Oncol (2009) 16(4):1043–50. doi:10.1245/s10434-009-0325-8
78. Killeen S, Wang J, Andrews E, Redmond H. Bacterial endotoxin enhances colorectal cancer cell adhesion and invasion through TLR-4 and NF-κB-dependent activation of the urokinase plasminogen activator system. Br J Cancer (2009) 100(10):1589–602. doi:10.1038/sj.bjc.6604942
79. Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest (2001) 107(3):241–6. doi:10.1172/JCI11991
80. Hatta Y, Koeffler HP. Role of tumor suppressor genes in the development of adult T cell leukemia/lymphoma (ATLL). Leukemia (2002) 16(6):1069–85. doi:10.1038/sj.leu.2402458
81. Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kappaB in p53-mediated programmed cell death. Nature (2000) 404(6780):892–7. doi:10.1038/35009130
82. Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, et al. High expression of toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer (2010) 102(5):908–15. doi:10.1038/sj.bjc.6605558
83. Rakoff NS, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science (2007) 317(5834):124–7. doi:10.1126/science.1140488
84. Lee SH, Hu LL, Gonzalez-Navajas J, Seo GS, Shen C, Brick J, et al. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat Med (2010) 16(6):665–70. doi:10.1038/nm.2143
85. Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene (2010) 29(9):1249–59. doi:10.1038/onc.2009.512
86. Wilkins JA, Sansom OJ. C-Myc is a critical mediator of the phenotypes of Apc loss in the intestine. Cancer Res (2008) 68(13):4963–6. doi:10.1158/0008-5472.CAN-07-5558
87. Fukata M, Abreu MT. Microflora in colorectal cancer: a friend to fear. Nat Med (2010) 16(6):639–41. doi:10.1038/nm0610-639
88. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell (1991) 66(3):589–600. doi:10.1016/0092-8674(81)90021-0
89. Goss KH, Groden J. Biology of the adenomatous polyposis coli tumor suppressor. J Clin Oncol (2000) 18(9):1967–79.
90. Opal SM, Esmon CT. Bench-to-bedside review: functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Crit Care (2002) 7(1):23. doi:10.1186/cc1854
91. Hennessy EJ, Parker AE, O’Neill LA. Targeting toll-like receptors: emerging therapeutics? Nat Rev Drug Discov (2010) 9(4):293–307. doi:10.1038/nrd3203
92. Slattery ML, Herrick JS, Bondurant KL, Wolff RK. Toll-like receptor genes and their association with colon and rectal cancer development and prognosis. Int J Cancer (2012) 130(12):2974–80. doi:10.1002/ijc.26314
93. Pimentel-Nunes P, Teixeira AL, Pereira C, Gomes M, Brandao C, Rodrigues C, et al. Functional polymorphisms of toll-like receptors 2 and 4 alter the risk for colorectal carcinoma in Europeans. Dig Liver Dis (2013) 45(1):63–9. doi:10.1016/j.dld.2012.08.006
94. Omrane I, Baroudi O, Kourda N, Bignon YJ, Uhrhammer N, Desrichard A, et al. Positive link between variant toll-like receptor 4 (Asp299Gly and Thr399Ile) and colorectal cancer patients with advanced stage and lymph node metastasis. Tumour Biol (2013) 35(1):545–51. doi:10.1007/s13277-013-1075-6
95. Mockenhaupt FP, Cramer JP, Hamann L, Stegemann MS, Eckert J, Oh NR, et al. Toll-like receptor (TLR) polymorphisms in African children: common TLR-4 variants predispose to severe malaria. Proc Natl Acad Sci U S A (2006) 103(1):177–82. doi:10.1073/pnas.0506803102
96. Garza-Gonzalez E, Bosques-Padilla FJ, Mendoza-Ibarra SI, Flores-Gutierrez JP, Maldonado-Garza HJ, Perez-Perez GI. Assessment of the toll-like receptor 4 Asp299Gly, Thr399Ile and interleukin-8-251 polymorphisms in the risk for the development of distal gastric cancer. BMC Cancer (2007) 7(1):70. doi:10.1186/1471-2407-7-70
97. Ferwerda B, Mccall MB, Verheijen K, Kullberg BJ, Van Der Ven AJ, Van Der Meer JW, et al. Functional consequences of toll-like receptor 4 polymorphisms. Mol Med (2008) 14(5–6):346–52. doi:10.2119/2007-00135.Ferwerda
98. Boraska Jelavic T, Barisic M, Drmic Hofman I, Boraska V, Vrdoljak E, Peruzovic M, et al. Microsatelite GT polymorphism in the toll-like receptor 2 is associated with colorectal cancer. Clin Genet (2006) 70(2):156–60. doi:10.1111/j.1399-0004.2006.00651.x
99. Li XX, Sun GP, Meng J, Li X, Tang YX, Li Z, et al. Role of toll-like receptor 4 in colorectal carcinogenesis: a meta-analysis. PLoS One (2014) 9(4):e93904. doi:10.1371/journal.pone.0093904
100. Santini D, Angeletti S, Ruzzo A, Dicuonzo G, Galluzzo S, Vincenzi B, et al. Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms in gastric cancer of intestinal and diffuse histotypes. Clin Exp Immunol (2008) 154(3):360–4. doi:10.1111/j.1365-2249.2008.03776.x
101. Zhao X, Kang S, Liu L, Zhang D. Correlation of Asp299Gly and Thr399Ile polymorphisms in toll-like receptor 4 gene with digestive cancer risk: a meta-analysis. Biomed Rep (2013) 1(2):294–302. doi:10.3892/br.2012.32
102. Castro FA, Forsti A, Buch S, Kalthoff H, Krauss C, Bauer M, et al. TLR-3 polymorphism is an independent prognostic marker for stage II colorectal cancer. Eur J Cancer (2011) 47(8):1203–10. doi:10.1016/j.ejca.2010.12.011
103. Tsilidis KK, Helzlsouer KJ, Smith MW, Grinberg V, Hoffman-Bolton J, Clipp SL, et al. Association of common polymorphisms in IL10, and in other genes related to inflammatory response and obesity with colorectal cancer. Cancer Causes Control (2009) 20(9):1739–51. doi:10.1007/s10552-009-9427-7
104. Guo Q, Zhu J, Xia B. Polymorphism of CD14 gene but not the mutation of TLR4 gene is associated with colorectal cancer in Chinese patients. J Gastroenterol Hepatol (2006) 21(1 Pt 1):92–7. doi:10.1111/j.1440-1746.2005.04156.x
105. Chen R, Luo FK, Wang YL, Tang JL, Liu YS. LBP and CD14 polymorphisms correlate with increased colorectal carcinoma risk in Han Chinese. World J Gastroenterol (2011) 17(18):2326–31. doi:10.3748/wjg.v17.i18.2326
106. Landi S, Gemignani F, Bottari F, Gioia-Patricola L, Guino E, Cambray M, et al. Polymorphisms within inflammatory genes and colorectal cancer. J Negat Results Biomed (2006) 5:15. doi:10.1186/1477-5751-5-15
107. Davoodi H, Seow HF. Variant toll-like receptor4 (Asp299Gly and Thr399Ile alleles) and toll-like receptor2 (Arg753Gln and Arg677Trp alleles) in colorectal cancer. Iran J Allergy Asthma Immunol (2011) 10(2):91–9. doi:010.02/ijaai.9199
108. Kutikhin AG, Yuzhalin AE, Volkov AN, Zhivotovskiy AS, Brusina EB. Correlation between genetic polymorphisms within IL-1B and TLR4 genes and cancer risk in a Russian population: a case-control study. Tumour Biol (2014) 35(5):4821–30. doi:10.1007/s13277-014-1633-6
109. Takahashi K, Sugi Y, Hosono A, Kaminogawa S. Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. J Immunol (2009) 183(10):6522–9. doi:10.4049/jimmunol.0901271
110. Govind CK, Ginsburg D, Hinnebusch AG. Measuring dynamic changes in histone modifications and nucleosome density during activated transcription in budding yeast. Methods Mol Biol (2012) 833:15–27. doi:10.1007/978-1-61779-477-3_2
111. Takahashi K, Sugi Y, Nakano K, Tsuda M, Kurihara K, Hosono A, et al. Epigenetic control of the host gene by commensal bacteria in large intestinal epithelial cells. J Biol Chem (2011) 286(41):35755–62. doi:10.1074/jbc.M111.271007
112. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr (2003) 133(7 Suppl):2485S–93S.
113. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem (2008) 19(9):587–93. doi:10.1016/j.jnutbio.2007.08.002
114. Vamadevan AS, Fukata M, Arnold ET, Thomas LS, Hsu D, Abreu MT. Regulation of toll-like receptor 4-associated MD-2 in intestinal epithelial cells: a comprehensive analysis. Innate Immun (2010) 16(2):93–103. doi:10.1177/1753425909339231
115. Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with toll-like receptor agonists and antagonists. Nat Med (2007) 13(5):552–9. doi:10.1038/nm1589
116. Akira S. Mammalian toll-like receptors. Curr Opin Immunol (2003) 15(1):5–11. doi:10.1016/S0952-7915(03)00005-0
117. Mackensen A, Galanos C, Engelhardt R. Treatment of cancer patients with endotoxin induces release of endogenous cytokines. Pathobiology (1991) 59(4):264–7. doi:10.1159/000163659
118. Engelhardt R, Mackensen A, Galanos C. Phase I trial of intravenously administered endotoxin (Salmonella abortus equi) in cancer patients. Cancer Res (1991) 51(10):2524–30.
119. Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautes-Fridman C, et al. Trial watch: experimental toll-like receptor agonists for cancer therapy. Oncoimmunology (2012) 1(5):699–716. doi:10.4161/onci.20696
120. Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science (2007) 316(5831):1628–32. doi:10.1126/science.1138963
121. Persing DH, Coler RN, Lacy MJ, Johnson DA, Baldridge JR, Hershberg RM, et al. Taking toll: lipid A mimetics as adjuvants and immunomodulators. Trends Microbiol (2002) 10(10):s32–7. doi:10.1016/S0966-842X(02)02426-5
123. Basith S, Manavalan B, Yoo TH, Kim SG, Choi S. Roles of toll-like receptors in cancer: a double-edged sword for defense and offense. Arch Pharm Res (2012) 35(8):1297–316. doi:10.1007/s12272-012-0802-7
124. D’Agostini C, Pica F, Febbraro G, Grelli S, Chiavaroli C, Garaci E. Antitumour effect of OM-174 and cyclophosphamide on murine B16 melanoma in different experimental conditions. Int Immunopharmacol (2005) 5(7–8):1205–12. doi:10.1016/j.intimp.2005.02.013
125. De Ridder M, Verovski VN, Chiavaroli C, Van Den Berge DL, Monsaert C, Law K, et al. The radiosensitizing effect of immunoadjuvant OM-174 requires cooperation between immune and tumor cells through interferon-gamma and inducible nitric oxide synthase. Int J Radiat Oncol Biol Phys (2006) 66(5):1473–80. doi:10.1016/j.ijrobp.2006.07.1381
126. Isambert N, Fumoleau P, Paul C, Ferrand C, Zanetta S, Bauer J, et al. Phase I study of OM-174, a lipid A analogue, with assessment of immunological response, in patients with refractory solid tumors. BMC Cancer (2013) 13:172. doi:10.1186/1471-2407-13-172
127. Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulieres D, et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol (2005) 23(27):6674–81. doi:10.1200/JCO.2005.13.011
Keywords: colorectal cancer, immune response, inflammation, ligand, toll-like receptor 4
Citation: Yesudhas D, Gosu V, Anwar MA and Choi S (2014) Multiple roles of toll-like receptor 4 in colorectal cancer. Front. Immunol. 5:334. doi: 10.3389/fimmu.2014.00334
Received: 13 April 2014; Paper pending published: 30 April 2014;
Accepted: 01 July 2014; Published online: 15 July 2014.
Edited by:
Anton G. Kutikhin, Russian Academy of Medical Sciences, RussiaReviewed by:
Amedeo Amedei, University of Florence, ItalyRajesh Kumar Sharma, University of Louisville, USA
Arseniy E. Yuzhalin, University of Oxford, UK
Copyright: © 2014 Yesudhas, Gosu, Anwar and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangdun Choi, Department of Molecular Science and Technology, Ajou University, Suwon 443-749, South Korea e-mail:c2FuZ2R1bmNob2lAYWpvdS5hYy5rcg==
†Dhanusha Yesudhas and Vijayakumar Gosu have contributed equally to this work.