 Theofilos Poutahidis
Theofilos Poutahidis Markus Kleinewietfeld
Markus Kleinewietfeld Susan E. Erdman
Susan E. Erdman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 07 April 2014
Sec. Cancer Immunity and Immunotherapy
Volume 5 - 2014 | https://doi.org/10.3389/fimmu.2014.00157
This article is part of the Research Topic Autoimmuno-Anti-Tumour Immunity (AATI) – Understanding the Immune Responses against “Self” & “Altered-self” View all 17 articles
It is recently shown that beneficial environmental microbes stimulate integrated immune and neuroendocrine factors throughout the body, consequently modulating regulatory T-lymphocyte phenotypes, maintaining systemic immune balance, and determining the fate of preneoplastic lesions toward regression while sustaining whole body good health. Stimulated by a gut microbiota-centric systemic homeostasis hypothesis, we set out to explore the influence of the gut microbiome to explain the paradoxical roles of regulatory T-lymphocytes in cancer development and growth. This paradigm shift places cancer prevention and treatment into a new broader context of holobiont engineering to cultivate a tumor-suppressive macroenvironment.
The neoplastic process is characterized by overwhelming complexity. Cancer is comprised of a genetically unstable population of cells that proliferate at an extraordinarily high rate. Millions of cancer deaths each year make it obvious that the battle against cancer is asymmetric, with humankind often being the weaker element (1). To date, cancer research efforts directly confront malignancy by targeting properties of individual cancer cells. In 2000, Hanahan and Weinberg described that most of the research on origins and treatment of cancer had just contributed toward “adding further layers of complexity to a scientific literature that is already complex almost beyond measure” (2).
In the same landmark paper, however, the authors were optimistic enough to predict groundbreaking upcoming advances in the conceptual rather than the technical level (2). They were proven right. One such advancement was the increased awareness for the importance of the tumor microenvironment in the etiopathogenesis of neoplasia (3, 4). We now know that initially transformed cells are much less autonomous in their growth than previously thought (5, 6). Among the microenvironment elements, immune cells and factors have emerged as fundamental players (4–6). Accumulating evidence suggests that tumor-associated inflammatory cell accumulation, whether overt or smoldering, could be viewed as a tumor-promoting event (7–9). These inflammatory responses enhance mutagenesis by oxidative DNA damage and shape the tumor stroma in favor of cancer cell survival and expansion (6, 10, 11).
Will this knowledge base in the field of inflammation, immunity, and cancer lead to new, highly effective, and biologically safe cancer immunotherapy modalities? We assert that the outcome will depend upon the philosophy and the strategic goals that will dominate the bench-to-bedside research. We propose that research in this field should focus upon stimulating systemic innate immune balance and adaptive immune resiliency, making the mammalian host more powerful to resist its cancer challenger. One possible approach utilizes gut microbiota or microbial antigens to stimulate beneficial immune cells. On the other hand, existing immunotherapy aims to selectively interrupt immune factors to better recognize and exterminate cancer cells (12–17), an approach that may ultimately lead to host instability. To further explain this point of view, we will refer to the recently discovered paradoxical roles of regulatory T-cells (TREG) in cancer (10, 14).
FOXP3+ CD4+ CD25+/high TREG are dominant cellular elements of the professional suppressor arm of the immune system and are important for orchestrating the control of peripheral immunological tolerance (18). The transcription factor FOXP3 is a fundamental regulator of TREG function in rodents and humans, and so far the most reliable phenotypic indicator of their identity. Recent studies on human TREG subpopulations, however, revealed that low but discernible levels of FOXP3 expression could be detected in non-suppressive TREG or even in activated effector T-cells. It is probable that this finding reflects the inherent plasticity of TREG; FOXP3+ cells co-expressing effector T-cell phenotypic markers or cytokines may be in stages of a progressive, epigenetically regulated, phenotypical, and functional shift process (14, 16, 19–22), ultimately favorable for healthful recovery of the host after environmental challenges. The role of TREG is central in preserving immune system homeostasis for health and the balance of beneficial inflammatory responses during infections while minimizing collateral tissue damage. In cancer, however, roles of TREG are traditionally considered to be negative (14–16, 23).
A large body of data suggest that TREG gather near tumors and suppress the anti-tumor inflammatory response, thus favoring cancer cell survival. To this end, tumor-associated TREG are thought as a major impediment of anti-tumor vaccines (13–16, 23). Clinical and experimental data suggest that tumor-associated TREG recognize both self and neoantigens expressed by tumor cells, counteracting antigen-specific effector T-cell responses. Consequently, immunotherapy strategies based on the vaccination with tumor-associated antigens fail to evoke an effective response against cancer cells due to the activation and expansion of tumor antigen-specific TREG (14–16). This potential interplay of TREG within tumors has been reviewed in detail elsewhere (12–16), and has led to the proposal of several anti-TREG regimens for cancer immunotherapy. These regimens aim to deplete TREG, inhibit their suppressive function, prevent their homing into tumor sites, or block their differentiation/proliferation (12–16).
Several of these TREG-targeting modalities have already been tested in the clinic, with mixed results (13, 16). Blocking TREG function by depleting the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) appears promising (24), due to the depletion of TREG from tumor tissues (25, 26). However, a similar regimen could lead to an opposite effect with the accumulation of TREG and CD8+ T-cells in tumors (27, 28). A phase III study of melanoma patients using a gp100 peptide vaccine with interleukin (IL)-2 administration led to equally promising results with discovery of TREG expansion in responding patients (29).
As the results of these trials are anticipated, the literature reveals contradictory evidence. Indeed, the studies associating high densities of tumor-associated cells expressing TREG markers including FOXP3 with a poor prognosis in several types of human cancers are now challenged by similar studies on the very same types of cancer showing the opposite outcome (30–34). The different CD8+:TREG ratios and the presence of FOXP3+ cell subsets of undetermined identity in the tumor microenvironment have been proposed as probable explanations (16). Indeed, data from animal models show under certain conditions of microbial priming that TREG not only protect but also alter the tumor microenvironment to induce remission of already established intestinal, mammary, and prostate cancers (35–41). The hypothesis that the composition of the different subsets of FOXP3+, which may include effector Foxp3+ cells, is intriguing (16). Indeed, it was previously shown that IFN-γ levels were increased during TREG-mediated tumor regression in mice (37). Further, feeding of probiotic microbes to mice induces systemic oxytocin secretion that shifts immunity toward IFN-γ and CD25 for improved wound healing capacity and systemic good health (42). A question subsequently arising is whether gut microbiota may be engineered to harness an anti-neoplastic FOXP3+ cell milieu (5, 10, 41).
Stimulated by a gut-centric systemic homeostasis hypothesis, we set out to explore and explain the paradoxical roles of TREG in cancer using several different mouse models of cancer and adoptive cell transfer methodologies (10). We found that TREG may suppress, promote, or have no effect in carcinogenesis depending upon their timing and prior exposure to gut bacterial antigens and presence of IL-10 (35–39, 41, 43, 44). Under some conditions, adoptive transfer of TREG rapidly led to apoptosis of emerging tumor cells (37, 45). Using as a model organism an opportunistic pathogen, Helicobacter hepaticus, commonly residing in the lower bowel of mice, we have shown in Rag2-deficient mice (otherwise lacking lymphocytes) that gut microbiota modulate inflammatory bowel disease and inflammation-associated colon cancer, a cancer process inhibited by properly functioning IL-10-dependent TREG (35, 36). Subsequently, by introducing H. hepaticus into the large bowel flora of mice lacking the APC tumor suppressor gene (ApcMin/+), we found that intestinal polypogenesis was greatly enhanced by bacteria and subsequently suppressed by immune-competent TREG. Furthermore, adenomas of infected ApcMin/+ mice progressed into adenocarcinoma, a transition atypical of polyps of aged-matched uninfected controls (38, 41). Interestingly, ApcMin/+ mice having H. hepaticus in their gut flora were prone to develop cancer in tissues distant from intestine, such as prostate and the mammary glands (40, 41, 43, 46, 47). H. hepaticus-induced tumorigenic events were inhibited by supplementation with TREG from immune-competent wild type donor mice.
A potent treatment to counteract these local and systemic H. hepaticus-induced tumorigenic events was supplementation with TREG in an IL-10-dependent manner (10, 36, 38–40, 44, 46, 48). Purified TREG exhibited greatest anti-cancer potency when taken from donor mice previously colonized with H. hepaticus. By contrast, TREG taken from donor mice without prior H. hepaticus exposure were ineffective, and in some cases actually enhanced tumorigenesis (10). Based on these results, we theorize that the tumor microenvironment is subject to systemic inflammatory events arising from environmental exposures in the gastrointestinal tract (Figure 1). This microbe-inducible pro-inflammatory condition contributes to tumor trophic signaling. Interestingly, bacterial antigen triggered IL-10-dependent activities in the GI-tract impart sustained protection from the aforementioned events, resulting in immune cell recruitment, including TREG, which, by being more potent in their anti-inflammatory roles, work locally and systemically to suppress sepsis, myeloid precursor mobilization, and inflammatory signaling important in extra-intestinal cancer evolution (10, 43). These systemic events comprise the tumor macroenvironment.
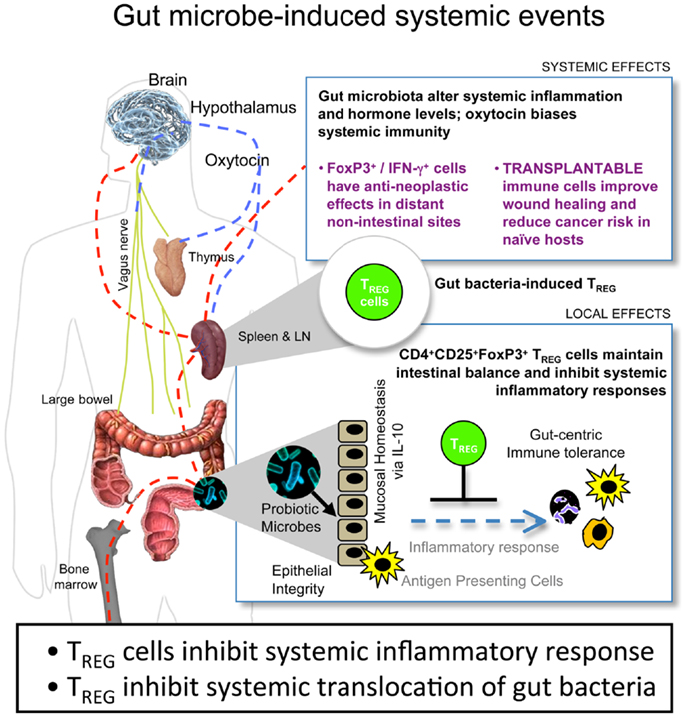
Figure 1. Gut bacteria–host crosstalk is continuous and reciprocal in the cancer macroenvironment. Beneficial microbes trigger IL-10-mediated GI-tract immune and neuronal networks that lower systemic inflammatory tone and up-regulate hypothalamic–hypophyseal targets, including oxytocin, constituting a gut–systemic immunity-endocrine-axis. In this way, microbiota stimulate CD4+ lymphocytes including regulatory T cells (TREG) that suppress, promote, or have no effect in carcinogenesis depending upon their timing and prior exposure to gut bacterial antigens and presence of interleukin (IL)-10. This places neoplastic development and growth into a new broader context of the holobiont (comprised of the mammalian host plus resident microbes) and the cancer macroenvironment, highlighting microbes that may be engineered for sustained good health.
The roles of intestinal microflora in promoting cancer development within the bowel have been well established (35, 49–52). Linking gut microbial flora and local and systemic effects that promote (38) or suppress (45) tumors throughout the body, expands this paradigm in a challenging manner. Recent findings show that gut flora imbalances considerably undermine the response to both immune (53, 54) and non-immune chemotherapeutic regimens, such as cisplatin and oxaliplatin (53).
These gut microbe-centric findings in mice are consistent with the “hygiene hypothesis,” according to which insufficient microbial exposures earlier in life predispose to allergies, autoimmune disorders, and uncontrollable inflammation-associated pathologies later in life. We have shown that the basic principles of this hypothesis may apply not only to auto-immunity, but also to neoplastic disease as well, and that TREG play a central role in this phenomenon (10, 41, 55). The ability of TREG to decrease risk for cancer and counteract established tumors depends upon microbe-triggered IL-10, which works to maintain immune system homeostasis and reinforce a protective anti-inflammatory, anti-neoplastic TREG phenotype (41). TREG display inherent phenotypic plasticity (10). Hygienic individuals with a weakened IL-10 and TREG feedback loop are prone to a re-direction of unstable resting peripheral TREG toward a T helper (Th)-17 pro-inflammatory process. As a result “hygienic” subjects are at higher risk to develop auto-immune diseases and cancer (10). It is tempting to postulate that this may explain why only a few people go on to develop cancer, while nearly everyone bears dysplastic and early neoplastic lesions throughout their body (56).
Depending on composition of gut microbiota, the immune system of mice may acquire different subclinical characteristics, even in the absence of overt inflammatory processes. The clinically silent immune system status may determine the risk of developing sporadic cancer in epithelia throughout the body. Further, we found that consuming beneficial probiotic bacteria led to the expansion of a Foxp3+ cell population in the periphery (42, 45, 57) conferring protection to diet-related and genetic predisposition to mammary cancer (45). Targeted oral challenge with such probiotic bacteria resulted in the activation of interrelated systemic inflammatory and metabolic pathways, either through blood circulation or via the vagus nerve (Figure 1). Consequently, there was an upregulation of systemic hormone levels, such as oxytocin, testosterone, and thyroxin. Oxytocin serves to sustain immune and integumentary homeostasis, biasing the immune system toward IL-10 and IFN-γ, without anergy, subsequently minimizing the deleterious systemic effects of IL-17 (57). This altered immune system and metabolic profile of mice imparted healthful phenotypes including shiny fur and youthful hair follicle cycling, accelerated skin wound healing capacity, and resistance to diet-induced obesity and senility (42, 47, 57, 58). Through tightly regulated immune activities, competent TREG permit brief beneficial host inflammatory responses to eliminate invading pathogens, and later inhibit chronic deleterious inflammatory tissue damage (43). The results of our wound healing assays further suggest that the probiotic microbe-induced enhancement of the TREG-dominated arm of the immune system did not compromise the ability of mice to respond to invading pathogens (42).
Adoptive cell transfer models offer mechanistic insight as these beneficial effects were isolated to bacteria-primed TREG (42, 47, 57–59). In fact, healthful phenotypes were entirely reproducible in naive recipient mice by the adoptive transfer of highly purified TREG derived from probiotic-fed cell donors (42, 57, 59). These results suggest gut microbe-induced crosstalk with the host in a continuous and reciprocal manner. The fate of preneoplastic and neoplastic lesions arising in epithelia throughout the body depends upon this macroenvironment at the whole organism level. Consequently, the tumor macroenvironment is defined as the “holobiont,” i.e., the mammalian organism plus the microbial symbionts it bears. The TREG population is a central player of the tumor macroenvironment connecting gut bacteria with reproductive fitness, youthful phenotypes, and anti-neoplastic properties.
Taken together, microbial engineering strategies using food-grade bacteria highlight alternative directions in cancer immunotherapy. Modulating beneficial TREG via diet is a biologically safe and efficient approach, originating from genetic programs that have been shaped during the millions of years of co-evolution of mammals with their gut bacteria symbionts. These attributes remain largely inactive in individuals with a modern lifestyle, Westernized dietary habits, and stringent hygiene practices. Awakening these latent TREG-mediated capabilities may provide an alternative avenue to reduce cancer risk at a population level for public health. The perspectives presented here should be considered as an alternative paradigm – not only for fighting cancer – but also for promoting overall good health and longevity.
Theofilos Poutahidis, Markus Kleinewietfeld, and Susan E. Erdman wrote the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by National Institutes of Health grants P30-ES002109 (pilot project award to Susan E. Erdman), U01 CA164337 (to Susan E. Erdman), and RO1CA108854 (to Susan E. Erdman).
2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi:10.1016/S0092-8674(00)81683-9
3. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer (2001) 1:46–54. doi:10.1038/35094059
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
5. Erdman S, Poutahidis T. Roles for inflammation and regulatory T cells in colon cancer. Toxicol Pathol (2010) 38:76–87. doi:10.1177/0192623309354110
6. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21:309–22. doi:10.1016/j.ccr.2012.02.022
7. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi:10.1016/S0140-6736(00)04046-0
8. Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature (2004) 431:405–6. doi:10.1038/431405a
9. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer (2004) 4:540–50. doi:10.1038/nrc1388
10. Erdman SE, Poutahidis T. Cancer inflammation and regulatory T cells. Int J Cancer (2010) 127:768–79. doi:10.1002/ijc.25430
11. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis (2009) 30:1073–81. doi:10.1093/carcin/bgp127
12. Curiel TJ. Regulatory T cells and treatment of cancer. Curr Opin Immunol (2008) 20:241–6. doi:10.1016/j.coi.2008.04.008
13. Whiteside TL. What are regulatory T cells (Treg) regulating in cancer and why? Semin Cancer Biol (2012) 22:327–34. doi:10.1016/j.semcancer.2012.03.004
14. Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res (2010) 107:57–117. doi:10.1016/S0065-230X(10)07003-X
15. Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer (2007) 7:880–7. doi:10.1038/nrc2250
16. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol (2014) 27C:1–7. doi:10.1016/j.coi.2013.12.005
17. Gajewski TF, Fuertes MB, Woo SR. Innate immune sensing of cancer: clues from an identified role for type I IFNs. Cancer Immunol Immunother (2012) 61:1343–7. doi:10.1007/s00262-012-1305-6
18. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
19. Roncador G, Brown PJ, Maestre L, Hue S, Martinez-Torrecuadrada JL, Ling KL, et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur J Immunol (2005) 35:1681–91. doi:10.1002/eji.200526189
20. Gavin MA, Torgerson TR, Houston E, Deroos P, Ho WY, Stray-Pedersen A, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A (2006) 103:6659–64. doi:10.1073/pnas.0509484103
21. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30:899–911. doi:10.1016/j.immuni.2009.03.019
22. Kleinewietfeld M, Hafler DA. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin Immunol (2013) 25:305–12. doi:10.1016/j.smim.2013.10.009
23. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10:942–9. doi:10.1038/nm1093
24. Hodi FS. Overcoming immunological tolerance to melanoma: targeting CTLA-4. Asia Pac J Clin Oncol (2010) 6(Suppl 1):S16–23. doi:10.1111/j.1743-7563.2010.01271.x
25. Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A (2008) 105:3005–10. doi:10.1073/pnas.0712237105
26. Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, et al. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci U S A (2008) 105:14987–92. doi:10.1073/pnas.0806075105
27. Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and Gm-Csf combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest (2006) 116:1935–45. doi:10.1172/JCI27745
28. Kavanagh B, O’Brien S, Lee D, Hou Y, Weinberg V, Rini B, et al. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood (2008) 112:1175–83. doi:10.1182/blood-2007-11-125435
29. Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med (2011) 364:2119–27. doi:10.1056/NEJMoa1012863
30. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol (2009) 27:186–92. doi:10.1200/JCO.2008.18.7229
31. Badoual C, Hans S, Fridman WH, Brasnu D, Erdman S, Tartour E. Revisiting the prognostic value of regulatory T cells in patients with cancer. J Clin Oncol (2009) 27:e5–6. Author reply e7.
32. Whiteside TL. Disarming suppressor cells to improve immunotherapy. Cancer Immunol Immunother (2012) 61:283–8. doi:10.1007/s00262-011-1171-7
33. Leffers N, Gooden MJ, De Jong RA, Hoogeboom BN, Ten Hoor KA, Hollema H, et al. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol Immunother (2009) 58:449–59. doi:10.1007/s00262-008-0583-5
34. Wilke CM, Wu K, Zhao E, Wang G, Zou W. Prognostic significance of regulatory T cells in tumor. Int J Cancer (2010) 127:748–58. doi:10.1002/ijc.25464
35. Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, et al. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am J Pathol (2003) 162:691–702. doi:10.1016/S0002-9440(10)63863-1
36. Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, et al. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res (2003) 63:6042–50.
37. Erdman SE, Sohn JJ, Rao VP, Nambiar PR, Ge Z, Fox JG, et al. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res (2005) 65:3998–4004. doi:10.1158/0008-5472.CAN-04-3104
38. Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, et al. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res (2006) 66:7395–400. doi:10.1158/0008-5472.CAN-06-0558
39. Poutahidis T, Haigis KM, Rao VP, Nambiar PR, Taylor CL, Ge Z, et al. Rapid reversal of interleukin-6-dependent epithelial invasion in a mouse model of microbially induced colon carcinoma. Carcinogenesis (2007) 28:2614–23. doi:10.1093/carcin/bgm180
40. Poutahidis T, Rao VP, Olipitz W, Taylor CL, Jackson EA, Levkovich T, et al. CD4+ lymphocytes modulate prostate cancer progression in mice. Int J Cancer (2009) 125:868–78. doi:10.1002/ijc.24452
41. Erdman SE, Rao VP, Olipitz W, Taylor CL, Jackson EA, Levkovich T, et al. Unifying roles for regulatory T cells and inflammation in cancer. Int J Cancer (2010) 126:1651–65. doi:10.1002/ijc.24923
42. Poutahidis T, Kleinewietfeld M, Smillie C, Levkovich T, Perrotta A, Bhela S, et al. Microbial reprogramming inhibits western diet-associated obesity. PLoS One (2013) 8:e68596. doi:10.1371/journal.pone.0068596
43. Rao VP, Poutahidis T, Fox JG, Erdman SE. Breast cancer: should gastrointestinal bacteria be on our radar screen? Cancer Res (2007) 67:847–50. doi:10.1158/0008-5472.CAN-06-3468
44. Erdman SE, Rao VP, Poutahidis T, Rogers AB, Taylor CL, Jackson EA, et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci U S A (2009) 106:1027–32. doi:10.1073/pnas.0812347106
45. Lakritz JR, Poutahidis T, Levkovich T, Varian BJ, Ibrahim YM, Chatzigiagkos A, et al. Beneficial bacteria stimulate host immune cells to counteract dietary and genetic predisposition to mammary cancer in mice. Int J Cancer (2013). doi:10.1002/ijc.28702
46. Rao VP, Poutahidis T, Ge Z, Nambiar PR, Horwitz BH, Fox JG, et al. Proinflammatory CD4+ CD45RB(hi) lymphocytes promote mammary and intestinal carcinogenesis in Apc(Min/+) mice. Cancer Res (2006) 66:57–61. doi:10.1158/0008-5472.CAN-05-3445
47. Poutahidis T, Cappelle K, Levkovich T, Lee CW, Doulberis M, Ge Z, et al. Pathogenic intestinal bacteria enhance prostate cancer development via systemic activation of immune cells in mice. PLoS One (2013) 8:e73933. doi:10.1371/journal.pone.0073933
48. Houghton J, Li H, Fan X, Liu Y, Liu JH, Rao VP, et al. Mutations in bone marrow-derived stromal stem cells unmask latent malignancy. Stem Cells Dev (2010) 19:1153–66. doi:10.1089/scd.2009.0439
49. Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology (2010) 138(2101–2114):e5. doi:10.1053/j.gastro.2010.01.058
50. Zhu Y, Michelle Luo T, Jobin C, Young HA. Gut microbiota and probiotics in colon tumorigenesis. Cancer Lett (2011) 309:119–27. doi:10.1016/j.canlet.2011.06.004
51. Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science (2012) 338:120–3. doi:10.1126/science.1224820
52. Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, et al. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity (2014) 40:140–52. doi:10.1016/j.immuni.2013.11.018
53. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (2013) 342:967–70. doi:10.1126/science.1240527
54. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science (2013) 342:971–6. doi:10.1126/science.1240537
55. Rook GA, Dalgleish A. Infection, immunoregulation, and cancer. Immunol Rev (2011) 240:141–59. doi:10.1111/j.1600-065X.2010.00987.x
57. Poutahidis T, Kearney SM, Levkovich T, Qi P, Varian BJ, Lakritz JR, et al. Microbial symbionts accelerate wound healing via the neuropeptide hormone oxytocin. PLoS One (2013) 8:e78898. doi:10.1371/journal.pone.0078898
58. Levkovich T, Poutahidis T, Smillie C, Varian BJ, Ibrahim YM, Lakritz JR, et al. Probiotic bacteria induce a ‘glow of health.’ PLoS One (2013) 8:e53867. doi:10.1371/journal.pone.0053867
Keywords: tumor macroenvironment, regulatory T-cells, cancer immunotherapy, inflammation and cancer, probiotic bacteria
Citation: Poutahidis T, Kleinewietfeld M and Erdman SE (2014) Gut microbiota and the paradox of cancer immunotherapy. Front. Immunol. 5:157. doi: 10.3389/fimmu.2014.00157
Received: 11 February 2014; Accepted: 25 March 2014;
Published online: 07 April 2014.
Edited by:
Fang-Ping Huang, Imperial College London, UKReviewed by:
Graham Robert Leggatt, University of Queensland, AustraliaCopyright: © 2014 Poutahidis, Kleinewietfeld and Erdman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan E. Erdman, Massachusetts Institute of Technology, 77 Mass Avenue, Cambridge, MA 02139, USA e-mail:c2VyZG1hbkBtaXQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.