 Marc-André Bellavance
Marc-André Bellavance Serge Rivest
Serge Rivest
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 March 2014
Sec. Inflammation
Volume 5 - 2014 | https://doi.org/10.3389/fimmu.2014.00136
This article is part of the Research Topic NeuroEndocrine Interactions in Immune Inflammatory Diseases View all 8 articles
In response to physiological and psychogenic stressors, the hypothalamic–pituitary–adrenal (HPA) axis orchestrates the systemic release of glucocorticoids (GCs). By virtue of nearly ubiquitous expression of the GC receptor and the multifaceted metabolic, cardiovascular, cognitive, and immunologic functions of GCs, this system plays an essential role in the response to stress and restoration of an homeostatic state. GCs act on almost all types of immune cells and were long recognized to perform salient immunosuppressive and anti-inflammatory functions through various genomic and non-genomic mechanisms. These renowned effects of the steroid hormone have been exploited in the clinic for the past 70 years and synthetic GC derivatives are commonly used for the therapy of various allergic, autoimmune, inflammatory, and hematological disorders. The role of the HPA axis and GCs in restraining immune responses across the organism is however still debated in light of accumulating evidence suggesting that GCs can also have both permissive and stimulatory effects on the immune system under specific conditions. Such paradoxical actions of GCs are particularly evident in the brain, where substantial data support either a beneficial or detrimental role of the steroid hormone. In this review, we examine the roles of GCs on the innate immune system with a particular focus on the CNS compartment. We also dissect the numerous molecular mechanisms through which GCs exert their effects and discuss the various parameters influencing the paradoxical immunomodulatory functions of GCs in the brain.
Any imbalances to an organism homeostasis elicit a complex stress response that involves the coordinated activation of functionally overlapping neuroendocrine and autonomic systems. Among these critical systems is the hypothalamic–pituitary–adrenal (HPA) axis, which is triggered by stressors of various sources (physical, emotional, immunological, etc.) to provoke the systemic release of glucocorticoids (GCs).
The activity of the HPA axis is regulated by multiple afferent sympathetic, parasympathetic, and limbic circuits (e.g., amygdala, hippocampus, and medial prefrontal cortex) innervating either directly or indirectly the paraventricular nucleus (PVN) of the hypothalamus. The PVN integrates converging stimulatory (catecholaminergic, glutamatergic, and serotonergic) or inhibitory (GABA-ergic) inputs, and thus represents a critical relay in the control of the HPA axis (1–3). The HPA axis is activated when secretory neurons of the medial parvocellular division of the PVN are stimulated, either directly or by relieving inhibitory inputs (Figure 1). As a result, corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) are both released in the portal circulation of the anterior pituitary gland. In turn, these neuropeptides trigger the secretion of adrenocorticotrophic hormone (ACTH) in the bloodstream by pituitary corticotrophs. ACTH then induces the production and the systemic release of GCs by the zona fasciculata of the adrenal cells (4–7).
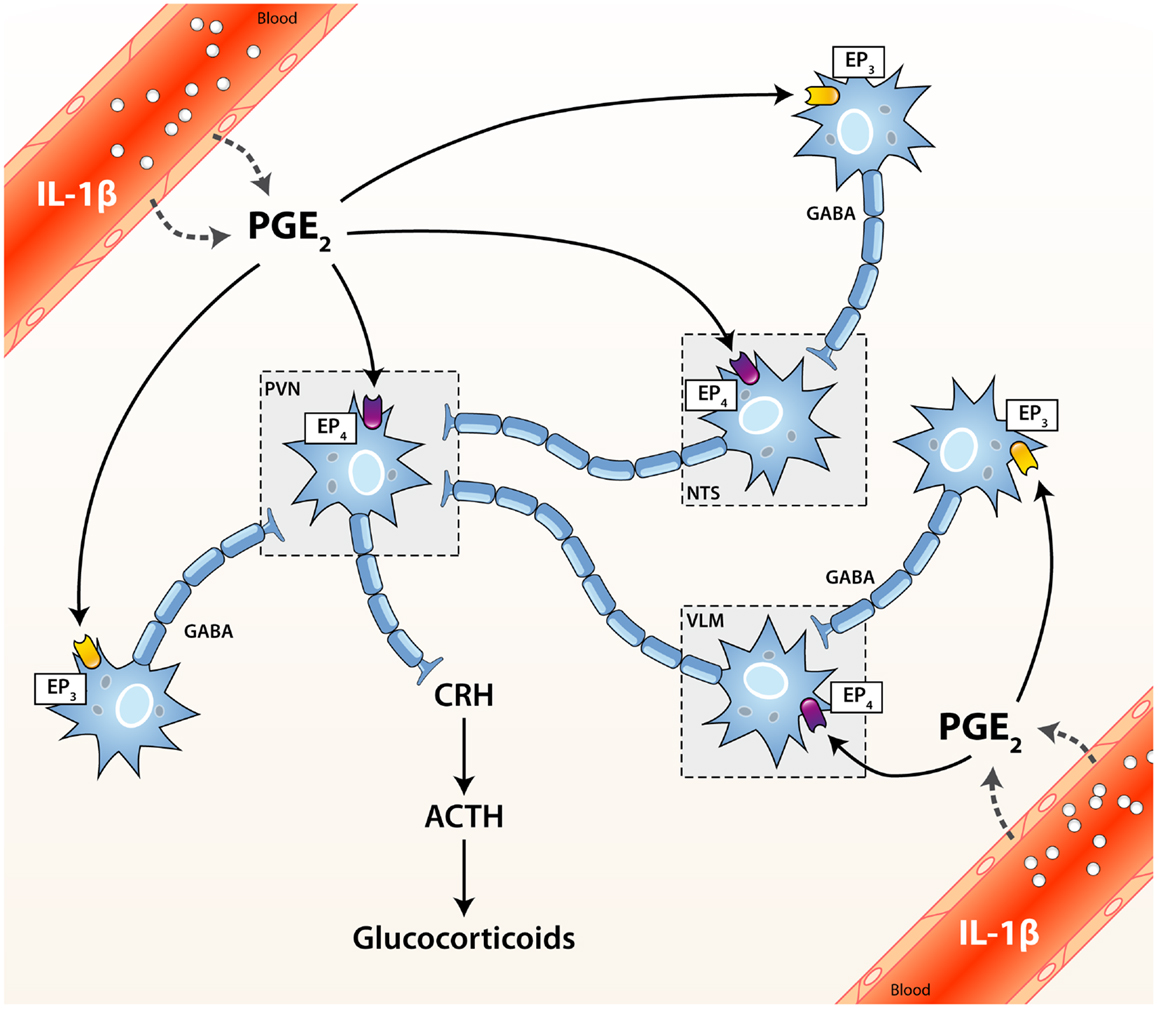
Figure 1. Activation cascade of the hypothalamic–pituitary–adrenal (HPA) axis by systemic immune stimuli. Integrated brain circuits trigger the parvocellular neurons of the PVN to release infundibular CRH, which stimulates the release of ACTH from corticotroph cells of the pituitary. ACTH reaches the bloodstream and finally induces the systemic release of GCs by the adrenals. PGE2 may activate or inhibit neurons through the EP4 and EP3 receptors, respectively. ACTH, adrenocorticotrophic hormone; CRH, corticotrophin releasing factor; EP1–4, PGE2 receptor subtypes; GABA, γ-aminobutyric acid (inhibitory); NTS, nucleus tractus solitarius (A2/C2 neurons); PGE2, prostaglandin of E2 type; PVN, paraventricular nucleus of the hypothalamus; VLM, ventrolateral medulla (A1/C1 neurons).
Under basal conditions, the HPA axis exhibits a continuous oscillatory activity characterized by circadian and ultraradian variations. GCs are thus secreted in a highly pulsatile fashion throughout a 24 h cycle, displaying greater mean levels during the awake phase (8). The circadian rhythm of the HPA axis is orchestrated by the suprachiasmatic nucleus (SCN) of the hypothalamus (9) and the oscillating release of GCs is believed to optimize stress responses. Interestingly, the ultraradian pulsatility of the HPA axis was recently associated with pulses of glucocorticoid receptor (GR)-mediated transcriptional regulation (10). Upon stress, the intensity and duration of the HPA response both depend on the specific nature of the encountered stressor (5, 11, 12). The precise circadian or ultraradian phase at which stress occurs also profoundly influences the systemic release of GCs, since higher levels are secreted when the challenge coincides with rising pulses (13).
Multiple non-exclusive pathways participate in the activation of the HPA axis upon cerebral or peripheral immune challenges. When an immunogenic insult takes place in the brain, various inflammatory mediators produced locally may trigger the HPA axis. In contrast, multiple routes convey stimulatory signals from the periphery to the HPA axis when a challenge occurs outside the CNS. In this scenario, circulating immunogenic or inflammatory factors may access and activate regulatory neuronal circuits (either directly or not) projecting to the PVN via the fenestrated endothelium of the circumventricular organs (CVOs) or a disrupted blood–brain barrier (BBB) (14, 15). Alternatively, circulating immune ligands may also bind their cognate receptor(s) anchored in the luminal membrane of endothelial cells of brain capillaries. Hence, they can simultaneously engage numerous transduction signaling pathways that disseminate the activating cues to the HPA axis through the parenchymal release of diverse inflammatory messengers. To this end, distinct lines of evidence support a critical role of MyD88, COX-2, microsomal prostaglandin E synthase (mPGES-1), and prostaglandin E2 (PGE2) in relaying peripheral stimulatory signals to the HPA axis. Although the exact cell types at play are still actively debated, endothelial cells are widely acknowledged as pivotal in this activating cascade. The contribution of perivascular cells (PVCs) however remains a controversial topic since they were identified either as a negligible (16) or substantial source of PGE2 (17) in murine models of systemic inflammation. In addition to COX-2, more recent findings also support a role for COX-1 in activating the HPA axis (18, 19). Finally, afferent fibers of the vagus nerve may also signal peripheral inflammation to the brain and thereby activate the HPA response (20, 21).
The precise regulation of the activity of the HPA axis is of the utmost importance since both an excessive or insufficient release of GCs entail severe detrimental metabolic and immunological effects. As a matter of fact, chronic exposure to GCs results in various adverse side effects such as osteoporosis, diabetes, hypertension, dyslipidemia, and even neurodegeneration (5, 22). On the other hand, a deficient or blunted HPA axis is commonly observed in the clinic in a wide range of autoimmune and inflammatory diseases. Likewise, disrupting the HPA axis surgically (through adrenalectomy) or pharmacologically (with GR antagonists) compromises the survival of normally resistant mice to septic shock (23–27). The magnitude and duration of the HPA response is thus tightly controlled by autoregulatory feedback loops involving the adrenals, pituitary, PVN, and upstream corticolimbic structures such as the hippocampus, amygdala, and medial prefrontal cortex (28). As a result, the HPA response is terminated through the same neuronal circuitry that mediates its activation [reviewed in Ref. (2)].
The anti-inflammatory and immunosuppressive actions of GCs were first unraveled by the pioneering work of Kendall, Reichstein, and Hench more than 70 years ago and were since exploited in the clinic to treat a plethora of allergic, autoimmune, inflammatory, and hematological disorders as well as for preventing allograft rejection (29, 30). GCs exert immunomodulatory functions by acting on practically every immune cell type, by virtue of the nearly ubiquitous expression of the GR. The cell-specific actions of GCs, which underlie the long-recognized anti-inflammatory and immunosuppressive effects of this steroid hormone, are briefly highlighted below.
Glucocorticoids strongly influence the phenotype, survival, and functions of monocytes and macrophages. GCs have long been recognized to increase the phagocytic potential of these critical effectors cells and thereby stimulate the clearance of foreign antigens, pathogens, inflammatory cells, cellular debris, and other potentially harmful elements (31, 32). The steroid hormone also suppresses immunostimulatory functions and efficiently abrogates the production of pro-inflammatory mediators (such as cytokines, chemokines, and reactive oxygen or nitrous species) through various synergistic genomic and non-genomic mechanisms (33). In doing so, GCs promote an anti-inflammatory phenotype and expand the migratory activity and survival of these myeloid cells (34, 35).
Glucocorticoids perform similar key functions in dendritic cells (DCs). In addition to regulating the maturation, survival, and motility of these antigen-presenting cells, GCs also hamper their immunogenic functions. Indeed, the end product of the HPA axis restricts the capacity of DCs to stimulate T cells by preventing the up-regulation of various co-stimulatory molecules, such as MHCII, B7.2 (CD86), and CD40 (33, 36, 37). GCs can also convert DCs to tolerogenic cells, which promote the production of regulatory T cells (38, 39). This immunosuppressive effect critically relies on the GC-mediated expression of glucocorticoid-induce leucine zipper (GILZ), since this transcription factor appears as both necessary and sufficient for the induction of a tolerogenic state (40, 41). Recent work also indicates that the co-repressor DC-SCRIPT participate to this conversion (37). Interestingly, GCs exert distinct actions in immature and mature DCs and a disparate expression of various isoforms of the GR was recently found to underlie these divergent effects (42).
Another salient outcome of GCs administration is neutrophilia. The steroid hormone expands the number of circulating neutrophils by (1) increasing their egress from the bone-marrow to the bloodstream and by (2) concomitantly hindering their transmigration to inflammatory sites by alleviating the expression of cell adhesion molecules (43, 44). Paradoxically, GCs were also shown to promote or attenuate neutrophil apoptosis, respectively through Annexin A1 (45) or Mcl-1 and XIAP (46).
In contrast to neutrophils, GCs reduce the levels of circulating T cells by enhancing their migration back to the bone-marrow and secondary lymphoid organs (47, 48). GCs also trigger T cells apoptosis, notably through the up-regulation of BIM (49, 50). The relative expression of distinct GR isoforms is also believed to dictate the susceptibility of T cells to the pro-apoptotic effects of GCs (50). Additionally, the steroid hormone potently represses the production of pro-inflammatory cytokines, more specifically of those promoting Th1 and Th17 polarization (33). Accordingly, the targeted disruption of the GR in T cells (GRlkc-Cre mice) produced hyperactive Th1 cells and increased mortality upon infection (51) while it precipitated the onset of disease in a murine model of multiple sclerosis (experimental autoimmune encephalomyelitis) (52). Complementarily, other lines of evidence suggest that GCs promote the differentiation of regulatory T cells (Treg), which are key suppressors of immune functions (53, 54). Finally, the steroid hormone may also interact directly with the signaling complex of the T cell receptor (TCR) after stimulation to inhibit its downstream transduction signaling pathways (55, 56). The actions of GCs in T cells thus heavily depend of the subtype targeted. Finally, GCs may also act on B cells and influence their survival, proliferation, and function (53, 57).
In addition to such prominent anti-inflammatory actions, a substantial body of knowledge now clearly indicates that GCs also have permissive and even stimulatory effects on immune processes. As a consequence, GCs are now more appropriately regarded as immunomodulators. The paradoxical actions of GCs are particularly evident in the CNS, where numerous lines of evidence either support a beneficial or detrimental role of the steroid hormone in various pathological contexts. In the next section, we aim to discuss the key functions assumed by the HPA axis and GCs in regulating the innate immune system of the brain in health and disease. We next dissect the multiple parameters defining the specific actions of GCs in these contexts.
The CNS was once regarded as an immune-privileged organ owing to the lack of lymphatic drainage, selective permeability of the BBB and apparent absence of immune responses. Significant advances in the last two decades have challenged this dogma and helped to considerably expand our understanding of the immune processes taking place in the CNS in health and disease. Robust inflammatory responses are elicited in neuropathological contexts such as infection, traumatic injury as well as autoimmune and neurodegenerative disorders by the resident innate immune cells of the brain, microglia. Dynamic research now aims at deciphering the crucial functions of microglia in preserving and restoring the brain homeostasis during immune challenges, injuries, and chronic diseases.
Microglia represent a heterogeneous cell population accounting for approximately 5–12% of total brain cells. They are distributed unevenly throughout the CNS, as grey matter and specific structures such as the hippocampus, basal ganglia, and substantia nigra typically all exhibit greater densities than the white matter (58–60). In resting conditions, microglial cells display a heavily ramified morphology and dynamically survey their immediate environment with highly motile but non-overlapping protrusions (61, 62). Upon encountering an endogenous or exogenous threat, microglia adopt an amoeboid morphology, show increased motility and promptly initiate an inflammatory response. In this context, activated microglia produce numerous inflammatory mediators and reactive species (oxygen or nitrous derived), which initially help to mobilize other immune cells and aim to restore homeostasis. Activated microglia also support neuronal survival and function by engulfing pathogens, cellular debris, or other neurotoxic entities and can synthesize various neurotrophic factors to promote tissue maintenance and repair (63, 64). However, the excessive or sustained activation of microglial cells (such as in chronic neurodegenerative diseases) usually produces significant inflammatory collateral damage and may hence fuel a vicious self-sustaining cycle driving further injury. Therefore, in order to preserve the fragile environment of the CNS many synergistic mechanisms are normally deployed to control microglia activity. Microglial cells represent prime targets of GCs in the CNS owing to a predominant expression of GR (65). Through these key innate immune cells, GCs therefore perform major regulatory functions on the innate immune system of the CNS in health and disease.
Owing that both endogenous and exogenous GC derivatives are highly lipophilic compounds, they easily diffuse through the BBB and act on nearly all cell types of the CNS. The bioavailability of endogenous and synthetic compounds can however diverge significantly according to three critical determinants: (1) the binding affinity to corticosteroid-binding globulin (CBG), (2) the uptake by various efflux pumps located at the BBB, and (3) the susceptibility to enzymatic metabolism. The endogenous bioactive form of GCs, namely cortisol in humans and corticosterone in rodents, is inactive while associated to plasma transport proteins such as serum albumin and CBG. CBG is a thermosensitive glycoprotein that binds up to 90% of circulating GCs in the bloodstream. The remaining 10% may also be associated to serum albumin. In consequence, the levels of free circulating GCs are usually about 5% (66, 67). Like most xenobiotics, synthetic GC analogs such as dexamethasone are efficiently expelled from the CNS by multidrug resistance (MDR) transporters expressed by the endothelial cells of the BBB (68). The efflux of free cortisol from the CNS is a major impediment and this phenomenon was suggested to underlie the preferential access of inactive GC metabolites to the brain.
Whereas plasma transport proteins and efflux pumps narrow the access of GCs to the brain, the activity of the steroid hormone is also regulated at the cellular level by the enzymatic interconversion of bioactive and inert GC species by 11β-hydroxysteroid dehydrogenases (11β-HSD). Briefly, 11β-HSD type 1 generates the bioactive form (cortisol or corticosterone) from inactive 11-keto derivatives while 11β-HSD type 2 catalyzes the opposite reaction. Interestingly, 11-keto derivatives do not bind to CBG in plasma nor are expelled by the efflux pumps of the BBB. As a result, 11-keto-GCs reach the CNS more readily than the bioactive form. 11β-HSD type 1 is widely expressed by both neurons and glial cells, whereas that of the type 2 isoform is more restricted (66, 68, 69). The distinct expression patterns of the two 11β-HSD enzymes in the CNS therefore also markedly influence GC signaling.
Collectively, these three pivotal parameters regulate GR signaling in the CNS by controlling the bioavailability of its ligands.
Glucocorticoids exert part of their biological effects by binding two proximate members of the nuclear receptor superfamily, namely the GR and the mineralocorticoid receptor (MR). Although GRs are ubiquitously expressed in the brain, they are most abundant in the PVN and the hippocampus. In contrast, MR expression appears mostly confined to a few limbic sites regulating salt appetite and autonomic outflow (70, 71). The binding affinity of cortisol is however 10-fold greater for the MR than the GR (66). MRs are thus heavily bound by basal/low levels of GCs while substantial ligation of GRs only occurs upon stress or when the highest ultraradian peaks are reached.
Like other nuclear receptors of its class, the GR is primarily composed of three characteristic domains: an N-terminal transactivation domain (NTD), a DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) (72). The human GR (hGR) is produced from a single gene (NR3C1, chromosome 5q31.3) and encompasses nine exons. The two best-characterized isoforms, namely GR-α and GR-β, arise from the alternative splicing of the exon 8 to distinct acceptor sites of exon 9. Alternative translation initiation sites further expand the variety of GR-α isoforms, producing height different proteins with a truncated NTD (GRα-A, GRα-B, GRα-C1, GRα-C2, GRα-C3, GRα-D1, GRα-D2, and GRα-D3). Finally, rearrangements in GR mRNA also yield three functionally distinct splice variants, namely GR-γ, GR-A, and GR-P (72, 73). The physiological significance of each GR isoform is beginning to emerge [elegantly reviewed in Ref. (74)], and remains best defined for the GR-α and β isoforms.
In the absence of ligand, GR-α is maintained in the cytoplasm but translocates to the nucleus upon ligation. In contrast, GR-β permanently resides in the nucleus where it selectively acts as a dominant negative inhibitor of GR-α (75). The relative abundance of the inhibitor GR-β, which normally represents only 1% of GR-α in the brain, is thus a key parameter in dictating GC responsiveness (76). Interestingly, pro-inflammatory cytokines were recently reported to increase the GR-β/GR-α ratio (77). Despite that endogenous or synthetic GC ligands do not bind to GR-β, previous work has nonetheless shown that its regulatory actions on transcription could be altered by the progesterone receptor (PR) as well as the GR antagonist mifepristone (RU486) (78). In the presence of other GR isoforms, GR-β appears to modulate (either positively or negatively) the transcription of a large set of genes that do not overlap with that of GR-α (77).
The non-liganded GR-α is sequestered in the cytoplasm by a multiprotein complex that may be composed of heat-shock protein (HSP) 90, 70, 56, or 40, as well as co-chaperones p23, p60, Src kinase, and immunophilins FKBP51, 52 (22, 75, 79). These cytoplasmic chaperones conceal the nuclear localization signal (NLS) of the steroid receptor and thereby preclude its shuttling to the nucleus. Importantly, these chaperones also maintain GR-α in a conformation state that is optimal for ligand binding. The binding of a single GC molecule to a GR (1:1 ratio) provokes a conformational change that leads to the dissociation of chaperones and unmasks the NLS to importin proteins. The liganded GR is then transported to the nucleus where it profoundly modulates gene expression through multiple genomic and non-genomic mechanisms.
Once in the nucleus, liganded GRs do not bind to DNA in a stable manner. They rather constantly shuttle between the nucleoplasm and GC-responsive elements (GRE) located in the promoter or enhancer regions of GC-responsive genes (80, 81). The variety of GRE is continuously expanding, but can nonetheless be divided into four broad categories: simple positive, composite, tethering, and the relatively novel simple negative GRE (nGRE) (Figure 2). Simple positive GREs (+GRE) represent imperfect palindromic sequences composed of inverted hexamers separated by a three base pair (bp) spacer. Each hexamer provides a binding interface for each monomer of GR homodimers. Simple GREs (+GRE) are deemed “positive” owing that they enable GR homodimers to stimulate gene expression (i.e., transactivation) through chromatin decondensation and recruitment of co-activators. GR-mediated transactivation typically induces the expression of potent signaling inhibitors (e.g., IκBα, MPK-1, IL-10, Annexin A1, GILZ, and SOCS proteins), which strongly interfere with salient immune signaling pathways such as those triggered by the Toll-like receptors (TLR) (82–85). Composite GREs (cGRE) represent chimeric sequences that are recognized by a GR monomer paired with another transcription factor (i.e., GR heterodimers). Tethering GREs (tGRE) stand out from other GRE types since they do not exhibit bindings motifs per se for the GR. They rather engage other transcription factor complexes that eventually recruit GRs through protein–protein interactions. As a result, tethered GRs do not physically interact with DNA at tGRE. Interestingly, both composite and tethering GREs enable direct transactivation or transrepression; the resulting effect on gene transcription is dictated by the transcription factors to which GRs are bound. For instance, tethering of a GR monomer was reported to increase the transcription of STAT3, STAT5, cAMP responsive element binding (CREB), and CCAAT/enhancer binding protein (C/EBP)-α responsive genes while inhibiting the activity of NF-κB, AP-1, activating transcriptions factors (ATFs), and IRF3 (72, 73, 86, 87). Additionally, the steric hindrance entailed by tethered GRs can interrupt gene transcription by hindering the recruitment and activity of the transcriptional machinery. In this line, very recent data point toward a role of GR:GRIP1 complexes in preventing the recruitment of PolII to initiation-controlled inflammatory genes such as IL-1α and IL-1β, and concomitantly promoting the accumulation of pause-inducing negative elongation factor, which precludes the release of PolII from the elongation block of genes like TNF-α, CCL2, and CCL3 (88). The fourth and last type is the simple nGRE. They are composed of two inverted repeats and are believed to be recognized by GR homodimers (89). In contrast to +GRE, GR activity at nGRE mediates the direct repression of transcription by recruiting the transrepressor nuclear receptor co-repressor 1 (NCoR) and silencing mediator of retinoid and thyroid hormone receptors (SMRT). Interestingly, nGRE were localized in many NF-κB and AP-1-responsive genes, which encode immune and inflammatory proteins. Finally, we note that through such genomic mechanisms, GCs regulate the expression and protein abundance of their own regulators (i.e., HES1) and that this feedback loop is mandatory for proper GC-mediated changes in gene expression (90).
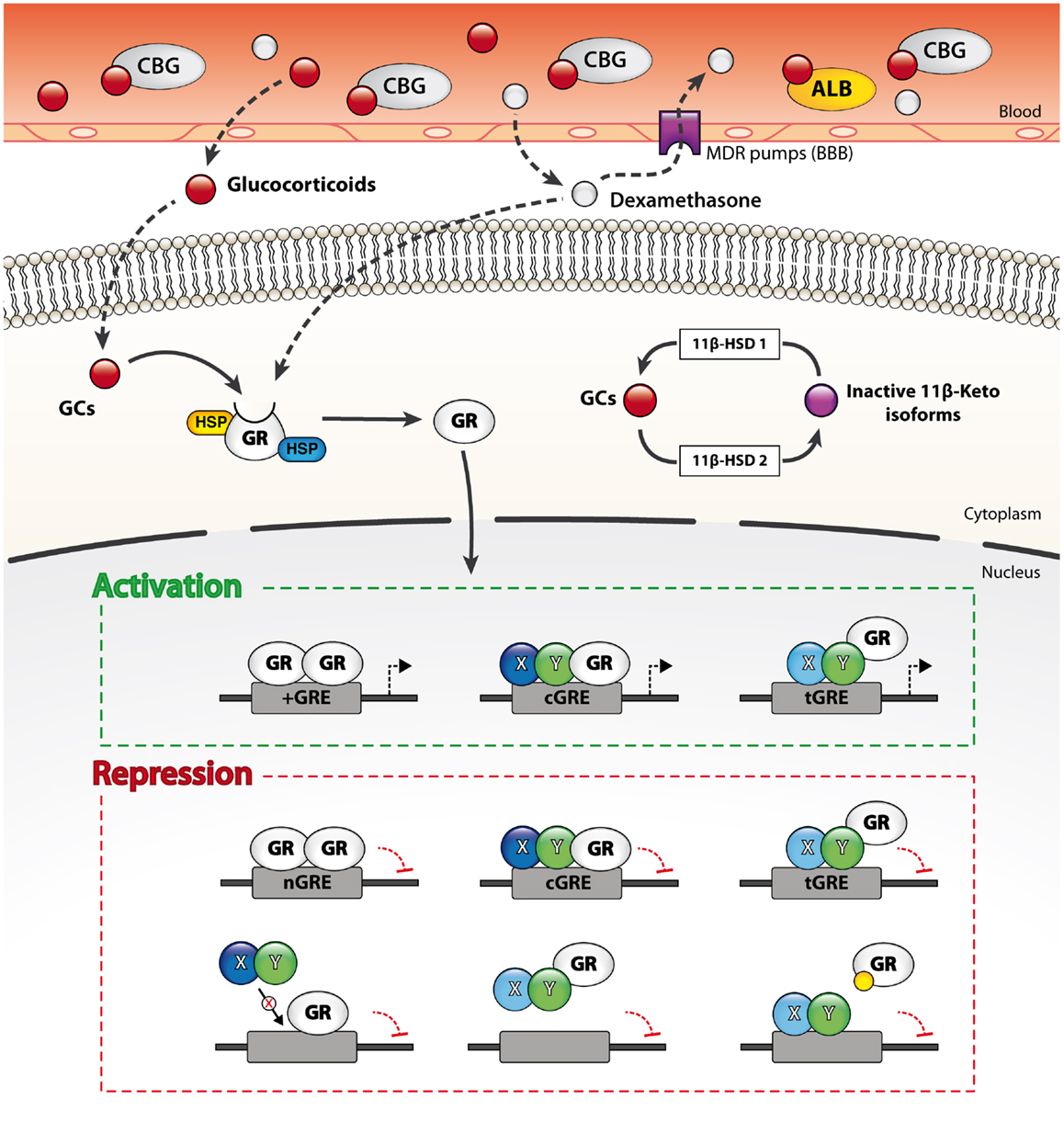
Figure 2. Genomic and non-genomic mechanisms through which GCs regulate gene transcription. Free circulating GCs easily diffuse through membranes such as the blood–brain barrier (BBB) and thus target both peripheral and CNS cells. The bioavailability of endogenous and exogenous GCs in the CNS is however limited at the organ level by efflux pumps expressed at the BBB and at the cellular level by enzymatic metabolism (11β-HSD enzymes). The unliganded GC receptor (GR) is sequestered in the cytoplasm by multiple chaperones. Ligation of the GR by a GC molecule (1:1 ratio) alters its conformation and results in the dissociation of the chaperones. The activated GR then translocates to the nucleus and dynamically modulate gene transcription through various mechanisms. Liganded GRs bind to four main types of GR-response elements (GREs). Activated GRs physically interact with DNA on simple (+GRE), negative (nGRE), and composite GREs (cGRE). The activated GR can also be recruited to other DNA-binding sequences (DBS) via protein–protein interactions (tGRE). Transactivation or transrepression activity of the GR is partly dictated by the type of GRE and its binding partners. Alternatively, GRs also regulate transcription through steric hindrance on DNA sites overlapping with GREs, by sequestering transcription factors from DNA and by competing for co-activators binding. Furthermore, liganded GRs may occupy other response elements by binding to overlapping GREs, sequester transcription factors from DNA and compete for co-activators.
In addition to their genomic actions, liganded GRs also impact on gene transcription through various non-genomic mechanisms. These mechanisms do not require de novo protein synthesis and thus underlie the more rapid actions of GCs. Prominent examples include mRNA destabilization, competition for co-activators [e.g., CREB-binding protein (CBP), p300, and GRIP], and interference with the binding of transcription factors to DNA (91–94). Complementarily, freed cytoplasmic chaperones may also perform anti-inflammatory actions following their dissociation from the activated GR. For instance, the immunophilin FKBP52 was recently identified as a gene-specific regulator of GR functions (95). It should be noted that other types of GRs, such as those embedded in the cytoplasmic or mitochondrial membranes, were also proposed to regulate transcriptional activity but the mechanistic details underlying their origin and function remain ill-defined (96–98). Although definite proof is still lacking, it was posited that the membrane-bound GR might be a variant of the cytosolic form potentially arising from alternative promoters, differential splicing, or post-translational modifications (99). Of particular interest, ligation of GRs anchored in membrane lipid rafts by dexamethasone was recently found to abate intercellular communication through gap junctions in neural progenitor cells (100). Such mechanism might also operate in immune cells and influence their function by modulating cell–cell interactions and the immune synapse. Additional non-genomic actions of the liganded GRs may include direct interactions with membrane (e.g., GPCRs, ion channels, TCR), and cytoplasmic proteins (MAPK and phospholipases) that indirectly impact on transcriptional activity (76, 79, 101). For instance, dexamethasone was shown to potentiate CXCR4 signaling and even synergize with its ligand CXCL12 in resting T cells, presumably through the CD45- and GR-dependent activation of Lck and other downstream kinases (102). Therefore, GCs trigger various non-genomic and genomic signaling pathways that can act in concert to shape the function of immune cells. Finally, GCs may also entail non-specific and GR-independent effects by altering the physicochemical properties of plasma and mitochondrial membranes at high concentrations (101, 103) and by modifying the composition (104) or the formation (105) of lipid rafts in various immune cells.
Several lines of GR mutant transgenic mice provided key insights about the functions of the GR in regulating the HPA axis and immune processes (106). The GRdim mouse, which carries a point mutation (A458T) preventing GR dimerization, is among the best characterized. Initial evidence suggested that classic transactivation by GRs was defective in these mice and that this mechanism was required for the full range of GC immunosuppressive actions (107, 108). This hypothesis is supported by prior work showing that repression of IL-1β, MIP-2, MCP-1, and IP-10 by GCs is hindered in macrophages isolated from GRdim mice (109, 110). Contrastingly, other findings indicate that in GRdim mice NF-κB and AP-1 target genes can still be silenced by GCs (107, 108), that the GC-inducible gene MKP-1 is normally expressed in macrophages (82) and that irritant dermatitis can successfully be treated with GCs (22). These contradictory data may arise from a residual transactivation activity of the GR in GRdim mice since many other amino acids were found to enable the dimerization of the receptor (111). The single point mutation A458T engineered in GRdim is thus unlikely to fully abrogate the formation of functional GR homodimers (30, 112, 113). Moreover, GRdim regulatory defects were recently shown to be tissue, cytokine, and time-dependent, which further complicate the interpretation of the dimerization-dependent genomic actions of the GR (114).
The targeted knock-out of the GR was also achieved in a variety of cell populations or types to investigate the cell-specific functions of GCs. For instance, in the GRlysMCre transgenic mice, GR expression is selectively reduced in macrophages, microglia, and other myeloid cells such as DCs, granulocytes, and mast cells. Upon MPTP intoxication (115) or intra-striatal injection of LPS (116), this strain exhibited greater neuronal loss, microglia reactivity, and expression of pro-inflammatory genes than age-matched controls. Of particular note, increased mortality was also observed in GRlysMCre mice compared to controls following a peripheral injection of LPS (109). The GRlysMCre mice therefore helped in uncovering the crucial actions of GR signaling in microglia and peripheral myeloid cells in the proper and timely regulation of the immune response in order to avoid secondary inflammatory damages. The role of GR signaling in endothelial cells was also recently tackled with two distinct transgenic lines. Using the Tie2-GRKO mice, one group recently revealed that GR activation in endothelial cells of brain vessels might be both necessary and sufficient to constrain the production of key structural proteins of the BBB following focal cerebral ischemia (117). Alternatively, the GREC KO mice displayed increased mortality, hemodynamic instability, and higher levels of inflammatory cytokines (TNF-α and IL-6) than controls during sepsis (118). Finally, the broad disruption of the GR allele in the CNS [GRNesCre (5)] and its targeted deletion in the forebrain [forebrain-specific GR knock-out; FBGRKO mice] (119), or the PVN all resulted in the disinhibition of the HPA axis (120). However, current evidence indicates that GR disruption in the forebrain (FBGRKO vs. WT) did not influence neuronal death or damaged area in the hippocampus following an excitotoxic insult, nor the infarct volume in a model of focal cerebral ischemia (117). Elucidating the impact of the targeted GR deletion in specific cell populations on central immune processes as well as survival of neurons and glial cells in different pathological contexts will undoubtedly help to further decipher the complex context and cell-specific functions of GCs in the brain.
A myriad of GR, epigenetic, and contextual parameters underlie the cell-, signal-, and gene-specific actions of GCs. As exemplified above for GR-α and GR-β, GR isoforms have unique transcriptional regulatory profiles. GC actions on gene expression are thus first influenced by the relative abundance of the numerous GR isoforms in a given cell or tissue. In similar fashion, the differential expression of transcription factors, co-activators, co-repressors, and other binding partners are critical in tailoring the actions of GCs through the GR (22).
The activation state of a target cell and the specific transduction signaling pathways triggered at a given time further define the actions of the GR. Signaling pathways can fine-tune GR activity through various post-translational modifications including phosphorylation, ubiquitination, sumoylation, and acetylation (72, 121). Through their own effects on transcription, chromatin, and epigenome, cell signaling pathways further shape GR effects. For instance, TLR signaling pathways are subjected to suppressive actions of GCs in a signal- and gene-specific manner. To this end, Ogawa et al. found that the liganded GR sequesters the co-activator p65 from IRF3 and IRF7 homodimers on interferon-stimulated response elements (IRSE) on a specific set of genes following TLR4 and TLR9, but not TLR3 signaling (122). In such context, regulatory actions of GCs thus appear to be specified by MyD88 signaling. The liganded GR was also found to prevent the recruitment of IRF3 (which then acts as a co-activator) to p65 on NF-κB target genes upon TLR4 stimulation by LPS (94, 123). It was also reported that dexamethasone inhibit JNK upon TLR3 (TRIF) or TLR9 (MyD88) activation through the GR, but that the concomitant activation of both signaling pathways (or TLR4) yields to the resistance of JNK to deactivation by GCs (124). The same group recently reported similar results for SOCS1, a critical regulator of type I IFN transduction signaling pathways (83).
Lastly, genome wide analyses unraveled that changes as subtle as the permutation of a single bp in GREs modifies the conformation of the liganded GR. Through structure alteration, such changes orchestrate interactions with cofactors as well as the regulatory actions of activated GRs on transcription (125, 126). GRE sequences therefore represent key allosteric modulators. The ever increasing variety of bona fide GREs reported in the literature implies that the interactions of liganded GRs and DNA are much more flexible than previously anticipated.
In addition to their long recognized and well characterized anti-inflammatory actions, accumulating evidence now indicate that GCs do not merely allow inflammatory responses to unfold but rather stimulate them under specific circumstances. Whether GCs play beneficial or detrimental roles in the CNS in health and diseases have been debated for decades, and the complex dichotomous and context-dependent actions of the steroid hormone certainly add to the confusion. The fact that GC actions evolve dynamically over time and the incomplete understanding of the parameters driving the CNS innate immune system toward tissue maintenance/repair or damage also warrant a careful interpretation of the literature. Here, we review experimental evidence supporting the bidirectional actions of GCs in the brain.
Substantial evidence established that GCs restrain and/or terminate the innate immune response in the CNS following either a peripheral or cerebral challenge. Almost 20 years ago, exacerbated levels of pro-inflammatory cytokines were found in the brain of adrenalectomized mice following a sub-cutaneous injection of LPS (127–130). GCs were also shown to prevent the production of pro-inflammatory mediatory in cultured microglia primed with LPS (131, 132). Glezer et al. have reported concordant results in vivo, by showing that dexamethasone suppresses LPS-induced NF-κB activity in the murine brain (133). More recently, Munhoz and colleagues demonstrated that a severe or mild deficiency in plasma corticosterone significantly enhanced NF-κB activation in the brain of rodents challenged with systemic LPS (134). Complementarily, the non-selective COX inhibitors ketorolac and indomethacin were found to potentiate the expression of NF-κB target genes in brain capillaries and parenchymal microglia during systemic inflammation (i.v. bolus of LPS, IL-1β, or TNF-α) by hampering the activation of the HPA axis and the subsequent release of GCs. Interestingly, this effect could be replicated by the administration of the GR antagonist RU486 (mifepristone) (135, 136).
Glucocorticoids also exert salient anti-inflammatory actions when the immunogenic insult is taking place within the CNS compartment. Accordingly, Nadeau et al. reported that the robust inflammatory response induced by an intracerebral injection of LPS could be abolished by a prior systemic administration of the endotoxin (137). Further investigation revealed that the immunosuppressive actions of peripheral LPS were mediated by plasma corticosterone and GR activation. Indeed, exogenous GCs mimicked whereas the GR antagonist RU486 nullified the anti-inflammatory effect of the systemic immune challenge. The physiological importance of the GC feedback in controlling innate immune responses in organs as sensitive as the brain was further illustrated in rodents and mice injected with RU486 before intracerebral LPS. Antagonizing the GR significantly exacerbated and prolonged the inflammatory response, which in turn provoked substantial neuronal death and even mortality (138, 139). Interestingly, these findings were replicated in GRlysMCre (vs. WT) mice challenged with an intra-striatal bolus of LPS (116), again supporting a pivotal of GR signaling in microglia in restraining the amplitude and duration of the immune response in this context.
The vast majority of GC pro-inflammatory actions in vivo were described in animal models of acute or chronic stress. As a matter of act, both types of stress elicit an HPA response and were shown to exacerbate salient features of inflammation in the CNS provided that they occur prior to peripheral or cerebral immune insults. For instance, acute stressors such as inescapable tailshocks (IS) were demonstrated to potentiate the expression of pro-inflammatory mediators in specific regions of the brain following the peripheral administration of LPS in rodents (140–142). Chronically stressed rodents exhibited enhanced NF-κB activity in multiple limbic regions during LPS-induced systemic inflammation (143). Chronically stressed animals injected either in the prefrontal cortex (144) or the hippocampus (145) with this endotoxin also displayed exacerbated microglia activation and tissue damage. Interestingly, in these studies the priming effects of stress (regardless of its chronicity) on the ensuing immune response could be abated with RU486 or by experimentally maintaining plasma GCs to basal levels in adrenalectomized animals. GR signaling was thus proposed to be essential for the cross-sensitization between stress and the inflammatory response to LPS. More specifically, exposure to high levels of GCs was suggested to prime the reactivity of microglial cells to a subsequent immune stimulation presumably by making the neuroimmune environment more permissive to inflammation, inducing GC resistance or blunting the HPA response (146). In support of this hypothesis, restraint stress and other physical stressors were shown to induce inflammatory mediators (e.g., iNOS, TNF-α, COX-2, PGE2, IL-1β, and CD14), reduce immunoregulatory proteins (e.g., CD200R) and trigger microglia proliferation in the rodent brain (147–152). In opposition, microglia GR signaling was recently found to suppress rather than bolster the priming effects of chronic stress on microglia reactivity to a subsequent intracerebral injection of LPS (116). Further investigation is needed to shed more light on the factors entailing these conflicting experimental observations about the specific role of microglial GR signaling the priming effect of stress.
Interestingly, GR ligation by endogenous GCs was also found to be required for the expression of several inflammatory genes. Notably, the pharmacological ablation of GR signaling with RU486 was found to hinder the LPS-mediated expression of various immune genes involved in host defense, such as IL-1β (153). Recent data also indicate that the concomitant signaling of GCs and damage-associated molecular patterns (DAMPs) accentuate the expression of pro-inflammatory mediators. In fact, the combined exposure of human and murine myeloid cells to dexamethasone and ATP following LPS was found to enhance the production of IL-1β, TNF-α, IL-6, and IL-10 (43). Synergistic effects of dexamethasone and ATP in the expression of the purinergic receptor P2Y2 and key adhesion molecules (e.g., VCAM and ICAM) were also reported (154). Together, these data suggest that GCs can exert pro-inflammatory actions in the context of acute cellular damage or death. Complementarily, dexamethasone and TNF-α were recently shown to coregulate a unique set of immune genes when combined (155). Therefore, as for the anti-inflammatory actions of GCs, the activation state and signaling context of a target cell also define its pro-inflammatory functions.
Numerous parameters were found to influence the dichotomous actions of GCs. Of particular interest is the timing of GC exposure relative to an immune challenge. Prior work established that acute stress or exogenous GCs can potentiate or conversely repress the same pro-inflammatory genes provided that they are respectively administered before or after LPS (140, 156). In another study, both physical and psychogenic stressors could abate LPS-induced neuroinflammation, as long as they occurred after the immune challenge (157). Timing of exposure to GCs following an immunogenic challenge thus appears to be a pivotal parameter dictating the opposite actions of the steroid hormone (158). The disparate susceptibility of distinct brain regions to innate immune events and GC signaling also represent decisive factors. For example, the prefrontal cortex and hippocampus were both found to be more sensitive than the hypothalamus to inflammation and the neurotoxic effects of GCs (142, 143, 159). Finally, other elements such as the length of exposure, dose, and route of administration as well as the type of synthetic GCs employed should be taken into account (160). In that regard, we noted that pharmacological compounds targeting the GR (agonists and antagonists) are invariably administered via peripheral routes in vivo and are rarely quantified in the CNS despite the distribution impediment caused by the BBB. Consequently, one must not exclude that significant immune and/or metabolic disturbances in the periphery may concomitantly influence the immune state of the brain. We also emphasize on the fact that pro-inflammatory markers such as the cytokines such as IL-1β and TNF-α also act in a context-dependent manner. Such cytokines can thus be harmful for the cerebral elements (138) but they also play beneficial roles in the inflammatory response (161–163). It should also be kept in mind that a pro-inflammatory environment does not automatically lead to collateral damages and that timing is again a key parameter here. While an exaggerated (magnitude or duration) inflammatory response can trigger neuronal injury and cell death, pro-inflammatory mediators may initially (i.e., acute phase) stimulate the clearance of debris, the recruitment of cellular reinforcements and program tissue remodeling. Despite complicating the overall picture, such factors must be carefully considered when interpreting the actions of GCs in the CNS.
A wealth of experimental and clinical data provides clear evidence that accurate signaling between the nervous, endocrine, and immune systems leading to a proper feedback (timing, amplitude, duration, sensitivity, etc.) by GCs is mandatory to avoid serious detrimental consequences for the brain elements following an immunogenic challenge. GCs induce important plastic changes in the brain and many of their effects, including those related to their priming and pro-inflammatory properties may play critical roles in the “yin and yang” effects of the innate immune reaction in the brain. In future studies, it will be critical to decipher the genomic and non-genomic functions underlying the contextual remodeling of the cerebral innate immune system by endogenous and synthetic GCs. Such research endeavor may unravel why GCs either succeed or fail in managing specific inflammatory and neuropathological diseases. Novel regulatory mechanisms of the HPA axis and GC/GR functions, such as those involving epigenetics or miRNAs, also represent promising avenues. Shedding more light on these intricacies will help to better dissect the beneficial and detrimental actions of GCs in the CNS and to develop new therapeutic strategies enabling to balance their paradoxical actions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Canadian Institutes in Health Research (CIHR) support this research. Marc-André Bellavance is supported by a Doctoral Fellowship from the Fonds de Recherche Québec – Santé (FRQS).
1. Lowry CA. Functional subsets of serotonergic neurones: implications for control of the hypothalamic-pituitary-adrenal axis. J Neuroendocrinol (2002) 14(11):911–23. doi:10.1046/j.1365-2826.2002.00861.x
2. Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci (2009) 10(6):397–409. doi:10.1038/nrn2647
3. Zhang Y, Damjanoska KJ, Carrasco GA, Dudas B, D’Souza DN, Tetzlaff J, et al. Evidence that 5-HT2A receptors in the hypothalamic paraventricular nucleus mediate neuroendocrine responses to (-)DOI. J Neurosci (2002) 22(21):9635–42.
4. Aguilera G. HPA axis responsiveness to stress: implications for healthy aging. Exp Gerontol (2011) 46(2–3):90–5. doi:10.1016/j.exger.2010.08.023
5. Howell MP, Muglia LJ. Effects of genetically altered brain glucocorticoid receptor action on behavior and adrenal axis regulation in mice. Front Neuroendocrinol (2006) 27(3):275–84. doi:10.1016/j.yfrne.2006.05.001
6. Turrin NP, Rivest S. Molecular and cellular immune mediators of neuroprotection. Mol Neurobiol (2006) 34(3):221–42. doi:10.1385/MN:34:3:221
7. Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol (2002) 20:125–63. doi:10.1146/annurev.immunol.20.082401.104914
8. Lightman SL, Conway-Campbell BL. The crucial role of pulsatile activity of the HPA axis for continuous dynamic equilibration. Nat Rev Neurosci (2010) 11(10):710–8. doi:10.1038/nrn2914
9. Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature (2002) 418(6901):935–41. doi:10.1038/nature00965
10. Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, et al. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol (2009) 11(9):1093–102. doi:10.1038/ncb1922
11. de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci (2005) 6(6):463–75. doi:10.1038/nrn1683
12. Lupien SJ, Mcewen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci (2009) 10(6):434–45. doi:10.1038/nrn2639
13. Windle RJ, Wood SA, Shanks N, Lightman SL, Ingram CD. Ultradian rhythm of basal corticosterone release in the female rat: dynamic interaction with the response to acute stress. Endocrinology (1998) 139(2):443–50. doi:10.1210/en.139.2.443
14. Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther (2011) 130(2):226–38. doi:10.1016/j.pharmthera.2011.01.014
15. Turrin NP, Rivest S. Unraveling the molecular details involved in the intimate link between the immune and neuroendocrine systems. Exp Biol Med (Maywood) (2004) 229(10):996–1006.
16. Gosselin D, Rivest S. MyD88 signaling in brain endothelial cells is essential for the neuronal activity and glucocorticoid release during systemic inflammation. Mol Psychiatry (2008) 13(5):480–97. doi:10.1038/sj.mp.4002122
17. Serrats J, Schiltz JC, García-Bueno B, van Rooijen N, Reyes TM, Sawchenko PE. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron (2010) 65(1):94–106. doi:10.1016/j.neuron.2009.11.032
18. Elander L, Engstrom L, Ruud J, Mackerlova L, Jakobsson PJ, Engblom D, et al. Inducible prostaglandin E2 synthesis interacts in a temporally supplementary sequence with constitutive prostaglandin-synthesizing enzymes in creating the hypothalamic-pituitary-adrenal axis response to immune challenge. J Neurosci (2009) 29(5):1404–13. doi:10.1523/JNEUROSCI.5247-08.2009
19. García-Bueno B, Serrats J, Sawchenko PE. Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. J Neurosci (2009) 29(41):12970–81. doi:10.1523/JNEUROSCI.2373-09.2009
20. Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol (2006) 6(4):318–28. doi:10.1038/nri1810
22. Kleiman A, Tuckermann JP. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol (2007) 275(1–2):98–108. doi:10.1016/j.mce.2007.05.009
23. Bertini R, Bianchi M, Ghezzi P. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J Exp Med (1988) 167(5):1708–12. doi:10.1084/jem.167.5.1708
24. Edwards CK, Yunger LM, Lorence RM, Dantzer R, Kelley KW. The pituitary gland is required for protection against lethal effects of Salmonella typhimurium. Proc Natl Acad Sci U S A (1991) 88(6):2274–7. doi:10.1073/pnas.88.6.2274
25. MacPhee IA, Antoni FA, Mason DW. Spontaneous recovery of rats from experimental allergic encephalomyelitis is dependent on regulation of the immune system by endogenous adrenal corticosteroids. J Exp Med (1989) 169(2):431–45. doi:10.1084/jem.169.2.431
26. Ramachandra RN, Sehon AH, Berczi I. Neuro-hormonal host defence in endotoxin shock. Brain Behav Immun (1992) 6(2):157–69. doi:10.1016/0889-1591(92)90015-G
27. Ruzek MC, Pearce BD, Miller AH, Biron CA. Endogenous glucocorticoids protect against cytokine-mediated lethality during viral infection. J Immunol (1999) 162(6):3527–33.
28. Hill MN, McLaughlin RJ, Pan B, Fitzgerald ML, Roberts CJ, Lee TT, et al. Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci (2011) 31(29):10506–15. doi:10.1523/JNEUROSCI.0496-11.2011
29. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci (2013) 34(9):518–30. doi:10.1016/j.tips.2013.07.003
30. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology (2013) 154(3):993–1007. doi:10.1210/en.2012-2045
31. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101(4):890–8. doi:10.1172/JCI1112
32. Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C, et al. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol (1999) 162(6):3639–46.
33. Franchimont D. Overview of the actions of glucocorticoids on the immune response: a good model to characterize new pathways of immunosuppression for new treatment strategies. Ann N Y Acad Sci (2004) 1024:124–37. doi:10.1196/annals.1321.009
34. Ehrchen J, Steinmüller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood (2007) 109(3):1265–74. doi:10.1182/blood-2006-02-001115
35. Vallelian F, Schaer CA, Kaempfer T, Gehrig P, Duerst E, Schoedon G, et al. Glucocorticoid treatment skews human monocyte differentiation into a hemoglobin-clearance phenotype with enhanced heme-iron recycling and antioxidant capacity. Blood (2010) 116(24):5347–56. doi:10.1182/blood-2010-04-277319
36. Elftman MD, Norbury CC, Bonneau RH, Truckenmiller ME. Corticosterone impairs dendritic cell maturation and function. Immunology (2007) 122(2):279–90. doi:10.1111/j.1365-2567.2007.02637.x
37. Hontelez S, Karthaus N, Looman MW, Ansems M, Adema GJ. DC-SCRIPT regulates glucocorticoid receptor function and expression of its target GILZ in dendritic cells. J Immunol (2013) 190(7):3172–9. doi:10.4049/jimmunol.1201776
38. Luther C, Adamopoulou E, Stoeckle C, Brucklacher-Waldert V, Rosenkranz D, Stoltze L, et al. Prednisolone treatment induces tolerogenic dendritic cells and a regulatory milieu in myasthenia gravis patients. J Immunol (2009) 183(2):841–8. doi:10.4049/jimmunol.0802046
39. Rutella S, Lemoli RM. Regulatory T cells and tolerogenic dendritic cells: from basic biology to clinical applications. Immunol Lett (2004) 94(1–2):11–26. doi:10.1016/j.imlet.2004.04.015
40. Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V, et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood (2006) 107(5):2037–44. doi:10.1182/blood-2005-07-2760
41. Hamdi H, Godot V, Maillot MC, Prejean MV, Cohen N, Krzysiek R, et al. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood (2007) 110(1):211–9. doi:10.1182/blood-2006-10-052506
42. Cao Y, Bender IK, Konstantinidis AK, Shin SC, Jewell CM, Cidlowski JA, et al. Glucocorticoid receptor translational isoforms underlie maturational stage-specific glucocorticoid sensitivities of dendritic cells in mice and humans. Blood (2013) 121(9):1553–62. doi:10.1182/blood-2012-05-432336
43. Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem (2011) 286(44):38703–13. doi:10.1074/jbc.M111.275370
44. Weber PS, Toelboell T, Chang LC, Tirrell JD, Saama PM, Smith GW, et al. Mechanisms of glucocorticoid-induced down-regulation of neutrophil L-selectin in cattle: evidence for effects at the gene-expression level and primarily on blood neutrophils. J Leukoc Biol (2004) 75(5):815–27. doi:10.1189/jlb.1003505
45. Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC, et al. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol (2012) 92(2):249–58. doi:10.1189/jlb.0112008
46. Saffar AS, Dragon S, Ezzati P, Shan L, Gounni AS. Phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase regulate induction of Mcl-1 and survival in glucocorticoid-treated human neutrophils. J Allergy Clin Immunol (2008) 121(2):492.e–8.e. doi:10.1016/j.jaci.2007.10.003
47. Besedovsky L, Born J, Lange T. Endogenous glucocorticoid receptor signaling drives rhythmic changes in human T-cell subset numbers and the expression of the chemokine receptor CXCR4. FASEB J (2013) 28(1):67–75. doi:10.1096/fj.13-237958
48. Fischer HJ, Schweingruber N, Lühder F, Reichardt HM. The potential role of T cell migration and chemotaxis as targets of glucocorticoids in multiple sclerosis and experimental autoimmune encephalomyelitis. Mol Cell Endocrinol (2013) 380(1–2):99–107. doi:10.1016/j.mce.2013.04.001
49. Molitoris JK, McColl KS, Swerdlow S, Matsuyama M, Lam M, Finkel TH, et al. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J Biol Chem (2011) 286(34):30181–9. doi:10.1074/jbc.M111.245423
50. Purton JF, Monk JA, Liddicoat DR, Kyparissoudis K, Sakkal S, Richardson SJ, et al. Expression of the glucocorticoid receptor from the 1A promoter correlates with T lymphocyte sensitivity to glucocorticoid-induced cell death. J Immunol (2004) 173(6):3816–24.
51. Kugler DG, Mittelstadt PR, Ashwell JD, Sher A, Jankovic D. CD4+ T cells are trigger and target of the glucocorticoid response that prevents lethal immunopathology in toxoplasma infection. J Exp Med (2013) 210(10):1919–27. doi:10.1084/jem.20122300
52. Wüst S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, et al. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol (2008) 180(12):8434–43.
53. Baschant U, Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol (2010) 120(2–3):69–75. doi:10.1016/j.jsbmb.2010.03.058
54. Tischner D, Reichardt HM. Glucocorticoids in the control of neuroinflammation. Mol Cell Endocrinol (2007) 275(1–2):62–70. doi:10.1016/j.mce.2007.03.007
55. Löwenberg M, Tuynman J, Bilderbeek J, Gaber T, Buttgereit F, van Deventer S, et al. Rapid immunosuppressive effects of glucocorticoids mediated through Lck and Fyn. Blood (2005) 106(5):1703–10. doi:10.1182/blood-2004-12-4790
56. Löwenberg M, Verhaar AP, Bilderbeek J, Marle Jv, Buttgereit F, Peppelenbosch MP, et al. Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN. EMBO Rep (2006) 7(10):1023–9. doi:10.1038/sj.embor.7400775
57. Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M, et al. The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev (2011) 10(6):305–10. doi:10.1016/j.autrev.2010.11.009
58. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci (2007) 8(1):57–69. doi:10.1038/nrn2038
59. Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci (2013) 7:44. doi:10.3389/fncel.2013.00044
60. Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol (2009) 9(6):429–39. doi:10.1038/nri2565
61. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci (2005) 8(6):752–8. doi:10.1038/nn1472
62. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (2005) 308(5726):1314–8. doi:10.1126/science.1110647
63. Hellwig S, Heinrich A, Biber K. The brain’s best friend: microglial neurotoxicity revisited. Front Cell Neurosci (2013) 7:71. doi:10.3389/fncel.2013.00071
64. Wolf Y, Yona S, Kim KW, Jung S. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci (2013) 7:26. doi:10.3389/fncel.2013.00026
65. Sierra A, Gottfried-Blackmore A, Milner TA, Mcewen BS, Bulloch K. Steroid hormone receptor expression and function in microglia. Glia (2008) 56(6):659–74. doi:10.1002/glia.20644
66. De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocr Rev (1998) 19(3):269–301. doi:10.1210/edrv.19.3.0331
67. Henley DE, Lightman SL. New insights into corticosteroid-binding globulin and glucocorticoid delivery. Neuroscience (2011) 180:1–8. doi:10.1016/j.neuroscience.2011.02.053
68. Wyrwoll CS, Holmes MC, Seckl JR. 11β-hydroxysteroid dehydrogenases and the brain: from zero to hero, a decade of progress. Front Neuroendocrinol (2011) 32(3):265–86. doi:10.1016/j.yfrne.2010.12.001
69. Gottfried-Blackmore A, Sierra A, Mcewen BS, Ge R, Bulloch K. Microglia express functional 11 beta-hydroxysteroid dehydrogenase type 1. Glia (2010) 58(10):1257–66. doi:10.1002/glia.21007
70. Joëls M, Karst H, DeRijk R, de Kloet ER. The coming out of the brain mineralocorticoid receptor. Trends Neurosci (2008) 31(1):1–7. doi:10.1016/j.tins.2007.10.005
71. Kolber BJ, Wieczorek L, Muglia LJ. Hypothalamic-pituitary-adrenal axis dysregulation and behavioral analysis of mouse mutants with altered glucocorticoid or mineralocorticoid receptor function. Stress (2008) 11(5):321–38. doi:10.1080/10253890701821081
72. Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem (2011) 286(5):3177–84. doi:10.1074/jbc.R110.179325
73. Beck IM, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev (2009) 30(7):830–82. doi:10.1210/er.2009-0013
74. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol (2013) 132(5):1033–44. doi:10.1016/j.jaci.2013.09.007
75. Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids (2010) 75(1):1–12. doi:10.1016/j.steroids.2009.09.002
76. Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol (2008) 4(10):525–33. doi:10.1038/ncprheum0898
77. Taniguchi Y, Iwasaki Y, Tsugita M, Nishiyama M, Taguchi T, Okazaki M, et al. Glucocorticoid receptor-beta and receptor-gamma exert dominant negative effect on gene repression but not on gene induction. Endocrinology (2010) 151(7):3204–13. doi:10.1210/en.2009-1254
78. Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Mol Cell Biol (2007) 27(6):2266–82. doi:10.1128/MCB.01439-06
79. Stahn C, Löwenberg M, Hommes DW, Buttgereit F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol Cell Endocrinol (2007) 275(1–2):71–8. doi:10.1016/j.mce.2007.05.019
80. McNally JG, Müller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science (2000) 287(5456):1262–5. doi:10.1126/science.287.5456.1262
81. Voss TC, Schiltz RL, Sung MH, Yen PM, Stamatoyannopoulos JA, Biddie SC, et al. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell (2011) 146(4):544–54. doi:10.1016/j.cell.2011.07.006
82. Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med (2006) 203(8):1883–9. doi:10.1084/jem.20060336
83. Bhattacharyya S, Zhao Y, Kay TW, Muglia LJ. Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc Natl Acad Sci U S A (2011) 108(23):9554–9. doi:10.1073/pnas.1017296108
84. D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, et al. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity (1997) 7(6):803–12. doi:10.1016/S1074-7613(00)80398-2
85. Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med (2006) 203(1):131–40. doi:10.1084/jem.20051794
86. Chinenov Y, Rogatsky I. Glucocorticoids and the innate immune system: crosstalk with the toll-like receptor signaling network. Mol Cell Endocrinol (2007) 275(1–2):30–42. doi:10.1016/j.mce.2007.04.014
87. De Bosscher K, Beck IM, Haegeman G. Classic glucocorticoids versus non-steroidal glucocorticoid receptor modulators: survival of the fittest regulator of the immune system? Brain Behav Immun (2010) 24(7):1035–42. doi:10.1016/j.bbi.2010.06.010
88. Gupte R, Muse GW, Chinenov Y, Adelman K, Rogatsky I. Glucocorticoid receptor represses proinflammatory genes at distinct steps of the transcription cycle. Proc Natl Acad Sci U S A (2013) 110(36):14616–21. doi:10.1073/pnas.1309898110
89. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell (2011) 145(2):224–41. doi:10.1016/j.cell.2011.03.027
90. Revollo JR, Oakley RH, Lu NZ, Kadmiel M, Gandhavadi M, Cidlowski JA. HES1 is a master regulator of glucocorticoid receptor-dependent gene expression. Sci Signal (2013) 6(304):ra103. doi:10.1126/scisignal.2004389
91. Almawi WY, Melemedjian OK. Negative regulation of nuclear factor-kappaB activation and function by glucocorticoids. J Mol Endocrinol (2002) 28(2):69–78. doi:10.1677/jme.0.0280069
92. Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol (2010) 120(2–3):76–85. doi:10.1016/j.jsbmb.2010.02.018
93. Flammer JR, Rogatsky I. Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol (2011) 25(7):1075–86. doi:10.1210/me.2011-0068
94. Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J (2006) 25(1):108–17. doi:10.1038/sj.emboj.7600919
95. Wolf IM, Periyasamy S, Hinds T, Yong W, Shou W, Sanchez ER. Targeted ablation reveals a novel role of FKBP52 in gene-specific regulation of glucocorticoid receptor transcriptional activity. J Steroid Biochem Mol Biol (2009) 113(1–2):36–45. doi:10.1016/j.jsbmb.2008.11.006
96. Groeneweg FL, Karst H, de Kloet ER, Joëls M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol (2012) 350(2):299–309. doi:10.1016/j.mce.2011.06.020
97. Haller J, Mikics E, Makara GB. The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Front Neuroendocrinol (2008) 29(2):273–91. doi:10.1016/j.yfrne.2007.10.004
98. Strehl C, Gaber T, Löwenberg M, Hommes DW, Verhaar AP, Schellmann S, et al. Origin and functional activity of the membrane-bound glucocorticoid receptor. Arthritis Rheum (2011) 63(12):3779–88. doi:10.1002/art.30637
99. Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, et al. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J (2004) 18(1):70–80. doi:10.1096/fj.03-0328com
100. Samarasinghe RA, Di Maio R, Volonte D, Galbiati F, Lewis M, Romero G, et al. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci U S A (2011) 108(40):16657–62. doi:10.1073/pnas.1102821108
101. Löwenberg M, Verhaar AP, van den Brink GR, Hommes DW. Glucocorticoid signaling: a nongenomic mechanism for T-cell immunosuppression. Trends Mol Med (2007) 13(4):158–63. doi:10.1016/j.molmed.2007.02.001
102. Ghosh MC, Baatar D, Collins G, Carter A, Indig F, Biragyn A, et al. Dexamethasone augments CXCR4-mediated signaling in resting human T cells via the activation of the Src kinase Lck. Blood (2009) 113(3):575–84. doi:10.1182/blood-2008-04-151803
103. Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M. Multiple actions of steroid hormones – a focus on rapid, nongenomic effects. Pharmacol Rev (2000) 52(4):513–56.
104. Van Laethem F, Liang X, Andris F, Urbain J, Vandenbranden M, Ruysschaert JM, et al. Glucocorticoids alter the lipid and protein composition of membrane rafts of a murine T cell hybridoma. J Immunol (2003) 170(6):2932–9.
105. Yamagata S, Tomita K, Sano H, Itoh Y, Fukai Y, Okimoto N, et al. Non-genomic inhibitory effect of glucocorticoids on activated peripheral blood basophils through suppression of lipid raft formation. Clin Exp Immunol (2012) 170(1):86–93. doi:10.1111/j.1365-2249.2012.04636.x
106. Beck IM, De Bosscher K, Haegeman G. Glucocorticoid receptor mutants: man-made tools for functional research. Trends Endocrinol Metab (2011) 22(8):295–310. doi:10.1016/j.tem.2011.03.009
107. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell (1998) 93(4):531–41. doi:10.1016/S0092-8674(00)81183-6
108. Tuckermann JP, Reichardt HM, Arribas R, Richter KH, Schütz G, Angel P. The DNA binding-independent function of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J Cell Biol (1999) 147(7):1365–70. doi:10.1083/jcb.147.7.1365
109. Kleiman A, Hübner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J (2012) 26(2):722–9. doi:10.1096/fj.11-192112
110. Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest (2007) 117(5):1381–90. doi:10.1172/JCI28034
111. Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, et al. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci U S A (2003) 100(24):13845–50. doi:10.1073/pnas.2336092100
112. Adams M, Meijer OC, Wang J, Bhargava A, Pearce D. Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol Endocrinol (2003) 17(12):2583–92. doi:10.1210/me.2002-0305
113. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood (2007) 109(10):4313–9. doi:10.1182/blood-2006-10-048215
114. Silverman MN, Mukhopadhyay P, Belyavskaya E, Tonelli LH, Revenis BD, Doran JH, et al. Glucocorticoid receptor dimerization is required for proper recovery of LPS-induced inflammation, sickness behavior and metabolism in mice. Mol Psychiatry (2013) 18(9):1006–17. doi:10.1038/mp.2012.131
115. Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol J-C, Lu L, et al. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci U S A (2011) 108:6632–7. doi:10.1073/pnas.1017820108
116. Carrillo-de Sauvage MÁ, Maatouk L, Arnoux I, Pasco M, Sanz Diez A, Delahaye M, et al. Potent and multiple regulatory actions of microglial glucocorticoid receptors during CNS inflammation. Cell Death Differ (2013) 20(11):1546–57. doi:10.1038/cdd.2013.108
117. Sorrells SF, Caso JR, Munhoz CD, Hu CK, Tran KV, Miguel ZD, et al. Glucocorticoid signaling in myeloid cells worsens acute CNS injury and inflammation. J Neurosci (2013) 33(18):7877–89. doi:10.1523/JNEUROSCI.4705-12.2013
118. Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci U S A (2013) 110(1):306–11. doi:10.1073/pnas.1210200110
119. Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, et al. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci U S A (2005) 102(2):473–8. doi:10.1073/pnas.0406458102
120. Jeanneteau FD, Lambert WM, Ismaili N, Bath KG, Lee FS, Garabedian MJ, et al. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc Natl Acad Sci U S A (2012) 109(4):1305–10. doi:10.1073/pnas.1114122109
121. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med (2005) 353(16):1711–23. doi:10.1056/NEJMra050541
122. Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell (2005) 122(5):707–21. doi:10.1016/j.cell.2005.06.029
123. McCoy CE, Carpenter S, Pålsson-McDermott EM, Gearing LJ, O’Neill LA. Glucocorticoids inhibit IRF3 phosphorylation in response to Toll-like receptor-3 and -4 by targeting TBK1 activation. J Biol Chem (2008) 283(21):14277–85. doi:10.1074/jbc.M709731200
124. Bhattacharyya S, Ratajczak CK, Vogt SK, Kelley C, Colonna M, Schreiber RD, et al. TAK1 targeting by glucocorticoids determines JNK and IkappaB regulation in Toll-like receptor-stimulated macrophages. Blood (2010) 115(10):1921–31. doi:10.1182/blood-2009-06-224782
125. Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science (2009) 324(5925):407–10. doi:10.1126/science.1164265
126. van Tilborg MA, Lefstin JA, Kruiskamp M, Teuben J, Boelens R, Yamamoto KR, et al. Mutations in the glucocorticoid receptor DNA-binding domain mimic an allosteric effect of DNA. J Mol Biol (2000) 301(4):947–58. doi:10.1006/jmbi.2000.4001
127. Butler LD, Layman NK, Riedl PE, Cain RL, Shellhaas J, Evans GF, et al. Neuroendocrine regulation of in vivo cytokine production and effects: I. In vivo regulatory networks involving the neuroendocrine system, interleukin-1 and tumor necrosis factor-alpha. J Neuroimmunol (1989) 24(1–2):143–53. doi:10.1016/0165-5728(89)90108-2
128. Goujon E, Parnet P, Cremona S, Dantzer R. Endogenous glucocorticoids down regulate central effects of interleukin-1 beta on body temperature and behaviour in mice. Brain Res (1995) 702(1–2):173–80. doi:10.1016/0006-8993(95)01041-9
129. Goujon E, Parnet P, Layé S, Combe C, Dantzer R. Adrenalectomy enhances pro-inflammatory cytokines gene expression, in the spleen, pituitary and brain of mice in response to lipopolysaccharide. Brain Res Mol Brain Res (1996) 36(1):53–62. doi:10.1016/0169-328X(95)00242-K
130. Masferrer JL, Seibert K, Zweifel B, Needleman P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc Natl Acad Sci U S A (1992) 89(9):3917–21. doi:10.1073/pnas.89.9.3917
131. Drew PD, Chavis JA. Inhibition of microglial cell activation by cortisol. Brain Res Bull (2000) 52(5):391–6. doi:10.1016/S0361-9230(00)00275-6
132. Tanaka J, Fujita H, Matsuda S, Toku K, Sakanaka M, Maeda N. Glucocorticoid- and mineralocorticoid receptors in microglial cells: the two receptors mediate differential effects of corticosteroids. Glia (1997) 20(1):23–37. doi:10.1002/(SICI)1098-1136(199705)20:1<23::AID-GLIA3>3.3.CO;2-U
133. Glezer I, Munhoz CD, Kawamoto EM, Marcourakis T, Avellar MC, Scavone C. MK-801 and 7-Ni attenuate the activation of brain NF-kappa B induced by LPS. Neuropharmacology (2003) 45(8):1120–9. doi:10.1016/S0028-3908(03)00279-X
134. Munhoz CD, Sorrells SF, Caso JR, Scavone C, Sapolsky RM. Glucocorticoids exacerbate lipopolysaccharide-induced signaling in the frontal cortex and hippocampus in a dose-dependent manner. J Neurosci (2010) 30(41):13690–8. doi:10.1523/JNEUROSCI.0303-09.2010
135. Blais V, Zhang J, Rivest S. In altering the release of glucocorticoids, ketorolac exacerbates the effects of systemic immune stimuli on expression of proinflammatory genes in the brain. Endocrinology (2002) 143(12):4820–7. doi:10.1210/en.2002-220598
136. Blais V, Turrin NP, Rivest S. Cyclooxygenase 2 (COX-2) inhibition increases the inflammatory response in the brain during systemic immune stimuli. J Neurochem (2005) 95(6):1563–74. doi:10.1111/j.1471-4159.2005.03480.x
137. Nadeau S, Rivest S. Endotoxemia prevents the cerebral inflammatory wave induced by intraparenchymal lipopolysaccharide injection: role of glucocorticoids and CD14. J Immunol (2002) 169(6):3370–81.
138. Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J Neurosci (2003) 23(13):5536–44.
139. Soulet D, Rivest S. Bone-marrow-derived microglia: myth or reality? Curr Opin Pharmacol (2008) 8(4):508–18. doi:10.1016/j.coph.2008.04.002
140. Frank MG, Miguel ZD, Watkins LR, Maier SF. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun (2010) 24(1):19–30. doi:10.1016/j.bbi.2009.07.008
141. Johnson JD, O’Connor KA, Deak T, Stark M, Watkins LR, Maier SF. Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun (2002) 16(4):461–76. doi:10.1006/brbi.2001.0638
142. Johnson JD, O’Connor KA, Hansen MK, Watkins LR, Maier SF. Effects of prior stress on LPS-induced cytokine and sickness responses. Am J Physiol Regul Integr Comp Physiol (2003) 284(2):R422–32.
143. Munhoz CD, Lepsch LB, Kawamoto EM, Malta MB, Lima Lde S, Avellar MC, et al. Chronic unpredictable stress exacerbates lipopolysaccharide-induced activation of nuclear factor-kappaB in the frontal cortex and hippocampus via glucocorticoid secretion. J Neurosci (2006) 26(14):3813–20. doi:10.1523/JNEUROSCI.4398-05.2006
144. de Pablos RM, Villarán RF, Argüelles S, Herrera AJ, Venero JL, Ayala A, et al. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci (2006) 26(21):5709–19. doi:10.1523/JNEUROSCI.0802-06.2006
145. Espinosa-Oliva AM, de Pablos RM, Villarán RF, Argüelles S, Venero JL, Machado A, et al. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol Aging (2011) 32(1):85–102. doi:10.1016/j.neurobiolaging.2009.01.012
146. Frank MG, Watkins LR, Maier SF. Stress-induced glucocorticoids as a neuroendocrine alarm signal of danger. Brain Behav Immun (2013) 33:1–6. doi:10.1016/j.bbi.2013.02.004
147. Blandino P, Barnum CJ, Solomon LG, Larish Y, Lankow BS, Deak T. Gene expression changes in the hypothalamus provide evidence for regionally-selective changes in IL-1 and microglial markers after acute stress. Brain Behav Immun (2009) 23(7):958–68. doi:10.1016/j.bbi.2009.04.013
148. Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun (2007) 21(1):47–59. doi:10.1016/j.bbi.2006.03.005
149. García-Bueno B, Madrigal JL, Pérez-Nievas BG, Leza JC. Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-gamma activation after stress in rats. Endocrinology (2008) 149(4):1969–78. doi:10.1210/en.2007-0482
150. Madrigal JL, Hurtado O, Moro MA, Lizasoain I, Lorenzo P, Castrillo A, et al. The increase in TNF-alpha levels is implicated in NF-kappaB activation and inducible nitric oxide synthase expression in brain cortex after immobilization stress. Neuropsychopharmacology (2002) 26(2):155–63. doi:10.1016/S0893-133X(01)00292-5
151. Madrigal JL, Moro MA, Lizasoain I, Lorenzo P, Fernández AP, Rodrigo J, et al. Induction of cyclooxygenase-2 accounts for restraint stress-induced oxidative status in rat brain. Neuropsychopharmacology (2003) 28(9):1579–88. doi:10.1038/sj.npp.1300187
152. O’Connor KA, Johnson JD, Hammack SE, Brooks LM, Spencer RL, Watkins LR, et al. Inescapable shock induces resistance to the effects of dexamethasone. Psychoneuroendocrinology (2003) 28(4):481–500. doi:10.1016/S0306-4530(02)00035-5
153. Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience (2007) 147(4):867–83. doi:10.1016/j.neuroscience.2007.02.055
154. Ding Y, Gao ZG, Jacobson KA, Suffredini AF. Dexamethasone enhances ATP-induced inflammatory responses in endothelial cells. J Pharmacol Exp Ther (2010) 335(3):693–702. doi:10.1124/jpet.110.171975
155. Lannan EA, Galliher-Beckley AJ, Scoltock AB, Cidlowski JA. Proinflammatory actions of glucocorticoids: glucocorticoids and TNFα coregulate gene expression in vitro and in vivo. Endocrinology (2012) 153(8):3701–12. doi:10.1210/en.2012-1020
156. Goujon E, Parnet P, Laye S, Combe C, Kelley KW, Dantzer R. Stress downregulates lipopolysaccharide-induced expression of proinflammatory cytokines in the spleen, pituitary, and brain of mice. Brain Behav Immun (1995) 9(4):292–303. doi:10.1006/brbi.1995.1028
157. Barnum CJ, Pace TW, Hu F, Neigh GN, Tansey MG. Psychological stress in adolescent and adult mice increases neuroinflammation and attenuates the response to LPS challenge. J Neuroinflammation (2012) 9:9. doi:10.1186/1742-2094-9-9
158. Fantuzzi G, Demitri MT, Ghezzi P. Differential effect of glucocorticoids on tumour necrosis factor production in mice: up-regulation by early pretreatment with dexamethasone. Clin Exp Immunol (1994) 96(1):166–9. doi:10.1111/j.1365-2249.1994.tb06247.x
159. Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci (2012) 13(1):22–37. doi:10.1038/nrn3138
160. Sorrells SF, Caso JR, Munhoz CD, Sapolsky RM. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron (2009) 64(1):33–9. doi:10.1016/j.neuron.2009.09.032
161. Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci (2001) 4(11):1116–22. doi:10.1038/nn738
162. Herx LM, Rivest S, Yong VW. Central nervous system-initiated inflammation and neurotrophism in trauma: IL-1 beta is required for the production of ciliary neurotrophic factor. J Immunol (2000) 165(4):2232–9.
Keywords: HPA axis, glucocorticoids, glucocorticoid receptor, inflammation, microglia, stress
Citation: Bellavance M-A and Rivest S (2014) The HPA – immune axis and the immunomodulatory actions of glucocorticoids in the brain. Front. Immunol. 5:136. doi: 10.3389/fimmu.2014.00136
Received: 09 January 2014; Paper pending published: 01 March 2014;
Accepted: 18 March 2014; Published online: 31 March 2014.
Edited by:
Oreste Gualillo, SERGAS (Servizo Galego de Saude), Santiago University Clinical Hospital, SpainReviewed by:
Giamila Fantuzzi, University of Illinois at Chicago, USACopyright: © 2014 Bellavance and Rivest. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serge Rivest, Faculty of medicine, Department of Molecular Medicine, Neuroscience Laboratory, CHU de Québec Research Center, Laval University, 2705 Laurier Blvd, Québec, QC G4V 1G2, Canada e-mail:c2VyZ2Uucml2ZXN0QGNyY2h1bC51bGF2YWwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.