

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 01 October 2013
Sec. Antigen Presenting Cell Biology
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00308
This article is part of the Research Topic Processing, editing, and presenting the MHC class II peptidome View all 21 articles
The adaptive immune response begins when CD4+ T cells recognize antigenic peptides bound to class II molecules of the Major Histocompatibility Complex (MHCII). The interaction between peptides and MHCII has been historically interpreted as a rigid docking event. However, this model has been challenged by the evidence that conformational flexibility plays an important role in peptide-MHCII complex formation. Thermodynamic analysis of the binding reaction suggests a model of complexation in which the physical-chemical nature of the peptide determines the variability in flexibility of the substates in the peptide-MHC conformational ensemble. This review discusses our understanding of the correlation between thermodynamics of peptide binding and structural features of the resulting complex as well as their impact on HLA-DM activity and on our ability to predict MHCII-restricted epitopes.
Upon immunization with a complex antigen, or challenge by a pathogen, an organism’s CD4+ T cells recognize antigen-derived peptides bound to MHC class II molecules (MHCII) exposed on the surface of antigen presenting cells (APCs). A critical feature of the elicited T cell response is the phenomenon of immunodominance (1). This refers to the evidence that, despite the diversity of the T cell repertoire, the co-expression of different MHCII alleles and the potential generation of a panoply of distinct peptides, a large proportion of the responding T cells tends to be specific for a few peptide determinants, known as immunodominant antigens. A relevant observation related to immunodominance is the evidence that complex antigens have many sequences that can bind to the MHC molecule and trigger a T cell response if exogenously administered as single peptides. These latter sequences, which can be recognized only after immunization with the peptide and not after processing of the full protein, are known as “cryptic.” Efforts have been made to understand the mechanisms behind these phenomena. Analysis of the antigen processing and presentation pathway indicates that elements of the epitope selection process are critical to the generation of the peptide repertoire presented to T cells (2). MHCII molecules are transported from the endoplasmic reticulum to the MHCII compartments (MIIC) as multimeric complexes with the chaperone protein invariant chain (Ii). Ii stabilizes the nascent MHCII and prevents binding of other endoplasmic reticulum-resident peptides. Upon arrival in the MIIC, the Ii molecule is cleaved by proteases, leaving a low-affinity peptide fragment named class II-associated invariant peptide (CLIP) in the MHCII binding groove. CLIP is then released to allow peptides derived by endosomal fragmentation of antigenic proteins to be selected for presentation. Evidence collected thus far in the analysis of immunodominance indicates that a significant correlation exists between affinity and kinetic stability of a peptide-MHCII dyad, its density on the surface of the priming APC and the selection of a responding T cell repertoire (1, 3, 4).
The multitude of peptides, which can be derived from a protein, includes a consistent number of low-affinity ligands for a given MHC allele, as well as peptides with intermediate affinity and few high-affinity ones. Thus, under an immunodominance-based model of T cell response, MHCII molecules must reach a thermodynamic equilibrium in which they are complexed with the highest affinity binders. Considering that the transit time of an MHCII through the MIIC is comparable to the dissociation rate of many low-intermediate affinity peptides, it is difficult to presuppose that the MHC-binder dyad is able to reach such equilibrium intrinsically. Rather, one would expect that the majority of MHC molecules is bound to low-affinity peptides that are in excess (including CLIP) as a consequence of a kinetic control of the selection process (5, 6). So what can possibly explain the evidence of immunodominance? One reasonable hypothesis would be the existence of mechanisms that can enhance the thermodynamic control of the peptide-binding process.
HLA-DM (DM) is a MIIC-resident MHCII-like molecule that plays an important role in MHCII-restricted epitope selection. In the presence of DM, CLIP removal from the newly synthesized MHCII is facilitated, and the presented peptide-MHCII repertoire is skewed in favor of kinetically stable, high-affinity peptide-MHCII complexes. Based on these well-known effects, DM could be considered the likeliest factor determining the thermodynamic equilibrium of the endosomal machinery. In consideration of the impact of binding energetics on the outcome of epitope selection, this article will review of the thermodynamics of peptide interaction with MHCII, its correlation to complex structure and how these relate to DM activity.
MHCII binds peptides in a groove defined by a β-sheet floor and two parallel helical sides. The groove is characterized by hydrophobic pockets and by a network of H-bonds that can form between MHCII side chains and the peptide backbone (7). MHCII show extensive polymorphism predominantly restricted to the peptide-binding groove. The contacts observed in available MHCII structures have helped cement an interpretation of MHCII-peptide-binding specificity based on hydrophobic pocket structure and charge matching (8). This interpretation reflects the historical model for explaining specificity of ligand-receptor interaction, namely the docking mechanism, which relies on structural rigidity and complementarity of shape (and possibly charge). The energetic contribution of each interaction – hydrophobic or H-bonding – has been evaluated by mutating individually single anchor residues or individually destabilizing specific H-bonds (9–11). Attempts have been made to predict peptide affinity by adding the energetic contribution to binding of each single interaction expected to be formed between the peptide and the MHCII. Indeed, several epitope prediction algorithms have been engineered on the assumption that peptide binding is a rigid, enthalpy-based process (12–16), but their specificity and sensitivity are questionable (17–19).
Biochemical and biophysical analyses have indicated an enhanced conformational heterogeneity or flexibility of the protein in the absence of a peptide (20–23). Several studies indicated that MHCII molecules undergo a conformational transition during peptide binding or exchange from a heterogenic state to a more rigid and stable state (21, 24, 25). The latter group of reports suggests that the mechanistic details of the peptide loading process are more complicated than those provided by a simple enthalpy-based docking model, and further analysis has confirmed this (26, 27). The ability to correlate MHCII and peptide structural rearrangement occurring during the interaction with the energetics of the binding process would provide important insights into the mechanisms controlling epitope selection. Whereas the literature concerning the thermodynamic properties of peptide-MHCII formation and their structural correlates is not extensive, most published observations indicate that complex formation can be interpreted as an instance of protein folding, and they will be reviewed in what follows.
The correlation between structural aspects of a peptide-MHCII complex and its thermodynamic features has been analyzed initially by thermal denaturation studies. Thermal stability of murine (28, 29) and human (20, 21, 30) MHCII, both empty and bound to different peptides at varied pH conditions, indicated that empty class II molecules have a different structure in terms of helicity as compared to bound structures. Moreover, binding of peptides with different length or sequence may result in complexes with different structure, and the derived thermodynamic parameters indicate that affinity of the peptide impacts the folding and unfolding of the complex.
The question of peptide binding as an instance of protein folding has been approached in recent times by probing cooperativity (31). Cooperativity in a multipoint ligand-receptor binding event is measured when one interaction is affected by other interactions. A general strategy to probe the occurrence of such a phenomenon is the mutant cycle approach (32). Mutant cycle analysis has been performed extensively in other systems to characterize transition states of folding and folding intermediates. This method consists of introducing multiple substitutions in the sequence of the reactants and assessing their binding parameters. If the effect on the binding free energy of the double (or multiple) mutation is not equal to the sum of effects of the single mutations, then the residues are coupled (cooperative). The first analysis of cooperativity with this strategy was performed on the I-Ad/HA (126–138) system (33, 34). The I-Ad MHCII protein binds peptides with high-affinity also in the absence of strong anchor, particularly relying on H-bonds. This first analysis indicated that the energetic contribution of a given anchor-pocket interaction is a function of the peptide sequence, suggesting that the binding energy is indeed distributed across the peptide, with a significant fraction sequestered in the N-terminal side of the complex. Whereas the H-bond network showed cooperativity, no detectable cooperativity between the anchor-pocket and H-bonding interactions was observed, even at a short distance, suggesting that for this allele, encapsulation of peptide side chains into MHCII pockets is independent and probably upstream of H-bond formation.
A similar strategy was applied to determine the extent of cooperativity the HLA-DR1 (DR1)/HA (306–319) system undergoes during complex formation or peptide release (35, 36). Several clusters of disruptive mutations have been applied to the peptide or the peptide and MHC up to a five-mutant construct, and they have evidenced cooperative effects in peptide binding to and release from MHCII. Due to the disruptive nature of the substitutions, cooperativity has been interpreted in terms of lack of folding (i.e., what is the effect of destabilizing a second interaction on the folding of the complex once a first interaction has been disrupted?). Cooperative effects impact peptide affinity or complex stability in an exponential fashion (35, 36). Differently than in the case of I-Ad, energetic coupling was observed between anchor/pocket interactions and H-bonds, when respectively located at opposite sides of the binding groove. A possible explanation for the different cooperativity in the two systems may be related to the evidence that the two alleles are differentially reliant on sequestration of peptide hydrophobic side chains into pockets for tight binding. This discrepancy, however, highlights the difficulty of finding a model of peptide-binding valid for all the class II alleles.
Analysis of cooperativity has indicated that, prior to the exponential increase phase, an energy window exists, in which cooperativity (lack of folding) cannot be measured (35, 36). Within this window, indicated as compensatory region, peptides with different chemistry can bind with relatively comparable free energy decrease, though the relative enthalpic and entropic contributions differ significantly. This phenomenon, known as (isothermal) entropy-enthalpy compensation, has been observed in several flexible binding systems, in which a decrease (or increase) in the ability of forming binding interactions (enthalpy) can be counterbalanced by the increase (or decrease) of search of conformational space (37).
Following the spectroscopic interpretation of the interplay between cooperativity and entropy-enthalpy compensation (38), important insights have been gained into the correlation between peptide-MHCII complex structure and thermodynamics (37). As the interaction between peptides and MHCII molecules can be considered a ligand-receptor multipoint binding, each single interaction leads to a local modification of both structure and energetics. Such local modification can be thermodynamically described for each point system, which is defined by all the atoms within the interacting core whose radius is determined by the structural features of the same protein. While peptide and MHCII molecule interact, each point system will be affected by other point systems (cooperativity) to an extent proportional to the flexibility of the molecules and the distance between the two points. Since the protein and in particular some regions of the binding groove fluctuate, then the effect of flexibility on energetic coupling of multiple point interactions is correlated to the range and frequency of fluctuations, which change as new interactions are established. Fluctuating interactions are associated with a component energy and entropy. The enthalpic term of binding energy describes the stored energy in the protein due to conformational change associated with the binding, whereas fluctuations of the entire protein manifest themselves in the conformational entropy term. As new point systems are engaged in interactions, the enthalpic contribution to free energy of binding increases. The conformational change associated with one interaction would facilitate formation of a new cluster of interaction (positive cooperativity). The entropic contribution to binding decreases, however, since any new interaction also restrains conformational flexibility; therefore, the net gain in binding energy by cooperativity is minimal/null. For this reason high-affinity peptides cannot be constructed just by adding more and more sources of binding. Indeed it has been shown that the same high-affinity peptide binds better in the absence of an H-bond, as compared to when the same H-bond is present (37).
Importantly, it was found that the compensatory mechanism is not absolute, that is increasing in conformational flexibility consequent to lack of viable interactions between peptide and MHCII cannot overcome extremely poor enthalpic contributions. This phenomenon was evidenced by identifying a peptide affinity limit above which cooperativity (as exaggerated lack of folding) increased exponentially upon addition of disruptive mutation to the complex (37).
Thus, within the compensatory region, different peptides may form a complex with MHCII displaying similar IC50 in equilibrium-based competition binding assays, or comparable Kd in titration calorimetry, but with different enthalpic and entropic contributions. The diversity in thermodynamic strategy adopted by the system will reflect in the range and frequency of the fluctuations of each point system and the structure of the complex overall. The consequence is that the macrostate of a complex whose formation is highly enthalpic and whose conformational mobility is greatly restrained will be a collection of similar peptide-MHCII dyads where most if not all point systems is engaged in interactions with minimal liability at every point in time. On the contrary, complexes whose formation is correlated to a smaller entropic penalty are a collection of dyads whose greater conformation mobility is attempting to optimize the available interactions leading, at the same time, to structural variability. This is why postulating that binding of different peptides to a given MHCII results in “equal” complexes from thermodynamic and possibly structural standpoints is not justified, even when the measured affinities are comparable.
DM function has been object of intense research in the past two decades, and we have recently gained important, though not conclusive, insights into its peptide editing activity. Other articles in this issue will review structure and editing function of DM, and only few aspects of DM activity will be highlighted here, which can be related to a thermodynamic/structural model of epitope selection.
Understanding DM function requires addressing several questions: (1) what renders a petide-MHCII complex susceptible to DM? (2) What is the effect of DM on the complex? (3) How does DM action result in selective binding of high-affinity peptides? Recent reports from different groups indicate that DM binds a short-lived MHCII intermediate whose N-terminal region features a specific conformation (39–42). This interaction seems to result in a stable, long-lived DM-DR complex after full dissociation of the prebound peptide has occurred (41). Still unclear is how the DM-labile conformation is generated. Historically, specific sources of binding energy have been indicated as responsible for generating DM-labile complexes (43–48), but now it appears that weakening of both H-bonds and “anchor-pocket” interactions results in susceptibility to DM (49–53). This latter observation would be consistent with a distributed model of peptide binding, where every position, to a different extent, affects the overall capacity of the peptide to bind, and the conformational flexibility of the complex. How does the overall binding property of the peptide determine DM susceptibility of the complex? One intriguing explanation would consider the interplay between compensatory mechanism and cooperativity as responsible for determining the probability by which the complex assumes a DM-labile conformation. Indeed, a complex whose formation is correlated to a smaller entropic penalty is expected to feature greater conformational mobility as compared to an isoenergetic complex whose formation requires a larger enthalpic contribution. As a consequence, the frequency increases at which regions within the N-terminal side of those complexes are disengaged from peptide interaction and are more amenable to DM. One observation in support of this hypothesis is the evidence that peptide-loaded MHCII conformers can be differentially susceptible to DM-mediated peptide release, and that the generation of the labile isomer is a function of the affinity of the bound peptide for DR (54), as well as the respective fraction of entropic contribution to the binding energy (37). Thus, when looking at the peptide release activity of DM, the thermodynamic strategy adopted by the peptide to bind seems to play a crucial role.
DM function needs to be considered in the context of peptide exchange, not only peptide release. The addition of an exchange peptide to the reaction is a confounding factor to the understanding of DM mechanism. Indeed it has been shown that exchange peptides are not simply replacing freshly dissociated peptides, but they intervene in, and possibly trigger the formation of a metastable intermediate that also includes the prebound peptide tethered to the MHCII (36). In particular, the role of the exchange peptide as “co-factor” in DM-mediated peptide exchange has been identified through its ability to convert DM-stable into DM-labile conformers (54).
For the active role played by the exchange peptide during a displacement reaction, the question of DM-mediated peptide exchange has been approached in terms of DM effect on the unfolding of the prebound peptide-MHCII complex and refolding around exchange peptide (36). When the analysis of cooperativity was performed in the presence of DM, no cooperativity could be observed in the release of the prebound peptide, contrasting what was observed for the intrinsic off-rate (35, 36). This evidence was interpreted as an indication that, once DM interacts with a complex, it promotes a dramatic disruption of the interactions between MHCII and the peptide, so that the typical coordinate unfolding of the intrinsic release cannot be measured. Interestingly, measuring cooperativity for the exchange peptide revealed that the latter needs to fold into the groove more efficiently than the prebound to displace it, and DM increases the energetic threshold that the exchange peptide has to overcome to displace the prebound peptide.
These results, along with structural (41) and molecular dynamic simulation analysis (55), supports a model in which DM interacts with complexes featuring a rearranged N-terminal and stabilizes the binding groove in a receptive state; DM shifts peptide exchange control from kinetic to thermodynamic. Moreover, DM interaction requires the engagement of MHCII residues, which would be otherwise elements of point system(s) with the peptide (39–42). Therefore, peptides are stably bound only when they effectively compete against DM to interact with DR residues for access to the N-terminal and if the overall binding energy is such to reduce the conformational lability. Such high-affinity, high-enthalpy binding would reverse the conformational rearrangement required for DM binding, thus leading to DM dissociation (Figure 1). Further studies are required to assess the correlation between thermodynamic signature of a peptide-MHCII complex, its conformational flexibility and susceptibility to DM-mediated peptide exchange. If confirmed, a similar mechanism would explain how the system reaches thermodynamic equilibrium with high-affinity binders in a timeframe compatible with epitope selection by the endosomal machinery.
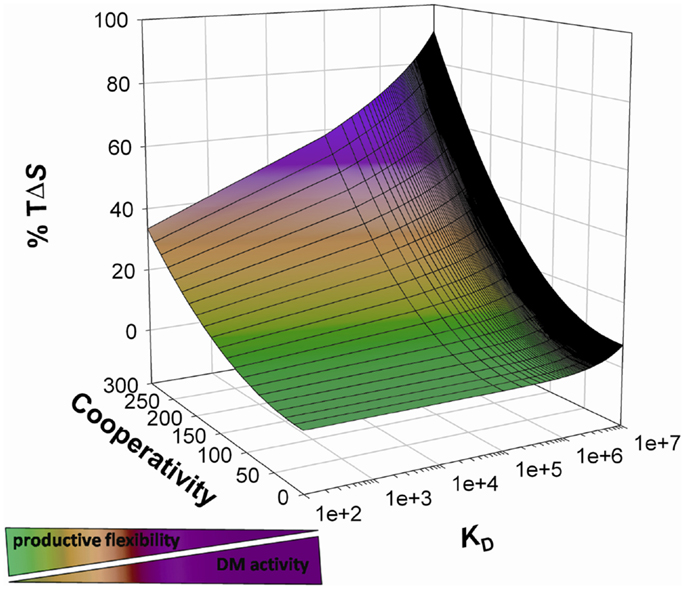
Figure 1. Thermodynamic model of peptide binding and DM action. The probability of a peptide binding to a given HLAII correlates with the probability of the interplay between cooperativity and compensation (here indicated as entropic contribution to free energy decrease) resulting in the optimization of all the available interactions. DM interacts with complexes featuring residual high entropy, promotes peptide release, and skews the presented peptide repertoire toward highly enthalpic complexes by increasing the effective free energy threshold for binding.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I wish to specially thank Dr. Jack Gorski for his remarkable mentorship and for his inspiring creative thinking. Funding for the research described here was provided by NIH grant R01AI63016 to Dr. Gorski. This work was supported by National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM103395, by the National Institute of Allergy and Infectious Diseases under Award Number 1R56AI098136, and by the Pfizer-sponsored Aspire Award Number WS1907326. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health or Pfizer. The author has no financial conflicts of interest.
1. Weaver JM, Sant AJ. Understanding the focused CD4 T cell response to antigen and pathogenic organisms. Immunol Res (2009) 45(2–3):123–43. doi: 10.1007/s12026-009-8095-8
2. Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol (2011) 11(12):823–36. doi:10.1038/nri3084
3. Leddon SA, Sant AJ. Generation of MHC class II-peptide ligands for CD4 T-cell allorecognition of MHC class II molecules. Curr Opin Organ Transplant (2010) 15(4):505–11. doi:10.1097/MOT.0b013e32833bfc5c
4. Yin L, Calvo-Calle JM, Dominguez-Amorocho O, Stern LJ. HLA-DM constrains epitope selection in the human CD4 T cell response to vaccinia virus by favoring the presentation of peptides with longer HLA-DM-mediated half-lives. J Immunol (2012) 189(8):3983–94. doi:10.4049/jimmunol.1200626
5. Elliott T, Williams A. The optimization of peptide cargo bound to MHC class I molecules by the peptide-loading complex. Immunol Rev (2005) 207:89–99. doi:10.1111/j.0105-2896.2005.00311.x
6. Schneeweiss C, Garstka M, Smith J, Hutt MT, Springer S. The mechanism of action of tapasin in the peptide exchange on MHC class I molecules determined from kinetics simulation studies. Mol Immunol (2009) 46(10):2054–63. doi:10.1016/j.molimm.2009.02.032
7. Nelson CA, Fremont DH. Structural principles of MHC class II antigen presentation. Rev Immunogenet (1999) 1(1):47–59.
8. Rammensee HG. Chemistry of peptides associated with MHC class I and class II molecules. Curr Opin Immunol (1995) 7(1):85–96. doi:10.1016/0952-7915(95)80033-6
9. Jardetzky TS, Gorga JC, Busch R, Rothbard J, Strominger JL, Wiley DC. Peptide binding to HLA-DR1: a peptide with most residues substituted to alanine retains MHC binding. EMBO J (1990) 9(6):1797–803.
10. Hill CM, Liu A, Marshall KW, Mayer J, Jorgensen B, Yuan B, et al. Exploration of requirements for peptide binding to HLA DRB1*0101 and DRB1*0401. J Immunol (1994) 152(6):2890–8.
11. Nelson CA, Viner N, Young S, Petzold S, Benoist C, Mathis D, et al. Amino acid residues on the I-Ak alpha-chain required for the binding and stability of two antigenic peptides. J Immunol (1996) 156(1):176–82.
12. Reche PA, Glutting JP, Zhang H, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics (2004) 56(6):405–19. doi:10.1007/s00251-004-0709-7
13. Altuvia Y, Margalit H. A structure-based approach for prediction of MHC-binding peptides. Methods (2004) 34(4):454–9. doi:10.1016/j.ymeth.2004.06.008
14. Schueler-Furman O, Altuvia Y, Sette A, Margalit H. Structure-based prediction of binding peptides to MHC class I molecules: application to a broad range of MHC alleles. Protein Sci (2000) 9(9):1838–46. doi:10.1110/ps.9.9.1838
15. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics (1999) 50(3–4):213–9. doi:10.1007/s002510050595
16. Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol (1999) 17(6):555–61. doi:10.1038/9858
17. Zhu S, Udaka K, Sidney J, Sette A, Aoki-Kinoshita KF, Mamitsuka H. Improving MHC binding peptide prediction by incorporating binding data of auxiliary MHC molecules. Bioinformatics (2006) 22(13):1648–55. doi:10.1093/bioinformatics/btl141
18. Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC Bioinformatics (2008) 9(Suppl 12):S22. doi:10.1186/1471-2105-9-S12-S22
19. Sieker F, May A, Zacharias M. Predicting affinity and specificity of antigenic peptide binding to major histocompatibility class I molecules. Curr Protein Pept Sci (2009) 10(3):286–96.
20. Zarutskie JA, Sato AK, Rushe MM, Chan IC, Lomakin A, Benedek GB, et al. A conformational change in the human major histocompatibility complex protein HLA-DR1 induced by peptide binding. Biochemistry (1999) 38(18):5878–87. doi:10.1021/bi983048m
21. Sato AK, Zarutskie JA, Rushe MM, Lomakin A, Natarajan SK, Sadegh-Nasseri S, et al. Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J Biol Chem (2000) 275(3):2165–73. doi:10.1074/jbc.275.3.2165
22. Carven GJ, Stern LJ. Probing the ligand-induced conformational change in HLA-DR1 by selective chemical modification and mass spectrometric mapping. Biochemistry (2005) 44(42):13625–37. doi:10.1021/bi050972p
23. Painter CA, Cruz A, Lopez GE, Stern LJ, Zavala-Ruiz Z. Model for the peptide-free conformation of class II MHC proteins. PLoS ONE (2008) 3(6):e2403. doi:10.1371/journal.pone.0002403
24. Sadegh-Nasseri S, Germain RN. A role for peptide in determining MHC class II structure. Nature (1991) 353(6340):167–70. doi:10.1038/353167a0
25. Sadegh-Nasseri S, Germain RN. How MHC class II molecules work: peptide-dependent completion of protein folding. Immunol Today (1992) 13(2):43–6. doi:10.1016/0167-5699(92)90131-P
26. Kasson PM, Rabinowitz JD, Schmitt L, Davis MM, McConnell HM. Kinetics of peptide binding to the class II MHC protein I-Ek. Biochemistry (2000) 39(5):1048–58. doi:10.1021/bi9921337
27. Joshi RV, Zarutskie JA, Stern LJ. A three-step kinetic mechanism for peptide binding to MHC class II proteins. Biochemistry (2000) 39(13):3751–62. doi:10.1021/bi9923656
28. Reich Z, Altman JD, Boniface JJ, Lyons DS, Kozono H, Ogg G, et al. Stability of empty and peptide-loaded class II major histocompatibility complex molecules at neutral and endosomal pH: comparison to class I proteins. Proc Natl Acad Sci U S A (1997) 94(6):2495–500. doi:10.1073/pnas.94.6.2495
29. Tobita T, Oda M, Morii H, Kuroda M, Yoshino A, Azuma T, et al. A role for the P1 anchor residue in the thermal stability of MHC class II molecule I-Ab. Immunol Lett (2003) 85(1):47–52. doi:10.1016/S0165-2478(02)00206-7
30. Sato AK, Sturniolo T, Sinigaglia F, Stern LJ. Substitution of aspartic acid at beta57 with alanine alters MHC class II peptide binding activity but not protein stability: HLA-DQ (alpha1*0201, beta1*0302) and (alpha1*0201, beta1*0303). Hum Immunol (1999) 60(12):1227–36. doi:10.1016/S0198-8859(99)00120-2
31. Horovitz A, Fersht AR. Co-operative interactions during protein folding. J Mol Biol (1992) 224(3):733–40. doi:10.1016/0022-2836(92)90557-Z
32. Horovitz A, Fersht AR. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J Mol Biol (1990) 214(3):613–7. doi:10.1016/0022-2836(90)90275-Q
33. McFarland BJ, Katz JF, Beeson C, Sant AJ. Energetic asymmetry among hydrogen bonds in MHC class II*peptide complexes. Proc Natl Acad Sci U S A (2001) 98(16):9231–6. doi:10.1073/pnas.151131498
34. McFarland BJ, Katz JF, Sant AJ, Beeson C. Energetics and cooperativity of the hydrogen bonding and anchor interactions that bind peptides to MHC class II protein. J Mol Biol (2005) 350(1):170–83. doi:10.1016/j.jmb.2005.04.069
35. Anderson MW, Gorski J. Cooperativity during the formation of peptide/MHC class II complexes. Biochemistry (2005) 44(15):5617–24. doi:10.1021/bi048675s
36. Ferrante A, Gorski J. Cooperativity of hydrophobic anchor interactions: evidence for epitope selection by MHC class II as a folding process. J Immunol (2007) 178(11):7181–9.
37. Ferrante A, Gorski J. Enthalpy-entropy compensation and cooperativity as thermodynamic epiphenomena of structural flexibility in ligand-receptor interactions. J Mol Biol (2012) 417(5):454–67. doi:10.1016/j.jmb.2012.01.057
38. Hunter CA, Tomas S. Cooperativity, partially bound states, and enthalpy-entropy compensation. Chem Biol (2003) 10(11):1023–32. doi:10.1016/j.chembiol.2003.10.009
39. Painter CA, Negroni MP, Kellersberger KA, Zavala-Ruiz Z, Evans JE, Stern LJ. Conformational lability in the class II MHC 310 helix and adjacent extended strand dictate HLA-DM susceptibility and peptide exchange. Proc Natl Acad Sci U S A (2011) 108(48):19329–34. doi:10.1073/pnas.1108074108
40. Guce AI, Mortimer SE, Yoon T, Painter CA, Jiang W, Mellins ED, et al. HLA-DO acts as a substrate mimic to inhibit HLA-DM by a competitive mechanism. Nat Struct Mol Biol (2013) 20(1):90–8. doi:10.1038/nsmb.2460
41. Pos W, Sethi DK, Call MJ, Schulze MS, Anders AK, Pyrdol J, et al. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell (2012) 151(7):1557–68. doi:10.1016/j.cell.2012.11.025
42. Anders AK, Call MJ, Schulze MS, Fowler KD, Schubert DA, Seth NP, et al. HLA-DM captures partially empty HLA-DR molecules for catalyzed removal of peptide. Nat Immunol (2011) 12(1):54–61. doi:10.1038/ni.1967
43. Weber DA, Evavold BD, Jensen PE. Enhanced dissociation of HLA-DR-bound peptides in the presence of HLA-DM. Science (1996) 274(5287):618–20. doi:10.1126/science.274.5287.618
44. Raddrizzani L, Bono E, Vogt AB, Kropshofer H, Gallazzi F, Sturniolo T, et al. Identification of destabilizing residues in HLA class II-selected bacteriophage display libraries edited by HLA-DM. Eur J Immunol (1999) 29(2):660–8. doi:10.1002/(SICI)1521-4141(199902)29:02<660::AID-IMMU660>3.0.CO;2-I
45. Sant AJ, Beeson C, McFarland B, Cao J, Ceman S, Bryant PW, et al. Individual hydrogen bonds play a critical role in MHC class II: peptide interactions: implications for the dynamic aspects of class II trafficking and DM-mediated peptide exchange. Immunol Rev (1999) 172:239–53. doi:10.1111/j.1600-065X.1999.tb01369.x
46. Marin-Esteban V, Falk K, Rotzschke O. “Chemical analogues” of HLA-DM can induce a peptide-receptive state in HLA-DR molecules. J Biol Chem (2004) 279(49):50684–90. doi:10.1074/jbc.M407598200
47. Narayan K, Chou CL, Kim A, Hartman IZ, Dalai S, Khoruzhenko S, et al. HLA-DM targets the hydrogen bond between the histidine at position beta81 and peptide to dissociate HLA-DR-peptide complexes. Nat Immunol (2007) 8(1):92–100. doi:10.1038/ni1414
48. Chou CL, Sadegh-Nasseri S. HLA-DM recognizes the flexible conformation of major histocompatibility complex class II. J Exp Med (2000) 192(12):1697–706. doi:10.1084/jem.192.12.1697
49. Bandyopadhyay A, Arneson L, Beeson C, Sant AJ. The relative energetic contributions of dominant P1 pocket versus hydrogen bonding interactions to peptide: class II stability: implications for the mechanism of DM function. Mol Immunol (2008) 45(5):1248–57. doi:10.1016/j.molimm.2007.09.011
50. Ferrante A, Anderson MW, Klug CS, Gorski J. HLA-DM mediates epitope selection by a “compare-exchange” mechanism when a potential peptide pool is available. PLoS ONE (2008) 3(11):e3722. doi:10.1371/journal.pone.0003722
51. Zhou Z, Callaway KA, Weber DA, Jensen PE. Cutting edge: HLA-DM functions through a mechanism that does not require specific conserved hydrogen bonds in class II MHC-peptide complexes. J Immunol (2009) 183(7):4187–91. doi:10.4049/jimmunol.0901663
52. Ferrante A, Gorski J. Cutting edge: HLA-DM-mediated peptide exchange functions normally on MHC class II-peptide complexes that have been weakened by elimination of a conserved hydrogen bond. J Immunol (2010) 184(3):1153–8. doi:10.4049/jimmunol.0902878
53. Belmares MP, Busch R, Wucherpfennig KW, McConnell HM, Mellins ED. Structural factors contributing to DM susceptibility of MHC class II/peptide complexes. J Immunol (2002) 169(9):5109–17.
54. Ferrante A, Gorski JA. Peptide/MHCII conformer generated in the presence of exchange peptide is substrate for HLA-DM editing. Sci Rep (2012) 2:386. doi:10.1038/srep00386
Keywords: antigen presentation, MHC class II, peptide binding, thermodynamics
Citation: Ferrante A (2013) Thermodynamics of peptide-MHC class II interactions: not all complexes are created equal. Front. Immunol. 4:308. doi: 10.3389/fimmu.2013.00308
Received: 30 July 2013; Paper pending published: 18 August 2013;
Accepted: 15 September 2013; Published online: 01 October 2013.
Edited by:
Lawrence J. Stern, University of Massachusetts Medical School, USAReviewed by:
Scheherazade Sadegh-Nasseri, Johns Hopkins University, USACopyright: © 2013 Ferrante. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Ferrante, Institute of Arctic Biology, 902 North Koyukuk Dr., P.O. Box 757000, Fairbanks, AK 99775-7000, USA e-mail:YWZlcnJhbnRlQGFsYXNrYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.