 Bridie J. Goggins1,2
Bridie J. Goggins1,2
- 1School of Biomedical Sciences and Pharmacy, University of Newcastle, Newcastle, NSW, Australia
- 2Hunter Medical Research Institute, New Lambton, NSW, Australia
- 3Royal Brisbane and Women’s Hospital, Brisbane, QLD, Australia
- 4Queensland Institute for Medical Research, Brisbane, QLD, Australia
Epithelial damage and loss of intestinal barrier function are hallmark pathologies of the mucosal inflammation associated with conditions such as inflammatory bowel disease. In order to resolve inflammation and restore intestinal integrity the mucosa must rapidly and effectively repair the epithelial barrier. Epithelial wound healing is a highly complex and co-ordinated process and the factors involved in initiating intestinal epithelial healing are poorly defined. In order for restitution to be successful there must be a balance between epithelial cell migration, proliferation, and differentiation within and adjacent to the inflamed area. Endogenous, compensatory epithelial signaling pathways are activated by the changes in oxygen tensions that accompany inflammation. These signaling pathways induce the activation of key transcription factors, governing anti-apoptotic, and proliferative processes resulting in epithelial cell survival, proliferation, and differentiation at the site of mucosal inflammation. In this review, we will discuss the primary processes involved in epithelial restitution with a focus on the role of hypoxia-inducible factor and epithelial integrins as mediators of epithelial repair following inflammatory injury at the mucosal surface.
Introduction
The successful healing of a mucosal wound requires the inter-related processes of inflammation, proliferation, granulation tissue formation, and tissue remodeling (1). These are highly regulated and over-lapping events with environmental stimulus from one event, dictating progression to the next (2). One important aspect of mucosal wound healing is the role of oxygen sensing in the tissue repair process, given the dramatic changes in tissue oxygen tension during inflammation and wounding, where oxygen tensions may decrease 10-fold at a wound site (3). Despite these changes most inflammatory wounds resolve and tissue homeostasis is restored. Thus, wounded and inflamed tissues may adapt to reduced oxygen availability at an inflammatory wound and retain the ability to repair, despite tissue hypoxia. Much of what we know about wound healing processes is derived from studies in dermal healing and there is relatively little known about mechanisms of mucosal wound healing, particularly at the intestinal mucosa, where normal oxygen tensions are low (4).
Mucosal Inflammation and Hypoxia
In the early stages of the initial mucosal insult, intestinal wounds are almost devoid of oxygen (3). This is a result of both the vascular damage occurring with injury and increased cellular oxygen demand at the wound. Infiltrating immune cells generate superoxide, combating infection, but greatly increasing oxygen demand (3, 5). In addition reparative processes such as cell proliferation and collagen production increase oxygen demand in the mucosal environment (6). Thus, the initial inflammatory response to mucosal damage promotes a state of chronic hypoxia within the microenvironment of the wound. This “inflammatory hypoxia” has been elegantly demonstrated in murine models of colitis. For instance, the 2,4,6-trinitrobenzenesulfonic acid (TNBS) model of murine colitis has been widely utilized to model inflammatory bowel diseases (IBDs) (7). Histologically these animals demonstrate profound vasculitis of the small submucosal vessels associated with mucosal inflammation, similar to observations in human tissues (8, 9). The chronic hypoxia and inflammation within the intestinal mucosa is associated with angiogenesis, further enhancing influx of inflammatory cells and endothelial dysfunction (10). Associated upregulation of collagen synthesis increases the risk of fibrosis, a key feature of chronic inflammatory disease potentially driven by chronic tissue hypoxia (11). Tissue hypoxia associated with inflammation has been demonstrated in animal models, through utilization of the characteristic reduction and binding of 2-nitroimidazole compounds, such as pimonidazole and EF5, to cellular thiol-containing proteins oxygen levels below 10 mmHg (12).
Animals with TNBS-induced colitis demonstrated dramatic levels of nitroimidazole retention associated with colitic lesions, both in superficial and in deeper submucosal regions of the mucosa (13, 14). This is in marked contrast to the superficial retention of nitroimidazoles observed in healthy animals. These findings, demonstrated in several other animal models, indicate that mucosal inflammation, such as that associated with models of mucosal inflammation likely result in significant tissue hypoxia, predominantly within the epithelium.
Molecular Signaling by Hypoxia
Oxygen is a key component in the generation of metabolic energy for all eukaryotic cells (15). Fluctuations in tissue oxygen supply (hypoxia) are common physiologic and pathophysiologic occurrences. These include frank vascular occlusion such as those occurring with stroke, tissue fibrosis, and the microvascular breakdown associated with chronic inflammation which also results in localized tissue hypoxia/ischemia. Alternatively, diminished oxygen delivery to tissues may occur in shock, hypotension, or in cases where the oxygen carrying capacity of blood is compromised [e.g., chronic obstructive pulmonary disease (COPD), carbon monoxide poisoning] (16, 17). Thus, mammalian cells have evolved compensatory mechanisms to adapt to tissue hypoxia (18). One such mechanism is the oxygen-sensing molecule; hypoxia-inducible factor (HIF), a transcription factor which functions as a global mechanism for adaptation to hypoxia (19).
Hypoxia-Inducible Factor Regulation and Cellular Oxygen Sensing
Hypoxia-inducible factor is a central regulatory transcription factor for hypoxia-induced gene expression, and serves as a sensitive and selective indicator of hypoxia (20–22). HIF is a heterodimeric nuclear protein made up of an α oxygen regulated and constitutively expressed β subunit (23, 24). Under conditions of normal tissue oxygen tensions (normoxia), the α subunit is continuously synthesized, and degraded through a cascade of events. The prolyl residues (402 and/or 564) on the α subunit undergo oxygen-dependent hydroxylation by the prolyl-4-hydroxylase (PHD) enzymes. PHDs, principally prolyl hydroxylase-2 (PHD2), target oxygen, and α-ketoglutarate as substrates to catalyze a dioxygenase reaction (22, 25), which facilitates hydroxylation of the hypoxia-inducible factor-1α (HIF-1α) subunit (26). This leads to binding of the von Hippel–Lindau (VHL) protein, which allows the recruitment of the ubiquitin ligase complex (22, 27) and targets HIF-1α for proteasomal degradation by the 26S proteasome (28). However, during periods of reduced oxygen availability (hypoxia), PHD2 activity is reduced due to substrate (oxygen) limitations. This allows stabilization of HIF-1α within the cytoplasm of the cell and translocation to the nucleus for dimerization with the HIF-1β subunit (29). Dimerization forms a transcriptionally functional HIF-αβ dimer, which then binds to cis-acting hypoxia response elements (HREs) in the promoter of target genes and recruits co-activator proteins (Figure 1A). As a result of this cascade, transcription of HIF target gene sequences to mRNA is increased (21, 30). However, this is not an all or nothing response, and HIF-1α stabilization is gradual and graded over the progression from mild to chronic hypoxia (31) (Figure 1B).
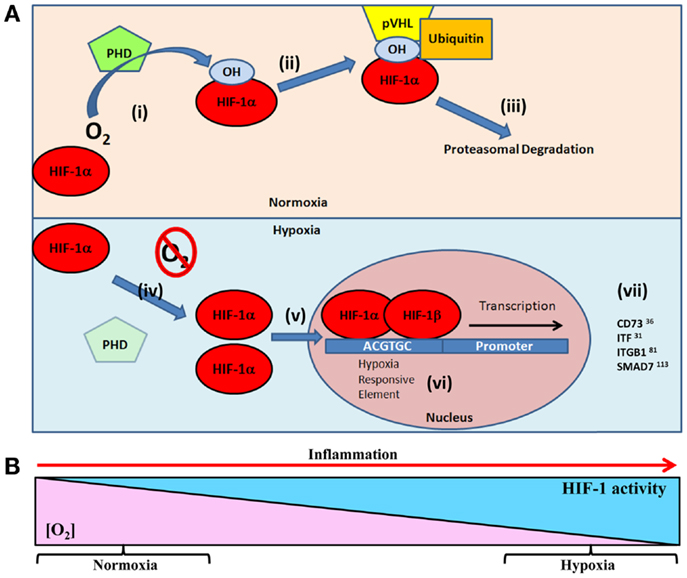
Figure 1. (A) Oxygen-dependent regulation of HIF-1α targets in epithelial restitution. Under normal oxygen tensions (normoxia) (i) prolyl hydroxylase (PHD) enzymes hydroxylate the HIF-1α subunit resident in the cellular cytoplasm (26). Hydroxylated HIF-1α facilitates, (ii) the binding of von Hippel–Lindau protein (pVHL) and subsequent recruitment of the ubiquitin ligase complex, (iii) targeting HIF-1α for 26S proteasomal degradation (28). Under conditions of reduced oxygen (hypoxia) (iv) the lack of oxygen substrate for PHD prevents hydroxylation of HIF-1α leading to (v) cytoplasmic accumulation and translocation to the cell nucleus (29). (vi) HIF-1α dimerizes with HIF-1β and binds to hypoxia responsive elements (5′-ACGTGC-3′) in the promoter of target genes (21, 30). (vii) This leads to transcription of HIF target genes involved in epithelial restitution. (B) As tissue oxygen levels gradually decline, HIF stabilization increases. This results in graded HIF stabilization during progressive hypoxia (31), such as the progression of inflammation.
Until very recently, most work with HIF focused on understanding the basic mechanisms by which HIF acts as a key mediator of the cellular hypoxic response, particularly in the context of carcinogenesis (17, 32). Solid tumors have been demonstrated to form hypoxic cores and adapt to this oxygen deficiency in order to maintain a proliferative state. However, recent studies reveal a potentially central role for HIF in endogenous protective and restorative pathways within a variety of inflammatory diseases, including respiratory distress syndrome, retinitis, diabetes, and arthritis (17).
HIF and Adaption to Mucosal Inflammation
Activation of HIF-1α due to the unavailability of oxygen has been widely shown to promote adaption to inflammation, primarily through an increase in mucosal barrier protection (4, 33, 34). Key to this protective response is the induction of genes involved in non-classical epithelial barrier function. These include genes that regulate the integrity of the mucous-gel layer; Mucin 1 and 3 (MUC1 and MUC3) (34, 35) and intestinal trefoil factor (ITF) (33), the epithelial xenobiotic drug efflux pump; (multi drug resistance protein 1, MDR1; P-glycoprotein) (36), leukocyte trafficking and clearance; CD55 (decay accelerating factor) (37), and cellular energy metabolism; CD73 (ecto-5′-nucleotidase) (38), and the adenosine A2B receptor (39). Thus, genes induced by HIF-1α support overall tissue integrity and include target proteins necessary for cellular, whole tissue, and whole animal adaptive responses to hypoxia (40, 41).
HIF and Mucosal Healing
Hypoxia-inducible factor regulates a diverse number of genes, many of which feeding back into processes critical for wound healing (31). While HIF signaling allows the tissue to adapt to, and protect against, inflammatory hypoxia, HIF also regulates the expression of genes that drive angiogenesis. As inflammatory damage to the tissue is a key driving factor in tissue hypoxia, it is unsurprising that HIF adaptive responses include restoration of the vascular oxygen supply. In particular, HIF regulates the expression of vascular endothelial growth factor A (VEGFA) and angiopoietins, which drive angiogenesis through endothelial mitosis and migration (42–45). As evidence of the role of HIF in wound healing, over-expression of HIF-1α improves wound healing in mouse models of diabetes (29, 46), a condition where impaired healing may lead to complications such as diabetic foot (47). Conversely suppression of HIF-1α expression results in dysfunctional wound healing and defective vascularization (48).
Hypoxia-inducible factor also regulates the induction of VEGF receptor Flt-1 (49, 50) and a range of vasomotor peptides, such as adrenomedullin (51) and endothelin-1 (52) which act to fine tune the angiogenic response, underpinning the importance of HIF signaling in the regulation of angiogenesis. The potential risks associated with angiogenesis in chronic inflammatory disease states such as CD include formation of a dysfunctional new vessel architecture and further recruitment of inflammatory cells. In the absence of fine tuning of the angiogenic response that remains functional in acute intestinal inflammation such as infectious colitis, the responses seen in CD lead to fibrosis, and the need for bowel resection (53).
In contrast to angiogenesis, little is known about how hypoxia and HIF signaling directly influences mucosal, epithelial wound healing at the molecular level. Moreover, while there is evidence of the importance of HIF in regulating keratinocyte re-epithelialization (54), there is far less understanding of how transcriptional amplification by hypoxia might be important in initiating mucosal wound healing responses.
Epithelial Wound Healing
Early in the healing process, epithelial cells adjacent to the mucosal wound lose polarity and convert into a migratory phenotype (55). The depolarized epithelial cells rapidly migrate into the denuded area and line the underlying matrix in order to re-establish a protective barrier (56). Once the barrier has been restored, epithelial cell proliferation begins and enterocyte numbers increase to resurface the wounded area (57). Proliferation occurs hours to days after injury, usually in the crypts near the damaged mucosal area. Finally, proliferation progenitor epithelial cells must differentiate into a specific lineage subset. Functionally, these encompass absorptive enterocytes or one of three secretory lineages cells (goblet, enteroendocrine, and paneth cells) (58). Once differentiated, IECs can restore the functional activities of the epithelial layer and intestinal homeostasis resumes (55).
Damage to the intestinal epithelial layer is a common pathology of the inflammation associated with diseases such as IBD (56). In order to suppress inflammation and restore normal intestinal homeostasis the mucosa must activate rapid repair mechanisms and restore epithelial defenses (59, 60). Even the most superficial injuries to the epithelium result in epithelial destruction and require healing (61). Successful epithelial repair requires a balance between epithelial restitution, proliferation, and differentiation within and adjacent to areas of mucosal damage (55, 62). Thus, for the resolution of inflammation to occur, the tissue must first halt the influx of luminal antigens through the damaged epithelium. Accordingly, the first phase of the wound healing process is restitution of the epithelial barrier.
Epithelial Restitution
Restitution is the rapid migration of epithelial cells adjacent to the wound/injury in order to reseal the damaged area. Migrating cells achieve restitution through a sequence of transient adherence to the extra-cellular matrix. This adherence is achieved via a collection of specialized basal structures that evolve from focal complexes followed by focal adhesions to fibrillar adhesions (63–67). Rapid migration of the epithelial cells adjacent to the wound, at the wound edge, marks the initiation of restitution, and this begins within minutes to hours of the injury occurring (56, 68). Epithelial cells surrounding the wound have the ability to rapidly migrate due to the loss of columnar polarity. These cells undergo extensive reorganization of their actin cytoskeleton (55, 69), losing their microvilli, and apical/basolateral orientation to adopt a flat morphology. The cells then re-polarize to induce migration, with polarization now defined from leading to trailing edge (70–72). Migration is dependant on F-actin-rich protrusions called lamellipodia at the leading edge, which enable transient adherence to the underlying matrix at focal adhesion complex sites (73, 74). This change in polarization and shape allows the cells to migrate rapidly to the injury site and attach via focal contacts (75), restoring barrier integrity (56, 68, 76). Restitution is a very rapid process and enables reconstitution of epithelial continuity much faster than could be achieved through proliferation of cells alone. However, proliferation is still required to ultimately restore the mucosal surface. Restitution is the central component to epithelial healing regardless of the cause or extent of the injury, as restitution ensures epithelial continuity is re-established (77).
HIF-Mediated Epithelial Restitution
In order to facilitate repair of the epithelial barrier, HIF directly targets a number of critical components for the epithelial wound healing process including energy metabolism and cell migration, both important processes in restitution (Figure 2). Induction of CD73 and glucose transporter 1 (GLUT-1), allow the cells to maintain energy metabolism in the hypoxic microenvironment of the mucosal wound (38, 78). Pre-epithelial barriers are augmented through the induction of mucins and ITF (33). ITF is a particularly noteworthy factor in mucosal wound healing, as it not only augments the barrier, through increased mucosal integrity via interactions with mucin glycoproteins (79), but also facilitates epithelial restitution. Both apically secreted and exogenous ITF accelerate epithelial cell migration into the wound area through pathways independent of transforming growth factor-β (TGF-β) signaling at the basolateral interface (68, 80). In addition ITF can prolong epithelial cell life at the site of a wound, through inhibition of apoptosis (81). Thus HIF-mediated induction of ITF, not only acts to protect a wounded mucosal surface, but also modulates epithelial restitution. However, a double-blind, randomized, placebo-controlled study, to examine the efficacy of supplementing conventional treatments with recombinant ITF for the treatment of mild-to-moderate ulcerative colitis, did not reveal any additional benefit above that of conventional therapies alone (82). This could be due to the enema method of ITF delivery, which may not facilitate sufficient ITF-epithelial interactions to mediate restitution. The study did not measure mucosal responses to local delivery of ITF to determine or confirm whether it was biologically active in the disease state in human subjects. Further studies will need to address this.
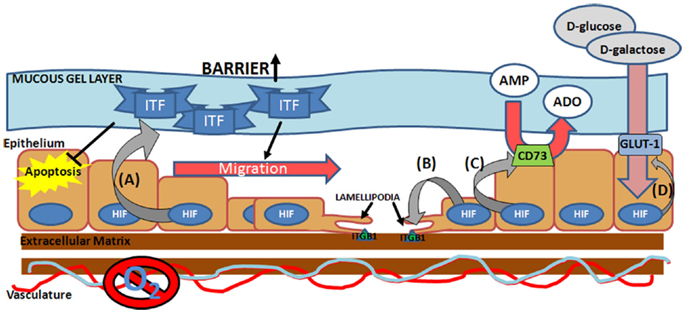
Figure 2. Hypoxia-inducible factor-mediated pro-restitution pathways. Hypoxia-inducible factor (HIF) activation at the site of the mucosal wound leads to induction of (A) epithelial intestinal trefoil factor (ITF) (33), which acts to increase mucosal barrier function, suppress epithelial apoptosis (127), and drive epithelial migration (80), (B) integrin β1 (ITGB1) (84), a lamellipodia protein critical for epithelial cell migration across the extra-cellular matrix, (C) ecto-5′-nucleotidase (CD73) (38), which facilitates conversion of AMP into adenosine (ADO), and (D) glucose transporter 1 (GLUT-1) (7, 8), which facilitates the transport of d-glucose and d-galactose across the plasma membrane.
Hypoxia-driven motility is associated with increased expression of lamellipodia proteins, increased expression of collagenase and decreased expression of laminin-5, the locomotion brake for keratinocytes (83). Our own studies have identified HIF-1α stabilization as a key promoter of integrin β1 (ITGB1), a critical mediator of lamellipodia binding, during wound healing in IBD (84). Here we demonstrated direct binding of HIF to the ITGB1 promoter, leading to increased expression of functional β1 integrin. In TNBS models of colitis, mucosal ITGB1 expression correlated directly with disease severity, revealing a correlation between mucosal hypoxia and ITGB1 expression in vivo. Interestingly, site-directed mutagenesis of the hypoxia responsive element (HRE) on the ITGB1 promoter did not completely abolish the HIF response, suggesting that secondary mechanisms may also be involved, for instance inhibitor of DNA binding-1 (Id-1) has been shown to induce ITGB1 in a HIF-dependent manner and Qiu et al. have speculated on whether Id-1 may regulate the ITGB1 promoter directly (85). Nevertheless, in subsequent studies examining the role of HIF-1α stabilization in mucosal response to colitis in murine models, we have demonstrated that stabilization of HIF-1α through prolyl hydroxylase inhibition (PHDi) results in earlier and increased epithelial ITGB1 expression, concurrent with accelerated mucosal healing and restitution of epithelial barrier function (86). Furthermore, the protective effect of the pharmacological HIF stabilization by PHDi is lost in conditional epithelial HIF deficient animals. Further evidence for the role of HIF in integrin-mediated wound healing has been shown in dermal healing models. HIF-1α silencing, led to decreased expression of Lamanin-322, α6 integrin and β1 integrin, and this was associated with impaired epidermal healing and dermis formation in both murine models and human keratinocyte cultures (87). This implicates an importance for HIF in aging and wound healing processes. Overall, these data suggest direct roles for both HIF-1α and ITGB1 in mediated mucosal wound healing.
Integrins as Mediators of Epithelial Wound Healing
A key factor in the co-ordination of restitution is the ability of cells to adhere to, and interact with the extra-cellular matrix. Integrins are critical mediators of these interactions and facilitate epithelial migration into the denuded mucosal wound. Integrins are a family of cell adhesion receptors responsible for mediating both cell-substratum and cell–cell adhesion (88). They exist as heterodimeric glycoproteins consisting of non-covalently bonded α and β subunits (89–91). Integrins provide essential links between the extra-cellular environment and intracellular signaling pathways. This makes them key regulators of cell behaviors such as cell survival, apoptosis, differentiation, migration, and transcriptional regulation (92), thus integrins are critical for processes in development, immune function, and wound healing.
In the context of wound healing and epithelial restitution Lotz et al. showed the functional importance of integrin heterodimers containing a β1-subunit, in particular α6β1 and α3β1 integrins in epithelial wound restitution (93). In T84 epithelial wounding models, cell migration was shown to be integrin-dependent, regulated by the expression of localized, specific integrins, and their cell matrix protein ligands. Monoclonal antibodies directed against functional epitopes on α3 and β1 integrins were found to inhibit wound closure completely, while functional blockade of α6 integrin also significantly inhibited wound closure. These heterodimers are differentially expressed within the process of restitution, with α6β1 integrin increased along the lateral surfaces of migrating cells, while α3β1 integrin expression localized to flattened cell surfaces and along wound and colony perimeters (93). These studies suggest a fundamental role of integrins, particularly α3β1 and α6β1, in epithelial restitution.
Inflammatory Signaling and Integrin-Mediated Restitution
Epithelial injury observed in patients with IBD is associated with the infiltration of inflammatory cells to the mucosa, which triggers an inflammatory cascade in the tissue causing the release of proinflammatory cytokines and, often, further tissue injury (94, 95). Secreted inflammatory cytokines can also directly influence the progression of epithelial restitution (96). For instance, interferon-γ (IFN-γ) has been identified as a key proinflammatory cytokine in IBD, with elevated levels observed in the mucosa of IBD patients (97). While extensive investigations have been conducted on the effects of IFN-γ on epithelial intercellular junctions and barrier properties (94, 98–100), there is a paucity of studies characterizing the effect of IFN-γ on the wound healing process. To address this, Tong et al. investigated the influence of IFN-γ on intestinal epithelial wound closure (96), examining epithelial cell migration in vitro. IFN-γ demonstrated clear inhibitory effects on epithelial migration, causing dysfunction of the F-actin-rich lamellipodia protrusions at the leading edge of the migrating cell. No difference in the average number of lamellipodia at the leading edge of cells was found between control and IFN-γ-treated monolayers (96). As IFN-γ is known to drive pathology in a number of mucosal inflammatory diseases, these findings may explain the impaired wound healing observed in mucosal disease, where IFN-γ alters lamellipodia formation and subsequently impairs cell migration.
Attachment of lamellipodia occurs at focal adhesion complex sites, and key components of these focal adhesion sites are integrin heterodimers (55). As migrating cells move, continuous attachments are formed to the extra-cellular matrix at the leading edge of the cell, in synchrony with rear edge detachments until the wound is resealed by intercellular focal contacts (73). Integrins contribute to this process through cycles of exocytosis and endocytosis of surface bound integrins. Integrin heterodimers are transported via endocytic vesicles to the cell surface at the leading edge where they can form new focal complexes (101, 102). The mechanism of IFN-γ mediated lamellipodia dysfunction appears to involve several key focal adhesion proteins. For instance, IFN-γ suppresses expression of vinculin, focal adhesion kinase, and paxillin (96). IFN-γ also reduces the deposits of intracellular β1 integrin in focal adhesions at the leading edge of migrating epithelial cells, while reducing the number of β1 integrin containing cellular vesicles overall. This change in integrin distribution is not a result of accelerated degradation or loss of integrin protein, but rather accelerated endocytosis of membrane integrins. Migrating cells treated with IFN-γ-shown broadly distributed clusters of integrins throughout the cell, rather than the accumulation of β1 integrin observed at the leading edge of the migrating control cells (96). These studies further highlight the fundamental role of integrin β1 in the wound healing process and the importance of integrin β1 localization at leading edge focal complexes and vesicular transport of β1 for cell migration and movement.
Studies by Glover et al. (103) have further characterized the pathogenic contribution of IFN-γ to inflammatory diseases such as IBD. Inflammation is associated with dramatic shifts in tissue metabolism due to immune cell recruitment to inflammatory wounds or lesions (104). Hypothesizing that inflammatory cytokine signaling may input into the hypoxic response to mucosal inflammation, Glover et al. investigated the effect of inflammatory mediators on HIF regulation in intestinal epithelial monolayers. Investigating a host of common inflammatory cytokines, including TNF-α, IL-4, PGE2, and IFN-γ in both normoxic and hypoxic cultures, IFN-γ demonstrated the ability to significantly repress HIF-1 transcriptional targets in both normoxic and hypoxic conditions. In contrast, HIF-1α mRNA expression showed moderate increases in expression in response to IFN-γ and further investigation demonstrated that attenuation of HIF activity is the result of selective repression of HIF-1β.
While HIF responses drive expression of both protective and reparative pathways, the expression of HIF-1α is concurrent with chronic mucosal inflammation, suggesting that in chronic inflammatory diseases such as IBD, the HIF response is not always sufficient to promote restitution. This may in part, be due to increased levels of mucosal IFN-γ associated with chronic inflammation (103). In vitro, IFN-γ was shown to repress the expression of the HIF-1β and dextran sodium sulfate (DSS) murine models of IBD showed an inverse correlation between IFN-γ and HIF-1β expression. This result is surprising, in that HIF-α subunits are generally considered to be the regulated components of HIF signaling, while HIF-β subunits are regarded as constitutively expressed (23). As many studies only examine the expression of HIF-α isoforms, the study by Glover et al. may offer a critically important explanation as to why chronic inflammation progresses despite the stabilization of HIF-1α, given that IFN-γ is involved in the pathogenesis of many mucosal diseases (105–107). In particular, inhibition of HIF signaling by IFN-γ could, hypothetically, significantly impair mucosal healing, through reduced expression of a number of cellular proteins, such as β1 integrin, critical for epithelial restitution, and wound healing.
Integrins and TGF-β-Mediated Pathways
Transforming growth factor-β is a pleiotropic cytokine and is critical to the regulation of cellular events involved in wound healing, including cell differentiation, proliferation, epithelial-mesenchymal transition, and cell migration. There is a strong degree of cross-talk between hypoxia and TGF-β (108), particularly the TGF-β1 isoform. Hypoxia has been shown to increase the transcription of TGF-β1 in dermal fibroblasts (109), while TGF-β1 may stabilize HIF-1α through selective inhibition of PHD2 (110). This inhibition is achieved through the downregulation of PHD2 gene expression via SMAD dependent pathways (111). SMAD proteins are intracellular TGF-β signal transducers that mediate the interaction between TGF-β receptor ligands and downstream nuclear responses (112). For instance, at the site of a wound, TGF-β1 mediated activation of SMAD2/3 complexes and subsequent interaction with SMAD4 leads to the formation of a SMAD transcription factor which drives cellular responses toward re-epithelialization (113–115).
While few studies have been conducted in the context of mucosal inflammation and healing, there is evidence to suggest convergence of HIF, TGF-β, and SMAD pathways in the co-ordinated regulation of epithelial restitution (Figure 3). For instance, studies in the hypoxic microenvironment of solid tumors have identified SMAD7 as a HIF-1α responsive gene (116). SMAD7 has been shown to be a potent inhibitor of TGF-β1 (116, 117) and thus may prevent TGF-mediated cell proliferation and anti-inflammatory signaling. However, SMAD7, is itself inhibited by integrin signaling, specifically by epithelial integrin heterodimers containing an integrin β1 (ITGB1) subunit (118). Reynolds et al. demonstrated that α3β1 integrin heterodimers inhibited SMAD7 and enhanced cutaneous re-epithelialization in murine models of wound healing (118). As both ITGB1 and SMAD7 are HIF responsive, it is feasible that they represent co-dependent modulators of TGF-β1 mediated wound healing. α3β1 integrins are expressed in depolarized intestinal epithelial cells, particularly around the wound edge (93), thus induction of these integrins may act to inhibit SMAD7, promote TGF-β1 signaling, and initiate intestinal epithelial wound closure. Interestingly, SMAD7 is overexpressed in the inflamed mucosa of IBD patients (117) and targeting of SMAD7 has shown efficacy in mouse models of colitis (119), while ITGB1 single nucleotide polymorphisms have been identified as a risk factor in IBD (120). Thus, dysfunction of the pathway by which HIF-induced ITGB1 inhibits SMAD7, and the subsequent elevation of TGF-β1, may lead to the progression of chronic inflammation instead of mucosal wound healing. Importantly, TGF-β1 plays a role in the pathogenesis of intestinal fibrosis in Crohn’s patients (95) and TGF-β1 codon 25 variants are associated with structuring (121). Whether this polymorphism represents a dysfunction ins SMAD7/ITGB1/TGF-β1 signaling is unknown, but as HIF-mediated ITGB1 drives fibroblast-collagen contraction in vitro, any dysfunction in TGF-β1 signaling is likely to interplay in this pathway.
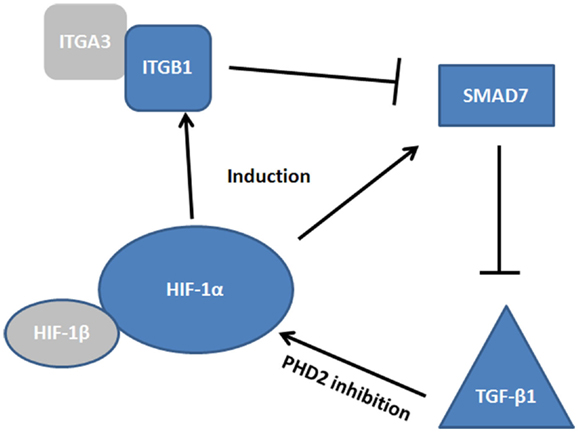
Figure 3. Convergence of HIF, TGF-β, and SMAD pathways. The complexity of the signaling cascade by which HIF regulates integrin β1 (ITGB1) and SMAD7 induction. At the site of the wound α3β1 integrin inhibits SMAD7, promoting TGF-β1 induction, which promotes restitution. How these factors interplay may be critical to our understanding of epithelial wound healing.
We may also consider that TGF-β1 acts to “fine tune” the HIF-1α response, as exogenous TGF-β1 enhances HIF-1α expression in hypoxic cells, while also increasing HIF-1α stabilization in normoxic conditions. As TGF-β1 does not affect transcription of HIF-1α itself, nor decrease degradation, it appears that this interplay occurs at the level of HIF-1α translation. This may occur independently of hypoxia, thus it is possible that TGF-β1 may act to “prime” the HIF response (122) in a manner similar to HIF-priming vasopeptides such as adrenomedullin (51).
Summary
While a role for oxygen in mediating wound healing has been recognized for decades (6, 123, 124), the importance of cellular oxygen sensing in cellular adaptive and reparative pathways is a relatively new area (3, 16, 125). Given the rapidly changing oxygen tensions in the mucosal wound, the role for hypoxia responsive pathways in processes such as epithelial restitution is unsurprising. Hypoxia seems to independently regulate several critical drivers of epithelial restitution that subsequently exhibit a high degree of interplay. The interactions between HIF, β1 integrin heterodimers, SMAD7, and TGF-β are complex and have not been fully elucidated. Crucially, much of our knowledge of these pathways come from models of dermal wound healing, where basal oxygen levels are markedly higher than that of intestinal mucosal tissues (126).
Therapeutically, wound healing pathways are an attractive target for mucosal disease. For instance, despite the successes of immunomodulators in the maintenance of IBD, up to 70% of IBD patients still require surgery to remove tissue damaged by repeated cycles of inflammatory damage and improper healing. Therapies aimed at modulating the healing process may reduce the need for these surgeries. Further elucidation of the pathways driving mucosal wound healing are therefore critically important, and may open the door for improved therapeutic strategies for the management of mucosal inflammatory disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This Authors acknowledge funding by National Health and Medical Research Council (NHMRC) project grant APP1021582.
References
1. Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res (2010) 89(3):219–29. doi:10.1177/0022034509359125
2. Rodriguez PG, Felix FN, Woodley DT, Shim EK. The role of oxygen in wound healing: a review of the literature. Dermatol Surg (2008) 34(9):1159–69. doi:10.1111/j.1524-4725.2008.34254.x
3. Tandara AA, Mustoe TA. Oxygen in wound healing – more than a nutrient. World J Surg (2004) 28(3):294–300. doi:10.1007/s00268-003-7400-2
4. Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med (Berl) (2007) 85(12):1295–300. doi:10.1007/s00109-007-0277-z
5. Steinbrecher UP. Role of superoxide in endothelial-cell modification of low-density lipoproteins. Biochim Biophys Acta (1988) 959(1):20–30. doi:10.1016/0005-2760(88)90145-2
6. Hunt TK, Zederfeldt B, Goldstick TK. Oxygen and healing. Am J Surg (1969) 118(4):521–5. doi:10.1016/0002-9610(69)90174-3
7. Kruschewski M, Foitzik T, Perez-Canto A, Hubotter A, Buhr HJ. Changes of colonic mucosal microcirculation and histology in two colitis models: an experimental study using intravital microscopy and a new histological scoring system. Dig Dis Sci (2001) 46:2336–43. doi:10.1023/A:1012334727509
8. Wakefield AJ, Dhillon AP, Rowles PM, Sawyer AM, Pitilo RM, Lewis AAM, et al. Pathogenesis of Crohn’s disease: multifocal gastrointestinal infarction. Lancet (1989) 2(8671):1057–62. doi:10.1016/S0140-6736(89)91078-7
9. Yokoyama K, Mitomi H, Kobayashi K, Katsumata T, Saigenji K, Okayasu I. Obliterative arteritis with nitric oxide synthase and HLA-DR expression in Crohn’s colitis. Hepatogastroenterology (2001) 48:401–7.
10. Danese S, Sans M, de la Motte C, Graziani C, West G, Phillips MH, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology (2006) 130(7):2060–73. doi:10.1053/j.gastro.2006.03.054
11. Carr ND, Pullan BR, Schofield PF. Microvascular studies in non-specific inflammatory bowel disease. Gut (1986) 27(5):542–9. doi:10.1136/gut.27.5.542
12. Evans SM, Hahn S, Pook DR, Jenkins WT, Chalian AA, Zhang P, et al. Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res (2000) 60:2018–24.
13. Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest (2004) 114(8):1098–106. doi:10.1172/JCI200421086
14. Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology (2008) 134(1):145–55. doi:10.1053/j.gastro.2007.09.033
15. Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev (1996) 76:839–85.
16. Semenza GL. HIF-1 and human disease: one highly involved factor. Genes Dev (2000) 14(16):1983–91.
17. Semenza GL, Agani F, Feldser D, Iyer N, Kotch L, Laughner E, et al. Hypoxia, HIF-1, and the pathophysiology of common human diseases. Adv Exp Med Biol (2000) 475:123–30. doi:10.1007/0-306-46825-5_12
18. Ratcliffe PJ, O’Rourke JF, Maxwell PH, Pugh CW. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J Exp Biol (1998) 201:1153–62.
19. Glover LE, Colgan SP. Hypoxia and metabolic factors that influence inflammatory bowel disease pathogenesis. Gastroenterology (2011) 140(6):1748–55. doi:10.1053/j.gastro.2011.01.056
20. Becker PM, Alcasabas A, Yu AY, Semenza GL, Bunton TE. Oxygen-independent upregulation of vascular endothelial growth factor and vascular barrier dysfunction during ventilated pulmonary ischemia in isolated ferret lungs. Am J Respir Cell Mol Biol (2000) 22(3):272–9. doi:10.1165/ajrcmb.22.3.3814
21. Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE (2007) 2007(407):cm8. doi:10.1126/stke.4072007cm8
22. Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell (2008) 30(4):393–402. doi:10.1016/j.molcel.2008.04.009
23. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A (1995) 92(12):5510–4. doi:10.1073/pnas.92.12.5510
24. Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem (1995) 270(3):1230–7. doi:10.1074/jbc.270.3.1230
25. Chowdhury R, Hardy A, Schofield CJ. The human oxygen sensing machinery and its manipulation. Chem Soc Rev (2008) 37(7):1308–19. doi:10.1039/b701676j
26. Baek JH, Mahon PC, Oh J, Kelly B, Krishnamachary B, Pearson M, et al. OS-9 interacts with hypoxia-inducible factor 1alpha and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1alpha. Mol Cell (2005) 17(4):503–12. doi:10.1016/j.molcel.2005.01.011
27. Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A (2000) 97(19):10430–5. doi:10.1073/pnas.190332597
28. Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem (1997) 272(36):22642–7. doi:10.1074/jbc.272.36.22642
29. Botusan IR, Sunkari VG, Savu O, Catrina AI, Grunler J, Lindberg S, et al. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci U S A (2008) 105(49):19426–31. doi:10.1073/pnas.0805230105
30. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem (1996) 271(51):32529–37. doi:10.1074/jbc.271.51.32529
31. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol (2004) 5(5):343–54. doi:10.1038/nrm1366
32. Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol (2000) 35:71–103. doi:10.1080/10409230091169186
33. Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med (2001) 193(9):1027–34. doi:10.1084/jem.193.9.1027
34. Louis NA, Hamilton KE, Canny G, Shekels LL, Ho SB, Colgan SP. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem (2006) 99(6):1616–27. doi:10.1002/jcb.20947
35. Mikami Y, Hisatsune A, Tashiro T, Isohama Y, Katsuki H. Hypoxia enhances MUC1 expression in a lung adenocarcinoma cell line. Biochem Biophys Res Commun (2009) 379(4):1060–5. doi:10.1016/j.bbrc.2009.01.002
36. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res (2002) 62(12):3387–94.
37. Louis NA, Hamilton KE, Kong T, Colgan SP. HIF-dependent induction of apical CD55 coordinates epithelial clearance of neutrophils. FASEB J (2005) 19(8):950–9. doi:10.1096/fj.04-3251com
38. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest (2002) 110(7):993–1002. doi:10.1172/JCI0215337
39. Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol (2009) 182(8):4957–64. doi:10.4049/jimmunol.0801324
40. Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumor angiogenesis. Nature (1998) 394:485–90. doi:10.1038/28867
41. Semenza GL. Regulation of erythropoietin production. New insights into molecular mechanisms of oxygen homeostasis. Hematol Oncol Clin North Am (1994) 8:863–84.
42. Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A (1997) 94:8104–9. doi:10.1073/pnas.94.15.8104
43. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol (1996) 16:4604–13.
44. Bosch-Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res (2007) 101(12):1310–8. doi:10.1161/CIRCRESAHA.107.153346
45. Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res (2003) 93(11):1074–81. doi:10.1161/01.RES.0000102937.50486.1B
46. Liu L, Marti GP, Wei X, Zhang X, Zhang H, Liu YV, et al. Age-dependent impairment of HIF-1alpha expression in diabetic mice: correction with electroporation-facilitated gene therapy increases wound healing, angiogenesis, and circulating angiogenic cells. J Cell Physiol (2008) 217(2):319–27. doi:10.1002/jcp.21503
47. Steed DL, Edington HD, Webster MW. Recurrence rate of diabetic neurotrophic foot ulcers healed using topical application of growth factors released from platelets. Wound Repair Regen (1996) 4(2):230–3. doi:10.1046/j.1524-475X.1996.40210.x
48. Zhang X, Liu L, Wei X, Tan YS, Tong L, Chang R, et al. Impaired angiogenesis and mobilization of circulating angiogenic cells in HIF-1alpha heterozygous-null mice after burn wounding. Wound Repair Regen (2010) 18(2):193–201. doi:10.1111/j.1524-475X.2010.00570.x
49. Konttinen YT, Mackiewicz Z, Povilenaite D, Sukura A, Hukkanen M, Virtanen I. Disease-associated increased HIF-1, alphavbeta3 integrin, and Flt-1 do not suffice to compensate the damage-inducing loss of blood vessels in inflammatory myopathies. Rheumatol Int (2004) 24(6):333–9. doi:10.1007/s00296-003-0379-z
50. Li H, Gu B, Zhang Y, Lewis DF, Wang Y. Hypoxia-induced increase in soluble Flt-1 production correlates with enhanced oxidative stress in trophoblast cells from the human placenta. Placenta (2005) 26(2–3):210–7. doi:10.1016/j.placenta.2004.05.004
51. MacManus CF, Campbell EL, Keely S, Burgess A, Kominsky DJ, Colgan SP. Anti-inflammatory actions of adrenomedullin through fine tuning of HIF stabilization. FASEB J (2011) 25(6):1856–64. doi:10.1096/fj.10-170316
52. Zhang W, Zhang Y, Wang W, Dai Y, Ning C, Luo R, et al. Elevated ecto-5’-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circ Res (2013) 112(11):1466–78. doi:10.1161/CIRCRESAHA.111.300166
53. Roifman I, Sun YC, Fedwick JP, Panaccione R, Buret AG, Liu H, et al. Evidence of endothelial dysfunction in patients with inflammatory bowel disease. Clin Gastroenterol Hepatol (2009) 7(2):175–82. doi:10.1016/j.cgh.2008.10.021
54. Elson DA, Ryan HE, Snow JW, Johnson R, Arbeit JM. Coordinate up-regulation of hypoxia inducible factor (HIF)-1alpha and HIF-1 target genes during multi-stage epidermal carcinogenesis and wound healing. Cancer Res (2000) 60(21):6189–95.
55. Polk DB, Frey MR. Mucosal restitution and repair. In: Physiology of the Gastrointestinal Tract, 5th Edn (2012). p. 1147–68.
56. Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol (2008) 14(3):348–53. doi:10.3748/wjg.14.348
57. Basson MD, Modlin IM, Flynn SD, Jena BP, Madri JA. Independent modulation of enterocyte migration and proliferation by growth factors, matrix proteins, and pharmacologic agents in an in vitro model of mucosal healing. Surgery (1992) 112(2):299–307.discussion 307-298,
58. Umar S. Intestinal stem cells. Curr Gastroenterol Rep (2010) 12(5):340–8. doi:10.1007/s11894-010-0130-3
59. Feil W, Lacy ER, Wong YM, Burger D, Wenzl E, Starlinger M, et al. Rapid epithelial restitution of human and rabbit colonic mucosa. Gastroenterology (1989) 97(3):685–701.
60. Iizuka M, Konno S. Wound healing of intestinal epithelial cells. World J Gastroenterol (2011) 17(17):2161–71. doi:10.3748/wjg.v17.i17.2161
61. Podolsky DK. Mucosal immunity and inflammation. V. Innate mechanisms of mucosal defense and repair: the best offense is a good defense. Am J Physiol (1999) 277(3 Pt 1):G495–9.
62. Moyer RA, Wendt MK, Johanesen PA, Turner JR, Dwinell MB. Rho activation regulates CXCL12 chemokine stimulated actin rearrangement and restitution in model intestinal epithelia. Lab Invest (2007) 87(8):807–17. doi:10.1038/labinvest.3700595
63. Morgan MR, Humphries MJ, Bass MD. Synergistic control of cell adhesion by integrins and syndecans. Nat Rev Mol Cell Biol (2007) 8(12):957–69. doi:10.1038/nrm2289
64. Laukaitis CM, Webb DJ, Donais K, Horwitz AF. Differential dynamics of alpha 5 integrin, paxillin, and alpha-actinin during formation and disassembly of adhesions in migrating cells. J Cell Biol (2001) 153(7):1427–40. doi:10.1083/jcb.153.7.1427
65. Partridge MA, Marcantonio EE. Initiation of attachment and generation of mature focal adhesions by integrin-containing filopodia in cell spreading. Mol Biol Cell (2006) 17(10):4237–48. doi:10.1091/mbc.E06-06-0496
66. Wiseman PW, Brown CM, Webb DJ, Hebert B, Johnson NL, Squier JA, et al. Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J Cell Sci (2004) 117(Pt 23):5521–34. doi:10.1242/jcs.01416
67. Zaidel-Bar R, Ballestrem C, Kam Z, Geiger B. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J Cell Sci (2003) 116(Pt 22):4605–13. doi:10.1242/jcs.00792
68. Dignass AU, Podolsky DK. Cytokine modulation of intestinal epithelial cell restitution: central role of transforming growth factor beta. Gastroenterology (1993) 105(5):1323–32. doi:10.1016/0016-5085(93)90136-Z
69. Hopkins AM, Pineda AA, Winfree LM, Brown GT, Laukoetter MG, Nusrat A. Organized migration of epithelial cells requires control of adhesion and protrusion through Rho kinase effectors. Am J Physiol Gastrointest Liver Physiol (2007) 292(3):G806–17. doi:10.1152/ajpgi.00333.2006
70. Lacy ER. Epithelial restitution in the gastrointestinal tract. J Clin Gastroenterol (1988) 10(Suppl 1):S72–7. doi:10.1097/00004836-198812001-00012
71. Buck RC. Ultrastructural features of rectal epithelium of the mouse during the early phases of migration to repair a defect. Virchows Arch B Cell Pathol Incl Mol Pathol (1986) 51(4):331–40. doi:10.1007/BF02899042
72. Feil W, Wenzl E, Vattay P, Starlinger M, Sogukoglu T, Schiessel R. Repair of rabbit duodenal mucosa after acid injury in vivo and in vitro. Gastroenterology (1987) 92(6):1973–86.
73. Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell (1996) 84(3):359–69. doi:10.1016/S0092-8674(00)81280-5
74. Horwitz AR, Parsons JT. Cell migration – movin’ on. Science (1999) 286(5442):1102–3. doi:10.1126/science.286.5442.1102
75. Sastry SK, Burridge K. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp Cell Res (2000) 261(1):25–36. doi:10.1006/excr.2000.5043
76. Dise RS, Frey MR, Whitehead RH, Polk DB. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol (2008) 294(1):G276–85. doi:10.1152/ajpgi.00340.2007
77. Dieckgraefe BK, Stenson WF, Alpers DH. Gastrointestinal epithelial response to injury. Curr Opin Gastroenterol (1996) 12(2):109–14. doi:10.1097/00001574-199603000-00001
78. Rademakers SE, Lok J, van der Kogel AJ, Bussink J, Kaanders JH. Metabolic markers in relation to hypoxia; staining patterns and colocalization of pimonidazole, HIF-1alpha, CAIX, LDH-5, GLUT-1, MCT1 and MCT4. BMC Cancer (2011) 11:167. doi:10.1186/1471-2407-11-167
79. Giraud ASX. Trefoil peptide and EGF receptor/ligand transgenic mice. Am J Physiol Gastrointest Liver Physiol (2000) 278(4):G501–6.
80. Dignass A, Lynch-Devaney K, Kindon H, Thim L, Podolsky DK. Trefoil peptides promote epithelial migration through a transforming growth factor beta-independent pathway. J Clin Invest (1994) 94(1):376–83. doi:10.1172/JCI117332
81. Kinoshita K, Taupin DR, Itoh H, Podolsky DK. Distinct pathways of cell migration and antiapoptotic response to epithelial injury: structure-function analysis of human intestinal trefoil factor. Mol Cell Biol (2000) 20(13):4680–90. doi:10.1128/MCB.20.13.4680-4690.2000
82. Mahmood A, Melley L, Fitzgerald AJ, Ghosh S, Playford RJ. Trial of trefoil factor 3 enemas, in combination with oral 5-aminosalicylic acid, for the treatment of mild-to-moderate left-sided ulcerative colitis. Aliment Pharmacol Ther (2005) 21(11):1357–64.
83. O’Toole EA, Marinkovich MP, Peavey CL, Amieva MR, Furthmayr H, Mustoe TA, et al. Hypoxia increases human keratinocyte motility on connective tissue. J Clin Invest (1997) 100(11):2881–91. doi:10.1172/JCI119837
84. Keely S, Glover LE, MacManus CF, Campbell EL, Scully MM, Furuta GT, et al. Selective induction of integrin beta1 by hypoxia-inducible factor: implications for wound healing. FASEB J (2009) 23(5):1338–46. doi:10.1096/fj.08-125344
85. Qiu J, Wang G, Hu J, Peng Q, Zheng Y. Id1-induced inhibition of p53 facilitates endothelial cell migration and tube formation by regulating the expression of beta1-integrin. Mol Cell Biochem (2011) 357(1–2):125–33. doi:10.1007/s11010-011-0882-6
86. Keely S, Campbell EL, Baird AW, Hansbro PM, Shalwitz RA, Kotsakis A, et al. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol (2013). doi:10.1038/mi.2013.29. [Epub ahead of print].
87. Rezvani HR, Ali N, Serrano-Sanchez M, Dubus P, Varon C, Ged C, et al. Loss of epidermal hypoxia-inducible factor-1alpha accelerates epidermal aging and affects re-epithelialization in human and mouse. J Cell Sci (2011) 124(Pt 24):4172–83. doi:10.1242/jcs.082370
89. Buck CA, Horwitz AF. Cell surface receptors for extracellular matrix molecules. Annu Rev Cell Biol (1987) 3:179–205. doi:10.1146/annurev.cellbio.3.1.179
90. Hynes RO. Integrins: a family of cell surface receptors. Cell (1987) 48(4):549–54. doi:10.1016/0092-8674(87)90233-9
91. Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science (1987) 238(4826):491–7. doi:10.1126/science.2821619
92. Schnapp LM. Adhesion, cell-matrix/integrins. In: Laurent GJ, Shapiro SJ, editors. Encyclopedia of Respiratory Medicine. Seattle: Academic Press (2006). p. 47–53.
93. Lotz MM, Nusrat A, Madara JL, Ezzell R, Wewer UM, Mercurio AM. Intestinal epithelial restitution. Involvement of specific laminin isoforms and integrin laminin receptors in wound closure of a transformed model epithelium. Am J Pathol (1997) 150(2):747–60.
94. Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol (2003) 171(11):6164–72.
95. Lawrance IC, Maxwell L, Doe W. Altered response of intestinal mucosal fibroblasts to profibrogenic cytokines in inflammatory bowel disease. Inflamm Bowel Dis (2001) 7(3):226–36. doi:10.1097/00054725-200108000-00008
96. Tong Q, Vassilieva EV, Ivanov AI, Wang Z, Brown GT, Parkos CA, et al. Interferon-gamma inhibits T84 epithelial cell migration by redirecting transcytosis of beta1 integrin from the migrating leading edge. J Immunol (2005) 175(6):4030–8.
97. Bisping G, Lugering N, Lutke-Brintrup S, Pauels HG, Schurmann G, Domschke W, et al. Patients with inflammatory bowel disease (IBD) reveal increased induction capacity of intracellular interferon-gamma (IFN-gamma) in peripheral CD8+ lymphocytes co-cultured with intestinal epithelial cells. Clin Exp Immunol (2001) 123(1):15–22. doi:10.1046/j.1365-2249.2001.01443.x
98. Schmitz H, Fromm M, Bentzel CJ, Scholz P, Detjen K, Mankertz J, et al. Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci (1999) 112(Pt 1):137–46.
99. Adams RB, Planchon SM, Roche JK. IFN-gamma modulation of epithelial barrier function. Time course, reversibility, and site of cytokine binding. J Immunol (1993) 150(6):2356–63.
100. Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest (1989) 83(2):724–7. doi:10.1172/JCI113938
101. Bretscher MS. Circulating integrins: alpha 5 beta 1, alpha 6 beta 4 and Mac-1, but not alpha 3 beta 1, alpha 4 beta 1 or LFA-1. EMBO J (1992) 11(2):405–10.
102. Pierini LM, Lawson MA, Eddy RJ, Hendey B, Maxfield FR. Oriented endocytic recycling of alpha5beta1 in motile neutrophils. Blood (2000) 95(8):2471–80.
103. Glover LE, Irizarry K, Scully M, Campbell EL, Bowers BE, Aherne CM, et al. IFN-gamma attenuates hypoxia-inducible factor (HIF) activity in intestinal epithelial cells through transcriptional repression of HIF-1beta. J Immunol (2011) 186(3):1790–8. doi:10.4049/jimmunol.1001442
104. Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol (1999) 66(6):889–900.
105. Hickey DK, Aldwell FE, Beagley KW. Oral immunization with a novel lipid-based adjuvant protects against genital Chlamydia infection. Vaccine (2010) 28(7):1668–72. doi:10.1016/j.vaccine.2009.12.010
106. Rabinovitch A, Suarez-Pinzon WL, Sorensen O, Bleackley RC, Power RF. IFN-gamma gene expression in pancreatic islet-infiltrating mononuclear cells correlates with autoimmune diabetes in nonobese diabetic mice. J Immunol (1995) 154(9):4874–82.
107. Southworth T, Metryka A, Lea S, Farrow S, Plumb J, Singh D. IFN-gamma synergistically enhances LPS signalling in alveolar macrophages from COPD patients and controls by corticosteroid-resistant STAT1 activation. Br J Pharmacol (2012) 166(7):2070–83. doi:10.1111/j.1476-5381.2012.01907.x
108. Sanchez-Elsner T, Ramirez JR, Sanz-Rodriguez F, Varela E, Bernabeu C, Botella LM. A cross-talk between hypoxia and TGF-beta orchestrates erythropoietin gene regulation through SP1 and Smads. J Mol Biol (2004) 336(1):9–24. doi:10.1016/j.jmb.2003.12.023
109. Falanga V, Qian SW, Danielpour D, Katz MH, Roberts AB, Sporn MB. Hypoxia upregulates the synthesis of TGF-beta 1 by human dermal fibroblasts. J Invest Dermatol (1991) 97(4):634–7. doi:10.1111/1523-1747.ep12483126
110. McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem (2006) 281(34):24171–81. doi:10.1074/jbc.M604507200
111. Han WQ, Zhu Q, Hu J, Li PL, Zhang F, Li N. Hypoxia-inducible factor prolyl-hydroxylase-2 mediates transforming growth factor beta 1-induced epithelial-mesenchymal transition in renal tubular cells. Biochim Biophys Acta (2013) 1833(6):1454–62. doi:10.1016/j.bbamcr.2013.02.029
112. Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature (1997) 390(6659):465–71. doi:10.1038/37284
113. Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol (2000) 1(3):169–78. doi:10.1038/35042034
114. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol (2012) 13(10):616–30. doi:10.1038/nrm3434
115. Ashcroft GS, Dodsworth J, van Boxtel E, Tarnuzzer RW, Horan MA, Schultz GS, et al. Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat Med (1997) 3(11):1209–15. doi:10.1038/nm1197-1209
116. Heikkinen PT, Nummela M, Jokilehto T, Grenman R, Kahari VM, Jaakkola PM. Hypoxic conversion of SMAD7 function from an inhibitor into a promoter of cell invasion. Cancer Res (2010) 70(14):5984–93. doi:10.1158/0008-5472.CAN-09-3777
117. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest (2001) 108(4):601–9. doi:10.1172/JCI12821
118. Reynolds LE, Conti FJ, Silva R, Robinson SD, Iyer V, Rudling R, et al. Alpha3beta1 integrin-controlled Smad7 regulates reepithelialization during wound healing in mice. J Clin Invest (2008) 118(3):965–74. doi:10.1172/JCI33538
119. Monteleone G, Caruso R, Pallone F. Role of Smad7 in inflammatory bowel diseases. World J Gastroenterol (2012) 18(40):5664–8. doi:10.3748/wjg.v18.i40.5664
120. Sim WH, Wagner J, Cameron DJ, Catto-Smith AG, Bishop RF, Kirkwood CD. Expression profile of genes involved in pathogenesis of pediatric Crohn’s disease. J Gastroenterol Hepatol (2012) 27(6):1083–93. doi:10.1111/j.1440-1746.2011.06973.x
121. Hume GE, Fowler EV, Lincoln D, Eri R, Templeton D, Florin TH, et al. Angiotensinogen and transforming growth factor beta1: novel genes in the pathogenesis of Crohn’s disease. J Med Genet (2006) 43(10):e51. doi:10.1136/jmg.2005.040477
122. Basu RK, Hubchak S, Hayashida T, Runyan CE, Schumacker PT, Schnaper HW. Interdependence of HIF-1alpha and TGF-beta/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am J Physiol Renal Physiol (2011) 300(4):F898–905. doi:10.1152/ajprenal.00335.2010
123. Hunt TK, Pai MP. The effect of varying ambient oxygen tensions on wound metabolism and collagen synthesis. Surg Gynecol Obstet (1972) 135(4):561–7.
124. Ninikoski J, Heughan C, Hunt TK. Oxygen tensions in human wounds. J Surg Res (1972) 12(2):77–82. doi:10.1016/0022-4804(72)90124-2
125. Gordillo GM, Sen CK. Revisiting the essential role of oxygen in wound healing. Am J Surg (2003) 186(3):259–63. doi:10.1016/S0002-9610(03)00211-3
126. Rezvani HR, Ali N, Nissen LJ, Harfouche G, de Verneuil H, Taieb A, et al. HIF-1alpha in epidermis: oxygen sensing, cutaneous angiogenesis, cancer, and non-cancer disorders. J Invest Dermatol (2011) 131(9):1793–805. doi:10.1038/jid.2011.141
Keywords: hypoxia, integrins, epithelial cells, wound healing, mucosal immunity, HIF-1a
Citation: Goggins BJ, Chaney C, Radford-Smith GL, Horvat JC and Keely S (2013) Hypoxia and integrin-mediated epithelial restitution during mucosal inflammation. Front. Immunol. 4:272. doi: 10.3389/fimmu.2013.00272
Received: 07 June 2013; Paper pending published: 11 July 2013;
Accepted: 27 August 2013; Published online: 11 September 2013.
Edited by:
Rajaraman D. Eri, University of Tasmania, AustraliaCopyright: © 2013 Goggins, Chaney, Radford-Smith, Horvat and Keely. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon Keely, School of Biomedical Sciences and Pharmacy, University of Newcastle, University Drive, Callaghan, Newcastle, 2308 NSW, Australia e-mail:c2ltb24ua2VlbHlAbmV3Y2FzdGxlLmVkdS5hdQ==