 Johannes Fessler1
Johannes Fessler1

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 06 August 2013
Sec. T Cell Biology
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00231
This article is part of the Research Topic How aging affects T lymphocyte-mediated immunity View all 10 articles
Age-related deviations of the immune system contribute to a higher likelihood of infections, cancer, and autoimmunity in the elderly. Senescence of T-lymphocytes is characterized by phenotypical and functional changes including the loss of characteristic T-cell surface markers, while an increase of stimulatory receptors, cytotoxicity as well as resistance against apoptosis is observed. One of the key mediators of immune regulation are naturally occurring regulatory T-cells (Tregs). Tregs express high levels of CD25 and the intracellular protein forkhead box P3; they exert their suppressive functions in contact-dependent as well as contact-independent manners. Quantitative and qualitative defects of Tregs were observed in patients with autoimmune diseases. Increased Treg activity was shown to suppress anti-tumor and anti-infection immunity. The effect of aging on Tregs, and the possible contribution of age-related changes of the Treg pool to the pathophysiology of diseases in the elderly are still poorly understood. Treg homeostasis depends on an intact thymic function and current data suggest that conversion of non-regulatory T-cells into Tregs as well as peripheral expansion of existing Tregs compensates for thymic involution after puberty to maintain constant Treg numbers. In the conventional T-cell subset, peripheral proliferation of T-cells is associated with replicative senescence leading to phenotypical and functional changes. For Tregs, different developmental stages were also described; however, replicative senescence of Tregs has not been observed yet.
The immune system combats against infectious agents and depletes damaged or transformed cells, whereas intact self-components are usually ignored. Nevertheless, clinical manifestations of autoimmunity occur in at least 5% of the general population. The exact causes of autoimmune diseases are elusive; however, genetic and environmental risk factors as well as an insufficient elimination of cells bearing autoreactive T-cell receptors (TCRs) in the thymus contribute to the evolvement of disease (1, 2). To prevent autoimmunity, tolerance mechanisms including clonal deletion, induction of apoptosis, or anergy of self-reactive T-cells are essential. In addition, regulatory T-cells (Tregs) were identified as sentinels of the immune response keeping aberrant/exaggerated immune reactions in balance. Several distinct T-cell subsets with regulatory function have been identified so far including natural Tregs, adaptive or induced Tregs (iTreg), type 1 regulatory T-cells (Tr1), T helper 3 cells (Th3), double-negative (dn) T-cells, γδ T-cells, and iNKT cells. In a number of autoimmune diseases a diminished prevalence and/or impaired function of Tregs were observed (3). As several autoimmune disorders (such as rheumatoid arthritis or vasculitis) occur more frequently in the elderly, the question arises whether aging is linked to quantitative and/or qualitative defects of the Treg pool (4–6).
In this review we summarize current data about the effects of aging on Tregs and highlight the possible mechanisms leading to senescence of Tregs.
Natural Tregs develop in the thymus through recognition of self-antigen presented by thymic epithelial or dendritic cells. For this process CD28 co-stimulation is required, whereas IL-2 and TGF-β are less important as indicated by knock-out mice models (7).
Today, there is still no consensus on the reliable identification of Tregs by flow cytometry. A variety of cell surface molecules have been proposed as specific Tregs markers such as glucocorticoid-induced tumor necrosis factor receptor (GITR), cytotoxic T-lymphocyte associated antigen-4 (CTLA-4), the co-receptors Neuropilin-1 and PD-1, the adhesion molecule CD62L, major histocompatibility complex (MHC) class II DR, or CD45 isoforms. The type I cytokine receptor CD127 is a negative marker of Tregs and the absence of this molecule is frequently used for Treg identification (8).
The forkhead transcription factor FoxP3 was proposed as the most specific marker of Tregs as FoxP3 expression is essential for Treg development and function (9): Tregs were unable to develop in a mouse receiving FoxP3-deficient progenitor cells from another animal (10) and retroviral expression of FoxP3 in human and murine T-cells enabled the conversion of non-regulatory naïve T-cells into a Treg-like phenotype with suppressive activity and surface expression of CD25 (9). A mutation of the FoxP3 gene in humans results in the fatal autoimmune syndrome IPEX (immune dysregulation, polyendocrinopathy, X-linked) (11). For experimental studies, however, FoxP3 appears not to be an optimal Treg marker because first, permeabilization of T-cells is necessary to stain FoxP3 and cells are thus not viable anymore and second, newer data indicate that human FoxP3 is up-regulated in activated T-cells without suppressive function as well (12).
The Ikaros family transcription factor Helios was proposed as an alternative indicator of human Tregs with a higher specificity compared to FoxP3. Recent data, however indicate that Helios is also up-regulated in activated non-regulatory T-cells (13). In summary, there is currently no specific marker of human Tregs available limiting the validity of studies investigating qualitative and/or quantitative changes of the Treg pool.
The mechanisms of Treg mediated immunosuppression are still unclear. Most likely, Tregs have multiple functions with direct and indirect inhibitory effects on antigen-presenting cells (APCs) and T-cells such as the following (14, 15): (a) expression of the surface molecule CTLA-4 directly suppressing the activity of T-cells, (b) indirect inhibition of effector cells by the induction of anti-inflammatory biochemical pathways in APC, (c) direct or indirect killing of effector cells and APCs, and/or (d) production of immunoregulatory cytokines such as TGF-β and IL-10 (16).
Interestingly, a recent study reported that human Tregs are able to induce senescence of naïve and memory responder T-cells in vitro and in vivo. The resulting senescent T-cell subset had an altered phenotype and revealed potent suppressive functions. The mechanisms leading to senescence of non-regulatory T-cells were not completely understood; however, the phosphorylation of p38 and ERK1/2 signaling pathways inhibiting naïve T-cell growth and cell-cycle regulation appeared to play a role (17).
A prevalence of approximately 0.6–15% out of the CD4+ T-cell pool has been reported for Tregs in healthy adults and mice (4, 18). The influence of aging on Treg prevalence in humans has been rarely studied so far and available reports suggest only minor changes of the circulating Treg pool through age (19). Higher proportions of Tregs were only found in cord blood samples suggesting a pivotal role of Tregs during homeostatic proliferation of naïve T-cells in the fetal life (20, 21). During the first 36 months of life Treg levels decline rapidly (22) and remain relatively stable thereafter.
Mouse studies showed increased Treg prevalences in lymphoid organs of aged compared to young animals, whereas frequencies in circulating blood and thymus were unchanged (23, 24). This finding led to the hypothesis that during aging Tregs accumulate in lymphoid tissues; hypothetically explaining the increased susceptibility to infections and reduced vaccine response in elderly animals. The accumulation of Tregs has further been observed in the skin of aged persons possibly resulting in a higher risk of skin cancer as Tregs reduce local anti-tumor immune responses (25–27).
In animals, Treg function seems to decrease with advancing age. The transfer of CD25+ Tregs from aged mice into young animals for example resulted in a lower suppression of delayed type hypersensitivity responses compared to the infusion of young Treg cells (23). Another study found that CD4+CD25high Tregs from aged animals less efficiently inhibited the proinflammatory activity of IL-17+ T-cells compared to Tregs from young mice (28). In human studies it was observed that Tregs from young and elderly individuals similarly inhibited the proliferation of responder cells whereas the production of the anti-inflammatory cytokine IL-10 was reduced in cells from the older group. The phenotype of Tregs including expression of CD25, FoxP3, IL-7Rα, or chemokine receptor expression, however, was unchanged (29). In conclusion Tregs from aged individuals are less efficient in preventing the occurrence of autoimmunity, while their number remains unaltered.
On the other hand, cancer and infections occur more commonly in the elderly suggesting increased Treg responses (see also above) (29–31). One mouse study found an increase of Treg prevalences in aged animals correlating with a defective tumor clearance. CD25-depletion restored the anti-cancer immune response (32). Similarly, CD25-depletion in aged mice reduced the lesion size in a Leishmania major infection model (24). Others reported that the depletion of Tregs with denileukin diftitox improved tumor-specific immunity only in young mice whereas tumor growth was unaffected in aged mice. This was explained by increased numbers of myeloid-derived suppressor cell (MDSC) in aged animals, and upon depletion of these cells tumor-specific immunity was restored (33).
In summary, current data on age-related changes of Treg prevalences and function are conflicting and do not completely explain the simultaneously increased risk of autoimmunity (suggesting lower Treg function), cancer, and infections (indicating increased Treg responses) in the elderly. Apart from the difficulty of a reliable identification of Tregs the possible accumulation of Tregs in lymphoid organs and/or tissues during aging might lead to an underestimation of the total Treg pool in current human studies. Future studies investigating tissue samples from immune-organs of elderly individuals would be desirable to better understand the role of Tregs in the pathogenesis of age-related diseases.
Development of natural Tregs in the thymus depends on a positive selection process including high affinity interactions of the TCR to cortically expressed host antigens. Thymic stromal lymphopoietin activated CD11c-positive dendritic cells (34), co-stimulatory molecules including CD28, PD-1, CD40L (35) as well as the cytokine IL-2 were all shown to be crucial for thymic Treg generation (36–38). Besides, the Nr4a nuclear receptors (involved in apoptosis, proliferation, DNA repair, inflammation, and others) were recently reported to contribute to Treg development. Mice lacking these receptors in T-cells were unable to produce Tregs and died early from systemic autoimmunity (39).
During aging a progressive degeneration of the thymus occurs leading to a substantial loss of its capacity to generate and export new T-cells (40, 41). Throughout middle age thymic epithelial space and the functional unit of thymopoiesis (and thus the production of T-cells) decline by approximately 3% per year until the age of 45 when only an irrelevant level of functional thymic tissue remains. The total number of T-cells in the periphery nevertheless is unchanged and peripheral mechanisms of T-cell renewal have to compensate for progressive thymic failure (42–44).
Parallel to the overall reduction of thymic T-cell output the production of thymically derived Tregs decreases with age (45). Alternative mechanisms such as increased surveillance of Tregs in the elderly (46) as well as peripheral Treg generation may compensate for the loss of thymic function to maintain a sufficient Treg pool (see Figure 1). Indeed, numerous studies indicate a possible conversion of non-regulatory CD4+CD25− T-cells into Tregs in vitro and in vivo (47, 48). Moreover, mouse studies showed that peripheral self-antigen-driven proliferation of Tregs is a thymus-independent mechanism to maintain Tregs (49–51). The proportion of conventional T-cells differentiating into Tregs as well as the relative contribution of homeostatic Treg proliferation to the overall Treg pool in elderly individuals are unknown.
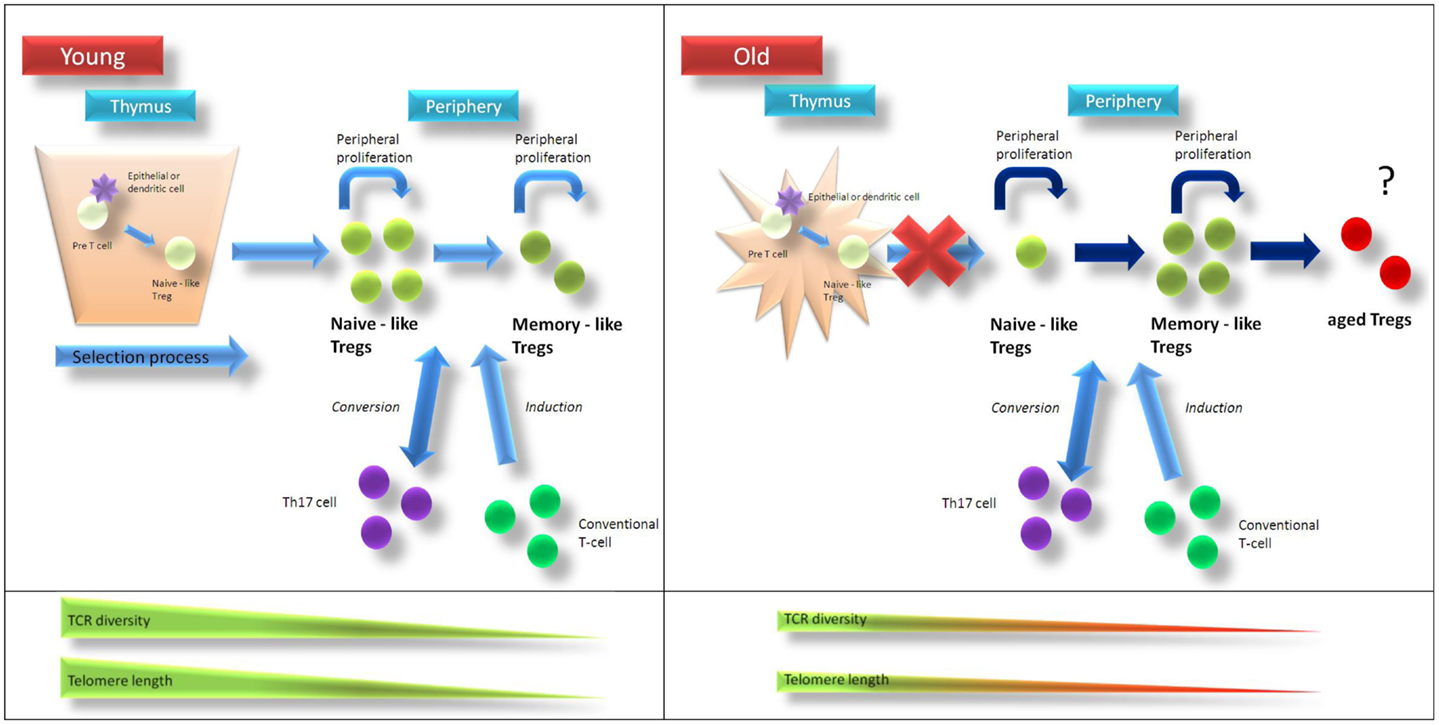
Figure 1. Age-related changes of Treg homeostasis. In young individuals Tregs are generated in the thymus and are released as “naïve-like” Tregs into circulation. After antigen-contact, Tregs develop into a “memory-like” phenotype. Treg homeostasis is supported by homeostatic proliferation of “naïve-like” and “memory-like” Tregs as well as conversion of non-regulatory T-cells into Tregs. Telomere length and T-cell receptor diversity is higher in naïve-like compared to memory-like Tregs. After puberty thymic function is progressively lost and in aged individuals homeostatic proliferation of existing Tregs as well as conversion of non-regulatory T-cells into Tregs compensate for thymic failure to maintain Treg pool. Due to ongoing homeostatic replication telomere length and T-cell receptor diversity of Tregs from elderly people are contracted compared to those from young individuals. Recurrent stimulation of Tregs might then lead to a status of “terminal-differentiation” with altered phenotype and function. Treg regulatory T-cell, TCR … T-cell receptor.
Peripheral mechanisms of T-cell renewal (particularly homeostatic expansion of existing Tregs) are probably not infinite. Normally, T-cells proliferate beyond the seventh decade of life. Thereafter, telomere lengths are usually contracted to levels known as the “Hayflick limit”. At this stage, non-regulatory T-cells do not proliferate anymore and undergo phenotypical and functional changes such as down-regulation of CD28 and acquisition of cytotoxic potential (4, 52, 53). Due to the fact that Tregs display even shorter telomeres than non-regulatory T-cells, it is conceivable that peripherally proliferating Tregs reach the “Hayflick limit” even earlier (54). Impaired Treg homeostasis may then result in immune dysfunction with increased risk of immune-mediated disorders.
In addition to the shortened telomere length, TCR diversity is also contracted to at least 100-fold in elderly individuals (55). This has been explained by the observation that homeostatic proliferation of T-cells is antigen dependent. Thus, T-cells with a high affinity TCR to self-antigens or antigens deriving from chronic virus infections have a survival advantage over other T-cells (42, 56). Given that similar mechanisms drive peripheral proliferation of non-regulatory T-cells and Tregs, a reduction of Treg TCR diversity (with a skew to certain antigens) can be expected in the elderly. Consequently, Tregs could mediate increased immunosuppression in response to specific self- (even if transformed) or viral antigens with increased incidence of malignancies and infections in the elderly. At the same time the reduced diversity of Tregs could result in decreased protection from autoimmunity (3).
Similar to the developmental stages known for non-regulatory T-cells (development form CD45RA+ naïve to CD45RO+ memory and finally to CD28− memory effector T-cells), different cellular subsets of Tregs were also observed. In humans, CD4+foxP3+ Tregs may have either a “naïve-like” phenotype characterized by the expression of CD25+CD45RA+ or a CD25hiCD45RO+ “memory-like” phenotype (54). In mice, naïve-like Tregs were characterized by the expression of CD25, CD62L, and CCR7 and by preferential homing to antigen-draining lymph nodes, where they were able to inhibit the induction of inflammation (10, 57, 58). Memory/effector-like Tregs (characterized by expression of CD29, CD44, ICOS, and LFA-1) migrated into non-lymphogenic tissues and sites of inflammation; a local down-regulation of immune reactions was shown (57, 58).
In humans, the highest prevalence of naïve-like Tregs were found in cord blood and it was assumed that these naïve-like Tregs are produced in the thymus (20, 59). The prevalence of memory-like Tregs increases rapidly during childhood and it was demonstrated that these memory-like Tregs have shorter telomeres and a lower content of TCR excision circles (Trecs) compared to naïve-like Tregs reflecting a longer replicative history (54). The mechanisms mediating the transition of a naïve-like Treg into a memory-like phenotype still have to be explored; however, it is believed that antigen experienced dendritic cells migrating to secondary lymphoid tissues are involved. Tregs proliferate upon stimulation with autologous immature and mature dendritic cells (54, 60). A low surface expression of CD45RB on memory-like Tregs further supports the hypothesis of an antigen-driven development of naïve-like Tregs. CD45RB is normally down-regulated after repeated antigen-contact (61).
Human adult peripheral blood usually contains both, naïve-like and memory-like Tregs. Parallel to the reduction of total naïve T-cells, the quantity of naïve-like Tregs declines with age whereas the prevalence of memory-like Tregs increases (29, 62). The total pool of circulating Tregs; however, remains unchanged as mentioned above (19). As naïve-like Tregs exhibit a higher proliferative potential in vitro compared to memory-like Tregs it can be expected that the capacity of the immune system to downregulate abnormal immune responses declines with age (54).
Replicative senescence of T-cells is a prominent feature of aging resulting from homeostatic proliferation and repetitive antigen exposure (63). The most important phenotypic feature of senescent T-cells is the loss of the type I transmembrane protein CD28, a major co-stimulatory molecule (64). From the functional perspective, non-regulatory CD28− T-cells produce large amounts of interferon γ, perforin, and granzyme B, providing them with the ability to lyse target cells (65). Another feature of CD28− T-cells is their longevity and persistence that can be explained by defects in the apoptotic pathway with upregulation of bcl-2 and Fas-associated death domain like IL-2-converting enzyme-like inhibitory protein (FLIP) (66, 67). Terminally differentiated T-cells also acquire new stimulatory receptors including killer cell immunoglobulin-like receptors (KIRs) and Toll-like receptors (TLRs) (68, 69). Thus, activation of CD4+CD28− T-cells no longer depends on professional antigen-presenting cells, rather it is promoted by stress molecules as well as bacterial and/or viral products (65). The frequency of terminally differentiated CD4+CD28− T-cells is increased in old individuals as well as in younger patients with autoimmune diseases such as rheumatoid arthritis or spondyloarthritis (70). Given that Tregs proliferate in the periphery to maintain the total Treg pool after thymic failure it is plausible to hypothesize that Tregs may undergo terminal-differentiation as well.
Interestingly, a proportion of Tregs from aged mice showed decreased expression of CD25 (46, 71). These CD25low Tregs occurred predominantly in the spleen (24) but had comparable functional properties to CD25+ Tregs. A similar CD4+CD25−foxP3+ Treg population has been observed in SLE patients. SLE patients are known to have a prematurely aged immune system (72) with accumulation of CD28−T-cells. A detailed characterization of CD4+CD25−FoxP3+ Tregs regarding the expression of naïve/memory T-cell markers or determination of telomere lengths was unfortunately not performed. Further evidence for the occurrence of Treg senescence was found in a study on healthy aged individuals reporting the occurrence of a CD8+CD25+ Treg population lacking CD28 expression. These regulatory cells shared phenotypic and functional features with CD4+ Tregs from the same population (73). The occurrence and possible characteristics of terminally differentiated CD4+ Tregs is an interesting issue that has to be investigated by future studies.
Accumulating evidence suggests age-associated changes of Treg prevalence and/or Treg function. Due to involution of thymus after puberty peripheral mechanisms including homeostatic proliferation of Tregs or conversion of non-regulatory T-cells into Tregs compensate for the decreasing generation of new Treg cells. However, these peripheral mechanisms are limited; this leads to altered composition of the Treg pool. Age-related changes of Tregs are suspected to increase the risk of autoimmunity, cancer, and infections in the elderly; however, the exact mechanisms are still poorly understood. Current studies are limited by the difficult identification of human Tregs and the uncertainty whether circulating Tregs reflect the total Treg pool or a cellular subset only. Future studies are required to investigate cellular senescence of Tregs and possible therapeutic approaches targeting Tregs in aged individuals.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Prof. Dr. Winfried Graninger (Medical University of Graz) for his critical review of the manuscript.
1. Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol (2004) 172(10):5967–72.
2. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet (2010) 42(6):508–14. doi:10.1038/ng.582
3. Dejaco C, Duftner C, Grubeck-Loebenstein B, Schirmer M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology (2006) 117(3):289–300. doi:10.1111/j.1365-2567.2005.02317.x
4. Dejaco C, Duftner C, Schirmer M. Are regulatory T-cells linked with aging? Exp Gerontol (2006) 41(4):339–45. doi:10.1016/j.exger.2006.01.008
5. Goronzy JJ, Weyand CM. Immune aging and autoimmunity. Cell Mol Life Sci (2012) 69(10):1615–23. doi:10.1007/s00018-012-0970-0
6. Hohensinner PJ, Goronzy JJ, Weyand CM. Telomere dysfunction, autoimmunity and aging. Aging Dis (2011) 2(6):524–37.
7. Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, et al. CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. J Exp Med (2002) 196(2):237–46. doi:10.1084/jem.20020590
8. Shevach EM. Regulatory/suppressor T cells in health and disease. Arthritis Rheum (2004) 50(9):2721–4. doi:10.1002/art.20500
9. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol (2005) 6(4):345–52. doi:10.1038/ni1178
10. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
11. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi:10.1038/83713
12. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood (2007) 110(8):2983–90. doi:10.1182/blood-2007-06-094656
13. Fessler J, Felber A, Duftner C, Dejaco C. Therapeutic potential of regulatory T cells in autoimmune disorders. BioDrugs (2013) 27(4):281–91. doi:10.1007/s40259-013-0026-5
14. Thornton AM, Piccirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol (2004) 34(2):366–76. doi:10.1002/eji.200324455
15. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. “Infectious” transplantation tolerance. Science (1993) 259(5097):974–7. doi:10.1126/science.8094901
16. Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev (2008) 223:371–90. doi:10.1111/j.1600-065X.2008.00637.x
17. Ye J, Huang X, Hsueh EC, Zhang Q, Ma C, Zhang Y, et al. Human regulatory T cells induce T-lymphocyte senescence. Blood (2012) 120(10):2021–31. doi:10.1182/blood-2012-03-416040
18. Ohkura N, Sakaguchi S. Regulatory T cells: roles of T cell receptor for their development and function. Semin Immunopathol (2010) 32(2):95–106. doi:10.1007/s00281-010-0200-5
19. Raynor J, Lages CS, Shehata H, Hildeman DA, Chougnet CA. Homeostasis and function of regulatory T cells in aging. Curr Opin Immunol (2012) 24(4):482–7. doi:10.1016/j.coi.2012.04.005
20. Wing K, Ekmark A, Karlsson H, Rudin A, Suri-Payer E. Characterization of human CD25+ CD4+ T cells in thymus, cord and adult blood. Immunology (2002) 106(2):190–9. doi:10.1046/j.1365-2567.2002.01412.x
21. Schonland SO, Zimmer JK, Lopez-Benitez CM, Widmann T, Ramin KD, Goronzy JJ, et al. Homeostatic control of T-cell generation in neonates. Blood (2003) 102(4):1428–34. doi:10.1182/blood-2002-11-3591
22. Sullivan KE, McDonald-McGinn D, Zackai EH. CD4(+) CD25(+) T-cell production in healthy humans and in patients with thymic hypoplasia. Clin Diagn Lab Immunol (2002) 9(5):1129–31.
23. Zhao L, Sun L, Wang H, Ma H, Liu G, Zhao Y. Changes of CD4+CD25+Foxp3+ regulatory T cells in aged Balb/c mice. J Leukoc Biol (2007) 81(6):1386–94. doi:10.1189/jlb.0506364
24. Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, et al. Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J Immunol (2008) 181(3):1835–48.
25. Agius E, Lacy KE, Vukmanovic-Stejic M, Jagger AL, Papageorgiou AP, Hall S, et al. Decreased TNF-alpha synthesis by macrophages restricts cutaneous immunosurveillance by memory CD4+ T cells during aging. J Exp Med (2009) 206(9):1929–40. doi:10.1084/jem.20090896
26. Green VL, Michno A, Stafford ND, Greenman J. Increased prevalence of tumour infiltrating immune cells in oropharyngeal tumours in comparison to other subsites: relationship to peripheral immunity. Cancer Immunol Immunother (2013) 62(5):863–73. doi:10.1007/s00262-013-1395-9
27. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer (2013) 108(4):914–23. doi:10.1038/bjc.2013.32
28. Sun L, Hurez VJ, Thibodeaux SR, Kious MJ, Liu A, Lin P, et al. Aged regulatory T cells protect from autoimmune inflammation despite reduced STAT3 activation and decreased constraint of IL-17 producing T cells. Aging Cell (2012) 11(3):509–19. doi:10.1111/j.1474-9726.2012.00812.x
29. Hwang KA, Kim HR, Kang I. Aging and human CD4(+) regulatory T cells. Mech Ageing Dev (2009) 130(8):509–17. doi:10.1016/j.mad
30. Tsaknaridis L, Spencer L, Culbertson N, Hicks K, LaTocha D, Chou YK, et al. Functional assay for human CD4+CD25+ Treg cells reveals an age-dependent loss of suppressive activity. J Neurosci Res (2003) 74(2):296–308. doi:10.1002/jnr.10766
31. Gregg R, Smith CM, Clark FJ, Dunnion D, Khan N, Chakraverty R, et al. The number of human peripheral blood CD4+ CD25high regulatory T cells increases with age. Clin Exp Immunol (2005) 140(3):540–6. doi:10.1111/j.1365-2249.2005.02798.x
32. Sharma S, Dominguez AL, Lustgarten J. High accumulation of T regulatory cells prevents the activation of immune responses in aged animals. J Immunol (2006) 177(12):8348–55.
33. Hurez V, Daniel BJ, Sun L, Liu AJ, Ludwig SM, Kious MJ, et al. Mitigating age-related immune dysfunction heightens the efficacy of tumor immunotherapy in aged mice. Cancer Res (2012) 72(8):2089–99. doi:10.1158/0008-5472.CAN-11-3019
34. Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, et al. Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature (2005) 436(7054):1181–5. doi:10.1038/nature03886
35. Paust S, Cantor H. Regulatory T cells and autoimmune disease. Immunol Rev (2005) 204:195–207. doi:10.1111/j.0105-2896.2005.00247.x
36. Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, et al. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol (2004) 172(2):834–42.
37. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol (2001) 2(4):301–6. doi:10.1038/86302
38. Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity (2010) 32(5):642–53. doi:10.1016/j.immuni.2010.04.012
39. Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, Kimura A, et al. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat Immunol (2013) 14(3):230–7. doi:10.1038/ni.2520
40. Simpson JG, Gray ES, Beck JS. Age involution in the normal human adult thymus. Clin Exp Immunol (1975) 19(2):261–5.
41. Berzins SP, Uldrich AP, Sutherland JS, Gill J, Miller JF, Godfrey DI, et al. Thymic regeneration: teaching an old immune system new tricks. Trends Mol Med (2002) 8(10):469–76. doi:10.1016/S1471-4914(02)02415-2
42. Goronzy JJ, Weyand CM. Thymic function and peripheral T-cell homeostasis in rheumatoid arthritis. Trends Immunol (2001) 22(5):251–5. doi:10.1016/S1471-4906(00)01841-X
43. Mackall CL, Bare CV, Granger LA, Sharrow SO, Titus JA, Gress RE. Thymic-independent T cell regeneration occurs via antigen-driven expansion of peripheral T cells resulting in a repertoire that is limited in diversity and prone to skewing. J Immunol (1996) 156(12):4609–16.
44. Steinmann GG, Klaus B, Muller-Hermelink HK. The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand J Immunol (1985) 22(5):563–75. doi:10.1111/j.1365-3083.1985.tb01916.x
45. Chiu BC, Stolberg VR, Zhang H, Chensue SW. Increased Foxp3(+) Treg cell activity reduces dendritic cell co-stimulatory molecule expression in aged mice. Mech Ageing Dev (2007) 128(11–12):618–27. doi:10.1016/j.mad.2007.09.002
46. Chougnet CA, Tripathi P, Lages CS, Raynor J, Sholl A, Fink P, et al. A major role for Bim in regulatory T cell homeostasis. J Immunol (2011) 186(1):156–63. doi:10.4049/jimmunol.1001505
47. Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25- T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol (2004) 173(12):7259–68.
48. Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci U S A (2005) 102(14):5126–31. doi:10.1073/pnas.0501701102
49. Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med (2003) 198(2):249–58. doi:10.1084/jem.20030315
50. Cozzo C, Larkin J III, Caton AJ. Cutting edge: self-peptides drive the peripheral expansion of CD4+CD25+ regulatory T cells. J Immunol (2003) 171(11):5678–82.
51. Fisson S, Darrasse-Jeze G, Litvinova E, Septier F, Klatzmann D, Liblau R, et al. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med (2003) 198(5):737–46. doi:10.1084/jem.20030686
52. Effros RB. From Hayflick to Walford: the role of T cell replicative senescence in human aging. Exp Gerontol (2004) 39(6):885–90. doi:10.1016/j.exger.2004.03.004
53. Goronzy JJ, Lee WW, Weyand CM. Aging and T-cell diversity. Exp Gerontol (2007) 42(5):400–6. doi:10.1016/j.exger.2006.11.016
54. Valmori D, Merlo A, Souleimanian NE, Hesdorffer CS, Ayyoub M. A peripheral circulating compartment of natural naive CD4 Tregs. J Clin Invest (2005) 115(7):1953–62. doi:10.1172/JCI23963
55. Naylor K, Li G, Vallejo AN, Lee WW, Koetz K, Bryl E, et al. The influence of age on T cell generation and TCR diversity. J Immunol (2005) 174(11):7446–52.
56. Weiss L, Donkova-Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood (2004) 104(10):3249–56. doi:10.1182/blood-2004-01-0365
57. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med (2004) 199(3):303–13. doi:10.1084/jem.20031562
58. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity (2005) 22(3):329–41. doi:10.1016/j.immuni.2005.01.016
59. Takahata Y, Nomura A, Takada H, Ohga S, Furuno K, Hikino S, et al. CD25+CD4+ T cells in human cord blood: an immunoregulatory subset with naive phenotype and specific expression of forkhead box p3 (Foxp3) gene. Exp Hematol (2004) 32(7):622–9. doi:10.1016/j.exphem.2004.03.012
60. Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and activation of regulatory T cells in the human fetus. Eur J Immunol (2005) 35(2):383–90. doi:10.1002/eji.200425763
61. Salmon M, Pilling D, Borthwick NJ, Viner N, Janossy G, Bacon PA, et al. The progressive differentiation of primed T cells is associated with an increasing susceptibility to apoptosis. Eur J Immunol (1994) 24(4):892–9. doi:10.1002/eji.1830240417
62. Seddiki N, Santner-Nanan B, Tangye SG, Alexander SI, Solomon M, Lee S, et al. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood (2006) 107(7):2830–8. doi:10.1182/blood-2005-06-2403
63. Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev (2005) 205:158–69. doi:10.1111/j.0105-2896.2005.00256.x
64. Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol (1993) 11:191–212. doi:10.1146/annurev.iy.11.040193.001203
65. Nakajima T, Schulte S, Warrington KJ, Kopecky SL, Frye RL, Goronzy JJ, et al. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation (2002) 105(5):570–5. doi:10.1161/hc0502.103348
66. Schirmer M, Vallejo AN, Weyand CM, Goronzy JJ. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28- T cells from rheumatoid arthritis patients. J Immunol (1998) 161(2):1018–25.
67. Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J Immunol (2000) 165(11):6301–7.
68. Namekawa T, Snyder MR, Yen JH, Goehring BE, Leibson PJ, Weyand CM, et al. Killer cell activating receptors function as costimulatory molecules on CD4+CD28null T cells clonally expanded in rheumatoid arthritis. J Immunol (2000) 165(2):1138–45.
69. Raffeiner B, Dejaco C, Duftner C, Kullich W, Goldberger C, Vega SC, et al. Between adaptive and innate immunity: TLR4-mediated perforin production by CD28null T-helper cells in ankylosing spondylitis. Arthritis Res Ther (2005) 7(6):R1412–20. doi:10.1186/ar1840
70. Dejaco C, Duftner C, Klauser A, Schirmer M. Altered T-cell subtypes in spondyloarthritis, rheumatoid arthritis and polymyalgia rheumatica. Rheumatol Int (2010) 30(3):297–303. doi:10.1007/s00296-009-0949-9
71. Nishioka T, Shimizu J, Iida R, Yamazaki S, Sakaguchi S. CD4+CD25+Foxp3+ T cells and CD4+CD25-Foxp3+ T cells in aged mice. J Immunol (2006) 176(11):6586–93.
72. Bonelli M, Savitskaya A, Steiner CW, Rath E, Smolen JS, Scheinecker C. Phenotypic and functional analysis of CD4+ CD25- Foxp3+ T cells in patients with systemic lupus erythematosus. J Immunol (2009) 182(3):1689–95.
Keywords: FOXP3, regulatory T-lymphocyte, aging, cellular senescence, thymus, suppressor cells
Citation: Fessler J, Ficjan A, Duftner C and Dejaco C (2013) The impact of aging on regulatory T-cells. Front. Immunol. 4:231. doi: 10.3389/fimmu.2013.00231
Received: 28 March 2013; Accepted: 22 July 2013;
Published online: 06 August 2013.
Edited by:
Dietmar Herndler-Brandstetter, Yale University School of Medicine, USAReviewed by:
Nikolai Petrovsky, Flinders Medical Centre, AustraliaCopyright: © 2013 Fessler, Ficjan, Duftner and Dejaco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Dejaco, Department of Rheumatology and Immunology, Medical University Graz, Auenbruggerplatz 15, A-8036 Graz, Austria e-mail:Y2hyaXN0aWFuLmRlamFjb0BnbXgubmV0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.