



95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 05 August 2013
Sec. T Cell Biology
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00221
This article is part of the Research Topic Investigating and harnessing T-cell functions with engineered immune receptors and their ligands. View all 21 articles
Recent early stage clinical trials evaluating the adoptive transfer of patient CD8+ T-cells re-directed with antigen receptors recognizing tumors have shown very encouraging results. These reports provide strong support for further development of the therapeutic concept as a curative cancer treatment. In this respect combining the adoptive transfer of tumor-specific T-cells with therapies that increase their anti-tumor capacity is viewed as a promising strategy to improve treatment outcome. The ex vivo genetic engineering step that underlies T-cell re-direction offers a unique angle to combine antigen receptor delivery with the targeting of cell-intrinsic pathways that restrict T-cell effector functions. Recent progress in genome editing technologies such as protein- and RNA-guided endonucleases raise the possibility of disrupting gene expression in T-cells in order to enhance effector functions or to bypass tumor immune suppression. This approach would avoid the systemic administration of compounds that disrupt immune homeostasis, potentially avoiding autoimmune adverse effects, and could improve the efficacy of T-cell based adoptive therapies.
Although there is still controversy over the role of the immune system in protecting the organism against the development of neoplasms in a natural setting (1) it is well accepted that artificial immunity can efficiently contain and even eradicate established tumors (2). Harnessing the anti-tumor potential of T-cells, and particularly CD8+ T-cells, is a promising approach for curative cancer treatment. Because of their relative ease of administration and documented low toxicities therapeutic vaccines that trigger T-cell responses are a very attractive approach. However, even though they efficiently induce antigen-specific immunity, the clinical benefit of cancer vaccines has so far been limited (3). In contrast adoptive cell therapies (ACT), where T-cells are modified ex vivo and re-infused in a patient’s circulation, are more difficult to implement and require important infrastructural investment. Yet a number of studies have now reported long-term remissions or tumor clearance (4–6), warranting further development of the therapeutic concept.
While conferring the immune system with the ability to recognize tumors through vaccination or ACT is a pre-requisite for the induction of efficient anti-tumor responses it is likely insufficient to achieve long-term clinical benefit in a majority of patients. An increasing body of evidence points to the necessity of combining different therapeutic approaches in order to improve treatment outcome (7, 8). For instance several small-molecule compounds that target oncogenic pathways also enhance tumor destruction by immune mechanisms, e.g., by sensitizing cancer cells to cytolysis (9, 10). The coordinated delivery of these compounds with immunotherapies is expected to improve clinical responses in an additive or even synergistic manner. Similarly the combination of immune-based therapies also holds great potential. Monoclonal antibodies (mAbs) blocking immune checkpoint receptors have recently emerged as promising therapeutics and many believe that the recent marketing authorization of Ipilimumab, targeting CTLA-4, heralds great strides in this area. Immune checkpoint receptor blocking agents are currently marketed or developed as single therapies but are expected to achieve maximal efficacy in combination with immune stimulatory approaches such as vaccination or ACTs (11, 12). Although generic treatment combinations will undoubtedly provide some degree of clinical benefit it is the prospect of developing personalized therapies tailored to individual needs that holds the greatest potential to improve clinical outcome in cancer therapy. The heterogenous nature of similar tumor histologies as well as individual genetic variability are believed to account for the varied response levels to generic treatments and the wider availability of prognostic tools should help define adequate treatment options that improve patient response. With respect to cancer vaccines or ACTs information about the nature of the immune checkpoint pathway(s) relevant to a tumor would be particularly useful in order to counteract immune suppression.
T-cell-based ACTs rely on the infusion in a patient’s circulation of ex vivo expanded tumor-infiltrating lymphocytes (TILs) or peripheral blood T-cells transduced with viral vectors expressing a tumor-specific antigen receptor. This engineering step offers the opportunity to transfer additional genetic material conferring T-cells with enhanced anti-tumor activity. Targeted genome editing relying on viral gene transfer could readily be combined with the delivery of antigen receptors at little additional cost in one unique therapeutic entity. This approach would avoid the drawbacks associated with combining treatment modalities of different nature requiring distinct administration regimens, e.g., cellular therapy and mAb injection. In addition cell-intrinsic disruption of immune checkpoints in tumor-specific T-cells is likely to display a better safety profile than the systemic administration of blocking agents. Recently developed gene targeting technologies such as zinc-finger proteins (ZFPs), transcription activator-like proteins (TALs), and RNA-guided endonucleases (RGENs) could thus be harnessed in order to silence the expression of T-cell-intrinsic genes that restrain their anti-tumor potential.
RNA interference (RNAi) often is the technique of choice to silence gene expression in somatic cells and lentivirus-mediated RNAi is a good option for sustained and efficient silencing. Most lentiviral RNAi systems express short-hairpin RNAs (shRNAs) from RNApolIII promoters, which drive high levels of transcription using precise initiation and termination sites. A recurrent problem of lentivirus-mediated RNAi, which is particularly salient in T-cells (13), is that the constant generation of shRNAs interferes with endogenous miRNA biogenesis and can result in the deregulation of gene expression (14, 15). This issue has prompted investigators to seek alternative methods to silence gene expression (16).
Recently developed genome editing technologies based on DNA-targeting proteins have the potential to revolutionize ACTs by offering convenient tools to alter gene expression. TAL effector-nucleases (TALENs) and ZFP-nucleases (ZFNs) effect complete gene knockout (Figure 1A) and are promising alternatives to RNAi for therapeutic applications (17–19). TALs are bacterial DNA-binding proteins consisting of near identical 34 amino-acid modules that bind one nucleotide with high affinity. The variable 12th and 13th amino-acids of TALs, called repeat-variable di-nucleotide confers base specificity (NN → G/A, NI → A, NG → T, NK → G, HD → C, and NS → A/T/C/G) and TAL arrays that target a nucleotide sequence can be generated by assembling individual modules (17, 20). ZFPs are eukaryotic DNA-binding proteins. Cys2-His2 fingers, which are used for genome editing, are the most common ZFP motif (21) and are each specific for a nucleotide triplet. Artificial ZFP domains that target specific DNA sequences, usually 9–18 nt long, can be constructed by assembling individual fingers (18). ZFPs and TALs have similar modular configurations but TALs can in theory target any stretch of nucleotides beginning with a thymidine whereas some structural incompatibilities between individual ZFP modules due to overlapping DNA-binding domains make the assembly of oligomeric ZFPs error-prone and narrow down the diversity of possible target DNA sequences. A successful and popular application of these technologies is the fusion of customized ZFPs or TALs to the catalytic domain of the restriction nuclease Fok1 (ZFNs and TALENs). Fok1 nucleases catalyze DNA double strand breaks (DSBs) when they dimerize (22). TALENs and ZFNs are therefore designed in pairs that target adjacent sequences on opposite DNA strands, thereby promoting Fok1 dimerization, separated by a spacer region where DNA cleavage occurs. Non-homologous end joining (NHEJ) repair of DSBs results in insertions or deletions at the DNA cleavage site (17, 23) and bi-allelic frameshift mutations that result in complete knockout occur at low frequencies (18, 24). The overall efficiency of the approach is sufficient to generate knockout cells following appropriate selection procedures. Of note TALEN design is more flexible as they can accommodate spacers of different lengths (25) whereas ZFNs strictly require 5–7 nt (26). Taking this into account, as well as structural constraints mentioned above, it is estimated that the frequency of target sequences is 1 in 500 bp for ZFNs and 1 in 35 bp for TALENs (20).
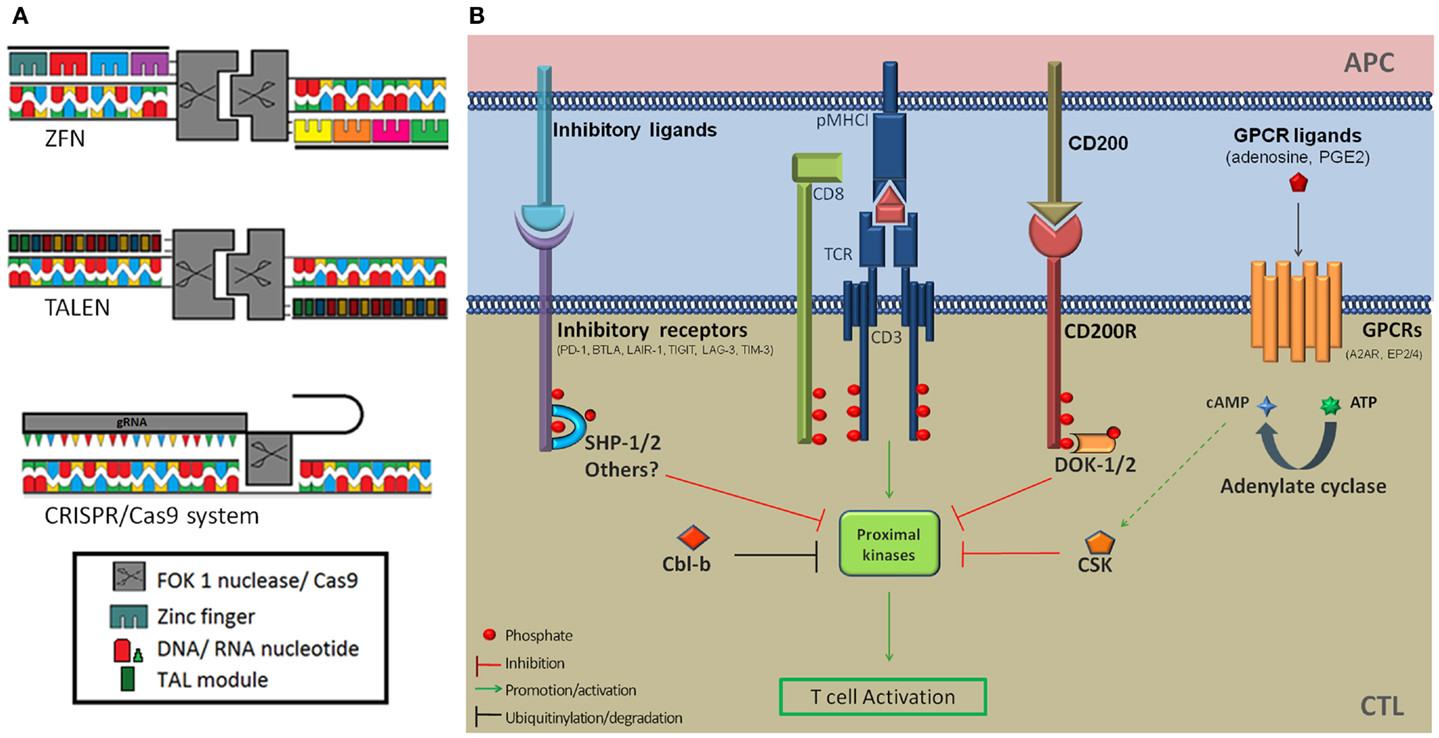
Figure 1. (A) Schematic diagram of the ZFN, TALEN, and CRISPR/Cas9 genome editing tools. (B) Inhibition of T-cell activation by immune checkpoint receptors and downstream signaling proteins. Several co-inhibitory receptors (PD-1, BTLA, and LAIR-1) inhibit T-cell signaling by recruiting the SHP-1 and/or SHP-2 tyrosine phosphatases at proximity of the TCR signaling complex via ITIMs and ITSMs. This results in the dephosphorylation of proximal kinases downstream of TCR triggering. In addition PD-1 ligation was shown to induce increased expression of the Cbl-b E3 ubiquitin ligase, which targets signaling molecules for degradation. Activation of the CD200R leads to the recruitment of DOK2 and RasGAP to its intra-cellular domain, resulting in the inhibition of downstream MAP kinases. The adenosine receptor 2A and PGE2 receptors EP2 and EP4 modulate T-cell activation through mobilization of the cAMP-PKAI-CSK pathway. CSK phosphorylates the inhibitory C-terminal tyrosine residue of Src kinases and negatively regulates TCR signaling. A2AR, adenosine A2a receptor; APC, antigen-presenting cell; BTLA-4, B- and T-lymphocyte attenuator; Cbl-b, casitas B-cell lymphoma; CTL, cytotoxic T-lymphocyte; DOK-1/2, docking protein 1/2; EP2/4, prostaglandin E receptor 2/4; Erk, extra-cellular signal regulated kinases; ITIM, immunoreceptor tyrosine-based inhibition motif; ITSM, immunoreceptor tyrosine-based switch motif; GPCR, G-protein coupled receptor; LAG-3, lymphocyte-activation gene 3; LAIR-1, leukocyte-associated immunoglobulin-like receptor 1; Lck, lymphocyte-specific protein tyrosine kinase; MHC1, major histocompatibility complex class 1; PD-1, programed death receptor 1; PD1-L1, programed death receptor 1-ligand 1; SHP-1, Src homology 2 domain containing protein tyrosine phosphatase; TCR, T-cell receptor; TIGIT, T-cell immunoreceptor with Ig and ITIM domains; TIM-3, T-cell immunoglobulin domain and mucin domain 3.
RNA-guided endonucleases provide a distinct and attractive alternative to genome editing compared with protein-guided nucleases. The functions of clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins as a system providing adaptive immunity to bacteria against bacteriophages (27, 28) was recently harnessed for genome engineering (29, 30). The Cas9 nuclease binds to a short complementary RNA (crRNA) providing DNA-targeting specificity and to a trans-activating crRNA (tracRNA), required for crRNA processing, expressed individually or combined as a chimeric guide RNA (gRNA) (Figure 1A). CRISPR-Cas9 systems displayed a cleavage efficiency comparable (31) or superior (32) to TALENs in human cells. The clear advantage of RGEN is that it can be easily adapted to target different genomic sequences by customizing the synthetic crRNA/gRNA delivered in combination with Cas9 (33). In comparison ZFNs and TALENs require extensive engineering and validation steps.
The delivery of genome editing agents to T-cells is a crucial aspect of their successful application to ACTs. Because nuclease-based genome editing relies on generating transmissible mutations, protein- or RNA-guided nucleases only need to be transiently expressed. In fact transient expression probably minimizes off-target DNA cleavage (34). Provasi et al. have used integration-deficient lentiviruses as well as adenoviruses in order to modify the genome of T-cells with ZFNs (35). Of note it was recently shown that, due to their very repetitive nature, TAL arrays were incompatible with efficient reverse transcription required for the delivery of genetic material using lentiviruses (36), thereby limiting the range of delivery methods for TALENs.
Crucially therapies based on T-cell genome editing have already entered clinical development. A phase II clinical trial based on preventing the expression of the CCR5 gene, acting as a co-receptor for HIV in CD4+ T-cells, using ZFNs (37) was recently initiated for the treatment of HIV/AIDS (NCT01252641). The safety results will be of huge importance for ZFN-based therapies and for genome editing in T-cells in general. Moreover a recent study provided proof of concept for the combination of TCR gene delivery with genome editing by using ZFNs specific for the endogenous constant TCR gene segments in order to prevent mispairing with ectopic TCR chains (35). The success of this approach provides a good rationale for wider applications of ZFN genome editing to T-cells.
Enhancing the anti-tumor potential of CD8+ T-cells through genome editing can be done in many ways. Here we will focus on disrupting the expression of genes that inhibit T-cell functions as a result of the suppressive activity of the tumor micro-environment. T-cell inhibitory pathways targeted by genome editing in the context cancer ACTs should meet several criteria. First, their mechanism of action should be strictly cell-intrinsic. Second, they should be relevant to effector T-cells as opposed to naive T-cells. For instance CTLA-4 does not meet these two criteria since it works at least partly by reducing the availability of co-stimulatory molecules on the surface of antigen-presenting cells during the priming of naive T-cells (38). Finally, since only anti-tumor T-cells are modified, one of the advantages of this approach is that it allows targeting ubiquitous suppressive pathways whose systemic blockade or inhibition might result in serious adverse effects. Because it is clinically validated the most obvious target is probably PDCD1: the gene encoding the co-inhibitory receptor PD-1. PD-1 is expressed on activated T-cells and its engagement by its two known ligands PD-L1 and PD-L2 inhibits proximal signaling events triggered by TCR stimulation through recruitment of the phosphatase SHP-2 (39) and increased expression of the E3 ubiquitin ligase Cbl-b (40), which impair key components of the TCR signaling cascade through dephosphorylation and proteasomal degradation (Figure 1B). High cellular expression levels of PD-1 are characteristic of exhausted CD8+ T-cells in chronic viral infections as well as TILs and correlate with impaired effector functions (41). Histological analyses have revealed that numerous tumor types express one or both PD-1 ligands (42, 43), prompting the targeting of this pathway in order to augment anti-tumor immunity. PD-1 blockade has shown promising objective response rates in a range of cancer indications and it is anticipated that PD-1 blocking agents will be approved for marketing authorization as mono-therapies. In addition these therapeutics are evaluated in combination with cancer vaccines, small-molecule signaling inhibitors, tumor-targeting mAbs, and cytokine therapy (http://clinicaltrials.gov/ct2/results?term=pd1&Search=Search). The combination of PD-1 blockade with these treatments, as well as with cancer ACTs, is expected to further enhance anti-tumor activity (11). Several other co-inhibitory receptors expressed by T-cells qualify as targets for gene editing coupled with antigen receptor delivery (Table 1; Figure 1B). In vivo and in vitro pre-clinical data strongly support the development of reagents targeting TIM-3 and LAG-3. Dual targeting of PD-1 and TIM-3 or LAG-3 with mAbs synergistically enhanced anti-tumor responses (44) and pre-clinical evaluations of a soluble Fc-LAG3 complex, which has now entered clinical development, were promising (45). Other targets are currently under similar evaluation procedures and might expand the list of druggable co-inhibitory receptors for cancer immunotherapy (Table 1; Figure 1B).
Table 1. Potential immune checkpoint receptor targets for genome editing in the context of cancer adoptive cellular therapies.
The presence of cognate ligands within the tumor micro-environment is a crucial aspect for targeting co-inhibitory receptors and other immune checkpoint receptors. In the case of PD-1 retrospective analysis of patient biopsies in the phase Ib clinical trial assessing the blocking mAb BMS-936558 showed that the objective response rate in patients whose tumors expressed PD-L1 reached 36% compared with 18% in the entire cohort and 0% among patients with PD-L1-negative tumors (46). These striking results highlight the importance of prognosis and patient stratification for the design of appropriate cancer immunotherapies based on PD-1 inhibition. Such a strong correlation is still to be established for other immune checkpoint receptors but it is tempting to speculate that similar principles are applicable. However, even though their relevance in tumor immunity is established, it is not entirely clear what the actual ligands for several co-inhibitory receptors are in the context of anti-tumor immunity. More fundamental and clinical investigations are required in order to unambiguously identify relevant inhibitory ligands and assess their presence in the tumor environment.
Several soluble regulatory mediators also act as immune checkpoints in anti-tumor immunity. For instance high levels of extra-cellular adenosine are found in the vicinity of many solid tumors because of the hypoxic environment, a well-known environmental factor promoting adenosine release. Suppressive adenosine is also generated through direct dephosphorylation of extra-cellular adenosine nucleotides by the cell-surface nucleotidases CD39 and CD73 expressed by regulatory T-cells (Tregs) and some tumors, e.g., ovarian carcinomas (47, 48). The adenosine receptor 2A (A2AR), belonging to the G-protein coupled receptor family (GPCRs), is expressed on T-cells and has been identified as a target for immunotherapy for over a decade (12, 49–51). The A2AR inhibits T-cell activation through the cAMP-PKAI-CSK pathway (Figure 1B) and pre-clinical in vivo models have shown that A2AR knock-down or antagonism in adoptively transferred T-cells dramatically increased anti-tumor immunity (50). Inhibiting adenosine-mediated immune suppression is therefore believed to be an efficient strategy for cancer immunotherapies. Yet because adenosine receptors are ubiquitously expressed and involved in many physiological processes, especially in neurotransmission, classical antagonistic approaches are likely to result in a number of side effects. Such systemic adverse effects could be avoided in the context of adoptive T-cell therapies through T-cell-intrinsic gene editing or the in vitro selection of desensitized and irresponsive T-cells (52). Similarly, prostaglandin E2 (PGE2) directly suppresses T-cell activation through the cAMP second messenger pathway in effector/memory CD8+ T-cells (53). Tumor-associated Tregs as well as colorectal cancer cells express high levels of immunosuppressive PGE2 (54). Interfering with EP2/EP4 receptors expression in T-cells may therefore enhance their anti-tumor potential.
A broader, possibly riskier, alternative to targeting individual immune checkpoint receptors would be interfering with the expression of downstream molecules conveying intra-cellular inhibitory signals. For instance several co-inhibitory receptors use the tyrosine phosphatases SHP-1 and/or SHP-2 to inhibit T-cell activation (Figure 1B). Inhibition of SHP-1/2 expression may therefore confer resistance to several checkpoint pathways used by tumors. Stromnes and colleagues reported that conditional knockout of SHP-1 in mature murine CD8+ T-cells improved effector cell functions and tumor clearance in an adoptive transfer setting similar to cancer ACTs without resulting in autoimmune toxicity, thereby providing a good rationale for such an approach (55).
T-cell based ACTs that rely on the re-infusion of patient T-cells expressing an artificial antigen receptor is an epitome of personalized medicine. These therapies require the identification of specific tumor antigens and/or patient HLA-type and would undoubtedly benefit from further prognostic analysis and subsequent treatment customization. Based on recent successes in cancer immunotherapy, immune checkpoint receptors that suppress T-cells represent a particularly attractive class of targets for such an approach. We believe that enhancing the anti-tumor potential of re-directed T-cells by targeting inhibitory pathways through genome editing can further improve the efficacy of cancer ACTs. Moreover cell-intrinsic inhibition of these pathways may display an advantageous safety profile compared with immune checkpoint blockade relying on the systemic administration of mAbs, recombinant proteins, or small molecules.
The field of genome editing is currently buzzing with new ideas and technologies that make the application of targeted gene knockout and gene correction a tantalizing prospect for the development of customized ACTs in regenerative medicine and immunotherapy. However, numerous potential pitfalls must be assessed in pre-clinical development. At this point ZFNs are the most advanced technology for genome editing and have already entered clinical development. However, their complicated and labor-intensive design, construction, and validation are major drawbacks for their widespread use in non-specialized laboratories, which often rely on commercial reagents. TALENs, on the other hand, are much more user-friendly and several toolboxes that allow investigators to generate custom reagents are available at low cost. With regard to T-cell based cancer ACTs, however, the incompatibility of TALENs with retroviral delivery, often used to express ectopic TCRs or CARs, might be problematic. Finally, CRISPR-Cas9 genome editing may prove to be the holy grail of genome editing but the current lack of hindsight on this technology does not allow concluding on its use yet. Thorough assessment of the respective advantages and drawbacks of each technology as well as pre-clinical feasibility and safety studies are warranted to validate genome editing applied to cancer ACTs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in. Science (2011) 331(6024):1565–70. doi:10.1126/science.1203486
2. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature (2011) 480:480–9. doi:10.1038/nature10673
3. Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunol Rev (2011) 239:27–44. doi:10.1111/j.1600-065X.2010.00979.x
4. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi:10.1056/NEJMoa1103849
5. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29:917–24. doi:10.1200/JCO.2010.32.2537
6. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17:4550–7. doi:10.1158/1078-0432.CCR-11-0116
7. Blank CU, Hooijkaas AI, Haanen JB, Schumacher TN. Combination of targeted therapy and immunotherapy in melanoma. Cancer Immunol Immunother (2011) 60:1359–71. doi:10.1007/s00262-011-1079-2
8. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer (2012) 12:237–51. doi:10.1038/nrc3237
9. Noh KH, Kang TH, Kim JH, Pai SI, Lin KY, Hung CF, et al. Activation of Akt as a mechanism for tumor immune evasion. Mol Ther (2009) 17:439–47. doi:10.1038/mt.2008.255
10. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res (2010) 70:5213–9. doi:10.1158/0008-5472.CAN-10-0118
11. Martinez Forero I, Okada H, Topalian SL, Gajewski TF, Korman AJ, Melero I. Workshop on immunotherapy combinations. Society for immunotherapy of cancer annual meeting Bethesda, November 3, 2011. J Transl Med (2012) 10:108. doi:10.1186/1479-5876-10-108
12. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
13. An DS, Qin FX, Auyeung VC, Mao SH, Kung SK, Baltimore D, et al. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther (2006) 14:494–504. doi:10.1016/j.ymthe.2006.05.015
14. Yi R, Doehle BP, Qin Y, Macara IG, Cullen BR. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA (2005) 11:220–6. doi:10.1261/rna.7233305
15. Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L, et al. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res (2007) 35:5154–64. doi:10.1093/nar/gkm543
16. Couto LB, High KA. Viral vector-mediated RNA interference. Curr Opin Pharmacol (2010) 10:534–42. doi:10.1016/j.coph.2010.06.007
17. Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc (2012) 7:171–92. doi:10.1038/nprot.2011.431
18. Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet (2010) 11:636–46. doi:10.1038/nrg2842
19. Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol (2013) 31:227–9. doi:10.1038/nbt.2501
20. Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res (2011) 39:e82. doi:10.1093/nar/gkr218
21. Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct (2000) 29:183–212. doi:10.1146/annurev.biophys.29.1.183
22. Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol (2009) 27:851–7. doi:10.1038/nbt.1562
23. Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJH, et al. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol (2013) 31:251–8. doi:10.1038/nbt.2517
24. Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature (2005) 435:646–51. doi:10.1038/nature03556
25. Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol (2011) 29:143–8. doi:10.1038/nbt.1755
26. Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics (2010) 186:757–61. doi:10.1534/genetics.110.120717
27. Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science (2008) 321:960–4. doi:10.1126/science.1159689
28. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (2012) 337:816–21. doi:10.1126/science.1225829
29. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (2013) 339:819–23. doi:10.1126/science.1231143
30. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science (2013) 339:823–6. doi:10.1126/science.1232033
31. Habib N, Hsu PD, Wu X, Jiang W, Luciano A, Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science (2013) 339:819–23. doi:10.1126/science.1231143
32. Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell (2013) 12:393–4. doi:10.1016/j.stem.2013.03.006
33. van der Oost J. Molecular biology. New tool for genome surgery. Science (2013) 339:768–70. doi:10.1126/science.1234726
34. Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol (2007) 25:1298–306. doi:10.1038/nbt1353
35. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med (2012) 18:807–15. doi:10.1038/nm.2700
36. Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res (2012) 41(5):e63. doi:10.1093/nar/gks1446
37. Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol (2008) 26:808–16. doi:10.1038/nbt1410
38. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi:10.1126/science.1202947
39. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192:1027–34. doi:10.1084/jem.192.7.1027
40. Karwacz K, Bricogne C, Macdonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med (2011) 3:581–92. doi:10.1002/emmm.201100165
42. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol (2012) 24:207–12. doi:10.1016/j.coi.2011.12.009
43. Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology (2012) 1:1223–5. doi:10.4161/onci.21335
44. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72:917–27. doi:10.1158/0008-5472.CAN-11-1620
45. Fougeray S, Brignone C, Triebel F. A soluble LAG-3 protein as an immunopotentiator for therapeutic vaccines: preclinical evaluation of IMP321. Vaccine (2006) 24:5426–33. doi:10.1016/j.vaccine.2006.03.050
46. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
47. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res (2010) 70:2245–55. doi:10.1158/0008-5472.CAN-09-3109
48. Häusler SF, Montalbán del Barrio I, Strohschein J, Anoop Chandran P, Engel JB, Hönig A, et al. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol Immunother (2011) 60:1405–18. doi:10.1007/s00262-011-1040-4
49. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414:916–20. doi:10.1038/414916a
50. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA (2006) 103:13132–7. doi:10.1073/pnas.0605251103
51. Sitkovsky M, Ohta A. Targeting the hypoxia-adenosinergic signaling pathway to improve the adoptive immunotherapy of cancer. J Mol Med (Berl) (2013) 91:147–55. doi:10.1007/s00109-013-1001-9
52. Ohta A, Kjaergaard J, Sharma S, Mohsin M, Goel N, Madasu M, et al. In vitro induction of T cells that are resistant to A2 adenosine receptor-mediated immunosuppression. Br J Pharmacol (2009) 156:297–306. doi:10.1111/j.1476-5381.2008.00019.x
53. Oberprieler NG, Lemeer S, Kalland ME, Torgersen KM, Heck AJ, Tasken K. High-resolution mapping of prostaglandin E2-dependent signaling networks identifies a constitutively active PKA signaling node in CD8+CD45RO+ T cells. Blood (2010) 116:2253–65. doi:10.1182/blood-2010-01-266650
54. Mosenden R, Tasken K. Cyclic AMP-mediated immune regulation – overview of mechanisms of action in T cells. Cell Signal (2011) 23:1009–16. doi:10.1016/j.cellsig.2010.11.018
55. Stromnes IM, Fowler C, Casamina CC, Georgopolos CM, McAfee MS, Schmitt TM, et al. Abrogation of SRC homology region 2 domain-containing phosphatase 1 in tumor-specific T cells improves efficacy of adoptive immunotherapy by enhancing the effector function and accumulation of short-lived effector T cells in vivo. J Immunol (2012) 189:1812–25. doi:10.4049/jimmunol.1200552
56. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
57. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
58. Turnis ME, Korman AJ, Drake CG, Vignali DA. Combinatorial immunotherapy: PD-1 may not be LAG-ing behind any more. Oncoimmunology (2012) 1:1172–4. doi:10.4161/onci.20593
59. Murphy KM, Nelson CA, Sedý JR. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol (2006) 6:671–81. doi:10.1038/nri1917
60. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol (2003) 4:670–9. doi:10.1038/ni944
61. Kretz-Rommel A, Qin F, Dakappagari N, Ravey EP, McWhirter J, Oltean D, et al. CD200 expression on tumor cells suppresses antitumor immunity: new approaches to cancer immunotherapy. Mian Yi Xue Za Zhi (2007) 178:5595–605.
62. Moreaux J, Veyrune JL, Reme T, De Vos J, Klein B. CD200: a putative therapeutic target in cancer. Biochem Biophys Res Commun (2008) 366:117–22. doi:10.1016/j.bbrc.2007.11.103
63. Pallasch CP, Ulbrich S, Brinker R, Hallek M, Uger RA, Wendtner C-M. Disruption of T cell suppression in chronic lymphocytic leukemia by CD200 blockade. Leuk Res (2009) 33:460–4. doi:10.1016/j.leukres.2008.08.021
64. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6:1245–52. doi:10.1038/ni1271
65. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. Mian Yi Xue Za Zhi (2011) 186:1338–42. doi:10.4049/jimmunol.1003081
66. Lebbink RJ, de Ruiter T, Adelmeijer J, Brenkman AB, van Helvoort JM, Koch M, et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J Exp Med (2006) 203:1419–25. doi:10.1084/jem.20052554
67. Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305). J Leukoc Biol (2008) 83:799–803. doi:10.1189/jlb.0907609
68. Mahic M, Yaqub S, Johansson CC, Taskén K, Aandahl EM. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. Mian Yi Xue Za Zhi (2006) 177:246–54.
69. Raskovalova T, Lokshin A, Huang X, Su Y, Mandic M, Zarour HM, et al. Inhibition of cytokine production and cytotoxic activity of human antimelanoma specific CD8+ and CD4+ T lymphocytes by adenosine-protein kinase A type I signaling. Cancer Res (2007) 67:5949–56. doi:10.1158/0008-5472.CAN-06-4249
Keywords: T-cells, genome editing, cancer, cell therapies, immune checkpoints
Citation: Lloyd A, Vickery ON and Laugel B (2013) Beyond the antigen receptor: editing the genome of T-cells for cancer adoptive cellular therapies. Front. Immunol. 4:221. doi: 10.3389/fimmu.2013.00221
Received: 18 April 2013; Accepted: 16 July 2013;
Published online: 05 August 2013.
Edited by:
Michael Sitkovsky, Northeastern University, USAReviewed by:
Balbino Alarcon, Consejo Superior de Investigaciones Cientificas, SpainCopyright: © 2013 Lloyd, Vickery and Laugel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno Laugel, Institute of Infection and Immunity, Cardiff University School of Medicine, Henry Wellcome Building, Heath Park, Cardiff, CF14 4XN, Wales, UK e-mail:bGF1Z2VsYmZAY2FyZGlmZi5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.