 Bruce M. Hall*
Bruce M. Hall* Giang T. Tran Nirupama D. Verma Karren M. Plain
Giang T. Tran Nirupama D. Verma Karren M. Plain Catherine M. Robinson Masaru Nomura and Suzanne J. Hodgkinson
Catherine M. Robinson Masaru Nomura and Suzanne J. Hodgkinson
- Immune Tolerance Laboratory, Medicine, University of New South Wales, Sydney, NSW, Australia
Antigen specific T regulatory cells (Treg) are often CD4+CD25+FoxP3+ T cells, with a phenotype similar to natural Treg (nTreg). It is assumed that nTreg cannot develop into an antigen specific Treg as repeated culture with IL-2 and a specific antigen does not increase the capacity or potency of nTreg to promote immune tolerance or suppress in vitro. This has led to an assumption that antigen specific Treg mainly develop from CD4+CD25−FoxP3− T cells, by activation with antigen and TGF-β in the absence of inflammatory cytokines such as IL-6 and IL-1β. Our studies on antigen specific CD4+CD25+ T cells from animals with tolerance to an allograft, identified that the antigen specific and Treg are dividing, and need continuous stimulation with specific antigen T cell derived cytokines. We identified that a variety of cytokines, especially IL-5 and IFN-γ but not IL-2 or IL-4 promoted survival of antigen specific CD4+CD25+FoxP3+ Treg. To examine if nTreg could be activated to antigen specific Treg, we activated nTreg in culture with either IL-2 or IL-4. Within 3 days, antigen specific Treg are activated and there is induction of new cytokine receptors on these cells. Specifically nTreg activated by IL-2 and antigen express the interferon-γ receptor (IFNGR) and IL-12p70 (IL-12Rβ2) receptor but not the IL-5 receptor (IL-5Rα). These cells were responsive to IFN-γ or IL-12p70. nTreg activated by IL-4 and alloantigen express IL-5Rα not IFNGR or IL-12p70Rβ2 and become responsive to IL-5. These early activated antigen specific Treg, were respectively named Ts1 and Ts2 cells, as they depend on Th1 or Th2 responses. Further culture of Ts1 cells with IL-12p70 induced Th1-like Treg, expressing IFN-γ, and T-bet as well as FoxP3. Our studies suggest that activation of nTreg with Th1 or Th2 responses induced separate lineages of antigen specific Treg, that are dependent on late Th1 and Th2 cytokines, not the early cytokines IL-2 and IL-4.
Historical Perspective
Immune tolerance results from a combination of deletion of antigen specific T and B cell clones, anergy, and suppression. Like all biological systems, immunity has in built self-regulation that prevents induction of destructive autoimmunity and controls or limits all immune effector responses against any antigen. While a variety of leukocytes can regulate, this review will focus only on CD4+ T regulatory cells (Treg).
Since the first description of suppressor T cells, the difference between non-antigen specific Treg that reside in thymus, bone marrow, and peripheral lymphoid tissues, and antigen specific Treg that are present mainly in spleen and tissues, has been appreciated (1–3). This division is consistent with natural Treg (nTreg) and antigen specific Treg. Early studies characterized CD8+ T suppressor cells, reviewed (4) but this work was discredited (5) and a common view was suppressor T cells did not exist, until the recognition of CD4+ Treg.
Antigen Specific CD4+CD25+ Treg
Alloantigen specific transplant tolerance was found in the mid 1980s to be mediated by CD4+ T cells not CD8+ T or B cells (6–8). In the early 1990s Waldman’s group found CD4+ T cells from host transplant tolerant animals infect adoptive hosts’ T cells to maintain alloantigen specific tolerance (9).
At that time, we observed that the CD4+ T cells that transferred antigen specific tolerance rapidly died in vitro (10–12). Death of antigen specific tolerance transferring CD4+ T cells could be prevented by both stimulation with specific antigen and cytokines provided at that time by supernatant from Concanavalin A stimulated spleen cells. This supernatant was a crude source of IL-2 (12), but is now known to contain a number of cytokines, as well as IL-2. This suggested that the CD4+ T cells that transfer transplant tolerance were activated cells that may depend on IL-2. We thus examined and found they expressed the IL-2 alpha receptor (CD25) (11). In 1990 we identified alloantigen specific tolerance transferring cells as CD25+ Class II MHC+CD45RC+CD4+ T cells (11). At that time CD25 was expressed by CD4+ T cells activated to effect rejection (13), thus we assumed the suppressor cells were derived from specific alloantigen activated CD4+ T cells. As IL-2 alone only partially sustained the capacity of tolerant CD4+ T cells to transfer antigen specific tolerance, we concluded other cytokines were required (12). Since we have systematically examined which cytokines are involved in the maintenance of antigen specific CD4+CD25+FoxP3+ Treg, and this is the focus of this review.
Natural Treg
We also found that normal animals have cells, particularly in thymus and bone marrow, that suppress immune responses in a non-antigen specific manner, and that adult thymectomy depletes these cells, leading to heightened immune responses (14) and greater susceptibility to autoimmunity (15). Alloantigen specific CD4+ T suppressor cells have a different tissue distribution, being greatest in spleen, less in lymph nodes, and not in thymus or bone marrow (7). Further, they do not re-circulate rapidly from blood to lymph, suggesting they re-circulated through peripheral somatic tissue not through lymphoid tissues (7), similar to memory T cells (16), and not like naïve T cells that re-circulate from blood through lymphoid tissues (17). These basic differences in the migration of antigen specific and nTreg can be used to distinguish these cell populations by cell surface markers that direct their migration pathways, reviewed (18).
Later, activated CD4+ T cell in normal animals that expressed CD25 and prevented autoimmunity in neonatal thymectomized mice were described (19). These CD4+CD25+ Treg suppressed in a non-antigen specific manner, and are known as nTreg. nTreg are thymus derived and express FoxP3 (20) that prevents IL-2 induction and induces CD25 expression. FoxP3 expression in mice is a marker of Treg, but in man activated CD4+ and CD8+ T cells transiently express FoxP3 (21) and can be induced to have prolonged expression of FoxP3 (22). IL-2 is essential for survival of nTreg in peripheral lymphoid tissues (23, 24). CD4+ T cell with high expression of CD25, are regulatory, whereas CD4+CD25lo T cells are not regulatory (25).
Natural Treg have low expression of CD127, the IL-7 receptor, which is highly expressed by effector lineage CD4+CD25− T cells (26), albeit activated CD4+ T cells (27), and T follicular helper cells (Tfh) also have low expression of CD127 (28). The survival of nTreg without an immune response is dependent on low levels of IL-2, whereas CD4+CD25− T cells depend upon IL-7 (29) not IL-2 for their survival without antigen activation. In the thymus IL-2 (30), not IL-7 (31) is critical for production of nTreg, although IL-7 plays a separate role in induction of nTreg in the thymus (32).
The CD4+CD25+FoxP3+ T cells are a heterogeneous group, and include naïve nTreg produced by the thymus, that have TCRs with increased affinity for self either due to thymic selection for self or expansion of self reactive clones in the periphery (33, 34). These naïve nTreg are polyclonal, with a wide repertoire of TCR. In normal immunological naïve hosts, some naïve nTreg, with TCR specific for autoantigens, may have contacted antigen and been activated or expanded, to increase the repertoire of autoreactive nTreg. In addition, especially in hosts with acquired immune tolerance, there may be CD4+CD25+ Treg reactive to foreign or alloantigens, that have been expanded and function as antigen specific Treg. These are no longer naïve nTreg. Hosts with established antigen specific tolerance may have a large population of activated Treg with TCR specific for the tolerated antigen that mediate this tolerance, as well as the normal naïve nTreg with a TCR repertoire for self as well as a limited repertoire for other foreign antigens.
Induction of Treg from CD4+CD25− T Cells
CD4+CD25− T cells can be activated by antigen in the absence of inflammatory cytokines, to antigen specific Treg. The first induced Treg (iTreg) described by Weiner are Th3 cells induced by TGF-β in oral tolerance, reviewed (35). Groux et al. described induction of antigen specific Treg by repeated culture of CD4+ T cells with antigen and IL-10, producing Tr1 cells that suppress via production of IL-10 and TGF-β (36). Tr1 and Th3 cell do not express CD25 or FoxP3 (35, 37).
Induced Treg are derived from peripheral CD4+ T cells that are stimulated by antigen and TGF-β in the absence of inflammation and inflammatory cytokines. These iTreg are induced to express FoxP3, albeit its expression is not stable as the Treg specific demethylation region (TSDR or CBS2) for FoxP3 is not demethylated (38). Both TGF-β which down regulates many genes, and FoxP3 expression which down regulates other genes, are required to induce iTreg from CD4+ T cells (39).
Most attempts to describe Treg oversimplify the complex nature of these cells in vivo, by describing all Treg as one type of cells, or dividing their description into nTreg and iTreg. nTreg remain non-antigen specific polyclonal Treg when cultured with IL-2 alone, whereas antigen specific nTreg are not expanded by IL-2. This and the small frequency of nTreg reactive to a specific antigen has led some to conclude that some, if not the majority, of antigen specific Treg reactive to foreign antigens may be derived from iTreg and not from activation of nTreg (40–43). The lack of a distinct surface marker to distinguish antigen specific Treg produced as iTreg from those derived from nTreg, makes determination of the precise contribution of nTreg and iTreg to states of induced tolerance difficult (44, 45).
This review will focus on antigen specific Treg induced from nTreg, not on iTreg. Most of the material presented is derived from murine models. In each section, murine results will be presented first, then any human data will be discussed. At the end of each section, any information on similar cells derived from iTreg will be briefly mentioned.
Our work on Treg has shown that differential cytokine receptor expression is key to the identification of different T cell subtypes, including nTreg (46). This differential expression of cytokine receptors can be used to identify and distinguish a large number of functionally distinct Treg populations and is the major focus of this review.
Are There Antigen Specific Treg?
Acquired or induced immune tolerance is antigen specific, as shown in allograft (6–8, 11) and autoimmune tolerance (47, 48). In autoimmunity induced tolerance is epitope specific (47, 48). The CD4+ T cells that transfer transplant tolerance are alloantigen specific (6–8, 11). Antigen specific Treg, not polyclonal nTreg, are needed to prevent autoimmunity including myelin basic protein induced EAE (49), type I diabetes (50–52), gastritis (53), and peptide specific Treg control EAE induced by that peptide (54).
Animals with tolerance to an antigen or allograft do not have a major increase in CD4+CD25+ T cells, which remain at ratios of approximately 1:10 to CD4+CD25− T cells (55, 56). As these antigen specific Treg represent a fraction of the CD4+CD25+ T cells, they suppress the immune response at ratios well below 1:10, whereas nTreg are required at non-physiological ratios of 1:1 to suppress in vivo (57) and in vitro (58, 59). Ratios of 1:1 have only transiently been achieved with IL-2/IL-2 mAb complexes where they can suppress pancreatic islet allograft rejection and autoimmunity (60). It has recently been appreciated that the number of nTreg that need to be produced for transfer to induce tolerance is impossibly large (61). Thus generation of antigen specific Treg from nTreg that suppress at ratios of <1:10 in an antigen specific manner would be highly desirable. We have described how such antigen specific Treg can be generated from naïve nTreg in vitro with 3–4 days of culture (46).
Is There More than One Antigen Specific Subset of Treg?
There is ample evidence that the pathways for activation of nTreg and iTreg are multiple and complex, producing antigen specific Treg that control different subpopulations of effector CD4+ T cells, including Th1, Th2, Th17, and Tfh cells. The generation of antigen specific Treg from either naïve nTreg or effector lineage CD4+CD25− T cells, is complex involving activation of antigen specific T cells with antigen in an environment of cytokines that promotes maturation and clonal expansion of these antigen specific Treg. The cytokines that induce these lineages differ and relate to the environment present at the location of activation.
Our hypotheses are that: (i) every phase of the immune response is regulated to some degree, and that Treg are integral to control of all immune responses. (ii) All normal immune response, both in vivo and in vitro, are associated with activation of a CD4+ Treg response. (iii) Treg activation is driven by the cytokines present, including those produced by activated effector T cells. (iv) The more advanced or aggressive the immune response, the more potent the Treg that are generated by the cytokines produced, to control the response. We propose there are several levels of regulation by different functional subclasses of CD4+ Treg that are induced and activated by the ambient cytokines. Some of these separate Treg lineages and types are described in Table 1.

Table 1. Subclasses of CD4+ T cells with regulatory function.
Why are Antigen Specific Treg Hard to Identify?
A key unanswered question is the relationship of naïve non-antigen specific Treg generally described as nTreg, to antigen specific Treg. In particular whether antigen specific Treg are derived from nTreg or a product of activation of effector lineage CD4+CD25− T cells, now known as iTreg (62). Whilst some conclude that antigen specific Treg are mainly iTreg, this review will examine the pathways by which nTreg can be activated to antigen specific Treg, raising the possibility that activation of nTreg may be the dominant source of antigen specific Treg.
Our thesis is based on our findings that antigen specific Treg die in vitro and in vivo, unless stimulated by specific antigen and cytokines produced by activated effector cells during immune response to the antigen (10–12). This makes identification of antigen specific Treg very difficult, unless they are re-exposed to specific antigen and the cytokines they depend upon. Further, antigen specific Treg do not require IL-2, and in fact may be killed by IL-2 (12). Thus most current protocols for the ex vivo expansion of nTreg will not promote antigen specific Treg.
Antigen Specific Treg Express Cell Surface Markers of Activated T Cells
Activated Treg express different cells surface markers to nTreg. As examples nTreg express CD45RA and are CD44lo, whereas activated Treg express markers of memory cells, being CD45RO+ and CD44hi. CD45RC is a marker of an activated Treg (11). Class II MHC is only expressed by activated Treg, and is a marker of these cells in man (63) and rats (11) but not in mice. nTreg express CD62L and re-circulate from blood to lymph, whereas activated Treg lose expression of CD62L and migrate through peripheral tissue not through lymphoid tissues in murine (64, 65) and humans (66). In naïve CD4+CD25+ Treg, CD62L+ not CD62L− Treg suppress GVHD (67, 68). Expression of CCR4 and CCR7, which facilitate migration to lymphoid tissues are expressed by nTreg but not antigen activated Treg (69). Activated Treg migrate to sites of inflammation and express E/P selection (70) and chemokine receptors (65, 71) that will direct them to the site of inflammation that they are programed to control (18). Thus, Treg effective against Th1 responses express CXCR3 (72), those effective against Th2 express CCR8 (73), those for Th17 express CCR6 (74), and those for Tfh express CXCR5 (75).
Activation of Treg to Express Transcription Factors and Cytokines of Th Lineages, Making Th-Like Treg That Suppress the Relevant Th Response
Cytokines normally associated with induction and function of Th1, Th2, Th17, and Tfh CD4+ T cells are now found to play a key role in the induction, maintenance, and function of activated Treg. Transcription factors that were considered the master regulators of Th responses, play an essential role in activated Treg function, including T-bet the Th1 transcription factor (76), GATA3 the Th2 transcription factor (77), and RORγt the Th17 transcription factor (78). There is plasticity in Th cell lineages, in that various lineages can at time express transcription factors and cytokines not classical for the lineage (79). Epigenetic modification of transcription factor genes and miRNA expression contribute to stability of a lineage, but this can be broken, discussed by O’Shea and Paul (79). CD4+CD25+FoxP3+ Treg can express Th effector lineage transcription factors, together with FoxP3, thereby retaining Treg capacity.
Activation of Treg in Association with Th1 Responses
In our studies, culture of nTreg with a specific alloantigen and either IL-2 or IL-4 induce antigen specific Treg within 3–4 days of culture (46). They suppress the capacity of naïve CD4+ T cells to proliferate in vitro to specific donor at 1:32–64 and to effect rejection of specific donor grafts at 1:10 (46), whereas nTreg only fully suppress at 1:1, both in vivo and in vitro (46, 57, 59). In an autoimmune model, antigen specific Treg were also induced in vitro by culture with specific autoantigen and IL-2 that prevented disease in vivo (unpublished results). No other Th1 or Th2 cytokines promote proliferation of nTreg, including IFN-γ, IL-12p70, IL-12p40, IL-5, IL-13, nor did TGF-β, and IL-10 (46).
With CD4+CD25+ T cells from animals with tolerance to a fully allogeneic graft, we found that IL-2 or IL-4 induces proliferation to self, specific donor, and third party alloantigen. Proliferation of these Treg to specific donor, and not to self or third party, is promoted by IFN-γ, IL-12p70, and IL-5, but not TGF-β, IL-12p40, IL-10, or IL-13 (Hall et al., unpublished data). These cytokines became candidates for the promotion of survival of alloantigen specific CD4+ Treg in vitro, where we had not yet identified the specific cytokines involved (12). We had shown that antibody blocking IFN-γ (12) IL-5 and TGF-β (55) does not prevent transfer and maintenance of tolerance by CD4+ T cells from tolerant animals, however. Polyclonal activation of nTreg was induced by self antigen and IL-2 or IL-4, and with an antigen proliferation of nTreg induced by IL-2 or IL-4 was further increased (46).
This led us to examine if there are two pathways for activation of antigen specific Treg, one promoted by Th1 cytokines and the other by Th2 cytokines (46). We identified separate pathways for Th1 and Th2, and called the early Th1 activated Treg, Ts1 cells, and the early Th2 activated Treg, Ts2. The characteristics of these cells are summarized in Table 2, which also shows that Ts1 and Ts2 cells are an intermediate step in the activation of antigen specific Treg, and that they can be further activated by late Th1 and Th2 cytokines to more potent Th1-like Treg (Figure 1) or Th2-like Treg (Figure 2).
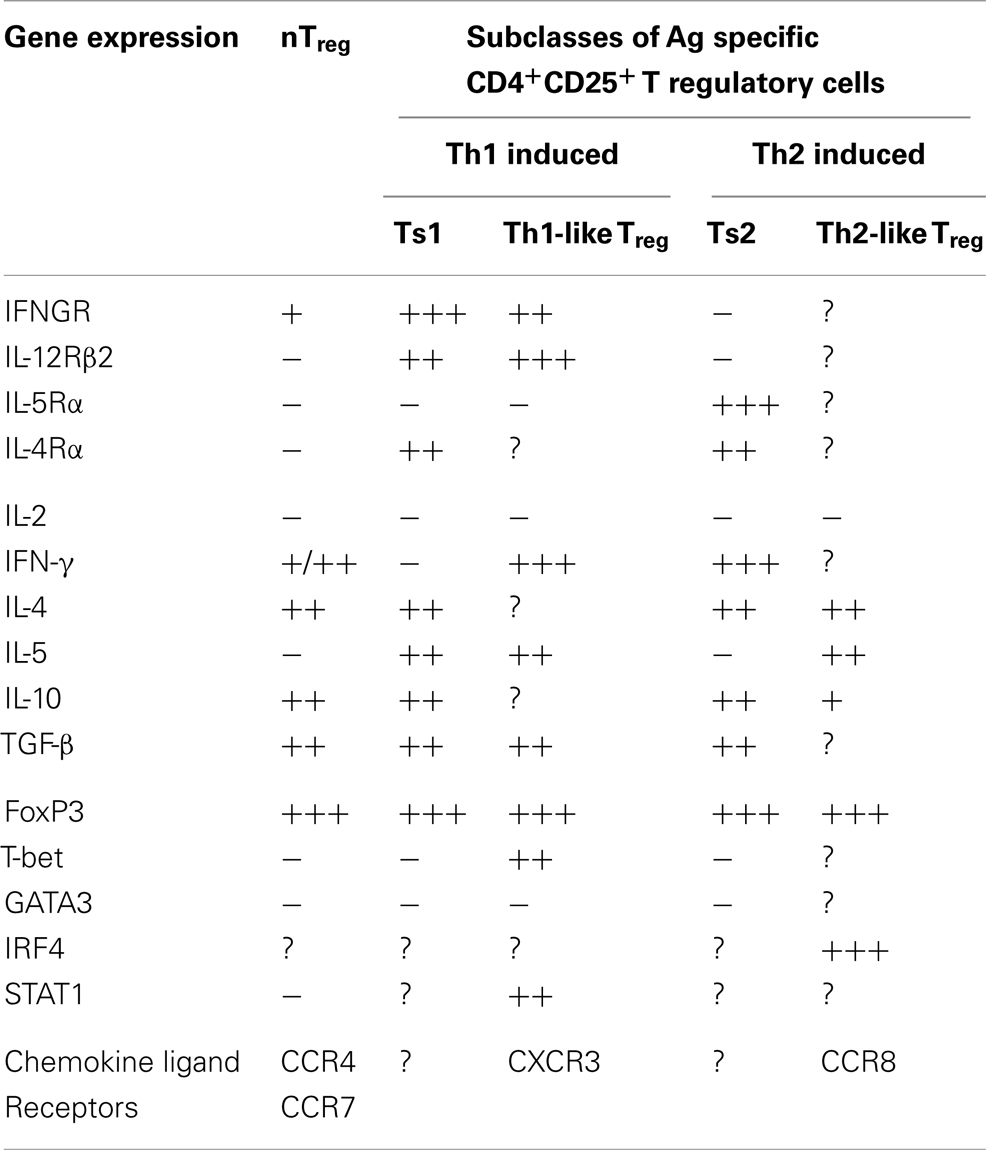
Table 2. Summarizes the differences in Th1 and Th2 activated Ag specific Treg and nTreg.
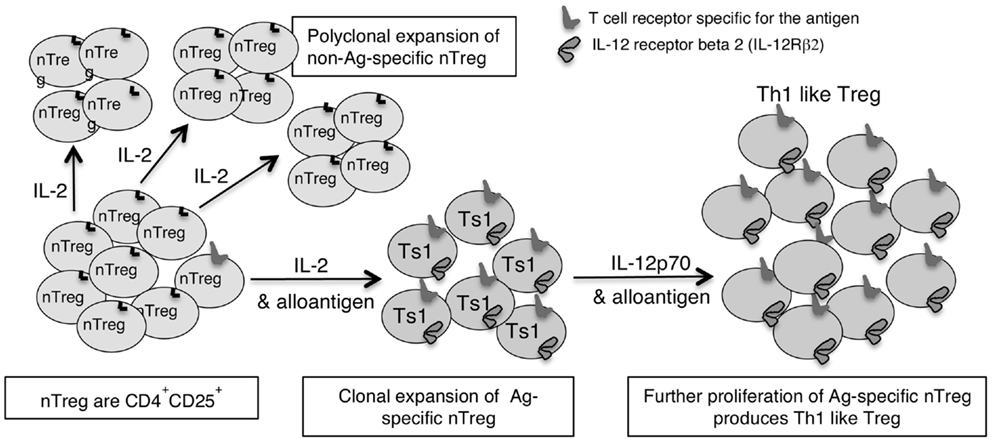
Figure 1. Shows how IL-2 without TCR engagement with specific Ag induces polyclonal expansion of nTreg. If antigen is present a minority population of nTreg that have TCR specific for antigen are activated to Ts1 by IL-2 and their specific antigen. Ts1 cells express IFNGR, IL-12Rβ2, IL-5, and FoxP3 but not IFN-γ, T-bet, or IL-2. The second step of activation of nTreg converts Ag specific Ts1 to Th1-like Treg and requires specific antigen and either IL-12 or IFN-γ in the absence of IL-2. The Ts1 are antigen specific Treg that continue to express FoxP3, CD25, and CD4, but also express IFNGR, IL-12Rβ2, T-bet, and IFN-γ. Ts1 cells have increased potency over nTreg of at least 10-fold that is antigen specific. Th1-like Treg have 100- to 1000-fold increased suppressor potency over nTreg.
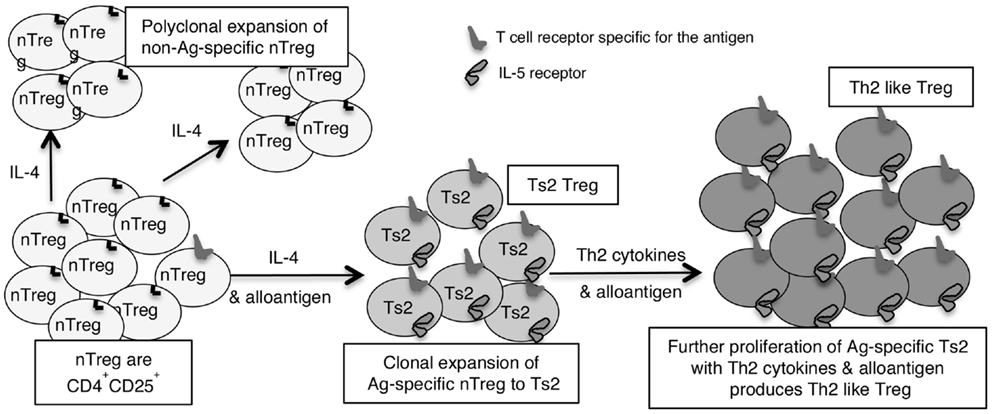
Figure 2. Shows how IL-4 without TCR engagement with specific Ag induces polyclonal expansion of nTreg. If antigen is present a minority population of nTreg that have TCR specific for antigen are activated to Ts2 by IL-4. Ts2 cells express IL-5Rα, IFN-γ, and FoxP3 but not IL-5, IFNGR, IL-12Rβ2, GATA3, T-bet, or IL-2. Ts2 cells have increased potency over nTreg of at least 10-fold that is antigen specific. The second step of activation of nTreg converts antigen specific Ts2 to Th2-like Treg and requires specific antigen and Th2 cytokines, probably IL-5. Th2-like Treg express IRF4 with FoxP3 and Th2 cytokines IL-4 and IL-5.
IL-2 and Antigen Activation of nTreg
In cultures of naïve CD4+CD25+FoxP3+ Treg with allo or autoantigen and IL-2, we found that within 2–4 days there was a change in phenotype of the cells, see Table 2. Their expression of mRNA for interferon-γ receptor (IFNGR) increases (46) and the receptor for IL-12p70 (IL-12Rβ2) is induced, whereas the receptor for IL-5 (IL-5Rα) is not induced. There is also enhanced expression of mRNA for IL-5 and reduced expression of IFN-γ. Other cytokine expression remains unchanged, with no IL-2, and similar expression of IL-4, IL-10, and TGF-β to that of fresh naïve nTreg. Foxp3 expression is maintained in the majority of cells, and there is no induction of T-bet or GATA3. These changes are not observed when nTreg are cultured with IL-2 and self antigen, suggesting these changes occur related to activation of antigen specific Treg. We called these cells Ts1 (46).
Ts1 cells are more potent than nTreg in suppression in vitro, as they fully suppress naïve CD4+ T cells proliferation in MLC at 1:32–1:64 (46), whereas nTreg only fully suppress MLC at 1:1 or greater (59). Evidence that antigen specific Treg are activated is that Ts1 cells suppress specific donor allograft rejection mediated by naïve CD4+ T cells at a ratio of 1:10 (46), whereas naïve nTreg only suppress rejection at 1:1 (57), and Ts1 cells do not suppress third party rejection at 1:10 (46). The animals where Ts1 suppressed rejection, develop tolerance to the allograft and after 150 days have CD4+CD25+FoxP3+ T cells that expressed IFNGR and IL-5, consistent with these Ts1 cells retaining their phenotype over a long period and being key to the maintenance of tolerance.
In other hosts with transplant tolerance, we identified CD4+CD25+FoxP3+ T cells that expressed IFNGR and IL-5, that in vitro respond to specific donor and not third party when IFN-γ is present (Hall et al., unpublished data). Further the capacity of tolerant CD4+ T cells to transfer tolerance is maintained in vitro by culture with specific donor and IFN-γ not IL-2 (Nomura et al., unpublished data). We suggest that these Ts1 maintain alloantigen specific tolerance but are dependent on production of IFN-γ by Th1 cells.
In an autoimmune model we have also generated antigen specific Ts1 cells in vitro by culture of nTreg with IL-2 and autoantigen. These Ts1 are induced to express IFNGR and IL-5, and suppressed the autoimmunity in an antigen specific manner (Tran et al., unpublished data).
We suggest induction of Ts1 cells is a key step in induction of antigen specific tolerance to Th1 responses. Ts1 would be promoted by the IFN-γ produced by an ongoing Th1 response, after they stop producing IL-2, which is an early Th1 cytokine. Ts1 cells may in part account for the paradoxical anti-inflammatory effects of IFN-γ, reviewed (80, 81).
IFN-γ and Activation of Antigen Specific Treg
IFN-γ is better known as a pro-inflammatory cytokine, but also has well described effects that control immune responses. IFN-γ directly inhibits Th2 and Th17 cell development, but promotes Th1 responses, including B cell isotype switching, macrophage activation, and cytotoxic T cell development. Activation of the Th1 lineage depends upon IFN-γ activating STAT1, which induces the Th1 transcription factor T-bet, which in turn regulates IFN-γ production by Th1 cells. Once CD4+ T cells are activated to a Th1 lineage, they cannot be converted to a Treg lineage (82). IFN-γ is key to CD8+ T cell mediated rejection (83, 84) and to allograft vasculopathy (85–87). IFN-γ also activates macrophages to M1 cells and promotes Ig switching to a complement fixing isotypes. IFN-γ promotes MHC class I and II expression on inflamed tissues such a during rejection (88). By induction of MHC class I, IFN-γ protects allografts from CD8+ T perforin/granzyme mediated rejection (84, 89–91).
IFN-γ can limit inflammation (92). IFNGR deficient mice have increased severity and reduced recovery from EAE (93, 94). IFN-γ induces iNOS to produce NO, which limits inflammation (95–98). IFN-γ treatment inhibits GVHD (99). CD8+ T cells deficient in IFN-γ mediate more severe GVHD, indicating IFN-γ produced by these cells inhibits the CD8+ T cell response by inhibiting proliferation and promoting cell death. CD8+CD45Rlo T cells induced to express IFN-γ, in turn induced indoleamine 2,3-dioxygenase (IDO), and accounts for promotion of indefinite allograft survival after blocking the CD40–CD40L interaction (100).
IFN-γ is also important in the generation and function of CD4+CD25+ Treg that mediate allograft tolerance (101) and prevents immune destruction of tumors (102). In vitro, IFN-γ promotes induction of alloantigen specific CD4+CD25+FoxP3+ Treg that prevent rejection (103). This work by Wood’s group in Oxford identifies that naïve CD4+ T cell cultured over a period of time in MLC supplemented with IFN-γ, produces antigen specific Treg that can prevent rejection (41, 103–107). Whether IFN-γ induces iTreg or expands nTreg or a combination of both is unclear. One possibility is that nTreg are initially activated by IL-2 produced by the activated CD4+CD25− T cells to induce antigen specific Ts1 cells, that in turn are activated by IFN-γ to expand and maintain the antigen specific Treg (as shown in Figure 1), while a variety of factors such as IFN-γ induction of NO or IDO by antigen presenting cells or IFN-γ promotion of antigen specific Treg may reduce the growth of the effector lineage. IFN-γ inhibits induction of iTreg from CD4+ T cells (82), whereas other report IFN-γ is key to induction of CD4+CD25− T cells to iTreg that suppress autoimmunity in IFN-γ deficient mice (108).
Th1-Like Treg
Th1-like Treg were first described in 2004 associated with a polarizing Th1 response to ovalbumin (109). Ovalbumin specific Treg are induced from CD4+CD25− T cells by mature CD8α+ DC that produced both IL-12 and IL-10 that are required to induce Th1-like Treg (109). These Th1-like Treg express both FoxP3 and the Th1 transcription factor T-bet, as well as ICOS, IFN-γ, and IL-10. The Th1-like Treg suppressed Th1 inflammation in vivo (109). In cancer, Th1-like Treg expressing FoxP3, helios, T-bet, IFN-γ, CXCR3 suppress Th1 responses and are associated with infiltrating Th1 effector cells, probably impairing tumor immunity (110). T-bet expression is required for full Treg function, as T-bet deficient nTreg do not fully control autoimmunity in FoxP3 deficient scurfy mice (72).
Treg induced by activation with a specific alloantigen become FoxP3+IFN-γ+ and suppress in an antigen specific manner (111). Human iTreg that express T-bet, IFN-γ, and CXCR3 are CD4+CD25+FoxP3+ T cells and suppress (112). Th1-like IFN-γ producing CD4+CD25+FoxP3+ Treg are present in the blood of multiple sclerosis and renal transplant patients during active immune responses (113, 114). Th1-like Treg can be induced by IFN-γ, IL-12, or IL-27 and each may be a separate lineage, albeit they all express FoxP3, T-bet, STAT1, IFN-γ but not IL-2.
IFN-γ Promotes Th1-Like Treg
Thymus derived nTreg activated in a Th1 environment initially by IL-2, can be further activated by IFN-γ inducing STAT1 to promote expression of the Th1 transcription factor T-bet (115). Absence of STAT1 results in impaired CD4+CD25+ Treg development and increases host susceptibility to autoimmunity (115). These STAT1/T-bet/FoxP3+ Treg control Th1 responses and express CXCR3, which promotes their migration to sites of Th1 inflammation (72). IFN-γ induces T-bet+CXCR3+ Treg that inhibit Th1 responses in the periphery (116). Collectively these studies confirm IFN-γ can act on Treg to increase their effectiveness in controlling Th1 responses, albeit excessive activation by IFN-γ can reduce their suppressive capacity and may convert them to effector Th1 cells.
IL-12 Promotes Th1-Like Treg
IL-12p70 is a hetero-dimer composed of p35 and p40 that is produced by APC not T cells (117). IL-12 is a pro-inflammatory cytokine that enhances Th1 (76, 118), cytotoxic CD8+T (119), and NK (120) cell responses by increasing IFN-γ (121).
IL-12p70 acts by binding to a high affinity receptor, which is a hetero-dimer of IL-12Rβ1 and IL-12Rβ2 (122), which when activated by IL-12p70 induces STAT4 and T-bet to stabilize the Th1 phenotype and IFN-γ production (123, 124). Resting T cells do not express high affinity IL-12Rβ2 (117), but both chains are up-regulated by TCR and CD28 stimulation, as well as by IL-2 and IFN-γ. IL-4 and IL-10 decrease expression of IL-12Rβ2.
Because IL-12p70 promotes induction of Th1 and cytotoxic T cell responses, it was predicted to amplify rejection and GVHD (125). Paradoxically, treatment with one dose of IL-12p70 at the time of bone marrow transfer inhibits fully allogeneic GVHD (126). Prevention of GVHD by IL-12p70 is dependent on donor IFN-γ (127) acting via Fas to inhibit donor T cell expansion (128). IL-12p70 treatment delays allograft rejection (98) and inhibits autoimmunity including uveitis (129) and EAE (130). The protective effects of IL-12p70 are associated with induction of IFN-γ and iNOS (129). Blocking IFN-γ or iNOS with L-NIL prevents IL-12p70 prolonging graft rejection (98). In other models IL-12 promotes autoimmunity (131–133).
IL-12p35−/− (134), IL-12Rβ2−/− (135), IFN-γ−/− (136), and IFNGR−/− (94) mice are more prone to type I diabetes and have reduced numbers of CD4+CD25+FoxP3+ Treg that are less suppressive in vitro (137). Some Treg express the IL-12Rβ2 (137). In a situation of an uncontrolled Th1 response, IL-12p70 induces Treg to express T-bet and with high IL-12p70 levels these Treg produce IFN-γ (138). These changes only occur when there is limited IL-2 (138).
In our studies, nTreg cultured with IL-2 and alloantigen (Ts1) expressed IL-12Rβ2 and proliferated with IL-12p70. Ts1 cells activated by specific antigen and IL-12p70 in the absence of IL-2 had greater capacity to suppress alloimmune responses in vitro at 1:1000 and in vivo at 1:100 (Verma et al., unpublished data). Further, these Ts1 cells cultured with IL-12p70 in the absence of IL-2, expressed mRNA for T-bet and IFN-γ. They continued to express CD25, FoxP3, and mRNA for IFNGR and IL-12Rβ2. Ts1 cultured with IL-2 and IL-12p70 did not express mRNA for T-bet or IFN-γ. The concept of how Th1 cytokines induce Ts1 cells that are activated to a specific antigen to express IFNGR and IL-12Rβ2, and the effects of IFN-γ and IL-12p70 on their further expansion of Ts1 to Th1-like Treg is illustrated in Figure 1.
Many of the anti-inflammatory effects of IL-12p70 are attributed to increased production of IFN-γ that in turn induces iNOS to produce NO (98) but this was not required for Th1-like Treg development in vitro. Our results suggested that Ts1 cells, express IL-12Rβ2, and that IL-12p70 directly promotes Treg proliferation and maturation of Ts1 to more potent Th1-like Treg similar to that described by others (72, 138).
IL-27 Promotes Th1-Like Treg
IL-27 is a member of the IL-12 family of hetero-dimers, that was thought to promote Th1 responses (139). A subset of CD4+CD25+ Treg express IL-27Rα (140) a receptor required to control excess inflammation during infection (141). IL-27 inhibits Th1, Th2, and Th17 by direct inhibition of cells and induction of T effectors to produce IL-10 (142, 143). IL-27 promotes T-bet and CXCR3 expression in Treg at mucosa sites (116). IL-27 produces specialized Treg that control immunity at sites of inflammation and these Treg appear to express IL-27 as well as IL-27Rα (116). For IL-27 iTreg to function, they must express IFNGR1 and IL-10 (116). The IL-27 induced Th1-like Treg express different genes to Th1-like Treg induced by IFN-γ (116).
IL-27 via the STAT1 pathway, promotes FoxP3 expression by STAT1 binding to the FoxP3 promoter region in iTreg (144).
Activation of Treg in Association with Th2 Responses
Dominance of Th2 responses (145–148) and Th2 cytokines IL-4 (148–150), IL-10 (151), and IL-13 (152), can protect against autoimmunity, but their effects are variable. Th2 cytokine expression is associated with prolongation of allograft survival in some models (153–158), including neonatal (159–161), and irradiation (162, 163) induced tolerance, but not in all models (164). Th2 cells transfer protection against chronic rejection (165) but do not directly mediate tolerance (166).
IL-4 Effects on nTreg and iTreg
IL-4 is key to the induction of Th2 responses by binding to the IL-4Rα and common gamma chain and inducing STAT6 signaling (167) which is required for GATA3 and Th2 cell induction (168). IL-4 makes Th2 cells resistant to Treg (169).
IL-4 also induces STAT6 in Treg and stabilizes expression of FoxP3 (169). GATA3 is essential for full expression of FoxP3 by nTreg and binds to a conserved element of the FoxP3 locus to enhance transcription of FoxP3 (170). GATA3 expression is required to maintain FoxP3 expression in nTreg (77). GATA3 binds to the CNS2 site of the Foxp3 promoter site as well as the Th2 locus, whereas in Th2 cells it only binds to the Th2 locus (77). This induction of GATA3 in nTreg is not via the IL-4/STAT6 pathway (171), whereas induction of GATA3 via the IL-4/STAT6 pathway in nTreg and iTreg (172) suppresses FoxP3 expression by binding to the FoxP3 promoter region (172).
GATA3 is induced in nTreg during inflammation, and sustains FoxP3 expression (171) especially in Treg at sites of low grade inflammation such as mucosa and skin. Absence of GATA3 in Treg results in a spontaneous inflammatory disorder and defective nTreg that gain a Th17 phenotype (77). Th1 polarizing conditions down regulate GATA3 in Th2 and Treg cells (77). GATA3 induced in nTreg in early inflammation inhibits induction of polarizing factors and generation of effector T cells from nTreg (171). This early induction of GATA3, is dependent upon IL-2 as it is enhanced by IL-2/anti-IL-2 mAb complexes and is absent in IL-2 deficient mice (171).
TGF-β inhibits T-bet expression (173) and GATA3 expression (174) in CD4+ T cells reducing Th1 and Th2 cell expansion, thereby favoring FoxP3 expression and iTreg development. On the other hand GATA3 inhibits FoxP3 expression in iTreg activated from CD4+ T cells by TGF-β (77) and diverts the cells to an IL-9 producing effector CD4+ T cell (175, 176). Thus IL-4 may promote nTreg, but inhibit induction of iTreg by promoting GATA3 induction, that down regulates FoxP3 expression. GATA3 is not expressed by RORγt or T-bet expressing Treg, nor by Th17 and Th1 cells (171).
IL-4 in culture prevents apoptosis of mice nTreg (177), but IL-4 does not induce proliferation of nTreg only inducing proliferation of CD4+CD45RBhiCD25−T cells (177). IL-4 enhances the capacity of nTreg to suppress IFN-γ induction in CD4+CD25− T cells (177). Others found IL-4 induces nTreg proliferation (178) and expression of CD25, FoxP3, and IL-4Rα (169, 177). In cultures, IL-4 induces proliferation of both CD4+CD25+ and CD4+CD25− T cells but promotes survival of CD4+CD25− T cells countering inhibition by nTreg (179).
IL-4 and Antigen Activation of nTreg
We found IL-4 and antigen in culture induced nTreg to antigen specific Treg (46, 56). This activation induces expression of the specific receptor for IL-5 (IL-5Rα) as well as for IL-4 (IL-4Rα) but not IFNGR or IL-12Rβ2, that we observe in cultures with IL-2 and an antigen (46). We call these antigen and Il-4 activated Treg, Ts2 cells (46). They continue to express FoxP3, but do not express GATA3, T-bet, or IL-2 (46). Ts2 cells features are summarized in Table 2. Ts2 cells have less expression of IL-5, enhanced expression of IFN-γ, and no change in expression of IL-4, IL-13, TGF-β, or IL-10 (46) (Table 2). These changes are not observed when nTreg were cultured with IL-4 and self antigen, suggesting they are due to activation of antigen specific Treg (see Figure 2).
Ts2 cells have increased potency of suppression in vitro as they fully suppressed naïve CD4+ T cells proliferation in MLC at 1:32 (46), whereas nTreg only fully suppress MLC at 1:1 or greater (59). Evidence that Ts2 cells are antigen specific Treg is that Ts2 cells suppress specific donor allograft rejection mediated by naïve CD4+ T cells at a ratio of 1:10 (46), whereas naïve nTreg only suppress rejection at 1:1 (57). Ts2 cells do not suppress third party rejection at 1:10 demonstrating the Ts2 cells are antigen specific (46). The animals restored with Ts2 cells to suppress rejection develop tolerance to the allograft and after 150 days have CD4+CD25+FoxP3+ T cells that expressed IL-5Rα and IFN-γ. These tolerant Treg proliferate in culture to specific donor, but not to self or third party alloantigen, if IL-5 is present (46). This is consistent with these alloantigen specific Treg retaining their phenotype over a long period and IL-5 being key to the maintenance of tolerance mediated by antigen specific CD4+CD25+FoxP3+ Treg.
In other hosts with transplant tolerance, we have identified CD4+CD25+FoxP3+ Ts2 cells that expressed IL-5Rα and IFN-γ, that in vitro responded to specific donor and not third party when IL-5 was present (unpublished). Alloantigen with IL-5, but not IL-4, promoted in vitro survival of transplant tolerance transferring alloantigen specific CD4+ T cells (Plain et al., unpublished data). We suggest that these Ts2 cells maintain alloantigen specific tolerance, albeit animals with tolerance can have both antigen specific Ts1 and Ts2 cells.
In an autoimmune model, we have also generated antigen specific Ts2 cells in vitro by culture of nTreg with IL-4 and autoantigen. These Ts2 cells are induced to express IL-5Rα and IFN-γ, not IFNGR, and IL-12Rβ2 (56).
Human CD4+CD25+CD127loFoxp3+ T cells cultured with antigen and IL-4 express IL-5Rα consistent with a human Ts2 cell (56).
We concluded that induction of Ts2 cells is a key step in induction of antigen specific tolerance to Th2 responses. Ts2 would be promoted by the IL-5 produced by an ongoing Th2 response, after the Th2 cells stop producing IL-4, an early Th2 cytokine.
IL-5 and Antigen Activation of nTreg
As IL-5Rα is not expressed by any other T cells subtype, and is mainly expressed by eosinophils and mast cells, and in rodents B cells, we proposed that IL-5 may be a therapy that could promote immune tolerance by activation and expansion of antigen specific Ts2 (56). Treatment with IL-5 delays neonatal heart allograft rejection and inhibit Th1 cytokine induction (180).
In an autoimmune demyelination model, IL-5 therapy given before disease onset prevents clinical disease and nerve demyelination. IL-5 therapy given after onset of disease, reduces clinical severity of disease and the number of demyelination nerves (56). This is associated with an increase in CD4+CD25+ Treg and these Treg express IL-5Rα. Further responses of these hosts Treg to the immunizing antigen are enhanced by adding IL-5 to cultures (56). The effect of IL-5 are abrogated by treatment with monoclonal antibodies to deplete CD25+ cells or to block IL-4, confirming that the nTreg of the host are activated by antigen and exposure to IL-4 produced in the immune response to the autoantigen (56). The IL-5 therapy promotes expansion of the IL-5Rα expressing antigen specific Ts2 cells (56). IL-5 therapy markedly reduces tissue inflammation and expression of mRNA for the Th1 cytokines IL-2 and TNF-α as well as the Th17 associated cytokine IL-17A. The Th2 cytokines IL-4 and IL-5 are not suppressed (56). This suggests that Ts2 cells may selectively suppress Th1 and Th17 responses, while sparing the Th2 response that produces the IL-4 and IL-5 required for the induction and expansion of Ts2 cells. Thus these Ts2 cells contribute to polarization of Th2 responses by suppressing Th1 and Th17 cells.
Human CD4+CD25+CD127loFoxP3+ Treg cultured with antigen and IL-4, but not IL-2, express IL-5Rα, suggesting IL-5 may promote these antigen specific Treg (56).
Th2-Like Treg
Th2-like Treg express the transcription factor Interferon regulatory factor-4 (IFR4) to control Th2 responses (73). IRF4 also promotes Th2 and Th17 (181) responses. IRF4 binds to the promoter region of FoxP3 and induces Treg to express IL-4 and IL-5 (73). Thus induction of IRF4 results in a Th2-like Treg. Antigen specific Th2-like Treg are induced in Th2 responses by IL-10 and ICOS/ICOS ligand interaction and secrete IL-10 and some IL-4 but not IL-13 (182). ICOS expressed on Treg promotes their expansion in sites of inflammation during parasitic infestation, whereas in lymphoid tissues ICOS promotes Th2 responses not Treg expansion (183).
During parasitic infestations, CD4+CD25+ Treg develop in parallel with the Th2 polarization and regulate the size of the immune response (184). These Th2 iTreg inhibit Th1 responses, thereby facilitating Th2 polarization (185, 186). The early immune response to parasites is markedly controlled by Treg (187). Persistence of parasitic infestation is due to CD4+CD25+ Treg (188, 189) and these hosts have expanded CD4+CD25+FoxP3+ Treg populations (190).
Chronic infestation with parasites is associated with dominance of Treg, which suppress Th1 and Th2 responses against the parasite (191, 192). Animals who fail to eliminate parasites have protective CCR8+CD4+CD25+ Treg producing IL-10 that regulates Th2 response (193). Transfer of CD4+CD25− T cells confer some protection against infestation, while transfer of activated CD4+CD25+FoxP3+CD103+ Treg impairs parasite clearance with greater effect than nTreg (194).
Animals with parasitic infections and an active Th2 response are resistant to the induction of autoimmunity (195, 196) through the effects of TGF-β (197) and have delayed allograft rejection (198–200). This suggests the Th2 milieu and possibly Th2 activated Treg protect these animal from Th1 and Th17 responses (201).
Multiple sclerosis patients with eosinophilia from parasitic infestation have markedly reduced episodes of relapses and new MRI lesions in brain associated with increased CD4+CD25+ Treg (202). Treatment of parasitic infestations leads to increased relapses and progression of multiple sclerosis with a reduction in Treg (203). Trials of therapeutic parasitic infestation are underway in inflammatory bowel disease (204) and MS (205). As parasitic infestation is associated with Th2 responses and production of IL-5, that induces eosinophilia, one possibility is that this IL-5 promotes antigen specific Ts2 cells to control autoimmunity.
A plausible hypothesis is that the evolution of the immune system was with persistent parasitic infestations and Th2 responses that inhibit innate and Th1/Th17 immunity (206). There is an increasing incidence of autoimmunity in the Western World where the parasitic infestation rate has markedly declined (206). Parasites induction of immune responses that promote Treg, possibly by production of IL-5, may also explain the reduced incidence of autoimmunity in populations that live closer to the equator and have poorer hygiene (206).
Our hypothesis is that persistent Th2 responses releasing IL-5 may through a by-stander effect promote expansion of activated antigen specific IL-5Rα+ Treg generated to new non-parasite antigens. We demonstrated that IL-5 was an essential growth factor for nTreg activated by IL-4 and these Ts2 cell reduce autoimmune injury (56). We propose that one of the beneficial effects of parasites may be the high IL-5 level produced by a chronic Th2 response, promotes IL-5Rα expressing antigen specific Ts2 cells to control autoimmunity and allograft rejection.
Activation of Treg in Association with Th17 Responses
Th17-Like Treg
T regulatory cells expressing both FoxP3 and IL-17 occur in mice and man (78, 207). IL-17 producing Treg are produced in the periphery not the thymus (78). STAT3, a transcription factor required for Th17 induction, is also required in Treg for induction and maintenance of FoxP3 expression induced by CD28 co-stimulation to produce iTreg (208). Specific deletion of STAT3 in Treg results in a fatal Th17 meditated colitis (209). It is proposed that STAT3 and FoxP3 together coordinate expression of a set of genes that specifically regulate Th17 effector T cells (209). STAT3 induces the receptors for IL-10, and for the pro-inflammatory cytokines IL-6 and IL-23 on Th17 cells and presumably on Treg associated with Th17 responses. IL-27 inhibits Treg via STAT3 (210). IL-10 at the site of inflammation can promote activated FoxP3+ Treg and FoxP3− Tr1 (211) and can directly inhibit Th17 and Th17/Th1 cells at the site of inflammation in colitis (212). This suggests that IL-10R is expressed by Th17, Th1/Th17 cells, as well as Th17-like Treg that suppress Th17.
Human peripheral blood and lymphoid tissue contain CD4+FoxP3+ Treg that express CCR6 and when activated produce IL-17. They express both FoxP3 and RORγt (78). These CD4+CD25+FoxP3+ cells, that produce IL-17, strongly inhibit CD4+ T cell proliferation, and could be cloned (78). Naïve CD4+FoxP3+CCR6−Treg that have their TCR stimulated in the presence of IL-1β, IL-2, IL-21, and IL-23 differentiate into IL-17 producing Treg (78). Human Treg that secrete IL-17A express the Th17 transcription factor RORγt (213). Both naive and memory Treg suppress Th17 cells and inhibit their production of IL-17 and IL-22, as well as their expression of CXCL8 (214).
CD4+CD25+FoxP3+ Treg expressing IL-17, that acquire IL-1R1 can be converted to Th17 cells by IL-1β (215). This group suggested the preferred route of induction of Th17 in man may be via activation of nTreg with lineage differentiating factors, such as activated APC, IL-1β, TGF-β, and IL-23 as well as IL-2 (74). They propose a new role for nTreg as precursors of Th17 effector cells. IL-2 therapy triggers conversion of Th17 producing FoxP3+ Treg to Th17 cells that do not express FoxP3 (216). The Th17 effectors, that no longer suppress, do not express FoxP3 or IL-1R1, but express CCR6; similar to a smaller population of Treg that express FoxP3 and IL-17 (74).
IL-21 synergizes with IL-2 to promote activation of effector CD4+ and CD8+ T cells but inhibits induction of iTreg when combined with IL-2 and TGF-β (217). Thus, there is evidence for activated Treg and iTreg being induced to suppress Th17 responses that use induction pathways, in part, shared with Th17 cells.
Activation of Treg in Association with Tfh Responses
Tfh-like Treg are specialized Treg that control germinal center expansion and autoimmune responses that are found in primary B cell follicles. These CD4+CD25+FoxP3+ T cells migrate to the T-B border areas of secondary lymphoid tissues, where they suppress Tfh dependent antibody responses by inhibiting both B cells and T cells (218, 219) These cells are CD4+CD25+FoxP3+ T cells that share transcription factors and cell surface phenotype with Tfh cells, including expression of the Tfh chemokine receptor CXCR5 (75, 219) and PD1 which is expressed by Tfh (75). The development of Tfh-like Treg is similar to Tfh cell development as it depends upon expression of the transcription factor Bcl-6 (75). Bcl-6 is a transcription factor that promotes Tfh and represses other Th lineages. They also express Blimp-1, which is repressed in B cells and Tfh that express Bcl-6 (75). Bcl-6 is a transcriptional repressor that promotes Tfh but represses other Th lineages. Bcl-6−/−Treg are selectively impaired at controlling Th2 responses, but not Th1 and Th17 responses, as Bcl-6 suppresses GATA3 and Th2 (220).
Both Tfh and Tfh-like Treg depend upon SAP, CD28, and B cells for their activation (75). Similar to Tfh cell induction, the Tfh-like Treg are induced by IL-21 and IL-6 and produce IL-21 with STAT3 expression. Tfh-like Treg are derived from nTreg and are not iTreg (75). Tfh-like Treg prevent over expansion of germinal centers and mediate tolerance in B cell responses.
Conclusion
This review sets out the evidence that nTreg are activated by cytokines released by the activation of CD4+CD25− T cells in all immune responses. It describes how the responsiveness of antigen activated nTreg changes during the immune response. Initially nTreg are activated by early cytokines such as IL-2 in Th1 and IL-4 in Th2 responses. With persistent active immune responses, the cytokines produced change. In late Th1 responses IFN-γ and IL-12p70, not IL-2 is produced, and these late Th1 cytokines further expand and activate IL-2 and antigen activated Ts1 cells. In late Th2 responses IL-5 and IL-13 are produced not IL-4. In late Th2 response IL-5 promotes IL-4 and antigen activated Ts2 cells.
Excessive amounts of these cytokines can further induce antigen specific Treg to express the transcription factor of the dominant inflammatory response, so that in Th1 responses T-bet and STAT1 are induced to Th1-like Treg that produce IFN-γ. In Th2 responses Treg express IRF4 and produce IL-5 and IL-4 to become Th2-like Treg. In Th17 responses activated Treg express RORγt and IL-17A to become Th17-like Treg, whereas in Tfh responses, Treg express Bcl-6, and IL-21 to become Tfh-like Treg. Each step of activation is associated with an increase in potency to suppress of the activated Treg, so that they can suppress at ratios of 1:10–1:1000, whereas nTreg only fully suppress at 1:1. These subsets are identifiable by expression of chemokine ligands, CXCR3 in Th1 responses, CCR8 in Th2 responses, CCR6 in Th17 responses, and CXCR5 in Tfh responses. Highly potent antigen specific Treg, with the potential to migrate to sites of tissue inflammation to control active destructive immune responses, has far reaching potential in therapy for allograft rejection, control of GVHD, and autoimmunity.
These activated Treg include antigen specific Treg and require specific antigenic stimulation and the relevant cytokines to promote their survival. The requirement for specific antigen and a restricted cytokine milieu makes study of these cells in vitro very difficult, unless the correct environment is created to promote their survival. Further, the expansion of enriched nTreg by repeated culture with IL-2 over more than a week, only expands nTreg and probably selects against antigen specific Treg as the cytokines required to sustain antigen specific Treg are absent and IL-2 prevents induction of Th1-like Treg.
It is now appreciated that the number of nTreg to control GVHD, graft rejection, or autoimmunity is impossibly large, as they need to be present at ratios of 1:1 or greater (221). Understanding the pathways for selective activation of antigen specific Treg from nTreg will allow growth of more potent Treg that suppress in a specific manner with smaller numbers of cells. This may be achieved by first culturing nTreg with IL-2 or IL-4, then with other cytokines, respectively IFN-γ or IL-12 and IL-5. The effector mechanisms of each subset or activated Treg also needs resolutions, as there are many effector mechanism other than inhibition of APC with CTLA4 and production of IL-10 and TGF-β, as reviewed (222).
Conflict of Interest Statement
Bruce M. Hall and Suzanne J. Hodgkinson hold patents related to T regulatory cells. The other co-authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The laboratory has received funding from NHMRC of Australia, a donation of Bob and Jack Ingham of Ingham Enterprises Liverpool NSW, UNSW funding, and Novartis CH. Bruce M. Hall and Suzanne J. Hodgkinson hold patents related to T regulatory cells.
References
1. Gershon RK, Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology (1970) 18:723–37.
4. Basten A, Fazekas de St Groth B. Special regulatory T-cell review: T-cell dependent suppression revisited. Immunology (2008) 123:33–9. doi:10.1111/j.1365-2567.2007.02772.x
5. Moller G. Do suppressor T cells exist? Scand J Immunol (1988) 27:247–50. doi:10.1111/j.1365-3083.1988.tb02344.x
6. Hall BM, Jelbart ME, Dorsch SE. Suppressor T cells in rats with prolonged cardiac allograft survival after treatment with cyclosporine. Transplantation (1984) 37:595–600. doi:10.1097/00007890-198406000-00014
7. Hall BM. Mechanisms maintaining enhancement of allografts. I. Demonstration of a specific suppressor cell. J Exp Med (1985) 161:123–33. doi:10.1084/jem.161.1.123
8. Hall BM, Jelbart ME, Gurley KE, Dorsch SE. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. Mediation of specific suppression by T helper/inducer cells. J Exp Med (1985) 162:1683–94. doi:10.1084/jem.162.5.1683
9. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. “Infectious” transplantation tolerance. Science (1993) 259:974–7. doi:10.1126/science.8094901
10. Pearce NW, Spinelli A, Gurley KE, Dorsch SE, Hall BM. Mechanisms maintaining antibody-induced enhancement of allografts. II. Mediation of specific suppression by short lived CD4+ T cells. J Immunol (1989) 143:499–506.
11. Hall BM, Pearce NW, Gurley KE, Dorsch SE. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. III. Further characterization of the CD4+ suppressor cell and its mechanisms of action. J Exp Med (1990) 171:141–57. doi:10.1084/jem.171.1.141
12. Pearce NW, Spinelli A, Gurley KE, Hall BM. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporin. V. Dependence of the CD4+ suppressor cell on the presence of alloantigen and cytokines, including interleukin-2. Transplantation (1993) 55:374–80. doi:10.1097/00007890-199302000-00027
13. Dallman MJ, Shiho O, Page TH, Wood KJ, Morris PJ. Peripheral tolerance to alloantigen results from altered regulation of the interleukin 2 pathway. J Exp Med (1991) 173:79–87. doi:10.1084/jem.173.1.79
14. Hall BM, Dorsch SE. Cells mediating allograft rejection. Immunol Rev (1984) 77:31–59. doi:10.1111/j.1600-065X.1984.tb00717.x
15. Cheng IKP, Dorsch SE, Hall BM. The regulation of autoantibody production in Heymann’s nephritis by T lymphocyte subsets. Lab Invest (1988) 59:780–8.
16. Hall BM, Dorsch S, Roser B. The cellular basis of allograft rejection in vivo. II. The nature of memory cells mediating second set heart graft rejection. J Exp Med (1978) 148:890–902. doi:10.1084/jem.148.4.890
17. Hall BM, Dorsch S, Roser B. The cellular basis of allograft rejection in vivo. I. The cellular requirements for first-set rejection of heart grafts. J Exp Med (1978) 148:878–89. doi:10.1084/jem.148.4.890
18. Campbell B. Phenotypic and functional specialization of FOXP3+ regulatory cells. Nat Rev Immunol (2011) 11:119–30. doi:10.1038/nri2916
19. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). J Immunol (1995) 155:1151–64.
20. Sakaguchi S. The origin of FOXP3-expressing CD4+ regulatory T cells: thymus or periphery. J Clin Invest (2003) 112:1310–2. doi:10.1172/JCI200320274
21. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol (2007) 19:345–54. doi:10.1093/intimm/dxm014
22. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther (2008) 16:194–202. doi:10.1038/sj.mt.6300341
23. Wolf M, Schimpl A, Hunig T. Control of T cell hyperactivation in IL-2-deficient mice by CD4+CD25- and CD4+CD25+ T cells: evidence for two distinct regulatory mechanisms. Eur J Immunol (2001) 31:1637–45. doi:10.1002/1521-4141(200106)31:6<1637::AID-IMMU1637>3.0.CO;2-T
24. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity (2002) 17:167–78. doi:10.1016/S1074-7613(02)00367-9
25. Wan YS, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature (2007) 445:766–70. doi:10.1038/nature05479
26. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med (2006) 203:1693–700. doi:10.1084/jem.20060468
27. Aerts NE, Dombrecht EJ, Ebo DG, Bridts CH, Stevens WJ, De Clerck LS. Activated T cells complicate the identification of regulatory T cells in rheumatoid arthritis. Cell Immunol (2008) 251:109–15. doi:10.1016/j.cellimm.2008.04.008
28. Lim HW, Kim CH. Loss of IL-7 receptor alpha on T cells defines terminally differentiated B cell-helping effector cells. J Immunol (2007) 179:7448–56.
29. Vella A, Teague TK, Ihle J, Kappler J, Marrack P. Interleukin 4 (IL-4) or IL-7 prevents the death of resting T cells: stat6 is probably not required for the effect of IL-4. J Exp Med (1997) 186:325–30. doi:10.1084/jem.186.2.325
30. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol (2007) 178:280–90.
31. Yu A, Malek TR. Selective availability of IL-2 is a major determinant controlling the production of CD4+ CD25+ Foxp3+ T regulatory cells. J Immunol (2006) 177:5115–21.
32. Vang KB, Yang J, Mahmud SA, Burchill MA, Vegoe AL, Farrar MA. IL-2, IL-7, and IL-15, but not thymic stromal lymphopoietin, redundantly govern CD4+ Foxp3+ regulatory T cell development. J Immunol (2008) 181:3285–90.
33. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+CD4+ T cell receptors. Immunity (2004) 21:267–77. doi:10.1016/j.immuni.2004.07.009
34. Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol (2006) 7:401–10. doi:10.1038/ni1318
35. Weiner H, da Cunha AP, Quintana F, Wu H. Oral tolerance. Immunol Rev (2011) 241:241–59. doi:10.1111/j.1600-065X.2011.01017.x
36. Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, et al. A CD4+ T-cell inhibits antigen-specific T-cell responses and prevents colitis. Nature (1997) 389:737–42. doi:10.1038/39614
37. Groux H. Type 1 T-regulatory cells: their role in the control of immune responses. Transplantation (2003) 75:8S–12. doi:10.1097/01.TP.0000067944.90241.BD
38. Baron U, Floess S, Wieczorek G, Baumann K, Grützkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur J Immunol (2007) 37:2378–89. doi:10.1002/eji.200737594
39. Regateiro FS, Chen Y, Kendal AR, Hilbrands R, Adams E, Cobbold SP, et al. Foxp3 expression is required for the induction of therapeutic tissue tolerance. J Immunol (2012) 189:3947–56. doi:10.4049/jimmunol.1200449
40. Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol (2007) 7:875–87. doi:10.1038/nri2189
41. Francis RS, Feng G, Tha-in T, Lyons IS, Wood KJ, Bushell A. Induction of transplantation tolerance converts potential effector T cells into graft-protective regulatory T cells. Eur J Immunol (2011) 41:726–38. doi:10.1002/eji.201040509
42. Wood KJ, Bushell A, Jones ND. Immunological unresponsiveness to alloantigen in vivo: a role for regulatory T cells. Immunol Rev (2011) 241:119–32. doi:10.1111/j.1600-065X.2011.01013.x
43. Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol (2012) 12:417–30. doi:10.1038/nri3227
44. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med (2012) 209:1713–22. doi:10.1084/jem.20120822
45. Lin X, Chen M, Liu Y, Guo Z, He XH, Brand D, et al. Advances in distinguishing natural from induced Foxp3+ regulatory T cells. Int J Clin Exp Pathol (2013) 6:116–23.
46. Verma ND, Plain KM, Nomura M, Tran GT, Robinson C, Boyd R, et al. CD4+CD25+ T cells alloactivated ex vivo by IL-2 or IL-4, become potent alloantigen specific inhibitors of rejection with different phenotypes, suggesting Th1 and Th2 responses activate by separate pathways. Blood (2009) 113:479–87. doi:10.1182/blood-2008-05-156612
47. Sakai K, Zamvil SS, Mitchell DJ, Hodgkinson S, Rothbard JB, Steinman L. Prevention of experimental encephalomyelitis with peptides that block interaction of T cells with major histocompatibility complex proteins. Proc Natl Acad Sci U S A (1989) 86:9470–4. doi:10.1073/pnas.86.23.9470
48. Smilek D, Wraith D, Hodgkinson SJ, Dwivedy S, Steinman L, McDevitt H. A single amino-acid change in a myelin-basic protein peptide confers the capacity to prevent rather than induce experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A (1991) 88:9633–7. doi:10.1073/pnas.88.21.9633
49. Hori S, Haury M, Coutinho A, Demengeot J. Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proc Natl Acad Sci U S A (2002) 99:8213–8. doi:10.1073/pnas.122224799
50. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199:1455–65. doi:10.1084/jem.20040139
51. Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, et al. Dendritic cell-expanded, islet-specific CD4+CD25+CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med (2007) 204:191–201. doi:10.1084/jem.20061631
52. Tonkin DR, He J, Barbour G, Haskins K. Regulatory T cells prevent transfer of type 1 diabetes in NOD mice only when their antigen is present in vivo. J Immunol (2008) 181:4516–22.
53. Huter EN, Stummvoll GH, Dipaolo RJ, Glass DD, Shevach EM. Cutting edge: antigen-specific TGF beta-induced regulatory T cells suppress Th17-mediated autoimmune disease. J Immunol (2008) 181:8209–13.
54. Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, et al. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol (2005) 174:6772–80.
55. Hall BM, Plain KM, Verma ND, Tran G, Boyd R, Robinson CM, et al. Transfer of allograft-specific tolerance requires CD4+CD25+ T cells, but not IL-4 or TGF-β and cannot induce tolerance to linked antigens. Transplantation (2007) 83:1075–84. doi:10.1097/01.tp.0000259553.66185.2f
56. Tran GT, Hodgkinson SJ, Carter NM, Verma ND, Plain KM, Boyd R, et al. Interleukin-5 (IL-5) promotes induction of antigen specific CD4+CD25+ T regulatory cells that suppress autoimmunity. Blood (2012) 119:4441–50. doi:10.1182/blood-2011-12-396101
57. Nomura M, Plain KM, Verma N, Robinson C, Boyd R, Hodgkinson SJ, et al. The cellular basis of cardiac allograft rejection. IX. Ratio of naive CD4+CD25+ T cells/CD4+CD25- T cells determines rejection or tolerance. Transpl Immunol (2006) 15:311–8. doi:10.1016/j.trim.2006.01.003
58. Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol (2000) 164:183–90.
59. Hall BM, Robinson CM, Plain KM, Verma ND, Carter N, Boyd R, et al. Studies on naïve CD4+CD25+ T cells inhibition of naïve CD4+CD25- T cells in mixed lymphocyte cultures. Transpl Immunol (2008) 18:291–300. doi:10.1016/j.trim.2007.09.002
60. Lee SY, Cho ML, Oh HJ, Ryu JG, Park MJ, Jhun JY, et al. Interleukin-2/anti-interleukin-2 monoclonal antibody immune complex suppresses collagen-induced arthritis in mice by fortifying interleukin-2/STAT5 signalling pathways. Immunology (2012) 137:305–16. doi:10.1111/imm.12008
61. Tang Q, Bluestone JA, Kang SM. CD4+Foxp3+ regulatory T cell therapy in transplantation. J Mol Cell Biol (2012) 4:11–21. doi:10.1093/jmcb/mjr047
63. Baecher-Allan C, Wolf E, Hafler DA. MHC class II expression identifies functionally distinct human regulatory T cells. J Immunol (2006) 176:4622–31.
64. Seddon B, Saoudi A, Nicholson M, Mason D. CD4+CD8- thymocytes that express L-selectin protect rats from diabetes upon adoptive transfer. Eur J Immunol (1996) 26:2702–8. doi:10.1002/eji.1830261123
65. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med (2004) 199:303–13. doi:10.1084/jem.20031562
66. Scotto L, Naiyer AJ, Galluzzo S, Rossi P, Manavalan JS, Kim-Schulze S, et al. Overlap between molecular markers expressed by naturally occurring CD4+CD25+ regulatory T cells and antigen specific CD4+CD25+ and CD8+CD28- T suppressor cells. Hum Immunol (2004) 65:1297–306. doi:10.1016/j.humimm.2004.09.004
67. Taylor PA, Panoskaltsis-Mortari A, Swedin JM, Lucas PJ, Gress RE, Levine BL, et al. L-selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood (2004) 104:3804–12. doi:10.1182/blood-2004-05-1850
68. Ermann J, Hoffmann P, Edinger M, Dutt S, Blankenberg FG, Higgins JP, et al. Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood (2005) 105:2220–6. doi:10.1182/blood-2004-05-2044
69. Tosello V, Odunsi K, Souleimanian NE, Lele S, Shrinkant P, Old LJ, et al. Differential expression of CCR7 defines two distinct subsets of human memory CD4+CD25+ Tregs. Clin Immunol (2008) 126:291–302. doi:10.1016/j.clim.2007.11.008
70. Siegmund K, Feuerer M, Siewert C, Ghani S, Haubold U, Dankof A, et al. Migration matters: regulatory T-cell compartmentalization determines suppressive activity in vivo. Blood (2005) 106:3097–104. doi:10.1182/blood-2005-05-1864
71. Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, et al. Altering the distribution of Foxp3+ regulatory T cells results in tissue-specific inflammatory disease. J Exp Med (2007) 204:1335–47. doi:10.1084/jem.20070081
72. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type-1 inflammation. Nat Immunol (2009) 10:595–602. doi:10.1038/ni.1731
73. Zheng Y, Chaudhry A, Kas A, Deroos P, Kim JM, Chu T-T, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature (2009) 458:351–6. doi:10.1038/nature07674
74. Valmori D, Raffin C, Raimbaud I, Ayyoub M. Human RORγt+ TH17 cells preferentially differentiate from naive FOXP3+ Treg in the presence of lineage-specific polarizing factors. Proc Natl Acad Sci U S A (2010) 107:19402–7. doi:10.1073/pnas.1008247107
75. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17:975–82. doi:10.1038/nm.2425
76. Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, et al. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science (2001) 282:1907–10. doi:10.1126/science.1059835
77. Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity (2011) 35:337–48. doi:10.1016/j.immuni.2011.08.012
78. Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A (2009) 106:4793–8. doi:10.1073/pnas.0900408106
79. O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327:1098–102. doi:10.1126/science.1178334
80. Hidalgo LG, Halloran PF. Role of IFN-gamma in allograft rejection. Crit Rev Immunol (2002) 22:317–49. doi:10.1615/CritRevImmunol.v22.i4.50
81. Wood KJ, Feng G, Wei B, Sawitzki B, Bushell AR. Interferon gamma: friend or foe? Transplantation (2007) 84:S4–5. doi:10.1097/01.tp.0000269115.60728.b1
82. Caretto D, Katzman SD, Villarino AV, Gallo E, Abbas AK. Cutting edge: the Th1 response inhibits the generation of peripheral regulatory T cells. J Immunol (2010) 184:30–4. doi:10.4049/jimmunol.0903412
83. Colantonio L, Iellem A, Sinigaglia F, D’Ambrosio D. Skin-homing CLA+ T cells and regulatory CD25+ T cells represent major subsets of human peripheral blood memory T cells migrating in response to CCL1/I-309. Eur J Immunol (2002) 32:3506–14. doi:10.1002/1521-4141(200212)32:12<3506::AID-IMMU3506>3.0.CO;2-#
84. Sis B, Famulski KS, Allanach KL, Zhu L-F, Halloran PF. IFN-γ prevents early perforin-granzyme-mediated destruction of kidney allografts by inducing donor class I products in the kidney. Am J Transplant (2007) 7:2301–10. doi:10.1111/j.1600-6143.2007.01947.x
85. Mitchell RN, Lichtman AH. The link between IFN-γ and allograft arteriopathy: is the answer NO? J Clin Invest (2004) 114:762–4. doi:10.1172/JCI200422927
86. Tellides G, Pober JS. Interferon-g axis in graft atherosclerosis. Circ Res (2007) 100:622–32. doi:10.1161/01.RES.0000258861.72279.29
87. Hart-Matyas M, Nejat S, Jordan JL, Hirsch GM, Lee TDJ. IFN-γ and Fas/FasL pathways cooperate to induce medial cell loss and neointimal lesion formation in allograft vasculopathy. Transplant Immunol (2010) 22:157–64. doi:10.1016/j.trim.2009.10.004
88. Hall BM, Bishop GA, Duggin GG, Horvath JS, Philips J, Tiller DJ. Increased expression of HLA-DR antigens on renal tubular cells in renal transplants: relevance to the rejection response. Lancet (1984) 2:247–51. doi:10.1016/S0140-6736(84)90297-6
89. Halloran PF, Afrouzian M, Ramassar V, Urmson J, Zhu LF, Helms LM, et al. Interferon-gamma acts directly on rejecting renal allografts to prevent graft necrosis. Am J Pathol (2001) 158:215–26. doi:10.1016/S0002-9440(10)63960-0
90. Halloran PF, Miller LW, Urmson J, Ramassar V, Zhu LF, Kneteman NM, et al. IFN-gamma alters the pathology of graft rejection: protection from early necrosis. J Immunol (2001) 166:7072–81.
91. Coley SM, Ford ML, Hanna SC, Wagener ME, Kirk AD, Larsen CP. IFN-γ dictates allograft fate via opposing effects on the graft and on recipient CD8 T cell responses. J Immunol (2008) 182:225–33.
92. Feuerer M, Eulenburg K, Loddenkemper C, Hamann A, Huehn J. Self-limitation of Th1-mediated inflammation by IFN-gamma. J Immunol (2006) 176:2857–63.
93. Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol (1996) 157:3223–7.
94. Willenborg DO, Fordham SA, Staykova MA, Ramshaw IA, Cowden WB. Interferon-gamma is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol (1999) 163:5278–86.
95. O’Brien NC, Charlton B, Cowden WB, Willenborg DO. Nitric oxide plays a critical role in the recovery of lewis rats from experimental autoimmune encephalomyelitis and the maintenance of resistance to reinduction. J Immunol (1999) 163:6841–7.
96. O’Brien NC, Charlton B, Cowden WB, Willenborg DO. Inhibition of nitric oxide synthase initiates relapsing remitting experimental autoimmune encephalomyelitis in rats, yet nitric oxide appears to be essential for clinical expression of disease. J Immunol (2001) 167:5904–12.
97. Staykova MA, Paridaen JT, Cowden WB, Willenborg DO. Nitric oxide contributes to resistance of the Brown Norway rat to experimental autoimmune encephalomyelitis. Am J Pathol (2005) 166:147–57. doi:10.1016/S0002-9440(10)62240-7
98. Verma ND, Boyd R, Robinson C, Plain KM, Tran GT, Hall BM. Interleukin-12p70 prolongs allograft survival by induction of interferon gamma and nitric oxide production. Transplantation (2006) 82:1324–33. doi:10.1097/01.tp.0000239519.56358.c1
99. Brok HPM, Heidt PJ, van der Meide PH, Zurcher C, Vossen JM. Interferon-gamma prevents graft-versus-host disease after allogeneic bone marrow transplantation in mice. J Immunol (1993) 151:6451–9.
100. Guillonneau C, Hill M, Hubert FX, Chiffoleau E, Hervé C, Li XL, et al. CD40Ig treatment results in allograft acceptance mediated by CD8CD45RC T cells, IFN-gamma, and indoleamine 2,3-dioxygenase. J Clin Invest (2007) 117:1096–106. doi:10.1172/JCI28801
101. Sawitzki B, Kingsley CI, Oliveira V, Karim M, Herber M, Wood KJ. IFN-gamma production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J Exp Med (2005) 201:1925–35. doi:10.1084/jem.20050419
102. Nishikawa H, Kato T, Tawara I, Ikeda H, Kuribayashi K, Allen PM, et al. IFN-γ controls the generation/activation of CD4+CD25+ regulatory T cells in antitumor immune response. J Immunol (2005) 175:4433–40.
103. Feng G, Wood KJ, Bushell A. Interferon-gamma conditioning ex vivo generates CD25+CD62L+Foxp3+ regulatory T cells that prevent allograft rejection: potential avenues for cellular therapy. Transplantation (2008) 86:578–89. doi:10.1097/TP.0b013e3181806a60
104. Feng G, Gao W, Strom TB, Oukka M, Francis RS, Wood KJ, et al. Exogenous IFN-gamma ex vivo shapes the alloreactive T-cell repertoire by inhibition of Th17 responses and generation of functional Foxp3+ regulatory T cells. Eur J Immunol (2008) 38:2512–27. doi:10.1002/eji.200838411
105. Feng G, Chan T, Wood KJ, Bushell A. Donor reactive regulatory T cells. Curr Opin Organ Transplant (2009) 14:69–80. doi:10.1097/MOT.0b013e32832c58f1
106. Feng A, Nadig SN, Bäckdahl L, Beck S, Francis RS, Schiopu A, et al. Functional regulatory T cells produced by inhibiting cyclic nucleotide phosphodiesterase type 3 prevent allograft rejection. Sci Transl Med (2011) 3:83ra40. doi:10.1126/scitranslmed.3002099
107. Feng G, Wood KJ, Bushell A. Regulatory T cell enrichment by IFN-γ conditioning. Methods Mol Biol (2011) 677:281–301. doi:10.1007/978-1-60761-869-0_20
108. Wang Z, Hong J, Sun W, Xu G, Li N, Chen X, et al. Role of IFN-g in induction of Foxp3 and conversion of CD4+CD25- T cells to CD4+ Tregs. J Clin Invest (2006) 116:2434–41.
109. Stock P, Akbari O, Berry G, Freeman GJ, Dekruyff RH, Umetsu DT. Induction of T helper type 1-like regulatory cells that express Foxp3 and protect against airway hyper-reactivity. Nat Immunol (2004) 5:1149–56. doi:10.1038/ni1122
110. Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunis K, et al. CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res (2012) 72:4351–60. doi:10.1158/0008-5472.CAN-12-0579
111. Stroopinsky D, Avivi I, Rowe JM, Avigan D, Katz T. Allogeneic induced human FOXP3+IFN-gamma+ T cells exhibit selective suppressive capacity. Eur J Immunol (2009) 39:2703–15. doi:10.1002/eji.200839097
112. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol (2011) 186:3918–26. doi:10.4049/jimmunol.1003099
113. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med (2011) 17:673–5. doi:10.1038/nm.2389
114. Daniel V, Sadeghi M, Wang H, Opelz G. CD4+CD25+Foxp3+IFN-γ+ human induced T regulatory cells are induced by interferon-γ and suppress alloresponses nonspecifically. Hum Immunol (2011) 72:699–707. doi:10.1016/j.humimm.2011.05.020
115. Nishibori T, Tanabe Y, Su L, David M. Impaired development of CD4+CD25+ regulatory T cells in the absence of STAT1: increased susceptibility to autoimmune disease. J Exp Med (2004) 199:25–34. doi:10.1084/jem.20020509
116. Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, et al. The cytokines interleukin 27 and interferon-γ promote distinct Treg cell populations required to limit infection-induced pathology. Immunity (2012) 37(23):511–23. doi:10.1016/j.immuni.2012.06.014
117. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol (2003) 3:133–46. doi:10.1038/nri1001
118. Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G, et al. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med (1993) 177:1199–204. doi:10.1084/jem.177.4.1199
119. Valenzuela J, Schmidt C, Mescher M. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J Immunol (2002) 169(12):6842–9.
120. Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med (1989) 170:827–45. doi:10.1084/jem.170.3.827
121. Manetti R, Gerosa F, Giudizi MG, Biagiotti R, Parronchi P, Piccinni MP, et al. Interleukin 12 induces stable priming for interferon gamma (IFN-gamma) production during differentiation of human T helper (Th) cells and transient IFN-gamma production in established Th2 cell clones. J Exp Med (1994) 179:1273–83. doi:10.1084/jem.179.4.1273
122. Gately MK, Renezetti LM, Magram J, Stern AS, Adorini L, Gubler U, et al. The interleukin-12-receptor system: role in normal and pathological responses. Annu Rev Immunol (1998) 16:495–521. doi:10.1146/annurev.immunol.16.1.495
123. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (1993) 260:547–9. doi:10.1126/science.8097338
124. Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature (1996) 382:174–7. doi:10.1038/382174a0
125. Williamson E, Garside P, Bradley JA, Mowat AM. IL-12 is a central mediator of acute-graft versus-host disease in mice. J Immunol (1996) 157:689–99.
126. Sykes M, Szot GL, Nguyen PL, Pearson DA. Interleukin-12 inhibits murine graft-versus-host disease. Blood (1995) 86:2429–38.
127. Yang YG, Dey BR, Sergio JJ, Pearson DA, Sykes M. Donor-derived interferon gamma is required for inhibition of acute graft-versus-host disease by interleukin 12. J Clin Invest (1998) 102:2126–35. doi:10.1172/JCI4992
128. Dey BR, Yang YG, Szot GL, Pearson DA, Sykes M. Interleukin-12 inhibits graft-versus-host disease through an Fas-mediated mechanism associated with alterations in donor T-cell activation and expansion. Blood (1998) 91:3315–22.
129. Tarrant TK, Silver PB, Wahlstern JL, Rizzo LV, Chan CC, Wiggert B, et al. Interleukin 12 protects from a T helper 1-mediated autoimmune disease, experimental autoimmune uveitis, through a mechanism involving interferon gamma, nitric oxide and apoptosis. J Exp Med (1999) 189:219. doi:10.1084/jem.189.2.219
130. Zhang GX, Yu S, Gran B, Li J, Iglienti I, Chen X, et al. Role of IL-12 receptor beta 1 in regulation of T cell response by APC in experimental autoimmune encephalomyelitis. J Immunol (2003) 171:4485–92.
131. Xu H, Rizzo LV, Silver PB, Caspi RR. Uveitogenicity is associated with a Th1-like lymphokine profile: cytokine-dependent modulation of early and committed effector T cells in experimental autoimmune uveitis. Cell Immunol (1997) 178:69–78. doi:10.1006/cimm.1997.1121
132. Yokoi H, Kato K, Kezuka T, Sakai J, Usui M, Yagita H, et al. Prevention of experimental autoimmune uveoretinitis by monoclonal antibody to interleukin-12. Eur J Immunol (1997) 27:641–6. doi:10.1002/eji.1830270310
133. Adorini L. Interleukin-12, a key cytokine in Th1-mediated autoimmune diseases. Cell Mol Life Sci (1999) 55:1610–25. doi:10.1007/s000180050400
134. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421:744–8. doi:10.1038/nature01355
135. Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol (2003) 170:2153–60.
136. Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol (1996) 26:1641–6. doi:10.1002/eji.1830260735
137. Zhao Z, Yu S, Fitzgerald DC, Elbehi M, Ciric B, Rostami AM, et al. IL-12R beta 2 promotes the development of CD4+CD25+ regulatory T cells. J Immunol (2008) 181:3870–6.
138. Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity (2009) 31:772–86. doi:10.1016/j.immuni.2009.10.001
139. Hall AO, Silver JS, Hunter CA. The immunobiology of IL-27. Adv Immunol (2012) 115:1–44. doi:10.1016/B978-0-12-394299-9.00001-1
140. Villarino A, Larkin JR, Saris CJ, Caton AJ, Lucas S, Wong T, et al. Positive and negative regulation of the IL-27 receptor during lymphoid activation. J Immunol (2005) 174(12):7684–91.
141. Villarino A, Hibbert L, Lieberman L, Wilson E, Mak T, Yoshida H, et al. The IL-27R (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity (2003) 19:645–55. doi:10.1016/S1074-7613(03)00300-5
142. Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, et al. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol (2007) 8(12):1363–71. doi:10.1038/ni1537
143. Yoshida H, Miyazaki Y. Regulation of immune responses by interleukin-27. Immunol Rev (2008) 226:234–47. doi:10.1111/j.1600-065X.2008.00710.x
144. Ouaked N, Mantel PY, Bassin C, Burgler S, Siegmund K, Akdis CA, et al. Regulation of the foxp3 gene by the Th1 cytokines: the role of IL-27-induced STAT1. J Immunol (2009) 182:1041–9.
145. Das MP, Nicholson LB, Greer JM, Kuchroo VK. Autopathogenic T helper cell type 1 (Th1) and protective Th2 clones differ in their recognition of the autoantigenic peptide of myelin proteolipid protein. J Exp Med (1997) 186:867–76. doi:10.1084/jem.186.6.867
146. Falcone M, Bloom BR. A T helper cell 2 (Th2) immune response against non-self antigens modifies the cytokine profile of autoimmune T cells and protects against experimental allergic encephalomyelitis. J Exp Med (1997) 185:901–7. doi:10.1084/jem.185.5.901
147. O’Garra A, Steinman L, Gijbels K. CD4+ T-cell subsets in autoimmunity. Curr Opin Immunol (1997) 9:872–83. doi:10.1016/S0952-7915(97)80192-6
148. Spicer ST, Ha H, Boyd RA, He XY, Carter N, Tran G, et al. IL-4 therapy prevents the development of proteinuria in active Heymann nephritis by inhibition of Tc1 cells. J Immunol (2001) 167:3725–33.
149. Racke MK, Bonomo A, Scott DE, Canella B, Levine A, Raine CS, et al. Cytokine-induced immune deviation as therapy for inflammatory autoimmune disease. J Exp Med (1994) 180:1961–6. doi:10.1084/jem.180.5.1961
150. Tran GT, Carter N, He XY, Spicer TS, Plain KM, Nicolls M, et al. Reversal of experimental allergic encephalomyelitis with non-mitogenic, non-depleting anti-CD3 mAb therapy with a preferential effect on Th1 cells that is augmented by IL-4. Int Immunol (2001) 13:1109–20. doi:10.1093/intimm/13.9.1109
151. Rott O, Fleischer B, Cash E. Interleukin-10 prevents experimental allergic encephalomyelitis in rats. Eur J Immunol (1994) 24:1434–40. doi:10.1002/eji.1830240629
152. Zaccone P, Phillips J, Conget I, Gomis R, Haskins K, Minty A, et al. Interleukin-13 prevents autoimmune diabetes in NOD mice. Diabetes (1999) 48:1522–8. doi:10.2337/diabetes.48.8.1522
153. Papp I, Wieder KJ, Sablinski T, O’Connell PJ, Milford EL, Strom TB, et al. Evidence for functional heterogeneity of rat CD4+ T cells in vivo. Differential expression of IL-2 and IL-4 mRNA in recipients of cardiac allografts. J Immunol (1992) 148:1308–14.
154. Kupiec-Weglinski JW, Wasowska B, Papp I, Schmidbauer G, Sayegh MH, Baldwin WMD, et al. CD4 mAb therapy modulates alloantibody production and intracardiac graft deposition in association with selective inhibition of Th1 lymphokines. J Immunol (1993) 151:5053–61.
155. Mottram PL, Han WR, Purcell LJ, McKenzie IFC, Hancock WW. Increased expression of IL-4 and IL-10 and decreased expression of IL-2 and interferon-gamma in long-surviving mouse heart allografts after brief CD4-monoclonal antibody therapy. Transplantation (1995) 59:559–65. doi:10.1097/00007890-199559040-00021
156. Sayegh MH, Akalin E, Hancock WW, Russell ME, Carpenter CB, Linsley PS, et al. CD28-B7 blockade after alloantigenic challenge in vivo inhibits Th1 cytokines but spares Th2. J Exp Med (1995) 181:1869–74. doi:10.1084/jem.181.5.1869
157. Takeuchi T, Ueki T, Sunaga S, Ikuta K, Sasaki Y, Li B, et al. Murine interleukin 4 transgenic heart allograft survival prolonged with down-regulation of TH1 cytokine mRNA in grafts. Transplantation (1997) 64:152–7. doi:10.1097/00007890-199707150-00026
158. Plain KM, Chen J, Merten S, He XY, Davidson C, Hall BM. Induction of specific tolerance to allografts in rats by therapy with non-mitogenic, non depleting anti-CD3 monoclonal antibody: association with Th2 cytokines not anergy. Transplantation (1999) 67:605–13. doi:10.1097/00007890-199902270-00020
159. Chen N, Field EH. Enhanced type 2 and diminished type 1 cytokines in neonatal tolerance. Transplantation (1995) 59:933–41. doi:10.1097/00007890-199504150-00002
160. Chen N, Gao Q, Field EH. Expansion of memory TH2 cells over TH1 cells in neonatal primed mice. Transplantation (1995) 60:1187–93. doi:10.1097/00007890-199512000-00002
161. Chen N, Gao Q, Field EH. Prevention of TH1 responses is critical for tolerance. Transplantation (1996) 61:1076–83. doi:10.1097/00007890-199604150-00016
162. Field EH, Rouse TM. Alloantigen priming after total lymphoid irradiation alters alloimmune cytokine responses. Transplantation (1995) 60:695–701. doi:10.1097/00007890-199510150-00014
163. Field EH, Rouse TM, Gao Q, Chang B. Associations between enhanced Th1/Th2 cytokine profile and donor T cell chimerism following total lymphoid irradiation. Hum Immunol (1997) 52:144–50. doi:10.1016/S0198-8859(96)00291-1
164. Plain KM, Boyd R, Verma N, Robinson C, Tran G, Hodgkinson SJ, et al. Transplant tolerance associated with a Th1 response and not broken by IL-4. IL-5 and TGF-beta blockade or Th1 cytokine administration. Transplantation (2007) 83:764–73. doi:10.1097/01.tp.0000256326.11647.2e
165. Waaga-Gasser AM, Grimm MR, Lutz J, Lange V, Lenhard SM, Aviles B, et al. Regulatory allospecific T cell clones abrogate chronic allograft rejection. J Am Soc Nephrol (2009) 20:820–30. doi:10.1681/ASN.2008020164
166. Hall BM, Fava L, Chen J, Plain KM, Boyd RA, Spicer ST, et al. Anti-CD4 monoclonal antibody-induced tolerance to MHC-incompatible cardiac allografts maintained by CD4+ suppressor T cells that are not dependent upon IL-4. J Immunol (1998) 161:5147–56.
167. Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, et al. Essential role of Stat6 in IL-4 signalling. Nature (1996) 380:627–30. doi:10.1038/380627a0
168. Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity (1996) 4:313–9. doi:10.1016/S1074-7613(00)80439-2
169. Pillemer BB, Qi Z, Melgert B, Oriss TB, Ray P, Ray A. STAT6 activation confers upon T helper cells resistance to suppression by regulatory T cells. J Immunol (2009) 183:155–63. doi:10.4049/jimmunol.0803733
170. Campbell DJ. Regulatory T cells GATA have it. Immunity (2011) 35:313–5. doi:10.1016/j.immuni.2011.09.002
171. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J Clin Invest (2011) 121:4503–15. doi:10.1172/JCI57456
172. Mantel PY, Kuipers H, Boyman O, Rhyner C, Ouaked N, Rückert B, et al. GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol (2007) 5:e329. doi:10.1371/journal.pbio.0050329
173. Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med (2002) 195:1499–505. doi:10.1084/jem.20012076
174. Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol (2000) 165:4773–7.
175. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3- effector T cells. Nat Immunol (2008) 9:1347–55. doi:10.1038/ni.1677
176. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta “reprograms” the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol (2008) 9:1341–6. doi:10.1038/ni.1659
177. Maerten P, Shen C, Bullens DM, Van Assche G, Van Gool S, Geboes K, et al. Effects of interleukin 4 on CD25+CD4+ regulatory T cell function. J Autoimmun (2005) 25:112–20. doi:10.1016/j.jaut.2005.04.001
178. Thornton AM, Piccirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol (2004) 34:366–76. doi:10.1002/eji.200324455
179. Pace L, Pioli C, Doria G. IL-4 modulation of CD4+CD25+ T regulatory cell-mediated suppression. J Immunol (2005) 174:7645–53.
180. He XY, Verma N, Chen J, Robinson C, Boyd R, Hall BM. IL-5 prolongs allograft survival by downregulating IL-2 and IFN-gamma cytokines. Transplant Proc (2001) 33:703–4. doi:10.1016/S0041-1345(00)02212-0
181. Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, et al. The development of inflammatory Th-17 cells requires interferon-regulatory factor 4. Nat Immunol (2007) 8:958–66. doi:10.1038/ni1500
182. Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, et al. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med (2002) 8:1024–32. doi:10.1038/nm745
183. Redpath SA, van der Werf N, Cervera AM, Macdonald AS, Gray D, Maizels RM, et al. ICOS controls Foxp3+ regulatory T-cell expansion, maintenance and IL-10 production during helminth infection. Eur J Immunol (2013) 43:705–15. doi:10.1002/eji.201242794
184. Taylor JJ, Mohrs M, Pearce EJ. Regulatory T cell responses develop in parallel to Th responses and control the magnitude and phenotype of the Th effector population. J Immunol (2006) 176:5839–47.
185. McKee AS, Pearce EJ. CD25+CD4+ cells contribute to Th2 polarization during helminth infection by suppressing Th1 response development. J Immunol (2004) 173:1224–31.
186. Whary MT, Fox JG. Th1-mediated pathology in mouse models of human disease is ameliorated by concurrent Th2 responses to parasite antigens. Curr Top Med Chem (2004) 4:531–8. doi:10.2174/1568026043451276
187. Blankenhaus B, Klemm U, Eschbach ML, Sparwasser T, Huehn J, Kühl AA, et al. Strongyloides ratti infection induces expansion of Foxp3+ regulatory T cells that interfere with immune response and parasite clearance in BALB/c mice. J Immunol (2011) 186:4295–305. doi:10.4049/jimmunol.1001920
188. Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature (2002) 420:502–7. doi:10.1038/nature01152
189. Taylor MD, Harris A, Babayan SA, Bain O, Culshaw A, Allen JE, et al. CTLA-4 and CD4+CD25+ regulatory T cells inhibit protective immunity to filarial parasites in vivo. J Immunol (2007) 179:4626–34.
190. McSorley HJ, Harcus YM, Murray J, Taylor MD, Maizels RM. Expansion of Foxp3+ regulatory T cells in mice infected with the filarial parasite Brugia malayi. J Immunol (2008) 181:6456–66.
191. Babu S, Blauvelt V, Kumaraswami V, Nutman TB. Regulatory networks induced by live parasites impair both Th1 and Th2 pathways in patent lymphatic filariasis: implications for parasitic persistence. J Immunol (2006) 176:3248–56.
192. Maizels RM, Balic A, Gomez-Escobar N, Nair M, Taylor MD, Allen JE. Helminth parasites – masters of regulation. Immunol Rev (2004) 201:89–116. doi:10.1111/j.0105-2896.2004.00191.x
193. Freeman CM, Chiu BC, Stolberg VR, Hu J, Zeibecoglou K, Lukacs NW, et al. CCR8 is expressed by antigen-elicited, IL-10-producing CD4+CD25+ T cells, which regulate Th2-mediated granuloma formation in mice. J Immunol (2005) 174:1962–70.
194. Rausch S, Huehn J, Kirchhoff D, Rzepecka J, Schnoeller C, Pillai S, et al. Functional analysis of effector and regulatory T cells in a parasitic nematode infection. Infect Immun (2008) 76:1908–19. doi:10.1128/IAI.01233-07
195. Tadokoro CE, Vallochi AL, Rios LS, Martins GA, Schlesinger D, Mosca T, et al. Experimental autoimmune encephalomyelitis can be prevented and cured by infection with Trypanosoma cruzi. J Autoimmun (2004) 23:103–15. doi:10.1016/j.jaut.2004.05.003
196. Gruden-Movsesijan A, Ilic N, Mostarica-Stojkovic M, Stosic-Grujicic S, Milic M, Sofronic-Milosavljevic L. Mechanisms of modulation of experimental autoimmune encephalomyelitis by chronic Trichinella spiralis infection in Dark Agouti rats. Parasite Immunol (2010) 32:450–9. doi:10.1111/j.1365-3024.2010.01207.x
197. Walsh KP, Brady MT, Finlay CM, Boon L, Mills KH. Infection with a helminth parasite attenuates autoimmunity through TGF-beta-mediated suppression of Th17 and Th1 responses. J Immunol (2009) 183:1577–86. doi:10.4049/jimmunol.0803803
198. Ledingham DL, McAlister V, Ehigiator HN, Giacomantonio C, Theal M, Lee TD. Prolongation of allograft survival by nematodes. Transplantation (1996) 61:184–9. doi:10.1097/00007890-199601270-00003
199. Liwski R, Zhou J, McAlister V, Lee TD. Prolongation of allograft survival by Nippostrongylus brasiliensis is associated with decreased allospecific cytotoxic T lymphocyte activity and development of T cytotoxic cell type 2 cells. Transplantation (2000) 69:1912–22. doi:10.1097/00007890-200005150-00029
200. Zeng Z, Ming C, Zhen Z, Sun R, Chen W. Can an allograft become harmonious with its recipient by analogous mechanisms to the long-term survival of a parasite in its host? Transplantation is similar to parasitism and chronic parasite infection can prolong allograft survival. Med Hypotheses (2005) 65:1047–50. doi:10.1016/j.mehy.2005.06.027
201. Maizels RM, Yazdanbakhsh M. T-cell regulation in helminth parasite infections: implications for inflammatory diseases. Chem Immunol Allergy (2008) 94:112–23. doi:10.1159/000154944
202. Correale J, Farez M. Association between parasite infection and immune responses in multiple sclerosis. Ann Neurol (2007) 61:97–108. doi:10.1002/ana.21067
203. Correale J, Farez MF. The impact of parasite infections on the course of multiple sclerosis. J Neuroimmunol (2011) 233:6–11. doi:10.1016/j.jneuroim.2011.01.002
204. Summers RW, Elliott DE, Urban JFJ, Thompson RA, Weinstock JV. Trichuris suis therapy for active ulcerative colitis: a randomized controlled trial. Gastroenterology (2005) 128:825–32. doi:10.1053/j.gastro.2005.01.005
205. Elliott DE, Weinstock JV. Helminthic therapy: using worms to treat immune-mediated disease. Adv Exp Med Biol (2009) 666:157–66. doi:10.1007/978-1-4419-1601-3_12
206. Jackson JA, Friberg IM, Little S, Bradley JE. Review series on helminths, immune modulation and the hygiene hypothesis: immunity against helminths and immunological phenomena in modern human populations: coevolutionary legacies? Immunology (2009) 126:18–27. doi:10.1111/j.1365-2567.2008.03010.x
207. Beriou G, Costantino CM, Ashley CW, Yang L, Kuchroo VK, Baecher-Allan C, et al. IL-17-producing human peripheral regulatory T cells retain suppressive function. Blood (2009) 113:4240–9. doi:10.1182/blood-2008-10-183251
208. Pallandre JR, Brillard E, Créhange G, Radlovic A, Remy-Martin JP, Saas P, et al. Role of STAT3 in CD4+CD25+FOXP3+ regulatory lymphocyte generation: implications in graft-versus-host disease and antitumor immunity. J Immunol (2007) 179:7593–604.
209. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science (2009) 326:986–91. doi:10.1126/science.1172702
210. Huber M, Steinwald V, Guralnik A, Brustle A, Kleemann P, Rosenplanter C, et al. IL-27 inhibits the development of regulatory T cells via STAT3. Int Immunol (2008) 20:223–34. doi:10.1093/intimm/dxm139
211. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity (2011) 34:566–78. doi:10.1016/j.immuni.2011.03.018
212. Huber S, Gagliani N, Esplugues E, O’Connor WJ, Huber FJ, Chaudhry A, et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3- and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity (2011) 34:554–65. doi:10.1016/j.immuni.2011.01.020
213. Ayyoub M, Deknuydt F, Raimbaud I, Dousset C, Leveque L, Bioley G, et al. Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the Th17 lineage-specific transcription factor RORgamma t. Proc Natl Acad Sci U S A (2009) 106:8635–40. doi:10.1073/pnas.0900621106
214. Crome SQ, Clive B, Wang AY, Kang CY, Chow V, Yu J, et al. Inflammatory effects of ex vivo human Th17 cells are suppressed by regulatory T cells. J Immunol (2010) 185:3199–208. doi:10.4049/jimmunol.1000557
215. Raffin C, Raimbaud I, Valmori D, Ayyoub M. Ex vivo IL-1 receptor type I expression in human CD4+ T cells identifies an early intermediate in the differentiation of Th17 from FOXP3+ naive regulatory T cells. J Immunol (2011) 187:5196–202. doi:10.4049/jimmunol.1101742
216. Leveque L, Deknuydt F, Bioley G, Old LJ, Matsuzaki J, Odunis K, et al. Interleukin 2-mediated conversion of ovarian cancer-associated CD4+ regulatory T cells into proinflammatory interleukin 17-producing helper T cells. J Immunother (2009) 32:101–8. doi:10.1097/CJI.0b013e318195b59e
217. Battaglia A, Buzzonetti A, Baranello C, Fanelli M, Fossati M, Catzola V, et al. IL-21 synergises with IL-2 to enhance TCR-induced human T cell proliferation and counteracts IL-2/TGF-β-induced regulatory T cell development. Immunology (2012) 139:109–20. doi:10.1111/imm.12061
218. Lim HW, Hillsamer P, Kim CH. Regulatory T cells can migrate to follicles upon T cell activation and suppress GC-Th cells and GC-Th cell-driven B cell responses. J Clin Invest (2004) 114:1640–9. doi:10.1172/JCI22325
219. Lim HW, Hillsamer P, Banham AH, Kim CH. Cutting edge: direct suppression of B cells by CD4+ CD25+ regulatory T cells. J Immunol (2005) 175:4180–3.
220. Sawant DV, Sehra S, Nguyen ET, Jadhav R, Englert K, Shinnakasu R, et al. Bcl6 controls the Th2 inflammatory activity of regulatory T cells by repressing Gata3 function. J Immunol (2012) 189:4759–69. doi:10.4049/jimmunol.1201794
221. Tang Q, Lee K. Regulatory T-cell therapy for transplantation: how many cells do we need? Curr Opin Organ Transplant (2012) 17:349–54. doi:10.1097/MOT.0b013e328355a992
222. Hall BM, Verma ND, Tran GT, Hodgkinson SJ. Distinct regulatory CD4+ T cell subsets; differences between naïve and antigen specific T regulatory cells. Curr Opin Immunol (2011) 23:1–7. doi:10.1016/j.coi.2011.07.012
223. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
224. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med (2000) 192:303–10. doi:10.1084/jem.192.2.303
225. Mantel PY, Schmidt-Weber CB. Transforming growth factor-beta: recent advances on its role in immune tolerance. Methods Mol Biol (2011) 677:303–38. doi:10.1007/978-1-60761-869-0_21
226. Collison LW, Pillai MR, Chaturvedi V, Vignali DA. Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J Immunol (2009) 182:6121–8. doi:10.4049/jimmunol.0803646
227. Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O’Shea JJ, et al. CD4+FoxP3+ regulatory T cells confer infectious tolerance in a TGF-β-dependent manner. J Exp Med (2008) 205:1975–81. doi:10.1084/jem.20080308
228. Del Prete G, De Carli M, Almerigogna F, Giudizi MG, Biagiotti R, Romagnani P. Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cells and inhibits their antigen specific proliferation and cytokine production. J Immunol (1993) 150:353–60.
229. Assenmacher M, Schmitz J, Radbruch A. Flow cytometric determination of cytokines in activated murine T helper lymphocytes: expression of interleukin-10 in interferon-gamma and interleukin-4-expression cells. Eur J Immunol (1994) 24:1097–101. doi:10.1002/eji.1830240513
Keywords: antigen specific Treg, nTreg, Th1-like Treg, Th2-like Treg, immune tolerance
Citation: Hall BM, Tran GT, Verma ND, Plain KM, Robinson CM, Nomura M and Hodgkinson SJ (2013) Do natural T regulatory cells become activated to antigen specific T regulatory cells in transplantation and in autoimmunity? Front. Immunol. 4:208. doi: 10.3389/fimmu.2013.00208
Received: 03 April 2013; Accepted: 08 July 2013;
Published online: 02 August 2013.
Edited by:
Eyad Elkord, United Arab Emirates University, UAE; University of Salford, UK; University of Manchester, UKReviewed by:
Dominique M. A. Bullens, KU Leuven, BelgiumJocelyne Demengeot, Instituto Gulbenkian de Ciencia, Portugal
Copyright: © 2013 Hall, Tran, Verma, Plain, Robinson, Nomura and Hodgkinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruce M. Hall, Immune Tolerance Laboratory, Medicine, University of New South Wales, Suite 206 National Innovation Centre, 4 Cornwallis Street, Australian Technology Park, Eveleigh, Sydney, NSW 1430, Australia e-mail:Yi5oYWxsQHVuc3cuZWR1LmF1