
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 June 2013
Sec. T Cell Biology
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00161
This article is part of the Research Topic How aging affects T lymphocyte-mediated immunity View all 10 articles
 Jana Raynor1 Allyson Sholl1 David R. Plas2 Philippe Bouillet3,4
Jana Raynor1 Allyson Sholl1 David R. Plas2 Philippe Bouillet3,4 Claire A. Chougnet1†
Claire A. Chougnet1† David A. Hildeman1*†
David A. Hildeman1*†
We and others have shown that regulatory T cells (Treg) accumulate dramatically with age in both humans and mice. Such Treg accrual contributes to age-related immunosenescence as they reduce the response to tumors and parasite infection. While we reported earlier that aged Treg have decreased expression of the pro-apoptotic molecule Bim and germline deletion of Bim promoted earlier accumulation of Treg, it remains unclear whether the effects of Bim are: (i) Treg intrinsic and (ii) dominant to other BH3-only pro-apoptotic molecules. Further, the mechanism(s) controlling Bim expression in aged Treg remain unclear. Here we show that Treg-specific loss of Bim is sufficient to drive Treg accrual with age and that additional loss of the downstream apoptotic effectors Bax and Bak did not exacerbate Treg accumulation. Further, our results demonstrate that a subpopulation of Treg expands with age and is characterized by lower expression of CD25 (IL-2Rα) and Bim. Mechanistically, we found that IL-2 levels decline with age and likely explain the emergence of CD25loBimlo Treg because Treg in IL-2−/− mice are almost entirely comprised of CD25loBimlo cells, and IL-2 neutralization increases CD25loBimlo Treg in both young and middle-aged mice. Interestingly, the Treg population in aged mice had increased expression of CD122 (IL-2/IL-15Rβ) and neutralization or genetic loss of IL-15 led to less Treg accrual with age. Further, the decreased Treg accrual in middle-aged IL-15−/− mice was restored by the additional loss of Bim (IL-15−/−Bim−/−). Together, our data show that aging favors the accrual of CD25lo Treg whose homeostasis is supported by IL-15 as IL-2 levels become limiting. These data have implications for manipulating Treg to improve immune responses in the elderly.
Aging is associated with declining immune function, which contributes to increased infectious diseases, increased cancer, and decreased vaccine efficacy in the elderly. The aging immune system is characterized by a progressive deterioration in the adaptive immune system, particularly affecting T cells (Linton and Dorshkind, 2004; Maue et al., 2009). Factors contributing to T cell dysfunction with age include: (i) thymic involution and decreased naïve T cell production (Sempowski et al., 2002; Hale et al., 2006); (ii) impaired TCR signaling and immune synapse formation (Miller et al., 1997; Miller, 2000; Tamir et al., 2000; Garcia and Miller, 2002); (iii) impaired T cell proliferation (Murasko et al., 1987; Haynes et al., 2005); (iv) CD8+ T cell clonal expansion driven by chronic infections (Khan et al., 2002; Ouyang et al., 2003; Clambey et al., 2005). While many of these effects are T cell intrinsic, recent work has found that regulatory T cell (Treg) frequencies increase dramatically with age in both mice and humans and may contribute substantially to impaired T cell responses in aged hosts (Valmori et al., 2005; Nishioka et al., 2006; Sharma et al., 2006; Lages et al., 2008; Agius et al., 2009).
Regulatory T cells, a specialized subset of CD4+FoxP3+ T cells, are known to control the intensity of immune responses through modulating the activation and function of both effector T cells and antigen-presenting cells (Miyara and Sakaguchi, 2007; Onishi et al., 2008). The functionality of aged Treg have not been well studied, however we have shown that aged FoxP3+ Treg have an equal or increased in vitro suppressive capacity compared to young Treg (Lages et al., 2008). In vivo, depletion of CD25+ Treg allowed for a more robust CD4+ T cell response against Leishmania major in aged mice, suggesting increased Treg in the aged can dampen effector T cell activation (Lages et al., 2008). Additionally, Treg accrual with age has been shown to inhibit anti-tumor responses (Sharma et al., 2006). Thus, aged Treg appear to be functional in vivo and Treg accrual may contribute significantly to immunosenescence in aging.
Many studies have looked at the factors involved in Treg homeostasis in young mice, particularly the γc cytokines IL-2, IL-7, and IL-15. The receptor for IL-2 is comprised of CD25 (IL-2Rα), CD122 (IL-2/15Rβ), and CD132 (IL-2/15/7Rγ). IL-2 shares the IL-2/15Rβ receptor with IL-15, and the γc receptor (CD132) with both IL-15 and IL-7. IL-2 is the dominant cytokine required for Treg survival and homeostasis, as the loss of IL-15 or IL-7 signaling does not substantially affect the frequency of CD4+ cells that are Treg when IL-2 is present (Burchill et al., 2007; Bayer et al., 2008; Vang et al., 2008). However, CD132 and to a lesser extent CD122-deficient mice have a more profound loss of Treg compared to IL-2 or CD25 deficient mice, suggesting that IL-15 and/or other γc cytokines also contribute to Treg homeostasis (Fontenot et al., 2005a). All of these studies examining the requirements for cytokine signaling in Treg development and survival have been done in young mice, and the role for the γc cytokines in aged Treg homeostasis is unclear.
Our previous study showed that Bim plays a major role in Treg homeostasis and that Bim levels decline significantly in aged Treg (Chougnet et al., 2011). Further germline deletion of Bim led to significantly faster accrual of Treg (Chougnet et al., 2011). Here, we found that Treg-specific loss of Bim was sufficient to drive Treg accrual and that Bim was the dominant pro-apoptotic molecule driving Treg accrual. Further, decreased Bim levels in aged Treg is reflected by decreased Bim mRNA and increased Bim turnover. Additionally, declining IL-2 levels with age resulted in reduced levels of CD25 and increased levels of CD122 which foster Treg dependence upon IL-15, which, in turn, functions to restrain the remaining Bim in aged Treg.
C57BL/6 mice were purchased from either Taconic Farms (Germantown, NY, USA) or the National Institutes of Aging colony located at Charles River Laboratories (Wilmington, MA, USA). B6.129P2-Il2tm1Hor/J (IL-2−/−) mice and their C57BL/6 controls were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). B6.SJL-Ptprca Pepcb/BoyJ mice on the C57BL/6 background were purchased from The Jackson Laboratory and aged in house. Bim−/− mice have been backcrossed to C57BL/6 mice for at least 20 generations. Bimf/f mice were generated at the Walter and Eliza Hall Institute as part of a collaborative effort with Dr. P. Bouillet. Briefly, a targeting vector was created by flanking coding exons 2, 3, and 4 of Bim with loxP sites. The vector was electroporated into C57BL/6 embryonic stem (ES) cells and homologously recombined ES cells were selected with hygromycin. The hygromycin cassette was removed by crossing the Bimf/f mice with B6.Cg-Tg(ACTFLPe)9205Dym/J (Jackson Labs) to generate a Bim floxed allele that could be crossed to tissue – specific cre transgenic mice to achieve tissue-specific deletion of Bim. Offspring from this cross were screened for removal of the Hygromycin cassette and maintenance of the conditional Bim allele. Mice were then bred to Cre-expressing mice and offspring screened for lack of the ACTFLPe allele. Lck-Cre Baxf/fBak−/− mice were a gift from Dr. S. Korsmeyer and were previously described (Takeuchi et al., 2005). IL-15-deficient mice on the C57BL/6 background were purchased from Taconic Farms, mated with the Bim−/− mice to generate IL-15−/− Bim−/− mice, and aged in house. FoxP3-IRES-DTR-GFP knock-in C57BL/6 mice (Kim et al., 2007) and FoxP3-Cre mice (Rubtsov et al., 2008) were a gift from Dr. A. Rudensky. FoxP3-IRES-DTR-GFP mice were aged in house. FoxP3-Cre mice were mated with Bimf/f mice and aged in house. Mice were housed under specific pathogen-free conditions. All animal protocols were reviewed and approved by our Institutional Animal Care and Use Committee.
Spleens, lymph nodes (inguinal, axillary, and brachial), and thymi were harvested and crushed through 100 μm filters (BD Falcon) to generate single-cell suspensions. About 1 × 106 cells were surface stained with Abs against CD4 (BD Biosciences, San Diego, CA, USA), CD25 (eBioscience, San Diego, CA, USA), CD44 (eBioscience), CD122 (Biolegend, San Diego, CA, USA), CD45.2 (eBioscience) and intracellularly for FoxP3 (eBioscience), Bim (Cell Signaling Technology, Beverly, MA, USA), and Ki67 (eBioscience). All intracellular stains were performed using the eBioscience FoxP3 staining protocol. For detection of Bim, secondary anti-rabbit IgG Abs were used (Jackson ImmunoResearch Laboratories, West Grove, PA, USA; Invitrogen, Carlsbad, CA, USA; or Cell Signaling Technology). Data were acquired on an LSRII flow cytometer (BD Biosciences), analyzed using FACSDiva software (BD Biosciences), and histogram overlays were prepared using FlowJo software (Tree Star).
Spleen cells from young (3–4 months) and old (19–23 months) FoxP3-IRES-DTR-GFP mice were enriched for CD4+ cells using the negative selection MACS CD4+ T cell Isolation Kit II (Miltenyi Biotec, Auburn, CA, USA) and stained with CD4 antibody. CD4+FoxP3GFP+ cells were then sorted by FACSAria (BD Biosciences), and>85% purity was obtained. About 5 × 105 cells were injected i.v. into young (2 months) or aged (15 months) C57BL/6 congenic CD45.1 recipient mice. The recipient mice were sacrificed either 1.5 or 10 days post-transfer, spleens were harvested, and single-cell suspensions were stained for CD4, CD45.2, CD25, and intracellularly for FoxP3. Cells were fixed with 4% methanol-free formaldehyde (Polysciences, Warrington, PA, USA), instead of the eBioscience FoxP3 fix-perm buffer, to better retain GFP within the cells.
IL-2 in vivo cytokine capture assay (IVCCA) was performed as described (Finkelman and Morris, 1999; Finkelman et al., 2003). Briefly, young (2–5 months), middle-aged (11–12 months), and old (>15 months) C57BL/6 mice were injected i.v. with 10 μg of biotinylated anti-IL-2 capture antibody (JES6-5H4-BD Biosciences), or with PBS as a control, and mice were bled 24 h later and serum was collected. Ninety-six-well Costar plates were coated overnight with anti-IL-2 JES61A12 (eBioscience) and then a luminescent ELISA was performed. For the cytokine:anti-cytokine mAb standard, 100 ng of recombinant mouse IL-2 (R&D) was incubated with 10 μg of the anti-IL-2 capture antibody (JES6-5H4) for 10 min, and 100 ng/ml aliquots were stored in −80° C freezer. The IL-2 concentration obtained from our PBS control was subtracted from our young and old samples to remove the ELISA background.
Anti-IL-2 Ab (clones S4B6 and Jes61A12) and rat IgG2A isotype control (2A3) were purchased from BioXcell (West Lebanon, NH, USA). Anti-IL-15 (M96) was a kind gift from Amgen (Seattle, WA, USA). For IL-2 neutralization: 3- and 12-month-old mice were injected i.p. with 170 μg of S4B6 and Jes61A12, or with isotype control (2A3), on days 0, 1, 2, 4, 6, and sacrificed on day 7 and spleens were harvested. For IL-15 neutralization: 3- and 12-month-old mice were injected i.p. with 25 μg of M96 on days 0, 2, 4, 6, and sacrificed on day 7. IL-15 neutralization was confirmed by assessing natural killer cells, which showed >60% deletion in the spleen (data not shown).
Splenocytes from either young (3 months) or old (22 months) C57BL/6 mice were cultured with or without cycloheximide (20 μM; Sigma-Aldrich, St. Louis, MO, USA) for 8 h at 37° C. Cells were stained for CD4, FoxP3, and Bim and analyzed by flow cytometry. Percent decrease in Bim expression was determined by comparing Bim expression between cells cultured with our without cycloheximide.
Spleen cells from young (3 months) and old (22 months) FoxP3-IRES-DTR-GFP mice were enriched for CD4+ cells using the negative selection MACS CD4+ T cell Isolation Kit II (Miltenyi Biotec) and stained with CD4 antibody. CD4+FoxP3GFP+ and CD4+FoxP3GFP− cells were then sorted by FACSAria (BD Biosciences) and >90% purity was obtained for both populations. RNA was isolated from cells using the RNeasy Mini Kit (Qiagen) and converted into cDNA using Superscript II Reverse Transcriptase (Invitrogen, CA, USA). The primers used for Bim were: 5′-ACAAACCCCAAGTCCTCCTT-3′ and 5′-GTTTCGTTGAACTCGTCTCC-3′; and for the internal control L19: 5′-CCTGAAGGTCAAAGGGAATGTG-3′ and 5′-GCTTTCGTGCTTCCTTGGTCT-3′. Quantitative real-time PCR was performed with Roche LightCycler 480 SYBRGreen 1 Master Mix using the Roche LightCycler 480 II instrument (Roche Diagnostics).
We have previously shown that Treg in aged mice have decreased expression of Bim, and Bim−/− mice accrued Treg more rapidly, together suggesting loss of Bim expression drives Treg accrual with age (Chougnet et al., 2011). Here we investigated this in more detail and found that the decrease in Bim expression with age is a progressive process (Figure 1A). The major loss in Bim expression is observed by 12 months (threefold; fourfold by 21 months), and most significantly within the CD4+FoxP3+ population, not in the CD4+FoxP3− population (Figure 1A) (Chougnet et al., 2011).
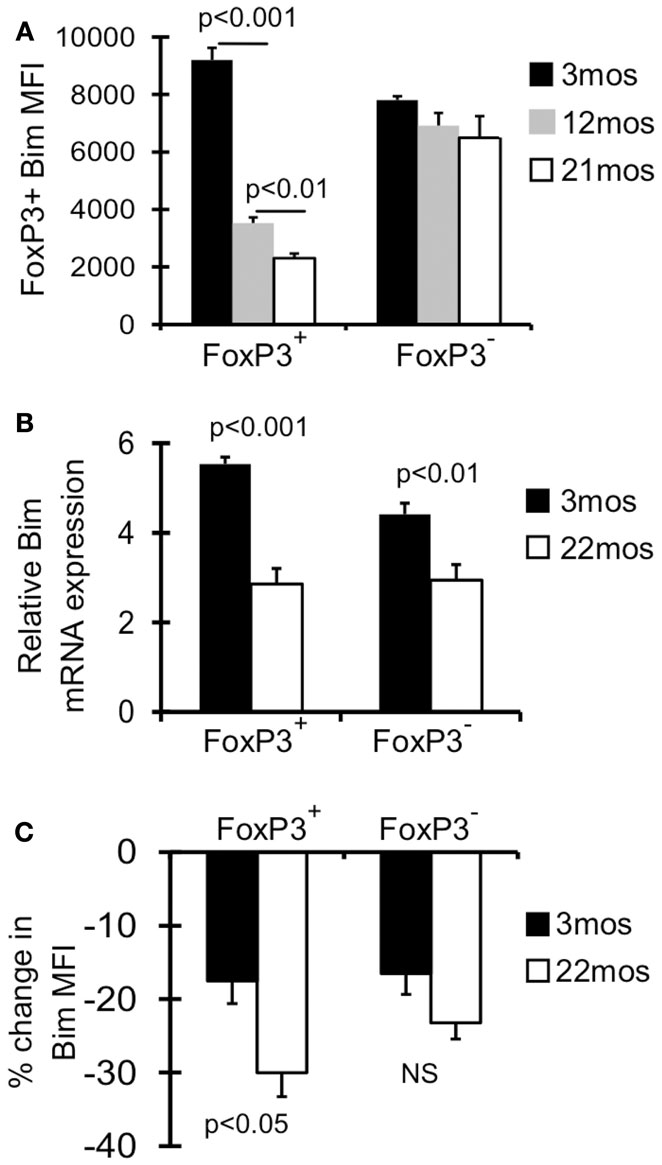
Figure 1. Bim levels in Treg are progressively decreased with age. (A) Splenocytes from 3 month (n = 4), 12 month (n = 4), and 21-month-old (n = 5) mice were stained with Abs against CD4, FoxP3, and Bim and analyzed by flow cytometry. Results show the average mean fluorescent intensity (MFI) of Bim in CD4+FoxP3+ T cells (±SE). The p values represent the difference between 3 and 12 months; 12 and 21 months (Student’s t test). (B) FoxP3GFP reporter mice were used to sort CD4+FoxP3GFP+ Treg, CD4+FoxP3GFP− non-Treg from young (3 months; n = 4) and old (22 months; n = 4) mice. Results show the relative expression of Bim mRNA in 3 and 22 months mice (±SE). The p values represent the difference between 3 and 22 months (Student’s t test). (C) Splenocytes from 3 months (n = 4) and 22 months (n = 3) mice were cultured with or without cycloheximide for 8 h at 37° C, and then stained for CD4, FoxP3, and Bim and analyzed by flow cytometry. Results show the percent decrease in Bim expression within CD4+FoxP3+ and CD4+FoxP3− cells cultured with cycloheximide (±SE). Results are representative of at least two independent experiments.
As Bim is subject to transcriptional, translational, and post-translational control, we next determined the relative contribution of these mechanisms. First, CD4+FoxP3GFP+ Treg and CD4+FoxP3GFP− non-Treg were sorted from young (3 months) and old (22 months) FoxP3GFP reporter mice (Kim et al., 2007). Treg cells from old mice showed significantly decreased Bim mRNA (∼2-fold), which was observed to a lesser extent in non-Treg (Figure 1B). Next, to examine Bim protein turnover, splenocytes from young and old mice were cultured with cycloheximide, an antibiotic that inhibits translation. Interestingly, old FoxP3+ Treg cells had significantly greater Bim turnover compared to young Treg (30 vs. 18%, respectively; Figure 1C). These data indicate that both decreased Bim transcription and increased Bim turnover likely contribute to the decreased Bim protein levels observed with age.
It remains unclear whether the effects of Bim on Treg accrual are Treg intrinsic. To address this question, we crossed FoxP3-Cre mice (Rubtsov et al., 2008) with mice that have loxP flanked Bim on both alleles (Bimf/f) (Figure 2A), and aged these FoxP3-Cre Bimf/f mice to 6 months. Bim staining in FoxP3-Cre+Bimf/f mice was comparable to Bim−/− mice, confirming efficient deletion of Bim (Figure 2B). There was a small fraction of CD4+FoxP3– cells with somewhat reduced expression of Bim, suggesting that some cells may have deleted one or both alleles of Bim (Figure 2B). However, we did not observe loss of Bim in non-T cell populations (i.e., CD11c+ or B220+ cells, data not shown). The frequency of Treg in FoxP3-Cre+Bimf/f mice was increased at 2 months of age, compared to either FoxP3-Cre– Bimf/f or Bim−/− mice (Figure 2C). This increase in Treg at 2 months of age may be due to increased thymic output in FoxP3-Cre+Bimf/f mice, as we see increased frequencies and numbers of FoxP3+ cells in the thymi of FoxP3-Cre+Bimf/f mice compared to FoxP3-Cre– Bimf/f (data not shown). Importantly, by 6 months of age the frequency of Treg in FoxP3-Cre+Bimf/f mice increased even further, reaching levels observed in Bim−/− mice (Figure 2C). These data show that Treg intrinsic loss of Bim is sufficient to promote Treg accumulation with age.
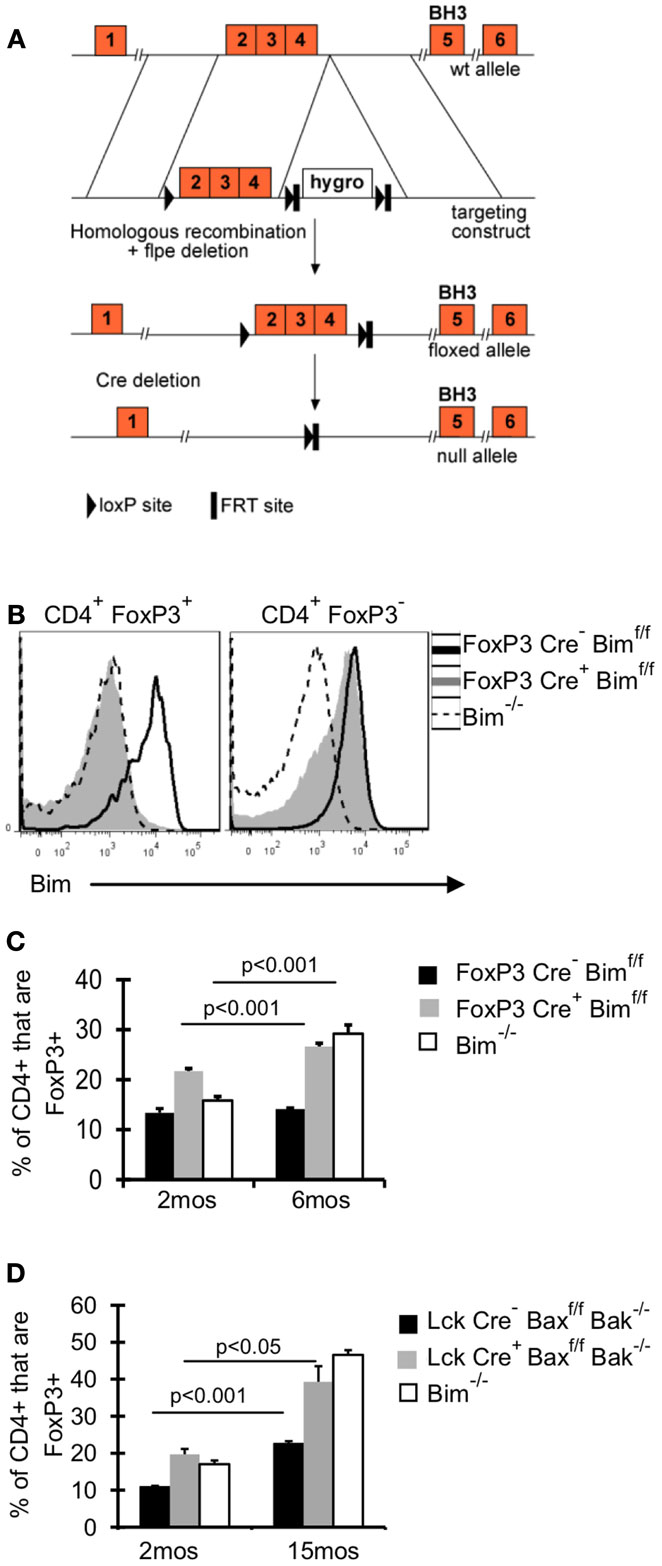
Figure 2. Bim is the dominant BH3-only molecule in Treg and its effects on Treg homeostasis are Treg intrinsic. (A) A conditional Bim allele was generated by targeting loxP sites upstream of exon 2 and downstream of exon 4. (B,C) Cells from the lymph nodes of 2- and 6-month-old FoxP3-Cre− Bimf/f, FoxP3-Cre+Bimf/f, and Bim−/− mice were stained for CD4, FoxP3, and Bim and analyzed by flow cytometry. (B) Histograms show the expression of Bim in CD4+FoxP3+ and CD4+FoxP3− T cells from 2-month-old mice. (C) Results show the frequency of CD4+ cells that are FoxP3+ in 2 month (n = 5/group) and 6-month-old mice (n = 3–4/group; ±SE). The p values represent the difference between 2 and 6 months (Student’s t test). Results are representative of two independent experiments. (D) Splenocytes from Lck-Cre−Baxf/fBak−/−(n = 3–5/group), Lck-Cre+Baxf/fBak−/−(n = 3–5/group), and Bim−/− (n = 3–4/group), mice were stained for CD4, FoxP3, and Bim and analyzed by flow. The Bim−/− mice were analyzed in an independent experiment. Results show the frequency of CD4+ cells that are FoxP3+ in 2- and 15-month-old mice (±SE). The p values represent the difference between 2 and 15 months (Student’s t test).
Although our data incriminate Bim as the major regulator of Treg accumulation with age, we addressed the contribution of other pro-apoptotic proteins in Treg survival, by using mice with T cell specific deletion of Bax and Bak, Lck-Cre+Baxf/fBak−/−mice. Bax and Bak are the downstream mediators of pro-apoptotic proteins, and the deletion of both genes eliminates apoptosis through the intrinsic pathway (Youle and Strasser, 2008). If other pro-apoptotic proteins were critical in limiting Treg survival, then Treg accrual in Lck-Cre+ mice would be significantly greater than that observed in Bim−/− mice. One caveat of comparing Treg accrual between Lck-Cre+Baxf/fBak−/−mice and Bim−/−mice is that non-Treg loss of Bim could affect Treg homeostasis; however, our data in FoxP3-Cre+Bimf/f show that Treg-specific loss of Bim is sufficient to drive accrual to levels similar to those observed in Bim−/− mice. T cell loss of Bax and Bak resulted in increased Treg accumulation at 15 months of age compared to Lck-Cre− mice (20% increase compared to 10% increase, respectively; Figure 2D). Importantly, the frequency of Treg was similar in 15-month-old Lck-Cre+Baxf/fBak−/−mice and age-matched Bim−/−mice, demonstrating that Bim is the major pro-apoptotic protein controlling Treg survival.
Old Bimlo Treg have increased survival ex vivo (Chougnet et al., 2011); however, it remains unclear if Treg with lower Bim expression emerge in vivo because they are afforded a selective survival advantage. To test this, we examined Bim levels in Lck-Cre+Baxf/fBak−/−Treg as their levels of Bim will be largely irrelevant to their survival. Notably, the levels of Bim were significantly increased in Lck-Cre+ mice compared to Lck-Cre− mice, indicating Treg are able to tolerate higher levels of Bim in the absence of Bax and Bak (Figure 3A). As expected, Bim levels in Treg from 15-month-old Lck-Cre− mice was significantly decreased compared to their 2-month-old counterparts (Figures 3A,B). Bim was also decreased in 15-month-old Lck-Cre+ mice (Figures 3A,B), but the fold-decrease in Bim expression in Lck-Cre− mice was greater than in Lck-Cre+ mice (threefold vs. twofold, respectively; Figure 3B). Together, these data suggest that Bim levels are controlled in Treg by both survival-dependent and independent mechanisms.
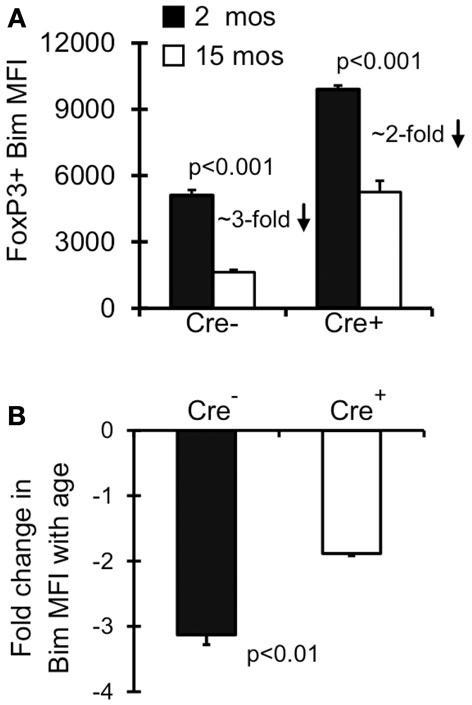
Figure 3. Both environment and survival contribute to the emergence of Bimlo Treg. Splenocytes from Lck-Cre−Baxf/fBak−/−(n = 3–5/group) and Lck-Cre+Baxf/fBak−/−(n = 3–5/group) mice were stained for CD4, FoxP3, and Bim and analyzed by flow. (A) Results show the Bim MFI in CD4+FoxP3+ cells (±SE). The p values represent the difference between 2 month-old and 15 month-old mice (Student’s t test). (B) Results show the fold change in CD4+FoxP3+ Bim expression in 15 month Lck-Cre−Baxf/fBak−/−and Lck-Cre+Baxf/fBak−/− mice compared to their 2 month counterparts. The p values represent the difference between Lck-Cre− and Lck-Cre+ (Student’s t test).
Treg are described in the literature as CD4+FoxP3+ CD25hi, however there is a substantial population of CD4+FoxP3+ CD25lo cells in young mice ( ∼10–20% of FoxP3+ cells; Figures 4A,B). Interestingly, this population of CD25lo Treg expands with age, comprising 50% of the Treg population by middle-aged (Figures 4A,B) (Nishioka et al., 2006; Lages et al., 2008). Importantly, while both CD25lo and CD25hi Treg expand with age, the relative increase in CD25lo Treg is much greater (Figure 4C). To assess whether the aged environment drives CD25lo Treg accumulation, we adoptively transferred young (3–4 months) and old (19–23 months) Treg into young (2 months) and aged (15 months) recipient mice (Figure 4D). At 1.5 and 10 days later, recipient mice were sacrificed and the frequency of CD25lo Treg from donors was assessed (Figure 4E). Interestingly, when young cells were placed into aged recipients, we observed an emergence of CD25lo Treg. Similarly, CD25lo Treg were enriched slightly if old Treg were transferred into aged mice. Increases in CD25lo Treg are likely not due to increased proliferation as there was an equivalent percentage of CD25lo and CD25hi Treg that were Ki67+ after transfer into aged mice (Figure A1A in Appendix). Further, the total numbers of CD25lo Treg were affected as the ratio of CD25hi to CD25lo Treg decreased by day 10 post-transfer (Figure A1B in Appendix), suggesting that CD25hi Treg may lose expression of CD25. On the other hand, when young or old Treg were placed into young donors, we did not see an enrichment of CD25lo Treg. Together, these data strongly suggest that environmental changes with age support the emergence of CD25lo Treg.
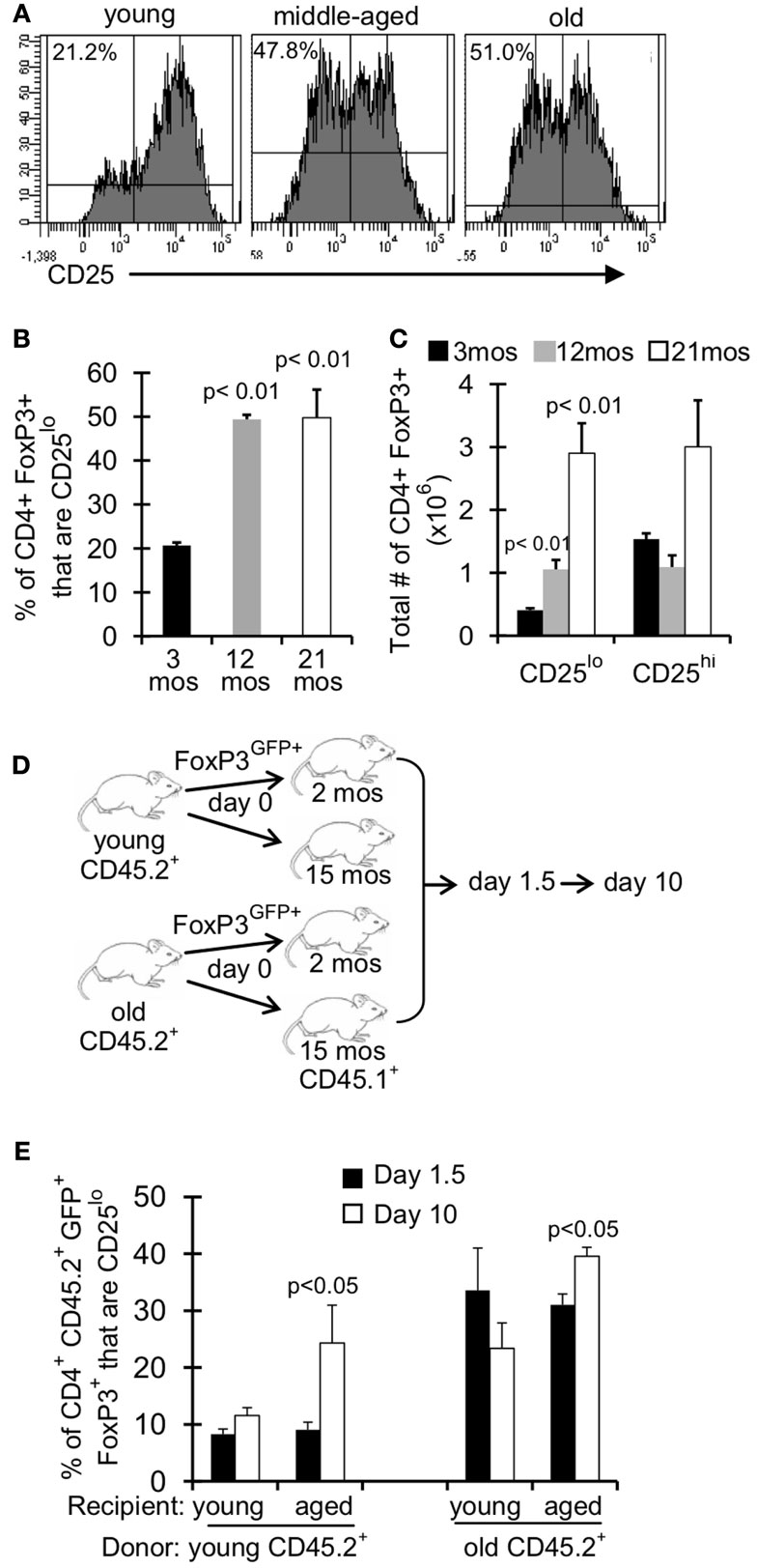
Figure 4. An aged environment promotes emergence of CD25lo Treg. (A–C) Splenocytes from young (3 months; n = 4), middle-aged (12 months; n = 4), and old (21 months; n = 5) mice were stained for CD4, FoxP3, and CD25 and analyzed by flow. (A) Histograms show the expression of CD25 on CD4+FoxP3+ cells. Percentages are the frequencies of CD4+FoxP3+ that are CD25lo. (B) Results show the frequency of CD4+FoxP3+ cells that are CD25lo (±SE). The p values represent the difference compared to 3-month-old mice (Student’s t test). (C) Results show the total number of CD4+FoxP3+ cells that are CD25lo or CD25hi. The p values represent the difference compared to 3-month-old mice (Student’s t test). Results are representative of at least three independent experiments. (D,E) CD4+FoxP3GFP+ T cells were sorted from splenocytes of young (3–4 months) and old (>18 months) CD45.2 FoxP3GFP reporter mice. About 5 × 105 CD4+FoxP3GFP+ cells were injected i.v. into young (2 months) and aged (15 months) congenic CD45.1 mice. Mice were sacrificed at day 1.5 (n = 3–5/group) and day 10 (n = 3–5/group) post-injection. Cells from the spleen were stained for CD4, FoxP3, CD25, and CD45.2 and analyzed by flow. (E) Results show the frequency of CD4+CD45.2+FoxP3+GFP+ cells that are CD25lo (±SE). The p values represent the difference between day 1.5 and 10 (Student’s t test).
IL-2 signaling is known to promote expression of its own receptor CD25 (IL-2Rα) (Liao et al., 2013), and could be critical for maintaining CD25hi Treg. As assessed in the serum, levels of IL-2 were significantly decreased in middle-aged and old mice compared to young mice (Figure 5A). Furthermore, Treg in IL-2−/− mice were comprised almost entirely of CD25lo cells (Figure 5B). Interestingly, CD25lo Treg were Bimlo at any age (Figure 5C), and this lower expression of Bim may promote their increased accrual with age. To assess whether IL-2 also controls Bim expression we measured Bim levels in Treg from IL-2−/− mice. Strikingly, Bim levels were significantly decreased in Treg from IL-2−/− mice (Figure 5D). This loss of Bim expression occurred in the periphery, as Bim levels in thymic Treg were similar between IL-2−/− and wild-type mice (data not shown). To independently test the role of IL-2 in maintaining CD25hiBimhi Treg, we neutralized IL-2 in young mice (3 months), which resulted in decreased expression of Bim and an enrichment of CD25lo Treg (Figures 5E,F). Also, the total number of Treg were not decreased after IL-2 neutralization, suggesting that CD25hi Treg lose expression of CD25 (data not shown). Taken together, these data show that IL-2 is critical for maintaining Treg with a CD25hiBimhi phenotype, both in young and aged mice. These data also suggest that reduced IL-2 with age promotes the emergence of CD25loBimlo cells.
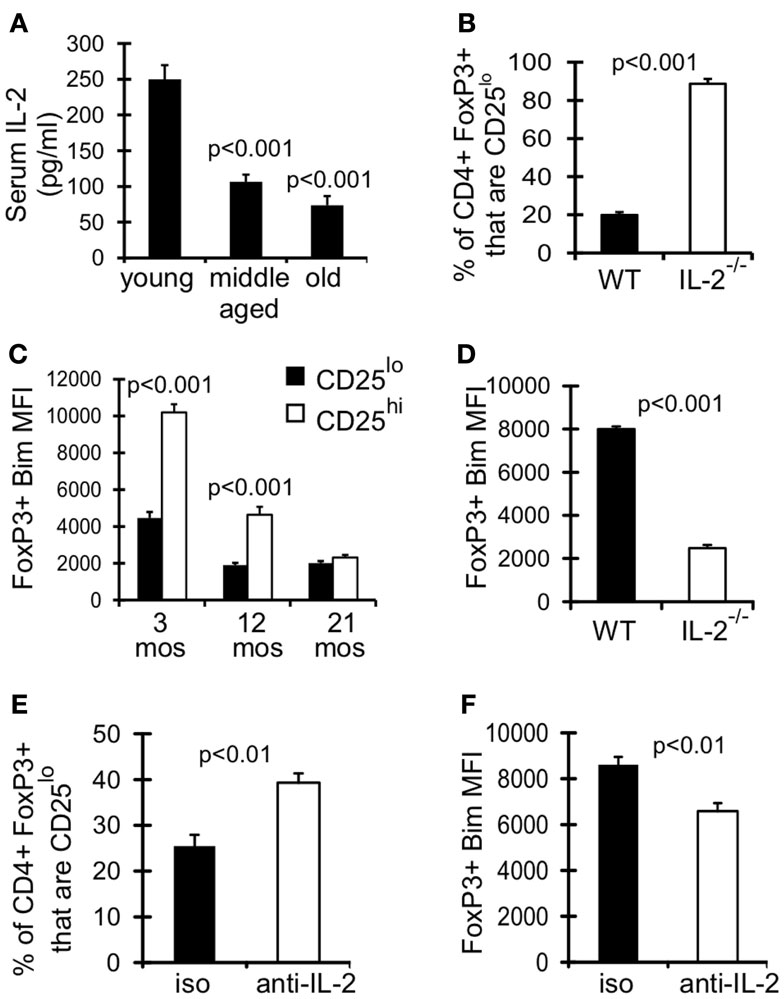
Figure 5. Declining IL-2 levels drives emergence of CD25loBimlo Treg. (A) Serum IL-2 was determined by IVCCA. Results show the average serum IL-2 levels from young (3–4 months; n = 10), middle-aged (11–12 months; n = 11), and old (>15 months; n = 9) mice (±SE). Results are representative of 3 independent experiments. The p values represent the difference compared to young (Student’s t test). (B,D) Splenocytes from 5-week-old C57BL/6 wild-type (WT; n = 4) and IL-2−/− (n = 3) mice were stained for CD4, FoxP3, CD25, and Bim and analyzed by flow. (B) Results show the frequency of CD4+FoxP3+ cells that are CD25lo (±SE) and are representative of two independent experiments. (C) Splenocytes from 3 month (n = 4), 12 month (n = 4), and 21-month-old (n = 5) mice were stained for CD4, FoxP3, CD25, and Bim and analyzed by flow. Results show the Bim MFI of CD4+FoxP3+CD25lo cells and CD4+FoxP3+CD25hi cells (±SE). The p values represent the difference between CD25lo and CD25hi Treg (Student’s t test). (D) Results show the Bim MFI of CD4+FoxP3+ Treg from WT and IL-2−/− mice (±SE) and are representative of two independent experiments. (E,F) Three-month-old mice were injected with IL-2 neutralizing antibodies or IgG isotype control (n = 4/group). Spleens were harvested at day 7 and stained for CD4, FoxP3, CD25, and Bim. (E) Results show the frequency of CD4+FoxP3+ cells that are CD25lo (±SE). (F) Results show the Bim MFI of CD4+FoxP3+ Treg (±SE).
We next examined the expression of receptors for other common γ cytokines which can function redundantly with IL-2 to support Treg homeostasis and survival in young mice (Fontenot et al., 2005a; Burchill et al., 2007; Bayer et al., 2008; Vang et al., 2008). Although the expression of IL-7Rα (CD127) increases with age, we previously showed that blocking CD127 signaling alone did not affect Treg survival in 12-month-old mice (Chougnet et al., 2011). IL-2Rβ (CD122) is a part of both the IL-2 and IL-15 receptor, and its expression was significantly upregulated on Treg by 12 months of age (Figure 6A). Additionally, CD122 expression was increased twofold on Treg in IL-2−/− mice (AVG MFI: WT = 1265; IL-2−/− = 2378; p < 0.001) (Figure 6B). We therefore assessed the role of IL-15 in Treg accumulation in middle-aged mice, as increased Treg CD122 expression and increased CD25lo Treg by 12 months of age may reflect a change in Treg cytokine dependency. While there was no difference in Treg frequency in young IL-15−/− and wild-type mice, the frequency of Treg was significantly reduced in IL-15−/− mice by 12 months of age (Figure 6C). Importantly, the additional loss of Bim (IL-15−/−Bim−/−), rescued the loss of Treg in middle-aged IL-15−/− mice (Figure 6C), suggesting that IL-15 supports Treg homeostasis in aged mice by combating Bim-mediated cell death.
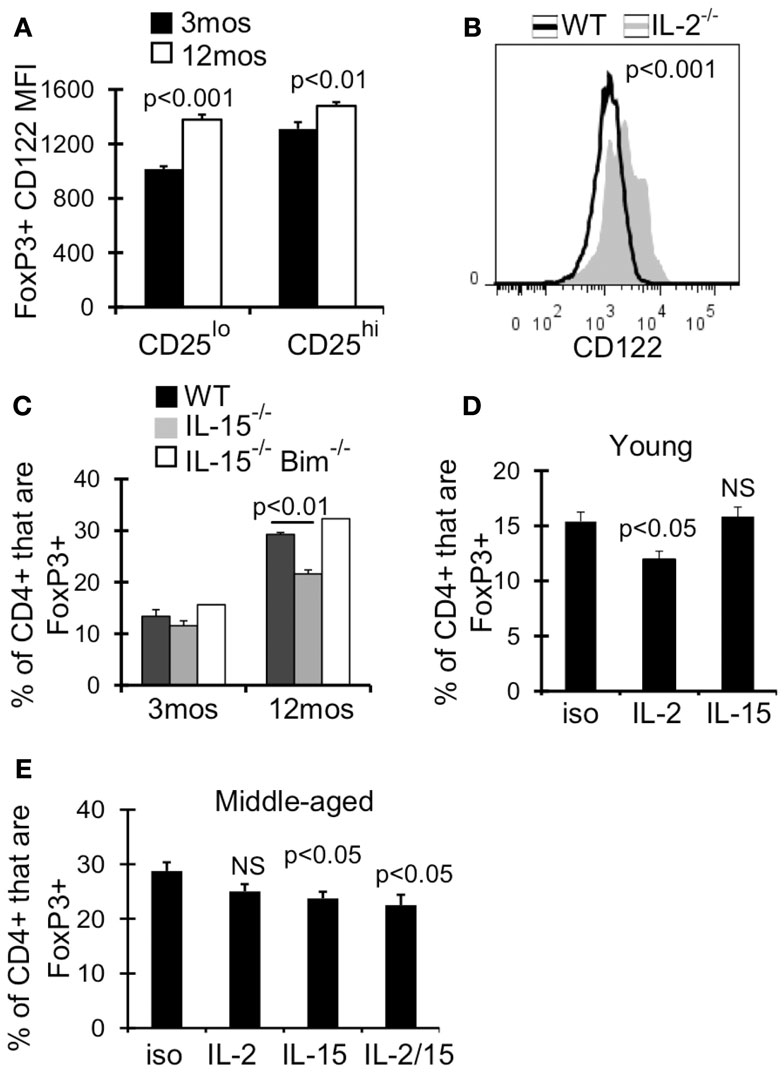
Figure 6. IL-15 antagonizes Bim to promote Treg homeostasis in middle-aged mice. Splenocytes from 3 month (n = 4) and 12-month-old (n = 4) mice (A) or 5-week-old WT (n = 4) and IL-2−/− (n = 3) mice (B) were stained for CD4, FoxP3, and CD122, and analyzed by flow. (A) Results show the MFI of CD122 on CD4+FoxP3+ cells (±SE). (B) Histograms show CD122 expression on CD4+FoxP3+ cells. The p value represents the difference between the average CD122 MFI from WT and IL-2−/− mice (Student’s t test). (C) Splenocytes from WT (n = 3–4/group), IL-15−/− (n = 3/group), and IL-15−/− Bim−/− (n = 2/group) were stained for CD4 and FoxP3 and analyzed by flow. Results show the frequency of CD4+ cells that are FoxP3+ (±SE) and are representative of two independent experiments. Young (3 months; n = 4/group) (D) and middle-aged (12 months; n = 5/group) (E) mice were injected with IL-2 and/or IL-15 neutralizing antibody or with IgG isotype control. Spleens were harvested at day 7 and stained for CD4, FoxP3, CD25, and Bim. (D,E) Results show the frequency of CD4+ cells that are FoxP3+ (±SE). The p values represent the difference between antibody treated and isotype control (Student’s t test).
To test whether IL-15 promotes Treg survival when IL-2 levels decline, we neutralized IL-2 and/or IL-15 in young and middle-aged mice. IL-2 neutralization in young mice (3 months) but not middle-aged mice (12 months) resulted in a significant loss of Treg (Figures 6D,E), supporting that IL-2 is less critical for Treg homeostasis in an aged environment. Conversely, IL-15 neutralization in middle-aged mice resulted in a significant loss of Treg, which was not observed in young mice (Figures 6D,E). Importantly, neutralization of IL-2 and IL-15 did not result in further loss of Treg compared to IL-15 neutralization alone (Figure 6E). Together, our data suggests that in an aging environment, IL-15 supports Treg survival in the face of declining IL-2.
Age-related immunosuppression is multi-factorial, encompassing cell-intrinsic defects within T and B cells, as well as population based defects such as a loss of naïve T cells and increased levels of regulatory T cells (Valmori et al., 2005; Nishioka et al., 2006; Sharma et al., 2006; Lages et al., 2008; Agius et al., 2009). Increased levels of Treg are observed in both mice and humans and functionally suppress T cell responses to both parasites (L. major) and tumors (Sharma et al., 2006; Lages et al., 2008). We also recently showed that Bim is a key controller of Treg homeostasis as Treg accrual is accelerated in Bim-deficient (Bim−/−) mice (Chougnet et al., 2011); although it was unclear if this was due to T cell specific effects of Bim in peripheral Treg. Here, we focused on further understanding the mechanisms controlling Treg accrual in aging.
First, the germline deletion of Bim results in loss of Bim from all tissues and it was possible that non-T cell expression of Bim controls Treg homeostasis. In particular, the increased lifespan of Bim-deficient dendritic cells (DC) (Chen et al., 2007) could contribute to Treg accrual as DC can promote Treg homeostasis and inducible Treg development (Yamazaki et al., 2003; Belkaid and Oldenhove, 2008). Second, Bim−/− thymocytes are resistant to negative selection (Bouillet et al., 2002) and such altered thymic development in Bim−/− mice could skew the levels of Treg in Bim−/− mice. By using FoxP3-Cre, which would likely not turn on until after cells have committed to the Treg lineage and largely at a point beyond negative selection (Fontenot et al., 2005b), we avoided the effects of Bim in other tissues and the effects of Bim on thymic development. Thus, our data clearly show that Treg-specific loss of Bim is sufficient to drive Treg accrual with age and strongly suggest that the normal decline of Bim expression seen in wild-type mice promotes Treg accrual in aging mice.
While our data support Bim as a major controller of Treg homeostasis, we could not exclude potentially redundant roles for other pro-apoptotic Bcl-2 family members. For instance, the additional loss of Puma in Bim-deficient mice enhances T cell survival, suggesting a redundant role for Puma with Bim in certain T cell contexts (Erlacher et al., 2006; Gray et al., 2012). Moreover, we have found that, like Bim, Puma levels are normally decreased as mice age (data not shown). However, mice singly deficient in either Bmf, Puma, Noxa, or Bad have no difference in Treg frequencies in young mice (Tischner et al., 2012), while young Bim-deficient mice have subtle, but significant increases in Treg (Zhan et al., 2011; Tischner et al., 2012). Further, our finding that Treg accrual was similar in aged Bim−/− mice and Lck-Cre+Baxf/fBak−/−mice strongly suggest that Bim is the main negative regulator of Treg homeostasis with age.
IL-2, on the other hand, is a major positive regulator of Treg homeostasis as IL-2-deficient mice have significantly reduced Treg (Fontenot et al., 2005a; Burchill et al., 2007; Barron et al., 2010). Although it has been accepted for many years that IL-2 levels decrease with age, this assumption was largely based upon a reduced production of IL-2 after in vitro stimulation of aged T cells (Thoman and Weigle, 1981). To the best of our knowledge, our study is the first demonstration that in vivo IL-2 levels are decreased in aged mice. We acknowledge that serum IL-2 levels may not fully reflect the in vivo bioavailability of IL-2, as IL-2 can bind to heparin sulfate on extracellular matrices (Wrenshall and Platt, 1999), but we did not observe substantially increased staining for IL-2 on spleen tissue from old mice (data not shown). We previously proposed that continued IL-2 signaling drives the expansion of Bimlo Treg, as exogenous IL-2 preferentially induced proliferation and expansion of Bimlo Treg (Chougnet et al., 2011). We note that these experiments were performed in young mice that were given supraphysiologic levels of IL-2. Given that we now know that IL-2 levels decline by middle-age, this model seems less likely. Further, our new data clearly show that neutralization of IL-2 and/or loss of IL-2 both lead to the accumulation of CD25lo Treg and decreased the levels of Bim within these cells. It does seem paradoxical that reduced levels of IL-2 in aged mice would be associated with Treg accrual. However, our and others data suggest that IL-2 and Bim antagonize one another to regulate Treg homeostasis. For example, the absence of Bim can restore Treg in IL-2-deficient mice (Barron et al., 2010) and we found that limiting or restricting IL-2 results in the accrual of Bimlo Treg. Thus, while it has been shown in cell lines that acute withdrawal of IL-2 leads to increased Bim expression (Stahl et al., 2002), chronic limiting IL-2 may select for Bimlo Treg. Further, we show that the lack of IL-15 reduces the age-related accumulation of Treg and that the additional loss of Bim restores Treg accrual. We did not observe an effect of IL-15 on Bim expression directly, but we cannot exclude that IL-15 acts to restrain the remaining levels of Bim through another mechanism (i.e., Bim phosphorylation and turnover, induction of another anti-apoptotic molecule). As IL-2 and IL-15 both signal through CD122 (IL-2/15Rβ) and CD132 (common γ chain), it is possible that the decline of IL-2 favors the emergence of CD25loBimlo Treg because these cells are selected to survive and are maintained by IL-2-independent mechanisms, such as IL-15 (Fontenot et al., 2005a; Burchill et al., 2007).
While IL-2 and IL-15 signal through the same receptors, they can have divergent effects on T cells. For example, both IL-2 and IL-15 can activate the STAT5 and PI-3K/AKT pathways (Waldmann, 2006). In CD8+ T cells, the magnitude of STAT5 activation by IL-2 and IL-15 is similar, although IL-2 drives more prolonged STAT5 activation (Castro et al., 2011). Further, while both IL-2 and IL-15 can activate PI-3K/AKT/mTOR, IL-2 drives both an increased magnitude and persistence of mTOR activation compared to IL-15 (Cornish et al., 2006; Castro et al., 2011). Recent work has shown that unrestrained activation of mTORC1 in T cells lead to significantly increased Bim expression (Yang et al., 2011). Thus, it is possible that the differential activation of mTORC1 by IL-2 and IL-15 may underlie Bim regulation in Treg. However, it remains to be determined whether or not the degree to which the mechanisms are operative in Treg cells.
Bim is also regulated at the transcriptional level by the FOXO family of transcription factors. FOXO transcription factors promote Bim expression in T cells in response to cytokine withdrawal (Stahl et al., 2002; Salih and Brunet, 2008). FOXO(s) are inhibited by PI-3K/AKT activation, which leads to FOXO phosphorylation and sequestration from the nucleus (Salih and Brunet, 2008). Consistent with this, we found that in vivo IL-2 administration to young mice resulted in accrual of Bimlo Treg (Chougnet et al., 2011). However, in aged mice we found decreased in vivo IL-2 levels and decreased Bim expression, which are inconsistent with a role for FOXO transcription factor control of Bim. Interestingly, we also find that FOXO1 and FOXO3a levels themselves are decreased in aged Treg (data not shown). As mentioned earlier, levels of Puma, another FOXO target are also decreased in aged Treg, raising the possibility that decreased levels of FOXO molecules may contribute to decreased expression of Bim.
Certainly, a survival advantage contributes to the accrual of Treg with low Bim expression as Treg with higher levels of Bim accumulate in mice whose Treg cannot undergo apoptosis (i.e., T cell-specific Bax/Bak-deficient mice). One factor that may promote selection of Bimlo Treg is the decreased expression of Bcl-2 and Mcl-1 observed in aged Treg (Chougnet et al., 2011). Indeed, Bcl-2 is the major anti-apoptotic reported to combat Bim-mediated death in T cells (Wojciechowski et al., 2007), and Bim and Bcl-2 have been shown to affect the expression of each other (Jorgensen et al., 2007). Further, we recently showed in CD8+ T cells that inhibition or loss of Bcl-2 selected for effector CD8+ T cells expressing low levels of Bim (Kurtulus et al., 2011). Thus, decreased expression of Bcl-2 with age may prompt the selection of Bimlo Treg. However, it is also clear that, relative to their young counterparts, Bim levels decline even in T cell-specific Bax/Bak-deficient mice, suggesting that levels of Bim may be controlled by non-survival related mechanisms as well. An alternative, and not mutually exclusive, explanation is that decreased Bim mRNA levels in aged Treg may be due to epigenetic modification of the Bim promoter. The Bim promoter is CpG rich, and there is evidence that Bim expression can be repressed through hypermethylation (Paschos et al., 2009; San Jose-Eneriz et al., 2009; De Bruyne et al., 2010; Richter-Larrea et al., 2010). Furthermore, increased CpG methylation is a trait observed in aged cells (Golbus et al., 1990; Issa, 2003). Thus, as epigenetic modifications can be inherited, Bim promoter methylation may aid in the prolonged survival of aged Treg. We are currently determining whether epigenetic and/or transcriptional repression affects Bim expression in aged Treg.
While the exact mechanisms leading to decreased Treg Bim expression with age are still unclear, progressive loss of Bim expression occurs in both CD25lo and CD25hi Treg and this loss is likely sufficient to drive accrual of both populations with age. Furthermore, it is likely the lower expression of Bim in CD25lo Treg that affords these cells a survival advantage over CD25hi Treg, especially when IL-2 becomes limiting, and promotes increased CD25lo Treg accrual. Indeed, not until IL-2 levels have declined do we begin to see the emergence of CD25lo Treg in middle-aged mice. Because IL-2 promotes the expression of CD25 (Liao et al., 2013), there is likely still enough bioavailable IL-2 present in aged mice to maintain CD25 expression on the accumulated CD25hi Treg population. Thus, it is likely that both reduced IL-2 signaling as well as other mechanism(s) (i.e., Bim promoter epigenetics) contribute to the control of Bim expression with age.
In this study, we show that Treg loss of Bim drives accrual with age, while IL-15 is critical for aged Treg survival in the face of declining IL-2. We have shown here that Treg accrual is driven by several non-mutually exclusive mechanisms, including: (i) decreased Bim transcription, (ii) decreased Bim protein half-life, (iii) selection of Bimlo Treg due to a survival advantage, and (iv) a switch in cytokine dependency (from IL-2 to IL-15) with age. Future studies will focus on the mechanisms driving decreased Bim gene expression and protein half-life (i.e., promoter epigenetics, AKT/FOXO pathway), which may expose potential therapeutic targets for manipulating Bim expression and Treg accrual. The ability to moderately manipulate Treg homeostasis and accrual, without inducing inflammation and autoimmune responses, may provide a potential therapy for enhancing immune competence in the elderly.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Drs. H. L. Grimes and J. Phelan for the aged Lck-Cre Baxf/fBak−/− mice. The authors thank the Hildeman and Chougnet labs for helpful suggestions and comments. This work was supported by Public Health Service Grant AG033057 (to David A. Hildeman and Claire A. Chougnet).
Agius, E., Lacy, K. E., Vukmanovic-Stejic, M., Jagger, A. L., Papageorgiou, A. P., Hall, S., et al. (2009). Decreased TNF-alpha synthesis by macrophages restricts cutaneous immunosurveillance by memory CD4+ T cells during aging. J. Exp. Med. 206, 1929–1940. doi:10.1084/jem.20090896
Barron, L., Dooms, H., Hoyer, K. K., Kuswanto, W., Hofmann, J., O’Gorman, W. E., et al. (2010). Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J. Immunol. 185, 6426–6430. doi:10.4049/jimmunol.0903940
Bayer, A. L., Lee, J. Y., De La Barrera, A., Surh, C. D., and Malek, T. R. (2008). A function for IL-7R for CD4+CD25+Foxp3+ T regulatory cells. J. Immunol. 181, 225–234.
Belkaid, Y., and Oldenhove, G. (2008). Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity 29, 362–371. doi:10.1016/j.immuni.2008.08.005
Bouillet, P., Purton, J. F., Godfrey, D. I., Zhang, L. C., Coultas, L., Puthalakath, H., et al. (2002). BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 415, 922–926. doi:10.1038/415922a
Burchill, M. A., Yang, J., Vogtenhuber, C., Blazar, B. R., and Farrar, M. A. (2007). IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 178, 280–290.
Castro, I., Yu, A., Dee, M. J., and Malek, T. R. (2011). The basis of distinctive IL-2- and IL-15-dependent signaling: weak CD122-dependent signaling favors CD8+ T central-memory cell survival but not T effector-memory cell development. J. Immunol. 187, 5170–5182. doi:10.4049/jimmunol.1003961
Chen, M., Huang, L., and Wang, J. (2007). Deficiency of Bim in dendritic cells contributes to overactivation of lymphocytes and autoimmunity. Blood 109, 4360–4367. doi:10.1182/blood-2006-11-056424
Chougnet, C. A., Tripathi, P., Lages, C. S., Raynor, J., Sholl, A., Fink, P., et al. (2011). A major role for Bim in regulatory T cell homeostasis. J. Immunol. 186, 156–163. doi:10.4049/jimmunol.1001505
Clambey, E. T., Van Dyk, L. F., Kappler, J. W., and Marrack, P. (2005). Non-malignant clonal expansions of CD8+ memory T cells in aged individuals. Immunol. Rev. 205, 170–189. doi:10.1111/j.0105-2896.2005.00265.x
Cornish, G. H., Sinclair, L. V., and Cantrell, D. A. (2006). Differential regulation of T-cell growth by IL-2 and IL-15. Blood 108, 600–608. doi:10.1182/blood-2005-12-4827
De Bruyne, E., Bos, T. J., Schuit, F., Van Valckenborgh, E., Menu, E., Thorrez, L., et al. (2010). IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood 115, 2430–2440. doi:10.1182/blood-2009-07-232801
Erlacher, M., Labi, V., Manzl, C., Bock, G., Tzankov, A., Hacker, G., et al. (2006). Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J. Exp. Med. 203, 2939–2951. doi:10.1084/jem.20061552
Finkelman, F., Morris, S., Orekhova, T., and Sehy, D. (2003). The in vivo cytokine capture assay for measurement of cytokine production in the mouse. Curr. Protoc. Immunol. Chapter 6, Unit 6.28. doi:10.1002/0471142735.im0628s54
Finkelman, F. D., and Morris, S. C. (1999). Development of an assay to measure in vivo cytokine production in the mouse. Int. Immunol. 11, 1811–1818. doi:10.1093/intimm/11.11.1811
Fontenot, J. D., Rasmussen, J. P., Gavin, M. A., and Rudensky, A. Y. (2005a). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat. Immunol. 6, 1142–1151. doi:10.1038/ni1263
Fontenot, J. D., Rasmussen, J. P., Williams, L. M., Dooley, J. L., Farr, A. G., and Rudensky, A. Y. (2005b). Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 22, 329–341. doi:10.1016/j.immuni.2005.01.016
Garcia, G. G., and Miller, R. A. (2002). Age-dependent defects in TCR-triggered cytoskeletal rearrangement in CD4+ T cells. J. Immunol. 169, 5021–5027.
Golbus, J., Palella, T. D., and Richardson, B. C. (1990). Quantitative changes in T cell DNA methylation occur during differentiation and ageing. Eur. J. Immunol. 20, 1869–1872. doi:10.1002/eji.1830200836
Gray, D. H., Kupresanin, F., Berzins, S. P., Herold, M. J., O’Reilly, L. A., Bouillet, P., et al. (2012). The BH3-only proteins Bim and Puma cooperate to impose deletional tolerance of organ-specific antigens. Immunity 37, 451–462. doi:10.1016/j.immuni.2012.05.030
Hale, J. S., Boursalian, T. E., Turk, G. L., and Fink, P. J. (2006). Thymic output in aged mice. Proc. Natl. Acad. Sci. U.S.A. 103, 8447–8452. doi:10.1073/pnas.0601040103
Haynes, L., Eaton, S. M., Burns, E. M., Randall, T. D., and Swain, S. L. (2005). Newly generated CD4 T cells in aged animals do not exhibit age-related defects in response to antigen. J. Exp. Med. 201, 845–851. doi:10.1084/jem.20041933
Issa, J. P. (2003). Age-related epigenetic changes and the immune system. Clin. Immunol. 109, 103–108. doi:10.1016/S1521-6616(03)00203-1
Jorgensen, T. N., McKee, A., Wang, M., Kushnir, E., White, J., Refaeli, Y., et al. (2007). Bim and Bcl-2 mutually affect the expression of the other in T cells. J. Immunol. 179, 3417–3424.
Khan, N., Shariff, N., Cobbold, M., Bruton, R., Ainsworth, J. A., Sinclair, A. J., et al. (2002). Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J. Immunol. 169, 1984–1992.
Kim, J. M., Rasmussen, J. P., and Rudensky, A. Y. (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8, 191–197. doi:10.1038/ni1428
Kurtulus, S., Tripathi, P., Moreno-Fernandez, M. E., Sholl, A., Katz, J. D., Grimes, H. L., et al. (2011). Bcl-2 allows effector and memory CD8+ T cells to tolerate higher expression of Bim. J. Immunol. 186, 5729–5737. doi:10.4049/jimmunol.1100102
Lages, C. S., Suffia, I., Velilla, P. A., Huang, B., Warshaw, G., Hildeman, D. A., et al. (2008). Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J. Immunol. 181, 1835–1848.
Liao, W., Lin, J. X., and Leonard, W. J. (2013). Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 38, 13–25. doi:10.1016/j.immuni.2013.01.004
Linton, P. J., and Dorshkind, K. (2004). Age-related changes in lymphocyte development and function. Nat. Immunol. 5, 133–139. doi:10.1038/ni1033
Maue, A. C., Yager, E. J., Swain, S. L., Woodland, D. L., Blackman, M. A., and Haynes, L. (2009). T-cell immunosenescence: lessons learned from mouse models of aging. Trends Immunol. 30, 301–305. doi:10.1016/j.it.2009.04.007
Miller, R. A. (2000). Effect of aging on T lymphocyte activation. Vaccine 18, 1654–1660. doi:10.1016/S0264-410X(99)00502-2
Miller, R. A., Garcia, G., Kirk, C. J., and Witkowski, J. M. (1997). Early activation defects in T lymphocytes from aged mice. Immunol. Rev. 160, 79–90. doi:10.1111/j.1600-065X.1997.tb01029.x
Miyara, M., and Sakaguchi, S. (2007). Natural regulatory T cells: mechanisms of suppression. Trends Mol. Med. 13, 108–116. doi:10.1016/j.molmed.2007.01.003
Murasko, D. M., Weiner, P., and Kaye, D. (1987). Decline in mitogen induced proliferation of lymphocytes with increasing age. Clin. Exp. Immunol. 70, 440–448.
Nishioka, T., Shimizu, J., Iida, R., Yamazaki, S., and Sakaguchi, S. (2006). CD4+CD25+Foxp3+ T cells and CD4+CD25-Foxp3+ T cells in aged mice. J. Immunol. 176, 6586–6593.
Onishi, Y., Fehervari, Z., Yamaguchi, T., and Sakaguchi, S. (2008). Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. U.S.A. 105, 10113–10118. doi:10.1073/pnas.0711106105
Ouyang, Q., Wagner, W. M., Wikby, A., Walter, S., Aubert, G., Dodi, A. I., et al. (2003). Large numbers of dysfunctional CD8+ T lymphocytes bearing receptors for a single dominant CMV epitope in the very old. J. Clin. Immunol. 23, 247–257. doi:10.1023/A:1024580531705
Paschos, K., Smith, P., Anderton, E., Middeldorp, J. M., White, R. E., and Allday, M. J. (2009). Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 5:e1000492. doi:10.1371/journal.ppat.1000492
Richter-Larrea, J. A., Robles, E. F., Fresquet, V., Beltran, E., Rullan, A. J., Agirre, X., et al. (2010). Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood 116, 2531–2542. doi:10.1182/blood-2010-02-268003
Rubtsov, Y. P., Rasmussen, J. P., Chi, E. Y., Fontenot, J., Castelli, L., Ye, X., et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558. doi:10.1016/j.immuni.2008.02.017
Salih, D. A., and Brunet, A. (2008). FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 20, 126–136. doi:10.1016/j.ceb.2008.02.005
San Jose-Eneriz, E., Agirre, X., Jimenez-Velasco, A., Cordeu, L., Martin, V., Arqueros, V., et al. (2009). Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur. J. Cancer 45, 1877–1889. doi:10.1016/j.ejca.2009.04.005
Sempowski, G. D., Gooding, M. E., Liao, H. X., Le, P. T., and Haynes, B. F. (2002). T cell receptor excision circle assessment of thymopoiesis in aging mice. Mol. Immunol. 38, 841–848. doi:10.1016/S0161-5890(01)00122-5
Sharma, S., Dominguez, A. L., and Lustgarten, J. (2006). High accumulation of T regulatory cells prevents the activation of immune responses in aged animals. J. Immunol. 177, 8348–8355.
Stahl, M., Dijkers, P. F., Kops, G. J., Lens, S. M., Coffer, P. J., Burgering, B. M., et al. (2002). The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J. Immunol. 168, 5024–5031.
Takeuchi, O., Fisher, J., Suh, H., Harada, H., Malynn, B. A., and Korsmeyer, S. J. (2005). Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc. Natl. Acad. Sci. U.S.A. 102, 11272–11277. doi:10.1073/pnas.0504783102
Tamir, A., Eisenbraun, M. D., Garcia, G. G., and Miller, R. A. (2000). Age-dependent alterations in the assembly of signal transduction complexes at the site of T cell/APC interaction. J. Immunol. 165, 1243–1251.
Thoman, M. L., and Weigle, W. O. (1981). Lymphokines and aging: interleukin-2 production and activity in aged animals. J. Immunol. 127, 2102–2106.
Tischner, D., Gaggl, I., Peschel, I., Kaufmann, M., Tuzlak, S., Drach, M., et al. (2012). Defective cell death signalling along the Bcl-2 regulated apoptosis pathway compromises Treg cell development and limits their functionality in mice. J. Autoimmun. 38, 59–69. doi:10.1016/j.jaut.2011.12.008
Valmori, D., Merlo, A., Souleimanian, N. E., Hesdorffer, C. S., and Ayyoub, M. (2005). A peripheral circulating compartment of natural naive CD4 Tregs. J. Clin. Invest. 115, 1953–1962. doi:10.1172/JCI23963
Vang, K. B., Yang, J., Mahmud, S. A., Burchill, M. A., Vegoe, A. L., and Farrar, M. A. (2008). IL-2, -7, and -15, but not thymic stromal lymphopoeitin, redundantly govern CD4+Foxp3+ regulatory T cell development. J. Immunol. 181, 3285–3290.
Waldmann, T. A. (2006). The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 6, 595–601. doi:10.1038/nri1901
Wojciechowski, S., Tripathi, P., Bourdeau, T., Acero, L., Grimes, H. L., Katz, J. D., et al. (2007). Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J. Exp. Med. 204, 1665–1675.
Wrenshall, L. E., and Platt, J. L. (1999). Regulation of T cell homeostasis by heparan sulfate-bound IL-2. J. Immunol. 163, 3793–3800.
Yamazaki, S., Iyoda, T., Tarbell, K., Olson, K., Velinzon, K., Inaba, K., et al. (2003). Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 198, 235–247. doi:10.1084/jem.20030422
Yang, K., Neale, G., Green, D. R., He, W., and Chi, H. (2011). The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat. Immunol. 12, 888–897. doi:10.1038/ni.2068
Youle, R. J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59. doi:10.1038/nrm2308
Zhan, Y., Zhang, Y., Gray, D., Carrington, E. M., Bouillet, P., Ko, H. J., et al. (2011). Defects in the Bcl-2-regulated apoptotic pathway lead to preferential increase of CD25 low Foxp3+ anergic CD4+ T cells. J. Immunol. 187, 1566–1577. doi:10.4049/jimmunol.1100027
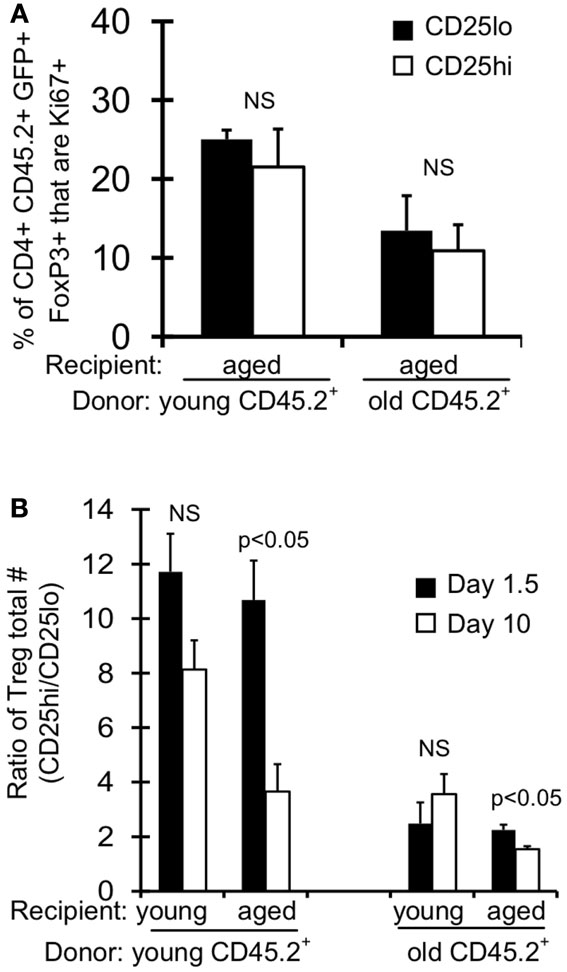
Figure A1. Absolute numbers and proliferation of Treg post-adoptive transfer. CD+FoxP3GFP+ T cells were sorted from splenocytes of young (3–4 months) and old (>18 months) CD45.2 FoxP3GFP reporter mice. About 5 × 105 CD4+FoxP3GFP+ cells were injected i.v. into young (2 months) and aged (15 months) congenic CD45.1 mice. Mice were sacrificed at day 1.5 (n = 3–5/group) and day 10 (n = 3–5/group) post-injection. Cells from the spleen were stained for CD4, CD25, CD45.2, and intracellularly for FoxP3 and Ki67 and analyzed by flow. (A) Results show the frequency of CD4+CD45.2+FoxP3+GFP+CD25lo and CD25hi cells that are Ki67+ on day 10 post-transfer into aged recipient mice (±SE). (B) Results show the numbers ratio of CD25hi to CD25lo CD4+CD45.2+FoxP3+GFP+ cells (±SE).
Keywords: CD25, Bim, IL-2, IL-15, aging, Treg
Citation: Raynor J, Sholl A, Plas DR, Bouillet P, Chougnet CA and Hildeman DA (2013) IL-15 fosters age-driven regulatory T cell accrual in the face of declining IL-2 levels. Front. Immunol. 4:161. doi: 10.3389/fimmu.2013.00161
Received: 16 April 2013; Accepted: 10 June 2013;
Published online: 24 June 2013.
Edited by:
Dietmar Herndler-Brandstetter, Yale University School of Medicine, USAReviewed by:
Andreas Villunger, Medical University Innsbruck, AustriaCopyright: © 2013 Raynor, Sholl, Plas, Bouillet, Chougnet and Hildeman. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: David A. Hildeman, Division of Cellular and Molecular Immunology, Department of Pediatrics, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, S5 Room 214, MLC 7038, 3333 Burnet Avenue, Cincinnati, OH 45229, USA e-mail:ZGF2aWQuaGlsZGVtYW5AY2NobWMub3Jn
†Claire A. Chougnet and David A. Hildeman have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.