


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 June 2013
Sec. Antigen Presenting Cell Biology
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00138
This article is part of the Research Topic The nature of activatory and tolerogenic dendritic cell-derived signal 2 View all 13 articles
Dendritic cells (DCs) initiate and control immune responses, participate in the maintenance of immunological tolerance and are pivotal players in the pathogenesis of autoimmunity. In patients with autoimmune disease and in experimental animal models of autoimmunity, DCs show abnormalities in both numbers and activation state, expressing immunogenic levels of costimulatory molecules and pro-inflammatory cytokines. Exogenous and endogenous danger signals activate DCs to stimulate the immune response. Classic endogenous danger signals are released, activated, or secreted by host cells and tissues experiencing stress, damage, and non-physiologic cell death; and are therefore referred to as damage-associated molecular patterns (DAMPs). Some DAMPs are released from cells, where they are normally sequestered, during necrosis (e.g., heat shock proteins, uric acid, ATP, HMGB1, mitochondria-derived molecules). Others are actively secreted, like Type I Interferons. Here we discuss important DAMPs in the context of autoimmunity. For some, there is a clear pathogenic link (e.g., nucleic acids and lupus). For others, there is less evidence. Additionally, we explore emerging danger signals. These include inorganic materials and man-made technologies (e.g., nanomaterials) developed as novel therapeutic approaches. Some nanomaterials can activate DCs and may trigger unintended inflammatory responses. Finally, we will review “homeostatic danger signals,” danger signals that do not derive directly from pathogens or dying cells but are associated with perturbations of tissue/cell homeostasis and may signal pathological stress. These signals, like acidosis, hypoxia, and changes in osmolarity, also play a role in inflammation and autoimmunity.
Dendritic cells (DCs) are specialized antigen-presenting cells (APCs) that help maintain peripheral tolerance and orchestrate the adaptive immune response by presenting both endogenous and exogenous antigens and producing immune modulatory factors, such as costimulatory/inhibitory molecules and cytokines (Banchereau et al., 2000; Hammer and Ma, 2013). DCs reside in virtually all the tissues of our body in a predominantly antigen-capturing state and maintain immunologic tolerance by routinely migrating to the draining lymph nodes and presenting self-antigens to lymphocytes in a tolerogenic manner (Steinman et al., 2003; Reis e Sousa, 2006). Upon stimulation, DCs undergo a process of maturation/activation, which allows them to present in an immunogenic fashion the antigens captured in the periphery and initiate immune responses (Cella et al., 1997; Blander and Medzhitov, 2004). Danger signals are molecules released during infections (Janeway, 1989; Medzhitov and Janeway, 1997b) and/or tissue damage and cellular stress (Matzinger, 1994, 1998, 2002; Gallucci and Matzinger, 2001; Land and Messmer, 2012). Danger signals activate DCs and stimulate both the innate and adaptive immune response (Box 1).
Box 1. Terms and definitions.
In order to help our readers, here we define the terms we use throughout this review:
Pathogen-associated molecular pattern: molecular structures common to bacteria, viruses, or other microorganism, like LPS, flagellin, and peptidoglycan, that are able to activate DCs. These molecules are recognized by pattern recognition receptors (PRRs) expressed on both immune and non-immune cells. Since PAMPs are also expressed by the normal flora of mammalian mucosas, they are also called Microbial Associate Molecular Patterns (MAMPs).
Damage-associated molecular pattern: endogenous molecular structures that are normally contained within the cell interior and hidden from the immune system, and are liberated upon tissue damage. Examples include ATP, HSPs, and HMGB1. These molecules are recognized by a number of receptors, including PRRs, and are capable of inducing inflammation and immune responses in the absence of infection.
Danger signal: a danger signal may be any substance or event that is able to activate DCs and therefore initiate immune responses. For the purposes of this review, we have split the danger signals into three categories shown below. They all have in common an association with cellular stress and the ability to activate DCs.
Classic danger signals: we use the term “classic” to refer to danger signals that are not emerging or homeostatic (defined below). The classic danger signals are molecular species either associated with pathogens (PAMPs) or directly derived from tissue injury damage-associated molecular patterns (DAMPs) as described above, or secreted by activated immune cells as amplifiers of the immune activation.
Emerging danger signals: we use the term “emerging” to turn attention to new fields of research and technology that may pose new challenges to the immune system. Specifically, emerging danger signals include inorganic materials and man-made technologies (e.g., nanomaterials) often used as novel therapeutic strategies. These materials have the potential to activate DCs directly or indirectly by inducing tissue damage and release of DAMPs.
Homeostatic danger signals: with the term “homeostatic” we aim to highlight a less appreciated category of endogenous danger signals. Homeostatic danger signals generally represent perturbations in tissue steady state often as a result of inflammation. These perturbations include (but are not limited to) hypoxia, changes in acidity, or osmolarity, and metabolic stress.
The recognition of tissue and cell damage – which may involve several types of innate immune cells beside DCs and cascades such as the complement – has been proposed to initiate multiple processes (Bianchi, 2007), including: (a) the development of inflammation, (b) the induction of innate effector functions leading to pathogen clearance, (c) the stimulation of adaptive immunity, which generates immunological memory for future encounters with the same antigen, and (d) the stimulation of tissue repair processes, necessary to reconstitute tissue integrity compromised by the infection or, in some cases, by the immune response itself (Stoecklein et al., 2012) (Figure 1).
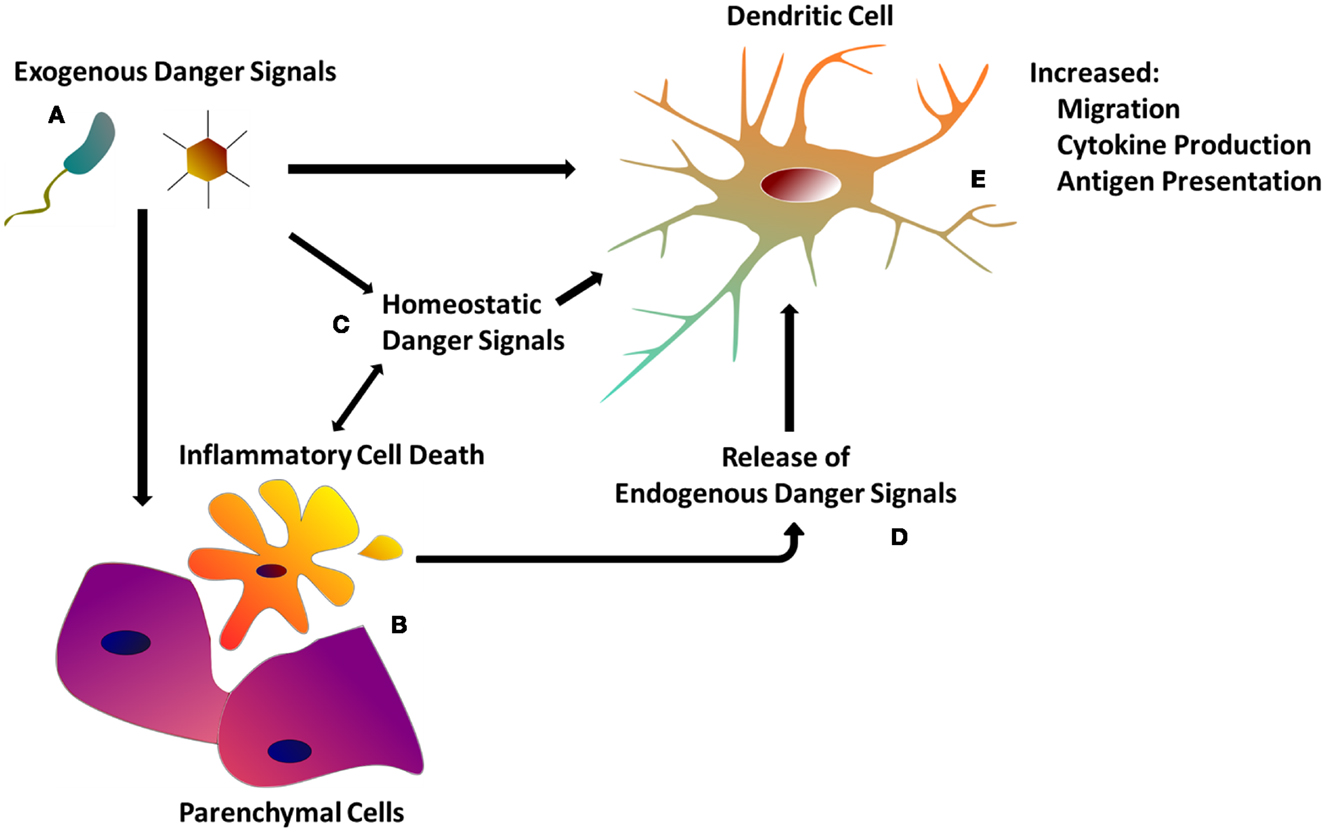
Figure 1. The dendritic cell response to danger. (A) Exogenous danger signals include pathogen-associated molecular patterns, as well as exogenous particulate matter. Exogenous danger signals activate DCs directly via pattern recognition receptors, or indirectly though tissue damage (B) and homeostatic perturbations (C). (B) Inflammatory cell death as a result of tissue injury or programed necrosis causes the release of endogenous danger signals (D) (see Table 1) which also activate DCs (see Table 1). (C) Homeostatic perturbations such as those found in inflamed tissue (e.g., decreased pH, hypoxia) may also act as endogenous danger signals, influencing DC immune functions. (E) DCs integrate both exogenous and endogenous danger signals in order to orchestrate the appropriate immune response.
Dendritic cells collaborate with other cells of the innate immune system to initiate what is often called “sterile inflammation and immunity;” that is, the activation of the innate and adaptive immune response in the absence of pathogens (Rock and Kono, 2008). Mechanisms of sterile inflammation are key to our understanding of clinical situations such as transplant rejection, tumor immunity, chronic inflammatory diseases, trauma-induced systemic inflammatory response syndrome (SIRS), and other inflammatory conditions like ischemia-reperfusion injury and atherosclerosis – in which immune responses develop when no overt infection is present. Autoimmune diseases, in which the adaptive immune system mounts an immune response against self-antigens, are also an important class of pathologies in which the role of infectious triggers for immunity is still controversial (Mills, 2011; Stranger and De Jager, 2012). The aim of this review is to analyze the danger signals that modulate DC functions and explore how those danger signals may contribute to the pathogenesis of autoimmunity.
The DC lineage comprises a complex population of several subsets – with recent advances demonstrating intricacies of gene transcription (Miller et al., 2012) and tissue localization (Gerner et al., 2012) that define each population. Indeed, DC subsets vary greatly in terms of lineage markers, cytokine production, and ability to stimulate different immune responses, largely depending on the tissue in which they reside or emigrate from [i.e., skin (Ginhoux et al., 2012) or gut (Rescigno, 2010)]. For simplicity, DCs are categorized as conventional DCs, strong APCs, or plasmacytoid DCs, the major producers of Type I Interferons (IFNs). DCs are either resident within the secondary lymphoid organs like lymph nodes and spleen, or migratory, coming into the lymph nodes from the peripheral tissues (Hammer and Ma, 2013).
The importance of DCs in the pathogenesis of autoimmune diseases is supported by the effects of their manipulation in experimental animal models. Indeed, the constitutive genetic depletion of DCs induces spontaneous development of autoimmunity characterized by autoantibodies against nuclear and tissue-specific antigens, multi-organ lymphocyte infiltration, and severe tissue damage, especially in the intestine (Ohnmacht et al., 2009). Therefore, DCs may be necessary for the establishment and maintenance of immunological self-tolerance (Reis e Sousa, 2006). Paradoxically, DCs also seem to be powerful inducers of autoimmunity. Mice in which DCs accumulate as a result of a DC specific defect in apoptosis also develop chronic lymphocyte activation and lupus-like systemic autoimmunity (Chen et al., 2006). Consequently, uncontrolled DC functions are sufficient to prime for autoimmune reactions. In many experimental models of autoimmunity, DCs accumulate in the secondary lymphoid organs and in the tissues targeted by the autoimmune process (Rosmalen et al., 2000; Kalled et al., 2001; Adachi et al., 2002; Colonna et al., 2006), although the causative mechanism for this accumulation remains unclear. Further support comes from studies demonstrating that the uptake of autoantibody-opsonized apoptotic cells increases DC presentation of self Ags in an inflammatory context (Frisoni et al., 2005). Moreover, in vivo priming with immunogenic (highly activated), self-antigen-loaded DCs induces, or accelerates autoimmunity (Bondanza et al., 2003; Eriksson et al., 2003), while the administration of tolerogenic DCs reduces disease and has been proposed as a potential therapeutic strategy in many models of autoimmune disease, such as type I diabetes (Feili-Hariri et al., 2002) and experimental autoimmune encephalomyelitis (EAE) (Menges et al., 2002; Toscano et al., 2010).
Dendritic cells may play a pathogenic role in autoimmunity by presenting self-antigens to T cells in an immunogenic fashion and by collaborating in the activation of autoreactive B cells. To do so, DCs have to be activated and express immunogenic costimulatory molecules and pro-inflammatory cytokines. Indeed, much evidence shows abnormally activated DC phenotypes in patients with different autoimmune diseases, as well as in murine models of autoimmunity (reviewed in Amodio and Gregori, 2012). In some autoimmune strains of mice, DCs show abnormalities even when generated in culture, far removed from the autoimmune microenvironment (i.e., DCs from young mice, before the onset of the disease) (Sriram et al., 2012). These results suggest a genetic defect intrinsic to DCs, leading to their excessive activation, possibly through an uncontrolled production of danger signals (Elkon and Stone, 2011). In other cases, abnormalities were present only in DCs in vivo or isolated ex vivo from diseased mice, therefore pointing to a direct association with the autoimmune process (Colonna et al., 2006; Melli et al., 2009) and suggesting that in some situations DC abnormalities are a consequence rather than the cause of the autoimmune environment. In either case, determining the role of danger signals in the activation of DCs is key to better understanding the pathogenesis of autoimmune disease. Knowledge of danger signals will also facilitate the identification of novel therapeutic approaches aimed at the prevention of autoreactive lymphocyte activation. For example, neutralization of stimuli that induce abnormal DC activation may be a far more physiologic strategy than relying on general suppression of the lymphocyte response, as is common with most current therapeutic protocols.
The term “danger signal” was originally proposed by Polly Matzinger to indicate endogenous molecules released by stressed or necrotic cells, which are able to activate DCs (Matzinger, 1994, 1998; Gallucci et al., 1999; Gallucci and Matzinger, 2001). Five years earlier, Janeway (1989) had theorized that the innate immune system becomes activated by conserved molecular species expressed by evolutionarily distant microorganisms. These features were called pathogen-associated molecular patterns (PAMPs) and were proposed to trigger PRRs present on host cells (Medzhitov and Janeway, 1997a,b). With his innovative theory, Janeway (1992) updated the classic self-non-self-discrimination model of immunity by theorizing that the immune system can distinguish between self and infectious non-self, i.e., the pathogen (Medzhitov et al., 1997; Poltorak et al., 1998). As our understanding of the biochemical basis of the PRRs (e.g., TLRs, NLRs, RIG-I) has improved, it has become clear that host factors, derived from damaged tissues and cells, signal through the same receptors, serving as “danger signals” to stimulate immunity and therefore allow the immune system to discriminate what is dangerous or damaged from what is not, as previously theorized by Matzinger (1994, 2002) (Figure 1). These endogenous danger signals are a subset of what Seong and Matzinger (2004) named “DAMPs,” analogous to the nomenclature of Janeway. Presently the term “danger signal” has a broad meaning and includes very different families of molecules that activate DCs: either exogenous molecules, such as the PAMPs; or endogenous molecules released, activated, or secreted by host cells and tissues undergoing stress, damage, and non-physiological cell death, namely DAMPs (Matzinger, 1998, 2002) (Figure 1; Table 1). We support this inclusive nomenclature as it conveys the idea that both pathogens and trauma/stress are inducers of tissue and cell damage, which results in a pathologic status that is required for the activation of the innate immune system.
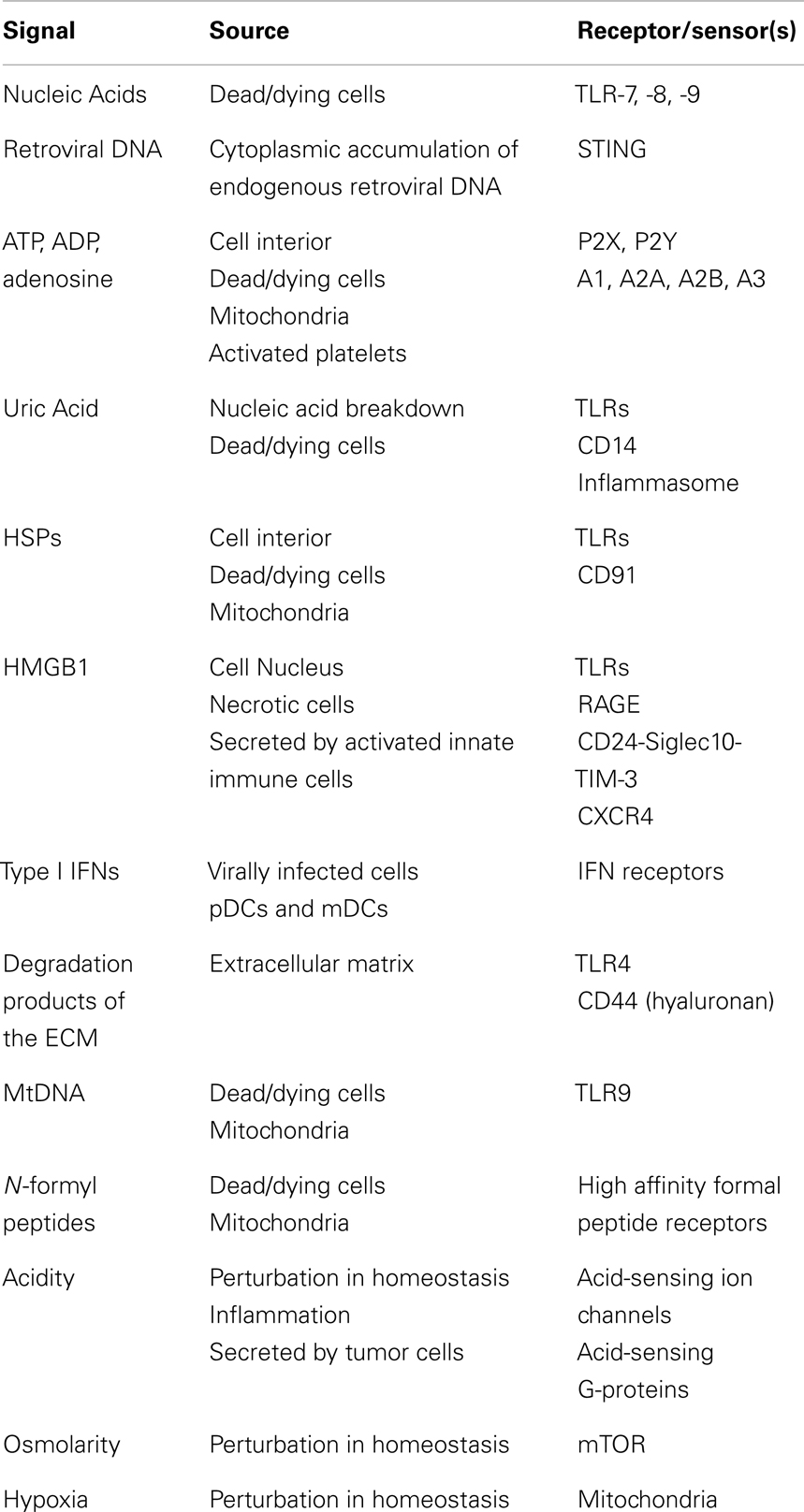
Table 1. Endogenous danger signals.
In the next section, we will review the DAMPs that activate DCs in autoimmunity, and we will then focus on danger signals that are less in the spotlight for immunologists, although already hypothesized in the original version of the Danger Model (Matzinger, 1994): the emerging and the homeostatic danger signals. Emerging danger signals are molecules that are newly developed by modern technology and that can activate DCs either by inducing cellular stress/damage or by attracting and carrying endogenous danger signals. Finally, we will review “homeostatic danger signals,” danger signals that do not derive directly from pathogens or dying cells but rather are associated with perturbations of tissue/cell homeostasis and may be considered signs of stress and non-physiological conditions. We will not review PAMPs since they have been subjects of many excellent reviews.
Classic endogenous danger signals are molecules that activate DCs (Table 1; Figure 1; Box 2). As DCs are central to antigen presentation and the initiation of the immune response, it was first shown that DCs respond to endogenous danger signals released from dying cells (Gallucci et al., 1999; Sauter et al., 2000), explaining the adjuvant effects of dying cells when co-injected with antigen in vivo (Gallucci et al., 1999; Shi et al., 2000, 2003).
Box 2. Primary vs. secondary endogenous danger signals.
Endogenous danger signals can be divided in two categories depending on the trigger and on the cellular source.
Primary endogenous danger signals: endogenous molecules that are normally contained within the cell interior or present in an inactive form, hidden from the immune system, and mostly performing non-immune functions. They are released upon tissue damage and are able to activate DCs by triggering a number of receptors, including PRRs. They are the first initiators of sterile immunity. Examples are listed in Table 1.
Secondary endogenous danger signals: endogenous molecules that are actively secreted by immune cells upon activation to mediate innate and adaptive immune activation in an autocrine and paracrine manner. They fuel and direct both sterile and non-sterile immunity. Secondary endogenous danger signals include lymphocyte-derived activators of DCs such as CD40-L (Ridge et al., 1998) and granulysin (Zitvogel and Kroemer, 2010); neutrophil-derived alarmins (Yang et al., 2009) and pro-inflammatory cytokines such as TNF-α, Type I IFNs, and HMGB1.
Endogenous danger signals can be classified as “primary” when secreted by stressed cells or released by dying cells, or as “secondary” when produced by activated immune cells (Gallucci and Matzinger, 2001) (Box 2). Some cytokines act as both primary and secondary danger signals, when they are secreted by damaged/dying cells (Box 2). Many primary endogenous danger signals are released from the interior of the cell when the cell loses plasma membrane integrity upon necrosis (Rock and Kono, 2008; Sims et al., 2010). In contrast, the process of apoptosis keeps these pro-inflammatory signals contained and can actively up-regulate anti-inflammatory mediators (Behrens et al., 2007).
We will next describe some of the most studied DAMPs in autoimmunity. Covering all possible endogenous danger signals is not the purpose of this review: we will discuss a few signals in detail with specific reference to their role in autoimmune diseases (Table 1).
Nucleic acids play a complex role in the pathogenesis of the autoimmune disease systemic lupus erythematosus (SLE). Nucleic acids are both the primary self-antigens and potent DAMPs, activating DCs through TLR7 and TLR9 and inducing the production of Type I IFNs (Barrat et al., 2005; Marshak-Rothstein and Rifkin, 2007). Nucleic acids often activate DCs either as part of an immune complex with autoantibodies (Leadbetter et al., 2002) or in complex with chaperones like heat shock proteins (HSP) and HMGB1. Oligonucleotides that inhibit the response to TLR7 and TLR9 ameliorate disease in lupus-prone mice (Barrat et al., 2007). Similarly the genetic deficiency of TLR7, or TLR7 and TLR9 combined prevent disease, while the single deficiency of TLR9 renders the disease worse, suggesting a double-edge effect of TLR9 (Santiago-Raber et al., 2009). In our hands, DCs from lupus-prone mice express constitutively high levels of Type I IFNs and IFN responsive genes and this IFN Signature was diminished by treatment with oligonucleotides inhibitory for TLR7 and TLR9, supporting a role for the response of DCs to nucleic acids in the excessive production of Type I IFNs in lupus (Sriram et al., 2012). The importance of TLR7 is further highlighted by the discovery that the Yaa translocation of a piece of chromosome X, containing 16 genes including TLR7, to the Y chromosome induces in male mice a severe lupus-like disease by creating a duplication of TLR7 (Pisitkun et al., 2006). Necrotic cells are a source of immunogenic nucleic acids (Eloranta et al., 2009) as well as neutrophils undergoing NETosis (Garcia-Romo et al., 2011).
A subset of PAMPs that can also be considered a form of special endogenous danger signals are endogenous viral elements (EVEs). EVEs are sections of viral DNA that have integrated into the mammalian genome during evolution (Stoye, 2012). The best characterized EVEs come from retroviruses, but may also derive from other viruses (Katzourakis and Gifford, 2010). Recent evidence demonstrates that defects in controlling retroviral elements are linked to autoimmunity. Indeed, mice deficient in the cytoplasmic three prime repair exonuclease 1 (Trex1) (formerly DNase III) express elevated IFN alpha (IFNα) levels in many organs and develop inflammatory myocarditis in the absence of viral infection (Morita et al., 2004). Trex1-deficient (Trex1−/−) murine embryonic fibroblasts accumulate endogenous retroviral DNA in the cytosol which induces excessive Type I IFN production via STING (Yang et al., 2007; Stetson et al., 2008; Gall et al., 2012). Loss-of-function mutations in Trex1 are present in patients with Aicardi–Goutières syndrome as well as other autoimmune diseases such as familial chilblain lupus (Lee-Kirsch et al., 2007a; Gunther et al., 2009) and SLE (Lee-Kirsch et al., 2007b; Hur et al., 2008). This evidence suggests that cytoplasmic DNA derived from endogenous retroviruses is an endogenous danger signal that, if not scavenged by Trex1, can trigger an intracellular nucleic acid-sensing machinery (Pichlmair and Reis e Sousa, 2007; Thompson et al., 2011), leading to excessive production of the pro-autoimmunogenic Type I IFNs (Elkon and Stone, 2011). Although DCs are activated by Type I IFNs (Gallucci et al., 1999), and might therefore become hyperactivated, and thus initiate the autoimmune process, phenotypic, or specific functional abnormalities of DCs in Trex1−/− mice or in patients with Aicardi–Goutières syndrome have not yet been reported.
Other forms of nucleic acids acting as danger signals are the intracellular single nucleotides ATP and UTP. Extracellular ATP concentration is typically kept very low (10 nM) by ectonucleotidases (Robson et al., 2006). Under pathologic conditions, however, local concentrations of ATP can be increased – when liberated from damaged tissue or when released by an active process such as platelet or phagocyte activation (Zeiser et al., 2011). These adenosine derivatives activate DCs (Marriott et al., 1999) by stimulating the high affinity purinergic receptors P2Z/P2X7 (Mutini et al., 1999). ATP and UTP activate the inflammasome in collaboration with TLRs (Mariathasan et al., 2004) and mediate at least some of the sterile inflammation induced by necrotic cells (Iyer et al., 2009; Aymeric et al., 2012). P2X7-activated DCs have been shown to be crucial for the induction of tumor immunity (Ghiringhelli et al., 2009), the lack of P2X7 signaling decreases the severity of EAE (Sharp et al., 2008), and oxidized ATP (oxATP), an inhibitor of the ATP receptor P2rx7, ameliorated autoimmune type I diabetes and autoimmune encephalitis in mice (Lang et al., 2010). Apoptotic cells can also release small amounts of ATP through the pannexin family of plasma membrane channels. At this low concentration, ATP binds low affinity purinergic receptors, including P2Y2 and P2X7 on phagocytes and recruits them to accelerate non-inflammatory clearance of apoptotic cells (Elliott et al., 2009). In a model of allergic lung inflammation, P2Y2 is required for myeloid DC and eosinophil migration, although this receptor does not seem to be involved in DC maturation (Muller et al., 2010). Therefore, the ATP molecule may represent an important DAMP, conserved from prokaryotes (Atarashi et al., 2008) to plants (Heil et al., 2012) to mammals (Aymeric et al., 2012), acting as an activation signal at high doses when released by damaged cells, while at lower doses it may act as a “find-me” signal that helps housekeeping clearance of apoptotic cells and prevents immune activation (Elliott et al., 2009).
Uric acid was one of the first DAMPs to be discovered and is partly responsible for the adjuvant properties of dying cells (Shi et al., 2003). Uric acid is a product of purine metabolism and is normally soluble inside cells. When released in the extracellular space uric acid precipitates and forms insoluble crystals of monosodium urate (MSU) capable of activating DCs in vitro and of acting as an adjuvant in vivo (Shi et al., 2003; Rock et al., 2010). Extracellular uric acid is highly inflammatory when it accumulates in joints to cause gout (Rock et al., 2013) or when it is released in large amounts during anti-cancer therapy causing tumor lysis syndrome (Muslimani et al., 2011). The inflammatory nature of uric acid, as well as of other crystals like silica and alum, is in part due to their ability to activate the inflammasome (Martinon et al., 2006) with the extracellular delivery of endogenous ATP, the stimulation of IL-1β, and other pro-inflammatory cytokines, and the induction of reactive oxygen species (ROS) (Schorn et al., 2010; Riteau et al., 2012). These crystals may activate the inflammasome by causing direct cell damage to phagocytes, including DCs, inducing phagolysosome rupture (Hornung et al., 2008). Indeed, membrane fragments of these organelles can activate several responses, including polyubiquitination, autophagy, and pyroptosis and may play a role in host defense against intracellular parasites (Hilbi, 2009). Excessive activation of the inflammasome has been strongly linked to a group of hereditary autoinflammatory diseases called cryopyrin associated periodic syndromes (Lamkanfi and Dixit, 2012) but it may also play a role in more classic autoimmune diseases such as multiple sclerosis (MS) as mice deficient for components of the inflammasome (Nlrp3 and ASC) are protected from the development of EAE (Gris et al., 2010; Shaw et al., 2010).
Heat shock proteins are the most abundant proteins in cells, responsible for maintaining the correct folding of nascent and misfolded proteins, and of proteins that must remain inactive for long periods. HSPs are up-regulated in response to tissue stressors such as elevated temperature, osmotic shock, and cytotoxic agents (Srivastava, 2002). During cellular necrosis, HSPs are released into the extracellular space where they activate the innate immune response by triggering TLRs (Vabulas et al., 2002). As HSPs are protein chaperones, they also actively participate in DC antigen processing and presentation to initiate adaptive immunity (Srivastava, 2002). Srivastava’s group showed that necrotic cells, but not apoptotic cells, release many HSPs such as HSP70, HSP90, calreticulin, and GP96 (Basu et al., 2000), which may activate immunity through the common receptor CD91 (Binder et al., 2000). In contrast, other HSPs, like the human heat shock protein 60, induce DC maturation and promote a Th1 phenotype via TLR4 (Flohe’ et al., 2003). Like HMGB1 and autoantibodies, HSPs may contribute to the development of autoimmunity not only as endogenous adjuvants but also as chaperones that physically deliver autoantigens to APCs (Biswas et al., 2006). This is the mechanism by which HSP70 promotes autoimmunity in a mouse model of diabetes (Liu et al., 2003; Millar et al., 2003). Furthermore, autoantibodies against HSPs are found in SLE patients and allelic variants of the Hsp70 genes are significantly associated with SLE (Furnrohr et al., 2010) and with several other autoimmune diseases such as MS (Ramachandran and Bell, 1995), Crohn’s disease (Debler et al., 2003), Grave’s disease (Ratanachaiyavong et al., 1991), and insulin-dependent diabetes mellitus (Pociot et al., 1993).
High mobility group box protein 1 (HMGB1) is a well-established endogenous danger signal (Lotze and Tracey, 2005), released by necrotic cells that have lost membrane integrity (Scaffidi et al., 2002) as well as secreted by phagocytic cells as a late mediator of inflammation (Gardella et al., 2002). HMGB1 normally has a nuclear localization and was originally described as a DNA binding protein (Javaherian et al., 1978; Bianchi et al., 1989), but a variety of other cellular functions have been attributed to HMGB1 largely depending on different post-translational modifications such as oxidation and hyperacetylation (Harris et al., 2012). Intracellular HMGB1 regulates gene transcription and autophagy (Tang et al., 2012). Extracellular HMGB1 acts as a classic DAMP when released by necrotic cells, or as a secondary danger signal when secreted by macrophages and DCs in response to LPS and TNF-α (Yang et al., 2005; Bianchi, 2007). Autocrine HMGB1 mediates DC activation and the ability to induce Th1 polarization (Dumitriu et al., 2005). The secretion of HMGB1 by monocytic cells is mediated by hyperacetylation of HMGB1, which localizes it to the cytoplasm where, upon a secondary signal, it is liberated into the extracellular compartment (Bonaldi et al., 2003).
HMGB1 is significantly increased in the serum of patients with brain and myocardial ischemia (Goldstein et al., 2006) and in septic patients in which it mediates the late phase of septic shock (Wang et al., 1999). HMGB1 is recognized by many receptors, like the receptor for advanced glycation end products (RAGE) (Kokkola et al., 2005) as well as by TLR2 and TLR4 (Park et al., 2004), CD24-Siglec10, and the recently reported TIM-3 (Chiba et al., 2012). When bound to the chemokine CXCL12, HMGB1 acts as a chemokine to recruit leukocytes to the site of inflammation by binding CXCR4 (Schiraldi et al., 2012). The shift from functioning as a chemokine to acting as a danger signal depends on its redox state: intracellular HMGB1 has three cysteines that are in a reduced state (all-thiol state); when it is secreted or released, the all-thiol HMGB1 can bind to CXCL12 and chemoattract unless two cysteines are oxidized, forming a disulfide bond and promoting HMGB1 danger signal functions (Yang et al., 2013). During apoptosis HMGB1 is kept intracellular, and uptake of apoptotic cells induces release of oxidized HMGB1 so that its pro-inflammatory activities are neutralized. Possible functions of this fully oxidized state are still under investigation (Yang et al., 2013).
Evidence suggests that HMGB1 participates in the pathogenesis of many autoimmune diseases like Rheumatoid Arthritis (RA), SLE, EAE, and diabetes (Zhang et al., 2009; Harris et al., 2012). For example, RA patients have increased levels of HMGB1 in the serum and synovial fluid that decreases upon therapy-induced amelioration of joint inflammation (Zetterstrom et al., 2008). Similarly, HMGB1 blockade ameliorates arthritis in animals, while its administration in the joints induces arthritis (Kokkola et al., 2003). In SLE patients, serum HMGB1 levels, as well as anti-HMGB1 autoantibody titers, positively correlate with disease activity (Jiang and Pisetsky, 2008; Abdulahad et al., 2011). HMGB1-containing nucleosomes from apoptotic cells have been shown to induce secretion of pro-inflammatory cytokines and expression of costimulatory molecules in macrophages and DCs and the administration of HMGB1-nucleosome complexes in mice induces lupus-like autoantibodies (Urbonaviciute et al., 2008; Urbonaviciute and Voll, 2011). In MS patients, extracellular HMGB1 is increased in the cerebrospinal fluid (CSF) and microglia and macrophages expressing cytosolic HMGB1 are increased in MS active lesions (Andersson et al., 2008). In mice, HMGB1 drives neuroinflammation in EAE and the inhibition of HMGB1 signaling with a neutralizing antibody ameliorates disease (Robinson et al., 2013). Clinical and experimental evidence also suggest a role for HMGB1 in the pathogenesis of type I diabetes. In diabetes-prone NOD mice, HMGB1 blockade significantly inhibited insulitis progression and delayed diabetes onset, decreased the number, and maturation of DCs in the pancreatic lymph nodes, and increased the number of regulatory T cells (Han et al., 2008). Once diabetes has developed, hyperglycemia can induce HMGB1 secretion (Yao and Brownlee, 2010; Dandona et al., 2013) and HMGB1 may contribute to the nephropathy and the vascular complications of diabetes by fueling inflammation and promoting tissue damage, especially in the kidney (Lin et al., 2012). Higher levels of serum HMGB1 were associated in type 1 diabetes patients with a higher risk of mortality, cardiovascular disease (Nin et al., 2012a), and kidney damage (Nin et al., 2012b).
In summary, HMGB1 is a powerful danger signal that plays a complex role in the pathogenesis of autoimmune diseases. Blockade of HMGB1 has thus far proven to be beneficial in ameliorating a number of types of experimental autoimmunity, making its therapeutic potential very promising. Further investigation is still needed to dissect the role of HMGB1 as an activator of DCs in terms of its multiple functions as a cytokine, chemokine, transcription regulator, and possible functions yet to be discovered.
Type I IFNs activate DCs in vitro and act as adjuvant in vivo (Gallucci et al., 1999). They are secreted by virally infected cells (Taniguchi and Takaoka, 2002) as a primary danger signal to alert both neighboring tissue cells and local immune cells to the presence of a viral infection. They are also secreted by immune cells as an amplifier of innate immunity (Liu, 2005). Type I IFNs (IFNs α and β) mediate the activation of DCs to promote adaptive immune responses such as cross-priming and isotype switching of responding murine B cells toward IgG2 (Theofilopoulos et al., 2005). Type I IFNs are particularly important in autoimmunity and especially in SLE in which an IFN Signature is present – an abnormally high expression of Type I IFN responsive genes in immune cells and tissues (Baechler et al., 2003; Bennett et al., 2003; Crow et al., 2003; Elkon and Stone, 2011). The hyper-activation of Type I IFNs may play a role in the early and most acute phases of disease because the IFN Signature is common in pediatric patients (Bennett et al., 2003) and in adults with central nervous system involvement and nephritis (Baechler et al., 2003). Supporting this notion, we have recently discovered that myeloid DCs and pDCs from lupus-prone Sle1,2,3 mice express an IFN Signature before the onset of autoimmunity. Even when generated in culture from bone marrow precursors, depleted of mature T and B cells, macrophages, and DCs, and thus, severed from the in vivo pro-autoimmune environment, DCs from Sle123 mice expressed an IFN Signature (Sriram et al., 2012). These results indicate that DCs are an independent cellular source of the pathogenic danger signals implicated in lupus (Elkon and Stone, 2011; Sriram et al., 2012). Since polymorphisms in genes that are part of the signaling pathway of Type I IFNs, such as IRF5, IRF7, and STAT4, are associated with a higher risk of developing SLE (Harley et al., 2009), the over-expression of Type I IFNs in these patients may be the result of the combination of genetic predisposition to produce high levels of Type I IFNs and exposure to danger signals that stimulate IFN production (Elkon and Stone, 2011; Niewold, 2011).
During tissue damage, not only are cells stressed and dying but components of the extracellular matrix (ECM) are disrupted leading to the production of low molecular weight degradation products such as hyaluronic acid (HA) and heparan sulfate, which have been shown to activate DCs and initiate inflammation (Termeer et al., 2002; Shirali and Goldstein, 2008; Brennan et al., 2012). Moreover, other components of the ECM such as fibrinogen, fibronectin extra-domain A (EDA), biglycan, and tenascin-C are up-regulated in response to tissue injury, and they all act as danger signals involved in the pathogenesis of RA (Goh and Midwood, 2011). Many of these molecules have been found increased in RA tissues; administration of danger signals, such as fibronectin EDA or tenascin-C, induces joint inflammation in vivo, while mice deficient in tenascin-C show a rapid resolution of inflammation (Goh and Midwood, 2011), indicating an important role for ECM molecules as initiator/amplifiers of the autoimmune process in RA.
The classic exogenous danger signals originate from pathogens (PAMPs), but exogenous particles not derived from microbes, such as silica, crystals, and nanoparticles can also induce DC activation and inflammatory disease. Indeed, it has long been known that inhalation of fine particulate material can cause inflammatory and fibrotic lung disease (i.e., pneumoconiosis). The inflammatory nature of non-living particulate matter, like crystals and nanoparticles, may stem in part from their hydrophobic character (Seong and Matzinger, 2004). Moreover, nanoparticles are preferentially taken up by phagocytes (Dobrovolskaia et al., 2008), including DCs, in which they cause destabilization and rupture of the phagolysosome (Hornung et al., 2008), leading to inflammasome activation (Cassel et al., 2008). Therefore, extracellular inorganic material may directly damage DCs, leading to inflammasome activation and DC maturation. This is the proposed mechanism of action for alum, a widely used adjuvant in human vaccines (Eisenbarth et al., 2008). Because of their immunogenic potential, it is important to consider the emerging use of nanomaterials to target DCs for novel therapeutics and their possible autoimmunogenic side effects.
The term nanoparticle may apply to almost any material with individual units at the nano scale. Nanomaterials can be generated from organic (e.g., liposomes, organic polymers) or inorganic materials (e.g., gold, silver) and engineered to display a molecule of interest or contain a therapeutic payload. Using nanoparticles and synthetic material systems, it may be possible to modulate the immune response through direct manipulation of DCs (Elamanchili et al., 2007).
The usefulness of pro-inflammatory nanoparticles has been demonstrated in a number of fields (Fadeel, 2012; Fadeel et al., 2012). For example, nanoparticle-based co-delivery of antigens and danger signals directly to DCs may be a promising strategy to direct anti-tumor immune responses (Kim and Mooney, 2011), as well as useful in next-generation vaccine adjuvants (Demento et al., 2009). However, because materials designed as next-generation therapies are sometimes capable of inducing DC-mediated inflammation similarly to PAMPs or DAMPs (Koike et al., 2008; Li et al., 2010), nanomaterials may raise concerns for safety (Dobrovolskaia and McNeil, 2007; Stern and McNeil, 2008). For example, nanoparticle debris from prosthetic alloys can activate the inflammasome (Caicedo et al., 2009), nanoparticles are known to modulate various forms of inflammatory cell death (Andón and Fadeel, 2013), and some nanomaterials are strong activators of complement (Salvador-Morales et al., 2006).
Additionally, it is now clear that nanoparticles do not stay “naked” when introduced in vivo – they become coated by biomolecules (e.g., proteins) to form what is termed a bio-corona (Fadeel, 2012). The composition and biophysical properties of the bio-corona is likely to have major implications for safety and effectiveness of future therapies (Aggarwal et al., 2009). The protein bio-corona first develops as a “soft” coat which eventually becomes a compact “hard” shell, becoming difficult to remove (Mohamed et al., 2012). Proteins within the hard coat are therefore aggregated and may become unfolded or modified (for example, citrullinated), possibly resulting in DCs recognizing altered self-antigen as danger signals, although evidence for this is still lacking (Fadeel, 2012).
The study of nanoparticle characteristics is also likely to uncover as yet unknown characteristics of danger signals. With the ability to control the characteristics and the delivery of the “danger signal,” we may better understand the immune response to those factors. For example, size of the nanoparticle seems to determine which DC populations interact with an injected particle: namely, larger particles require peripheral DCs for transport into the draining lymph nodes, whereas smaller particles act on lymph node resident cells directly (Manolova et al., 2008). Large particles also tend to be more inflammatory, with smaller particles often eliciting no reaction at all (Fadeel, 2012), possibly because larger sized particles more potently destabilize the phagolysosome.
Although most studies focus on the pro-inflammatory nature of particulate matter, some nanomaterials may suppress immune function (Mitchell et al., 2009; Zolnik et al., 2010) or can be engineered to evade the innate immune system (Moghimi, 2002). Indeed, gold has anti-inflammatory properties that have been exploited for many years in treatment for RA and gold nanoparticles are being engineered as novel treatments for a number of inflammatory diseases (Ulbrich and Lamprecht, 2010). Recently, Yeste et al. used gold nanoparticles to generate tolerogenic DCs in a murine model of EAE by delivering a tolerogenic compound in combination with oligodendrocyte antigen to DCs. These DCs in turn induced the generation of regulatory T cells which mitigated disease progression (Yeste et al., 2012).
Although gold may have some anti-inflammatory properties and may be useful as a delivery system, it also has many toxic side effects, earning itself a black box warning and precluding it as a practical therapy. Indeed, heavy metals can cause autoimmune-like syndromes (Schiraldi and Monestier, 2009) and therefore should be implemented cautiously as therapeutics. However, it is important to note that nanoparticles are being developed which are both biocompatible and biodegradable. For example, liposomes have been used as a delivery system to successfully treat experimental murine arthritis (Metselaar et al., 2003; Sethi et al., 2013), and biodegradable nanoparticles have been used to localize and target T cells (Fahmy et al., 2007). Nanoparticles coated with glycans have been used to target antigen to DCs (García-Vallejo et al., 2013), but the therapeutic use of DC-targeted nanoparticles in spontaneous autoimmunity remains largely unexplored (Ulbrich and Lamprecht, 2010). Nanomaterials therefore represent a promising strategy for next-generation therapy in the autoimmune diseases and an important area for future investigation to insure their safety and efficacy.
So far, we have briefly described the DAMPs investigated under the classic paradigm – cell stress/death from tissue damage, possibly induced by trauma, infections or foreign materials, provides endogenous danger signals, which propagate the immune response (Figure 1). But a number of investigations suggest the existence of endogenous danger signals that do not derive from damaged or dying cells. Rather, they are signals that alert immune cells, and possibly non-immune cells, about perturbations in the steady state of the cellular microenvironment, and we refer to these as “homeostatic” danger signals (Figure 2). Indeed, the immune system is closely tied to metabolic homeostasis (reviewed in Odegaard and Chawla, 2013; Pearce and Pearce, 2013). These homeostatic danger signals may include conditions often characteristic of chronically inflamed tissues such as localized acidosis, changes in osmolarity, decreased oxygen tension (hypoxia), oxidative stress with ROS, and other metabolic disturbances – all possible causes of cellular stress as hypothesized in the original Danger Model (Matzinger, 1994). Homeostatic changes also result from bacterial growth and acute tissue damage, further supporting the notion that cells may sense homeostatic changes as danger. Some of these homeostatic perturbations directly activate DCs, because DCs have specific sensors for these perturbations, while others lead to expression of more classic DAMPs: an example of the latter is the up-regulation of HSP like HSP70 by DCs in response to hyperthermia (40 °C), resulting in up-regulation of costimulatory molecules, pro-inflammatory cytokines, and T cell stimulation (Knippertz et al., 2011).
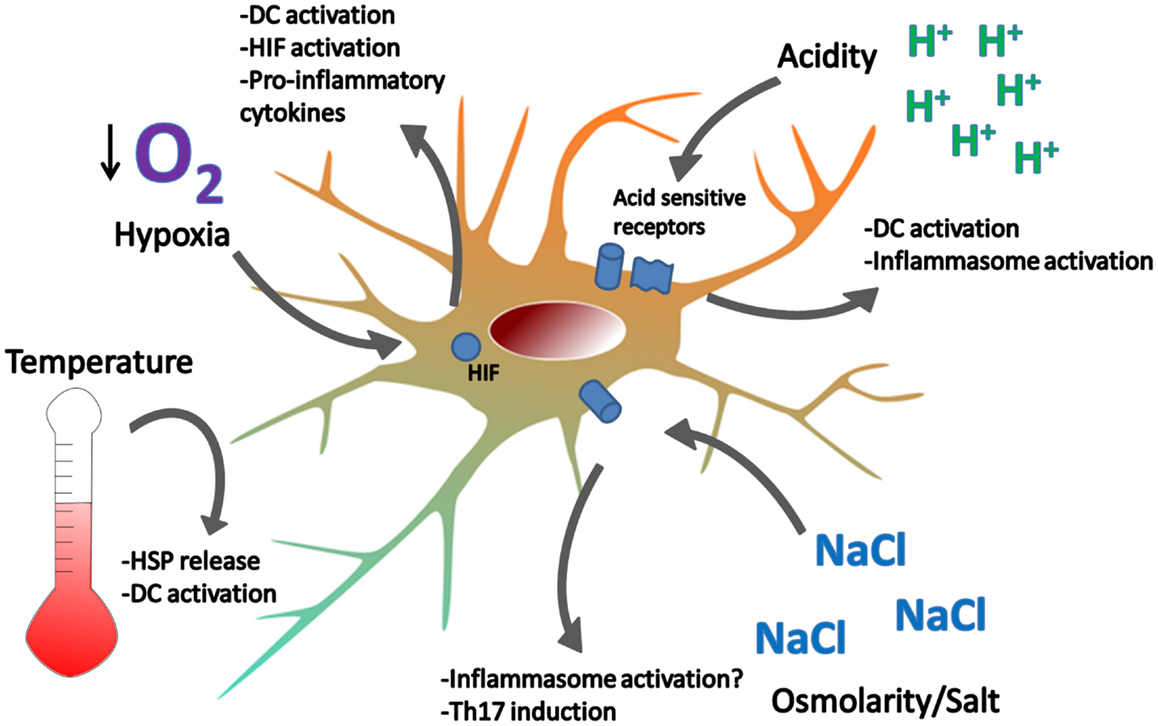
Figure 2. Examples of homeostatic danger signals. Localized perturbations in homeostasis are common in infection, ischemia-reperfusion injury, and inflammation associated with autoimmune disease. Extreme temperatures can cause DC activation and cell death. Heat induces the release of HSPs which can activate DC. Hypoxia is sensed by HIF and leads to DC activation, NF-kB signaling, and cytokine production. Acidity is sensed by acid-sensing ion channels and acid-sensing G-proteins, leading to DC activation. Hypotonicity activates the inflammasome in macrophages and high salt concentrations promote Th17 responses. How DCs respond to changes in osmolarity remains unclear.
Physiologic pH is normally held within relatively narrow range (pH 7.35–7.45). During localized inflammation (e.g., infection, arthritic joint) or tissue ischemia (e.g., lupus nephritis, MS lesions) extracellular pH can reach pH values as low as 5.5 (Treuhaft and McCarty, 1971; Nedergaard et al., 1991; Simmen and Blaser, 1993).
A significant amount of research on cell functions in acidic conditions comes from the cancer field. Tumor cells tend to use glycolysis for energy generation even when there is adequate oxygen supply – the Warburg effect (Cardone et al., 2005). The inefficiency of glycolytic metabolism, the active secretion of acid, and the poor perfusion in solid tumors is thought to contribute to an acidic microenvironment. Acidic microenvironments increase the invasiveness and metastatic potential of some cancers as a result of remodeling of the ECM (Martinez-Zaguilan et al., 1996; Rofstad et al., 2006) and activation of inflammatory processes that have been shown to promote tumor survival (Grivennikov, 2013).
Several studies have indicated that acidity might act as a danger signal. For example, extracellular acidification can promote activation of the inflammasome and secretion of IL-1β by monocytes (Jancic et al., 2011) and seems to activate neutrophils through the activation of PI3K-Akt and ERK pathways (Trevani et al., 1999; Martinez et al., 2006). Additionally, non-immune cells may also produce inflammatory cytokines in response to extracellular acidification (Shime et al., 2008; Ichimonji et al., 2010) or may alter cell death programs (Jouan-Lanhouet et al., 2012).
Acidity in the extracellular microenvironment was initially proposed to inhibit DC differentiation (Gottfried et al., 2006), but some work suggests that DCs are instead activated by acidosis (Martinez et al., 2006). Other evidence has shown that acidic environments might improve antigen uptake and antigen presentation in DCs, although the mechanisms are still unclear (Vermeulen et al., 2004). Recently, acid-induced activation of DCs was found to be in part due to acid-sensing ion channels (Tong et al., 2011), but our understanding of DC responses to protons remains limited (Lardner, 2001). Low pH may also be sensed by proton-sensing G protein-coupled receptors (Seuwen et al., 2006), as seen in responses from synovial cells (Christensen et al., 2005; Tomura et al., 2005). Acidity is a common feature of inflammation found in many autoimmune diseases (e.g. within the arthritic joint, nephritic kidney, ischemic lesions of MS). However, to our knowledge, it is still unclear whether pathways triggered by acidity are involved in DC immune functions in the context of autoimmune diseases.
Cell volume expansion, as a result of extracellular hypotonicity, may also act as a danger signal. In response to hypotonic osmotic challenge lymphocytes first swell, then undergo a process termed regulatory volume decrease (RVD) (Garber and Cahalan, 1997; Cahalan et al., 2001). A reciprocal process occurs in hypertonic environments called regulatory volume increase (RVI). RVD is largely the result of the activity of potassium and chloride channels, with efflux of KCl giving way to water efflux as a result of decreased intracellular osmolarity. In some cases KCl efflux also leads to activation of other ion channels and cellular depolarization, thus having major implications in fundamental cellular processes like metabolism and cell death. Indeed, a similar process is seen during apoptosis. Apoptotic cell volume decrease seems to be due to the activation of the same potassium and chloride channels (Maeno et al., 2000, 2006). Cellular responses resulting from homeostatic signals are complicated by the observation that ion channels form complexes with integrins and may signal together (Arcangeli and Becchetti, 2006).
Earlier studies demonstrated that hypotonicity influence immune cell function, for example in neutrophil shedding of L-selectin (Kaba and Knauf, 2001). Mammalian target of rapamycin (mTOR), in conjunction with mitochondrial metabolism, may also be an important sensor for osmotic stress, as well as other homeostatic signals (Desai et al., 2002; Kwak et al., 2012). DCs are able to pinocytose, uptake fluids from the extracellular compartment as a way to sample for antigens and danger signals. This process is very efficient in immature/resting DCs which can uptake fluids up to one thousand fold their volume (Sallusto et al., 1995). This large flow of fluids requires a tight control of the DC volume, and therefore DCs express aquaporins for this purpose (de Baey and Lanzavecchia, 2000). Inhibition of aquaporins leads to sudden increase in cell volume, which may ultimately result in DC cell death. The idea that hypotonicity may be regarded as a danger signal has recently gained credence with the finding that hypotonic solutions activate the NLRP3 inflammasome in macrophages via the efflux of K+ and Cl− ions and RVD (Compan et al., 2012). The role of alterations of cell volume in DC physiopathology remains largely unclear.
In contrast to hypotonicity, high salt concentrations do not seem to activate inflammasome activation, as hypertonic solutions are not able to induce caspase-1-dependent release of cytokines IL-18 and IL-1β (Compan et al., 2012). However, high salt concentrations may promote development of other pro-inflammatory conditions such as those associated with T helper 17 cells (Th17). Osmotic shock due to high salt was known to induce the production of IL-8 from human peripheral blood mononuclear cells (Shapiro and Dinarello, 1995), but recently two groups have demonstrated that high salt concentrations may promote pathogenic Th17 responses (Kleinewietfeld et al., 2013; Wu et al., 2013) and a high salt diet exacerbates EAE (Wu et al., 2013). These studies suggest that elevated salt concentrations, similar to levels found in tissues of animals on high salt diets, may exacerbate autoimmune reactions. It is still unclear how high salt concentrations affect DCs in vivo in the context of autoimmunity.
Decreased oxygen tension is another characteristic disturbance of inflamed or damaged tissue (Nizet and Johnson, 2009). Innate immune cells are thought to be better than adaptive immune cells at surviving in such low oxygen conditions (Sica et al., 2011), and a number of signaling pathways are relatively well known (Gloire et al., 2006; Semenza, 2009). In normoxic conditions, the α subunits of hypoxia-inducible factor (HIF), HIF-1α, and HIF-2α, are constantly produced and promptly degraded. Under hypoxic conditions, HIF-α accumulates, forms a dimer with the constitutively expressed HIF-1β subunit within the nucleus, and regulates transcription via hypoxia-responsive elements (HREs). Important gene targets are involved in adaptive response to hypoxia and include glycolytic enzymes, erythropoietin, and vascular endothelial growth factor.
A number of lines of evidence suggest that hypoxia acts as a homeostatic danger signal. Low oxygen levels leads to the generation of ROS, affecting both metabolism and mediators of inflammation. Furthermore, it is well established that HIF-1α signaling is closely tied to NF-kB signaling. In neutrophils, survival in hypoxic microenvironments, such as those found in infected tissue, is mediated by HIF-1α-dependent NF-kB activation (Walmsley et al., 2005). In a number of cell types, including DCs, hypoxia-induced HIF-1α prompts expression of TLR2 and TLR6 (Kuhlicke et al., 2007). Additionally, macrophages with a conditional deficiency in HIF-1α are compromised in their ability to clear bacteria (Cramer et al., 2003; Peyssonnaux et al., 2005), and TLR ligands like LPS may induce the HIF-NF-kB axis even under normoxic conditions (Spirig et al., 2010). Indeed, hypoxia-independent HIF induction may occur via other mechanisms, including in response to bacterial siderophores (Hartmann et al., 2008). Finally, hypoxia is also known to modulate cytokine production via HIF expression, inducing production of pro-inflammatory cytokines (Jeong et al., 2003; Nizet and Johnson, 2009) and enhancing the production of IFN-β in response to viral infection (Hwang et al., 2006).
Studies of DC responses to hypoxia are at times conflicting. It is unclear if hypoxia in itself activates DCs (Jantsch et al., 2008; Wang et al., 2010), and hypoxia may enhance (Jantsch et al., 2008) or inhibit (Mancino et al., 2008) LPS-induced costimulatory molecule expression. However, hypoxia does seem to augment DC production of pro-inflammatory cytokines, particularly in response to LPS (Jantsch et al., 2008; Mancino et al., 2008; Blengio et al., 2013). Similar activation has been demonstrated in human DCs associated with the inflamed joints of patients with juvenile idiopathic arthritis (Bosco et al., 2011).
HIF-1α expression is associated with a number of inflammatory and autoimmune diseases, including within the synovium of RA patients (Hollander et al., 2001) and in the muscle from patients with dermatomyositis and systemic sclerosis (Konttinen et al., 2004). HIF-1α is important for the development of B cells, with HIF-1α deficiency in chimeric mice leading to autoimmunity (Kojima et al., 2002). To our knowledge, the role of HIF signaling in DCs in the context of autoimmunity remains relatively unexplored.
As a number of excellent reviews on mitochondria-derived danger signals have recently been published (Arnoult et al., 2011; Krysko et al., 2011; Galluzzi et al., 2012; Rath and Haller, 2012), we can only briefly touch on this important subject. Mitochondria are key in both the recognition and propagation of danger signals and we discuss them last because, as we describe in this section, mitochondria belong to more than one category of danger signal. As ancient symbionts, mitochondria harbor a number of molecules that have bacterial origin and therefore can be considered PAMPs. At the same time, these molecules are released from the cell interior upon cell stress/death and therefore they act as DAMPs and propagate the inflammatory response. Mitochondrial-derived danger signals include mitochondrial DNA (mtDNA), which contains CpG motifs capable of stimulating TLR9, N-formyl peptides which mimic bacterial peptides, and ATP as described above (Arnoult et al., 2011). The double nature of mitochondria as PAMPs and DAMPs brings the models of Janeway and Matzinger together (Seong and Matzinger, 2004), showing how the immune system co-evolved with his “enemy within” (Zhang et al., 2010). Moreover, as mitochondria are the cell power-house, they are key players in programs of cell death and in controlling homeostatic perturbations (Desai et al., 2002; Galluzzi et al., 2012).
Mitochondrial-derived danger signals are associated with the inflammatory response upon tissue damage and have been implicated in the inflammatory response to trauma. Severe traumatic injury may result in SIRS, a life threatening condition in which extensive tissue injury leads to systemic inflammation and shock reminiscent of sepsis (Stoecklein et al., 2012). Indeed, trauma patients who have sustained major tissue injury have elevated levels of circulating mtDNA which contributes to the resultant sterile shock (Zhang et al., 2010).
Mitochondrial-derived CpGs are also an important danger signal in autoimmune disease. For example, injecting mtDNA into the joints of mice induces myeloid cell-mediated arthritis, but injection of nuclear DNA does not (Collins et al., 2004). The authors of this study also found mtDNA in the synovial fluid of patients with RA, but not from health controls (Collins et al., 2004). Recently, a mitochondrial-derived protein related to HMGB1, called mitochondrial transcription factor A, has been found to synergize with CpG to induce the production of Type I IFNs by pDCs (Julian et al., 2012).
Furthermore, as mitochondria are central to metabolism and cellular energy production, they represent a key sensor of homeostatic perturbations (Desai et al., 2002) and are known to regulate multiple cell death programs – linking homeostatic signals to cell death and release of danger signals (Galluzzi et al., 2012). Importantly, mitochondrial control of cell death includes the execution of both apoptosis and regulated necrosis, the latter a form of inflammatory cell death now known to propagate the immune response to pathogens (Vandenabeele et al., 2010). Regulated necrosis leads to the liberation of many danger signals, including those we have outlined in this review.
Perturbations in homeostasis or mitochondrial function also lead to the generation of ROS, which are an important danger signals in their own right. ROS are normal byproducts of respiration, but may also function as signaling molecules and antimicrobial agents under controlled conditions (Valko et al., 2007); as well as a danger signals under pathologic conditions (Galluzzi et al., 2012). Cells defend themselves against toxic ROS buildup using a multitude of antioxidants and enzymes including superoxide dismutase and catalase (Dröge, 2002). Superoxide dismutase converts oxygen radicals to hydrogen peroxide and oxygen; and catalase converts hydrogen peroxide to water and oxygen. However, when oxidative stress overwhelms these antioxidant mechanisms the result is activation of cell death programs (Galluzzi et al., 2012). In the context of autoimmune diseases like SLE, increased ROS may contribute to pathogenesis via modification of self-antigens, thereby increasing their auto-reactivity, and by promoting cell death resulting in the release of DAMPs (Perl et al., 2004; Oates and Gilkeson, 2006).
In addition to being a source and sensor of danger signals, mitochondrial dysfunction may directly influence the course of autoimmune and autoinflammatory diseases. This seems to be due to the participation of mitochondria in regulating the inflammasome and cell death. Indeed, inflammasome activation in the context of monogenic autoinflammatory diseases has been suggested to cause mitochondrial dysfunction (Escames et al., 2011). Mitochondrial dysfunction and subsequent cell death, in response to oxidative stress, may have a predominant role in the propagation of MS (Witte et al., 2010). Mitochondria from T cells of SLE patients also seem to be deregulated – having an elevated baseline transmembrane potential, predisposing them to necrotic cell death (Gergely et al., 2002; Perl et al., 2012) and other metabolic disturbances (Wu et al., 2012). Continued investigation of mitochondria at the cross-roads between metabolism and inflammation will very likely be important in our understanding of autoimmunity.
The immune response is often described as a double-edge sword. Too little immunity leads to uncontrolled infections, while too much immunity results in inflammatory damage, autoinflammatory disease, and some forms of autoimmunity. The delicate balance needed to keep immunity within this narrow range is the result of complex biochemical interactions. A threshold likely exists whereby tonic factors suppress noise in the system and other factors amplify or regulate immunologic signals in order to have a quick and efficient immune response (Germain, 2012). Danger signals may be both initiators of the immune response, as well as amplifiers of the immune response as a result of tissue injury and cell death. Indeed, inflammatory cell death seems to be required for proper pathogen clearance (Cho et al., 2009; Upton et al., 2010). Another layer of complexity comes in the fact that some molecules do not always act as danger signals, but rather serve other functions (e.g., HMGB1 acts as a chemokine, HSP as a chaperone, mitochondria as a power-house) and may even inhibit the immune response or promote tissue repair (Castiglioni et al., 2011; Stocki and Dickinson, 2012).
Danger signals are therefore diverse in form and function. Indeed, there are a number of conceptual frameworks in which danger signals are characterized, all of which have been extremely useful (Janeway, 1989; Matzinger, 1994). Here we have outlined, to the best of our ability, both the classic, emerging and homeostatic danger signals that influence DC biology. Although we describe some non-classic because they do not directly derive from pathogens or dead/dying cells, they are in fact well established. Man-made technologies (nanomaterials) are simply a novel form of particulate matter, which has been known for many years to induce inflammatory disease. Furthermore, the notion that homeostatic perturbations (e.g., osmotic stress, oxidative stress) act as danger signals is supported by a large body of literature (See “Mitochondria”). These data suggest that the immune system, set to recognize tissue/cell damage, can use sensors for basic physical and chemical perturbations of the tissue microenvironment to detect early causes of damage, even before the release of DAMPs. While much is now know about DC responses to exogenous and endogenous danger signals, many questions remain unresolved. Indeed, we have discussed signals as if they act independently, but we have little information on how signals interact with each other to control the immune response. Future studies will determine how the sum of these functions influences the development of autoimmunity.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank those who provided vigorous discourse and constructive feedback during the writing of this review: Drs. Roberto Caricchio, Philip Cohen, Doina Ganea, Joseph Gallo, Sergei Grivennikov, Laurie Kilpatrick, Polly Matzinger, Marc Monestier, and the members of the DC Lab Marita Chakhtoura, Jun Xu and Dr. Uma Sriram. We express a particular thank you to Polly Matzinger for her continuous inspiration and challenge.
Abdulahad, D. A., Westra, J., Bijzet, J., Limburg, P. C., Kallenberg, C. G., and Bijl, M. (2011). High mobility group box 1 (HMGB1) and anti-HMGB1 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Arthritis Res. Ther. 13, R71. doi:10.1186/ar3332
Adachi, Y., Taketani, S., Toki, J., Ikebukuro, K., Sugiura, K., Oyaizu, H., et al. (2002). Marked increase in number of dendritic cells in autoimmune-prone (NZW x BXSB)F1 mice with age. Stem Cells 20, 61–72. doi:10.1634/stemcells.20-1-61
Aggarwal, P., Hall, J. B., McLeland, C. B., Dobrovolskaia, M. A., and Mcneil, S. E. (2009). Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 61, 428–437. doi:10.1016/j.addr.2009.03.009
Amodio, G., and Gregori, S. (2012). Dendritic cells a double-edge sword in autoimmune responses. Front. Immunol. 3:233. doi:10.3389/fimmu.2012.00233
Andersson, A., Covacu, R., Sunnemark, D., Danilov, A., Dal Bianco, A., Khademi, M., et al. (2008). Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J. Leukoc. Biol. 84, 1248–1255. doi:10.1189/jlb.1207844
Andón, F. T., and Fadeel, B. (2013). Programmed cell death: molecular mechanisms and implications for safety assessment of nanomaterials. Acc. Chem. Res. 46, 733–742.
Arcangeli, A., and Becchetti, A. (2006). Complex functional interaction between integrin receptors and ion channels. Trends Cell Biol. 16, 631–639. doi:10.1016/j.tcb.2006.10.003
Arnoult, D., Soares, F., Tattoli, I., and Girardin, S. E. (2011). Mitochondria in innate immunity. EMBO Rep. 12, 901–910. doi:10.1038/embor.2011.157
Atarashi, K., Nishimura, J., Shima, T., Umesaki, Y., Yamamoto, M., Onoue, M., et al. (2008). ATP drives lamina propria T(H)17 cell differentiation. Nature 455, 808–812. doi:10.1038/nature07240
Aymeric, L., Apetoh, L., Ghiringhelli, F., Tesniere, A., Martins, I., Kroemer, G., et al. (2012). Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 70, 855–858. doi:10.1158/0008-5472.CAN-09-3566
Baechler, E. C., Batliwalla, F. M., Karypis, G., Gaffney, P. M., Ortmann, W. A., Espe, K. J., et al. (2003). Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. U.S.A. 100, 2610–2615. doi:10.1073/pnas.0337679100
Banchereau, J., Briere, F., Caux, C., Davoust, J., Lebecque, S., Liu, Y. J., et al. (2000). Immunobiology of dendritic cells. Annu. Rev. Immunol. 18, 767–811. doi:10.1146/annurev.immunol.18.1.767
Barrat, F. J., Meeker, T., Chan, J. H., Guiducci, C., and Coffman, R. L. (2007). Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur. J. Immunol. 37, 3582–3586. doi:10.1002/eji.200737815
Barrat, F. J., Meeker, T., Gregorio, J., Chan, J. H., Uematsu, S., Akira, S., et al. (2005). Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 202, 1131–1139. doi:10.1084/jem.20050914
Basu, S., Binder, R. J., Suto, R., Anderson, K. M., and Srivastava, P. K. (2000). Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 12, 1539–1546. doi:10.1093/intimm/12.11.1539
Behrens, E. M., Sriram, U., Shivers, D. K., Gallucci, M., Ma, Z., Finkel, T. H., et al. (2007). Complement receptor 3 ligation of dendritic cells suppresses their stimulatory capacity. J. Immunol. 178, 6268–6279.
Bennett, L., Palucka, A. K., Arce, E., Cantrell, V., Borvak, J., Banchereau, J., et al. (2003). Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197, 711–723. doi:10.1084/jem.20021553
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5. doi:10.1189/jlb.0306164
Bianchi, M. E., Beltrame, M., and Paonessa, G. (1989). Specific recognition of cruciform DNA by nuclear protein HMG1. Science 243, 1056–1059. doi:10.1126/science.2922595
Binder, R. J., Han, D. K., and Srivastava, P. K. (2000). CD91: a receptor for heat shock protein gp96. Nat. Immunol. 1, 151–155. doi:10.1038/77835
Biswas, C., Sriram, U., Ciric, B., Ostrovsky, O., Gallucci, S., and Argon, Y. (2006). The N-terminal fragment of GRP94 is sufficient for peptide presentation via professional antigen-presenting cells. Int. Immunol. 18, 1147–1157. doi:10.1093/intimm/dxl049
Blander, J. M., and Medzhitov, R. (2004). Regulation of phagosome maturation by signals from toll-like receptors. Science 304, 1014–1018. doi:10.1126/science.1096158
Blengio, F., Raggi, F., Pierobon, D., Cappello, P., Eva, A., Giovarelli, M., et al. (2013). The hypoxic environment reprograms the cytokine/chemokine expression profile of human mature dendritic cells. Immunobiology 218, 76–89. doi:10.1016/j.imbio.2012.02.002
Bonaldi, T., Talamo, F., Scaffidi, P., Ferrera, D., Porto, A., Bachi, A., et al. (2003). Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 22, 5551–5560. doi:10.1093/emboj/cdg516
Bondanza, A., Zimmermann, V. S., Dell’Antonio, G., Dal Cin, E., Capobianco, A., Sabbadini, M. G., et al. (2003). Cutting edge: dissociation between autoimmune response and clinical disease after vaccination with dendritic cells. J. Immunol. 170, 24–27.
Bosco, M. C., Pierobon, D., Blengio, F., Raggi, F., Vanni, C., Gattorno, M., et al. (2011). Hypoxia modulates the gene expression profile of immunoregulatory receptors in human mature dendritic cells: identification of TREM-1 as a novel hypoxic marker in vitro and in vivo. Blood 117, 2625–2639. doi:10.1182/blood-2010-06-292136
Brennan, T. V., Lin, L., Huang, X., Cardona, D. M., Li, Z., Dredge, K., et al. (2012). Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood 120, 2899–2908. doi:10.1182/blood-2011-07-368720
Cahalan, M. D., Wulff, H., and Chandy, K. G. (2001). Molecular properties and physiological roles of ion channels in the immune system. J. Clin. Immunol. 21, 235–252. doi:10.1023/A:1010958907271
Caicedo, M. S., Desai, R., Mcallister, K., Reddy, A., Jacobs, J. J., and Hallab, N. J. (2009). Soluble and particulate Co-Cr-Mo alloy implant metals activate the inflammasome danger signaling pathway in human macrophages: a novel mechanism for implant debris reactivity. J. Orthop. Res. 27, 847–854. doi:10.1002/jor.20826
Cardone, R. A., Casavola, V., and Reshkin, S. J. (2005). The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 5, 786–795. doi:10.1038/nrc1713
Cassel, S. L., Eisenbarth, S. C., Iyer, S. S., Sadler, J. J., Colegio, O. R., Tephly, L. A., et al. (2008). The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. U.S.A. 105, 9035–9040. doi:10.1073/pnas.0803933105
Castiglioni, A., Canti, V., Rovere-Querini, P., and Manfredi, A. A. (2011). High-mobility group box 1 (HMGB1) as a master regulator of innate immunity. Cell Tissue Res. 343, 189–199. doi:10.1007/s00441-010-1033-1
Cella, M., Engering, A., Pinet, V., Pieters, J., and Lanzavecchia, A. (1997). Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature 388, 782–787. doi:10.1038/42030
Chen, M., Wang, Y. H., Wang, Y., Huang, L., Sandoval, H., Liu, Y. J., et al. (2006). Dendritic cell apoptosis in the maintenance of immune tolerance. Science 311, 1160–1164. doi:10.1126/science.1122545
Chiba, S., Baghdadi, M., Akiba, H., Yoshiyama, H., Kinoshita, I., Dosaka-Akita, H., et al. (2012). Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 13, 832–842. doi:10.1038/ni.2376
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. doi:10.1016/j.cell.2009.05.037
Christensen, B. N., Kochukov, M., Mcnearney, T. A., Taglialatela, G., and Westlund, K. N. (2005). Proton-sensing G protein-coupled receptor mobilizes calcium in human synovial cells. Am. J. Physiol. Cell Physiol. 289, C601–C608. doi:10.1152/ajpcell.00039.2005
Collins, L., Hajizadeh, S., Holme, E., Jonsson, I.-M., and Tarkowski, A. (2004). Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. leukoc. Biol. 75, 995–1000. doi:10.1189/jlb.0703328
Colonna, L., Dinnall, J. A., Shivers, D. K., Frisoni, L., Caricchio, R., and Gallucci, S. (2006). Abnormal costimulatory phenotype and function of dendritic cells before and after the onset of severe murine lupus. Arthritis Res. Ther. 8, R49. doi:10.1186/ar1911
Compan, V., Baroja-Mazo, A., López-Castejón, G., Gomez, A. I., Martínez, C. M., Angosto, D., et al. (2012). Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37, 487–500. doi:10.1016/j.immuni.2012.06.013
Cramer, T., Yamanishi, Y., Clausen, B. R. E., Förster, I., Pawlinski, R., Mackman, N., et al. (2003). HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657. doi:10.1016/S0092-8674(03)00154-5
Crow, M. K., Kirou, K. A., and Wohlgemuth, J. (2003). Microarray analysis of interferon-regulated genes in SLE. Autoimmunity 36, 481–490. doi:10.1080/08916930310001625952
Dandona, P., Ghanim, H., Green, K., Sia, C., Abuaysheh, S., Kuhadiya, N., et al. (2013). Insulin infusion suppresses while glucose infusion induces Toll-like receptors and high-mobility group-B1 protein expression in mononuclear cells of type 1 diabetes patients. Am. J. Physiol. Endocrinol. Metab. 304, 8. doi:10.1152/ajpendo.00566.2012
de Baey, A., and Lanzavecchia, A. (2000). The role of aquaporins in dendritic cell macropinocytosis. J. Exp. Med. 191, 743–748. doi:10.1084/jem.191.4.743
Debler, J., Schiemann, U., Seybold, U., Mussack, T., Landauer, N., Ladurner, R., et al. (2003). Heat-shock protein HSP70-2 genotypes in patients with Crohn’s disease: a more severe clinical course with intestinal complications in presence of the PstI-polymorphism. Eur. J. Med. Res. 8, 120–124.
Demento, S. L., Eisenbarth, S. C., Foellmer, H. G., Platt, C., Caplan, M. J., Mark Saltzman, W., et al. (2009). Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine 27, 3013–3021. doi:10.1016/j.vaccine.2009.03.034
Desai, B. N., Myers, B. R., and Schreiber, S. L. (2002). FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc. Natl. Acad. Sci. U.S.A. 99, 4319–4324. doi:10.1073/pnas.261702698
Dobrovolskaia, M. A., Aggarwal, P., Hall, J. B., and Mcneil, S. E. (2008). Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol. Pharm. 5, 487–495. doi:10.1021/mp800032f
Dobrovolskaia, M. A., and McNeil, S. E. (2007). Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2, 469–478. doi:10.1038/nnano.2007.223
Dröge, W. (2002). Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95. doi:10.1152/physrev.00018.2001
Dumitriu, I. E., Baruah, P., Valentinis, B., Voll, R. E., Herrmann, M., Nawroth, P. P., et al. (2005). Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 174, 7506–7515. doi:10.1038/nature06939
Eisenbarth, S. C., Colegio, O. R., O’Connor, W., Sutterwala, F. S., and Flavell, R. A. (2008). Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126. doi:10.1038/nature06939
Elamanchili, P., Lutsiak, C. M. E., Hamdy, S., Diwan, M., and Samuel, J. (2007). “Pathogen-mimicking” nanoparticles for vaccine delivery to dendritic cells. J. Immunother. 30, 378–395. doi:10.1097/CJI.0b013e31802cf3e3
Elkon, K. B., and Stone, V. V. (2011). Type I interferon and systemic lupus erythematosus. J. Interferon Cytokine Res. 31, 803–812. doi:10.1089/jir.2011.0045
Elliott, M. R., Chekeni, F. B., Trampont, P. C., Lazarowski, E. R., Kadl, A., Walk, S. F., et al. (2009). Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286. doi:10.1038/nature08296
Eloranta, M. L., Lovgren, T., Finke, D., Mathsson, L., Ronnelid, J., Kastner, B., et al. (2009). Regulation of the interferon-alpha production induced by RNA-containing immune complexes in plasmacytoid dendritic cells. Arthritis Rheum. 60, 2418–2427. doi:10.1002/art.24686
Eriksson, U., Ricci, R., Hunziker, L., Kurrer, M. O., Oudit, G. Y., Watts, T. H., et al. (2003). Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 9, 1484–1490. doi:10.1038/nm960
Escames, G., López, L. C., García, J. A., García-Corzo, L., Ortiz, F., and Acuña-Castroviejo, D. (2011). Mitochondrial DNA and inflammatory diseases. Hum. Genet. 131, 161–173. doi:10.1007/s00439-011-1057-y
Fadeel, B. (2012). Clear and present danger? Engineered nanoparticles and the immune system. Swiss Med. Wkly. 142:w13609. doi:10.4414/smw.2012.13609
Fadeel, B., Pietroiusti, A., and Shvedova, A. (2012). Adverse Effects of Engineered Nanomaterials: Exposure, Toxicology, and Impact on Human Health. Boston: Academic Press.
Fahmy, T. M., Fong, P. M., Park, J., Constable, T., and Saltzman, W. M. (2007). Nanosystems for simultaneous imaging and drug delivery to T cells. AAPS J. 9, E171–180. doi:10.1208/aapsj0902019
Feili-Hariri, M., Falkner, D. H., and Morel, P. A. (2002). Regulatory Th2 response induced following adoptive transfer of dendritic cells in prediabetic NOD mice. Eur. J. Immunol. 32, 2021–2030. doi:10.1002/1521-4141(200207)32:7<2021::AID-IMMU2021>3.0.CO;2-J
Flohe’, S. B., Bruggemann, J., Lendemans, S., Nikulina, M., Meierhoff, G., Flohe’, S., et al. (2003). Human heat shock protein 60 induces maturation of dendritic cells versus a Th1-promoting phenotype. J. Immunol. 170, 2340–2348.
Frisoni, L., Mcphie, L., Colonna, L., Sriram, U., Monestier, M., Gallucci, S., et al. (2005). Nuclear autoantigen translocation and autoantibody opsonization lead to increased dendritic cell phagocytosis and presentation of nuclear antigens: a novel pathogenic pathway for autoimmunity? J. Immunol. 175, 2692–2701.
Furnrohr, B. G., Wach, S., Kelly, J. A., Haslbeck, M., Weber, C. K., Stach, C. M., et al. (2010). Polymorphisms in the Hsp70 gene locus are genetically associated with systemic lupus erythematosus. Ann. Rheum. Dis. 69, 1983–1989. doi:10.1136/ard.2009.122630
Gall, A., Treuting, P., Elkon, K. B., Loo, Y. M., Gale, M. Jr., Barber, G. N., et al. (2012). Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131. doi:10.1016/j.immuni.2011.11.018
Gallucci, S., Lolkema, M., and Matzinger, P. (1999). Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5, 1249–1255. doi:10.1038/15200
Gallucci, S., and Matzinger, P. (2001). Danger signals: SOS to the immune system. Curr. Opin. Immunol. 13, 114–119. doi:10.1016/S0952-7915(00)00191-6
Galluzzi, L., Kepp, O., and Kroemer, G. (2012). Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788. doi:10.1038/nrm3479
Garber, S. S., and Cahalan, M. D. (1997). Volume-regulated anion channels and the control of a simple cell behavior. Cell. Physiol. Biochem. 7, 229–241. doi:10.1159/000154877
Garcia-Romo, G. S., Caielli, S., Vega, B., Connolly, J., Allantaz, F., Xu, Z., et al. (2011). Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra20. doi:10.1126/scitranslmed.3001201
García-Vallejo, J., Ambrosini, M., Overbeek, A., Van Riel, W., Bloem, K., Unger, W., et al. (2013). Multivalent glycopeptide dendrimers for the targeted delivery of antigens to dendritic cells. Mol. Immunol. 53, 387–397. doi:10.1016/j.molimm.2012.09.012
Gardella, S., Andrei, C., Ferrera, D., Lotti, L. V., Torrisi, M. R., Bianchi, M. E., et al. (2002). The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 3, 995–1001. doi:10.1093/embo-reports/kvf198
Gergely, P. Jr., Grossman, C., Niland, B., Puskas, F., Neupane, H., Allam, F., et al. (2002). Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 46, 175–190. doi:10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
Germain, R. N. (2012). Maintaining system homeostasis: the third law of Newtonian immunology. Nat. Immunol. 13, 902–906. doi:10.1038/ni.2404
Gerner, M. Y., Kastenmuller, W., Ifrim, I., Kabat, J., and Germain, R. N. (2012). Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 37, 364–376. doi:10.1016/j.immuni.2012.07.011
Ghiringhelli, F., Apetoh, L., Tesniere, A., Aymeric, L., Ma, Y., Ortiz, C., et al. (2009). Activation of the NLRP3 inflammasome in dendritic cells induces IL-1Œ ≤,Äí dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178. doi:10.1038/nm.2028
Ginhoux, F., Ng, L. G., and Merad, M. (2012). Understanding the murine cutaneous dendritic cell network to improve intradermal vaccination strategies. Curr. Top. Microbiol. Immunol. 351, 1–24. doi:10.1007/82_2010_115
Gloire, G., Legrand-Poels, S., and Piette, J. (2006). NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 72, 1493–1505. doi:10.1016/j.bcp.2006.04.011
Goh, F. G., and Midwood, K. S. (2011). Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis. Rheumatology (Oxford) 51, 7–23. doi:10.1093/rheumatology/ker257
Goldstein, R. S., Gallowitsch-Puerta, M., Yang, L., Rosas-Ballina, M., Huston, J. M., Czura, C. J., et al. (2006). Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock 25, 571–574. doi:10.1097/01.shk.0000209540.99176.72
Gottfried, E., Kunz-Schughart, L. A., Ebner, S., Mueller-Klieser, W., Hoves, S., Andreesen, R., et al. (2006). Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107, 2013–2021. doi:10.1182/blood-2005-05-1795
Gris, D., Ye, Z., Iocca, H. A., Wen, H., Craven, R. R., Gris, P., et al. (2010). NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 185, 974–981. doi:10.4049/jimmunol.0904145
Grivennikov, S. I. (2013). Inflammation and colorectal cancer: colitis-associated neoplasia. Semin. Immunopathol. 35, 229–244. doi:10.1007/s00281-012-0352-6
Gunther, C., Meurer, M., Stein, A., Viehweg, A., and Lee-Kirsch, M. A. (2009). Familial chilblain lupus – a monogenic form of cutaneous lupus erythematosus due to a heterozygous mutation in TREX1. Dermatology (Basel) 219, 162–166. doi:10.1159/000222430
Hammer, G. E., and Ma, A. (2013). Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu. Rev. Immunol. 31, 743–791. doi:10.1146/annurev-immunol-020711-074929
Han, J., Zhong, J., Wei, W., Wang, Y., Huang, Y., Yang, P., et al. (2008). Extracellular high-mobility group box 1 acts as an innate immune mediator to enhance autoimmune progression and diabetes onset in NOD mice. Diabetes 57, 2118–2127. doi:10.2337/db07-1499
Harley, I. T., Kaufman, K. M., Langefeld, C. D., Harley, J. B., and Kelly, J. A. (2009). Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nat. Rev. Genet. 10, 285–290. doi:10.1038/nrg2571
Harris, H. E., Andersson, U., and Pisetsky, D. S. (2012). HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol 8, 195–202. doi:10.1038/nrrheum.2011.222
Hartmann, H., Eltzschig, H. K., Wurz, H., Hantke, K., Rakin, A., Yazdi, A. S., et al. (2008). Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology 134, 756–767. doi:10.1053/j.gastro.2007.12.008
Heil, M., Ibarra-Laclette, E., Adame-Alvarez, R. M., Martinez, O., Ramirez-Chavez, E., Molina-Torres, J., et al. (2012). How plants sense wounds: damaged-self recognition is based on plant-derived elicitors and induces octadecanoid signaling. PLoS ONE 7:e30537. doi:10.1371/journal.pone.0030537
Hilbi, H. (2009). Bacterial jailbreak sounds cellular alarm: phagosome membrane remnants trigger signaling. Cell Host Microbe 6, 102–104. doi:10.1016/j.chom.2009.08.001
Hollander, A. P., Corke, K. P., Freemont, A. J., and Lewis, C. E. (2001). Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 44, 1540–1544. doi:10.1002/1529-0131(200107)44:7¡1540::AID-ART277.3.0.CO;2-7
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856. doi:10.1038/ni.1631
Hur, J. W., Sung, Y. K., Shin, H. D., Park, B. L., Cheong, H. S., and Bae, S. C. (2008). TREX1 polymorphisms associated with autoantibodies in patients with systemic lupus erythematosus. Rheumatol. Int. 28, 783–789. doi:10.1007/s00296-007-0509-0
Hwang, I. I. L., Watson, I. R., Der, S. D., and Ohh, M. (2006). Loss of VHL confers hypoxia-inducible factor (HIF)-dependent resistance to vesicular stomatitis virus: role of HIF in antiviral response. J. Virol. 80, 10712–10723. doi:10.1128/JVI.01014-06
Ichimonji, I., Tomura, H., Mogi, C., Sato, K., Aoki, H., Hisada, T., et al. (2010). Extracellular acidification stimulates IL-6 production and Ca(2+) mobilization through proton-sensing OGR1 receptors in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 299, L567–L577. doi:10.1152/ajplung.00415.2009
Iyer, S. S., Pulskens, W. P., Sadler, J. J., Butter, L. M., Teske, G. J., Ulland, T. K., et al. (2009). Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 106, 20388–20393. doi:10.1073/pnas.0908698106
Jancic, C. C., Cabrini, M., Gabelloni, M. L., Rodríguez Rodrigues, C., Salamone, G., Trevani, A. S., et al. (2011). Low extracellular pH stimulates the production of IL-1Œ ≤ by human monocytes. Cytokine 57, 258–268. doi:10.1016/j.cyto.2011.11.013
Janeway, C. A. Jr. (1992). The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Today 13, 11–16. doi:10.1016/0167-5699(92)90198-G
Janeway, C. J. (1989). Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 54, 1–13. doi:10.1101/SQB.1989.054.01.003
Jantsch, J., Chakravortty, D., Turza, N., Prechtel, A. T., Buchholz, B. R., Gerlach, R. G., et al. (2008). Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 180, 4697–4705.
Javaherian, K., Liu, J. F., and Wang, J. C. (1978). Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science 199, 1345–1346. doi:10.1126/science.628842
Jeong, H.-J., Chung, H.-S., Lee, B.-R., Kim, S.-J., Yoo, S.-J., Hong, S.-H., et al. (2003). Expression of proinflammatory cytokines via HIF-1alpha and NF-kappaB activation on desferrioxamine-stimulated HMC-1 cells. Biochem. Biophys. Res. Commun. 306, 805–811. doi:10.1016/S0006-291X(03)01073-8
Jiang, W., and Pisetsky, D. S. (2008). Expression of high mobility group protein 1 in the sera of patients and mice with systemic lupus erythematosus. Ann. Rheum. Dis. 67, 727–728. doi:10.1136/ard.2007.074484
Jouan-Lanhouet, S., Arshad, M. I., Piquet-Pellorce, C., Martin-Chouly, C., Le Moigne-Muller, G., Van Herreweghe, F., et al. (2012). TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 19, 2003–2014. doi:10.1038/cdd.2012.90
Julian, M., Shao, G., Bao, S., Knoell, D., Papenfuss, T., Vangundy, Z., et al. (2012). Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J. immunol. 189, 433–443. doi:10.4049/jimmunol.1101375
Kaba, N. K., and Knauf, P. A. (2001). Hypotonicity induces L-selectin shedding in human neutrophils. Am. J. Physiol. Cell Physiol. 281, C1403–C1407.
Kalled, S. L., Cutler, A. H., and Burkly, L. C. (2001). Apoptosis and altered dendritic cell homeostasis in lupus nephritis are limited by anti-CD154 treatment. J. Immunol. 167, 1740–1747.
Katzourakis, A., and Gifford, R. J. (2010). Endogenous viral elements in animal genomes. PLoS Genet. 6:e1001191. doi:10.1371/journal.pgen.1001191
Kim, J., and Mooney, D. J. (2011). In vivo modulation of dendritic cells by engineered materials: towards new cancer vaccines. Nano Today 6, 466–477. doi:10.1016/j.nantod.2011.08.005
Kleinewietfeld, M., Manzel, A., Titze, J., Kvakan, H., Yosef, N., Linker, R., et al. (2013). Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496, 518–522. doi:10.1038/nature11868
Knippertz, I., Stein, M. F., Dorrie, J., Schaft, N., Muller, I., Deinzer, A., et al. (2011). Mild hyperthermia enhances human monocyte-derived dendritic cell functions and offers potential for applications in vaccination strategies. Int. J. Hyperthermia 27, 591–603. doi:10.3109/02656736.2011.589234
Koike, E., Takano, H., Inoue, K.-I., Yanagisawa, R., and Kobayashi, T. (2008). Carbon black nanoparticles promote the maturation and function of mouse bone marrow-derived dendritic cells. Chemosphere 73, 371–376. doi:10.1016/j.chemosphere.2008.05.054
Kojima, H., Gu, H., Nomura, S., Caldwell, C. C., Kobata, T., Carmeliet, P., et al. (2002). Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha-deficient chimeric mice. Proc. Natl. Acad. Sci. U.S.A. 99, 2170–2174. doi:10.1073/pnas.052706699
Kokkola, R., Andersson, A., Mullins, G., Ostberg, T., Treutiger, C. J., Arnold, B., et al. (2005). RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 61, 1–9. doi:10.1111/j.0300-9475.2005.01534.x
Kokkola, R., Li, J., Sundberg, E., Aveberger, A. C., Palmblad, K., Yang, H., et al. (2003). Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 48, 2052–2058. doi:10.1002/art.11161
Konttinen, Y. T., Mackiewicz, Z., Povilenaite, D., Sukura, A., Hukkanen, M., and Virtanen, I. (2004). Disease-associated increased HIF-1, alphavbeta3 integrin, and Flt-1 do not suffice to compensate the damage-inducing loss of blood vessels in inflammatory myopathies. Rheumatol. Int. 24, 333–339. doi:10.1007/s00296-003-0379-z
Krysko, D. V., Agostinis, P., Krysko, O., Garg, A. D., Bachert, C., Lambrecht, B. N., et al. (2011). Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164. doi:10.1016/j.it.2011.01.005
Kuhlicke, J., Frick, J. S., Morote-Garcia, J. C., Rosenberger, P., and Eltzschig, H. K. (2007). Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like receptors TLR2 and TLR6 during hypoxia. PLoS ONE 2:e1364. doi:10.1371/journal.pone.0001364
Kwak, D., Choi, S., Jeong, H., Jang, J.-H., Lee, Y., Jeon, H., et al. (2012). Osmotic stress regulates mammalian target of rapamycin (mTOR) complex 1 via c-Jun N-terminal Kinase (JNK)-mediated Raptor protein phosphorylation. J. Biol. Chem. 287, 18398–18407. doi:10.1074/jbc.M111.326538
Lamkanfi, M., and Dixit, V. M. (2012). Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161. doi:10.1146/annurev-cellbio-101011-155745
Land, W. G., and Messmer, K. (2012). The danger theory in view of the injury hypothesis: 20 years later. Front. Immunol. 3:349. doi:10.3389/fimmu.2012.00349
Lang, P., Merkler, D., Funkner, P., Shaabani, N., Meryk, A., Krings, C., et al. (2010). Oxidized ATP inhibits T-cell-mediated autoimmunity. Eur. J. Immunol. 40, 2401–2408. doi:10.1002/eji.200939838
Lardner, A. (2001). The effects of extracellular pH on immune function. J. Leukoc. Biol. 69, 522–530.
Leadbetter, E. A., Rifkin, I. R., Hohlbaum, A. M., Beaudette, B. C., Shlomchik, M. J., and Marshak-Rothstein, A. (2002). Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607. doi:10.1038/416603a
Lee-Kirsch, M. A., Chowdhury, D., Harvey, S., Gong, M., Senenko, L., Engel, K., et al. (2007a). A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J. Mol. Med. (Berl.) 85, 531–537. doi:10.1007/s00109-007-0199-9
Lee-Kirsch, M. A., Gong, M., Chowdhury, D., Senenko, L., Engel, K., Lee, Y. A., et al. (2007b). Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39, 1065–1067. doi:10.1038/ng2091
Li, A., Qin, L., Zhu, D., Zhu, R., Sun, J., and Wang, S. (2010). Signalling pathways involved in the activation of dendritic cells by layered double hydroxide nanoparticles. Biomaterials 31, 748–756. doi:10.1016/j.biomaterials.2009.09.095
Lin, M., Yiu, W., Wu, H., Chan, L. Y., Leung, J., Au, W., et al. (2012). Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J. Am. Soc. Nephrol. 23, 86–102. doi:10.1681/ASN.2010111210
Liu, B., Dai, J., Zheng, H., Stoilova, D., Sun, S., and Li, Z. (2003). Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc. Natl. Acad. Sci. U.S.A. 100, 15824–15829. doi:10.1073/pnas.2635458100
Liu, Y. J. (2005). IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 23, 275–306. doi:10.1146/annurev.immunol.23.021704.115633
Lotze, M. T., and Tracey, K. J. (2005). High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342. doi:10.1038/nri1594
Maeno, E., Ishizaki, Y., Kanaseki, T., Hazama, A., and Okada, Y. (2000). Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 9487–9492. doi:10.1073/pnas.140216197
Maeno, E., Shimizu, T., and Okada, Y. (2006). Normotonic cell shrinkage induces apoptosis under extracellular low Cl conditions in human lymphoid and epithelial cells. Acta Physiol (Oxf.) 187, 217–222. doi:10.1111/j.1748-1716.2006.01554.x
Mancino, A., Schioppa, T., Larghi, P., Pasqualini, F., Nebuloni, M., Chen, I. H., et al. (2008). Divergent effects of hypoxia on dendritic cell functions. Blood 112, 3723–3734. doi:10.1182/blood-2008-02-142091
Manolova, V., Flace, A., Bauer, M., Schwarz, K., Saudan, P., and Bachmann, M. F. (2008). Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 38, 1404–1413. doi:10.1002/eji.200737984
Mariathasan, S., Newton, K., Monack, D., Vucic, D., French, D., Lee, W., et al. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218. doi:10.1038/nature02664
Marriott, I., Inscho, E. W., and Bost, K. L. (1999). Extracellular uridine nucleotides initiate cytokine production by murine dendritic cells. Cell. Immunol. 195, 147–156. doi:10.1006/cimm.1999.1531
Marshak-Rothstein, A., and Rifkin, I. R. (2007). Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu. Rev. Immunol. 25, 419–441. doi:10.1146/annurev.immunol.22.012703.104514
Martinez, D., Vermeulen, M., Trevani, A. A., Ceballos, A., Sabatté, J., Gamberale, R., et al. (2006). Extracellular acidosis induces neutrophil activation by a mechanism dependent on activation of phosphatidylinositol 3-kinase/Akt and ERK pathways. J. Immunol. 176, 1163–1171.
Martinez-Zaguilan, R., Seftor, E. A., Seftor, R. E., Chu, Y. W., Gillies, R. J., and Hendrix, M. J. (1996). Acidic pH enhances the invasive behavior of human melanoma cells. Clin. Exp. Metastasis 14, 176–186. doi:10.1007/BF00121214
Martinon, F., Pétrilli, V., Mayor, A., Tardivel, A., and Tschopp, J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. doi:10.1038/nature04516
Matzinger, P. (1994). Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12, 991–1045. doi:10.1146/annurev.immunol.12.1.991
Matzinger, P. (1998). An innate sense of danger. Semin. Immunol. 10, 399–415. doi:10.1006/smim.1998.0143
Matzinger, P. (2002). The danger model: a renewed sense of self. Science 296, 301–305. doi:10.1126/science.1071059
Medzhitov, R., and Janeway, C. A. Jr. (1997a). Innate immunity: the virtues of a nonclonal system of recognition. Cell 91, 295–298. doi:10.1016/S0092-8674(00)80412-2
Medzhitov, R., and Janeway, C. A. Jr. (1997b). Innate immunity: impact on the adaptive immune response. Curr. Opin. Immunol. 9, 4–9. doi:10.1016/S0952-7915(97)80152-5
Medzhitov, R., Preston-Hurlburt, P., and Janeway, C. A. Jr. (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397. doi:10.1038/41131
Melli, K., Friedman, R. S., Martin, A. E., Finger, E. B., Miao, G., Szot, G. L., et al. (2009). Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J. Immunol. 182, 2590–2600. doi:10.4049/jimmunol.0803543
Menges, M., Rossner, S., Voigtlander, C., Schindler, H., Kukutsch, N. A., Bogdan, C., et al. (2002). Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J. Exp. Med. 195, 15–21. doi:10.1084/jem.20011341
Metselaar, J. M., Wauben, M. H., Wagenaar-Hilbers, J. P., Boerman, O. C., and Storm, G. (2003). Complete remission of experimental arthritis by joint targeting of glucocorticoids with long-circulating liposomes. Arthritis Rheum. 48, 2059–2066. doi:10.1002/art.11140
Millar, D. G., Garza, K. M., Odermatt, B., Elford, A. R., Ono, N., Li, Z., et al. (2003). Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat. Med. 9, 1469–1476. doi:10.1038/nm962
Miller, J. C., Brown, B. D., Shay, T., Gautier, E. L., Jojic, V., Cohain, A., et al. (2012). Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 13, 888–899. doi:10.1038/ni.2370
Mills, K. H. (2011). TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 11, 807–822. doi:10.1038/nri3095
Mitchell, L. A., Lauer, F. T., Burchiel, S. W., and Mcdonald, J. D. (2009). Mechanisms for how inhaled multiwalled carbon nanotubes suppress systemic immune function in mice. Nat. Nanotechnol. 4, 451–456. doi:10.1038/nnano.2009.151
Moghimi, S. M. (2002). Chemical camouflage of nanospheres with a poorly reactive surface: towards development of stealth and target-specific nanocarriers. Biochim. Biophys. Acta 1590, 131–139. doi:10.1016/S0167-4889(02)00204-5
Mohamed, B. M., Verma, N. K., Davies, A. M., Mcgowan, A., Crosbie-Staunton, K., Prina-Mello, A., et al. (2012). Citrullination of proteins: a common post-translational modification pathway induced by different nanoparticles in vitro and in vivo. Nanomed. 7, 1181–1195. doi:10.2217/nnm.11.177
Morita, M., Stamp, G., Robins, P., Dulic, A., Rosewell, I., Hrivnak, G., et al. (2004). Gene-targeted mice lacking the Trex1 (DNase III) 3′ →5′DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 24, 6719–6727. doi:10.1128/MCB.24.15.6719-6727.2004
Muller, T., Robaye, B., Vieira, R. P., Ferrari, D., Grimm, M., Jakob, T., et al. (2010). The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy 65, 1545–1553. doi:10.1111/j.1398-9995.2010.02426.x
Muslimani, A., Chisti, M. M., Wills, S., Nadeau, L., Zakalik, D., Daw, H., et al. (2011). How we treat tumor lysis syndrome. Oncology (Williston Park, N.Y.) 25, 369–375.
Mutini, C., Falzoni, S., Ferrari, D., Chiozzi, P., Morelli, A., Baricordi, O. R., et al. (1999). Mouse dendritic cells express the P2×7 purinergic receptor: characterization and possible participation in antigen presentation. J. Immunol. 163, 1958–1965.
Nedergaard, M., Kraig, R. P., Tanabe, J., and Pulsinelli, W. A. (1991). Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 260, R581–R588.
Niewold, T. (2011). Interferon alpha as a primary pathogenic factor in human lupus. J. Interferon Cytokine Res. 31, 887–892. doi:10.1089/jir.2011.0071
Nin, J., Ferreira, I., Schalkwijk, C., Jorsal, A., Prins, M., Parving, H. H., et al. (2012a). Higher plasma high-mobility group box 1 levels are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: a 12 year follow-up study. Diabetologia 55, 2489–2493. doi:10.1007/s00125-012-2622-1
Nin, J., Ferreira, I., Schalkwijk, C., Prins, M., Chaturvedi, N., Fuller, J., et al. (2012b). Serum high-mobility group box-1 levels are positively associated with micro- and macroalbuminuria but not with cardiovascular disease in type 1 diabetes: the EURODIAB Prospective Complications Study. Eur. J. Endocrinol. 166, 325–332. doi:10.1530/EJE-11-0662
Nizet, V., and Johnson, R. S. (2009). Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 9, 609–617. doi:10.1038/nri2607
Oates, J. C., and Gilkeson, G. S. (2006). The biology of nitric oxide and other reactive intermediates in systemic lupus erythematosus. Clin. Immunol. 121, 243–250. doi:10.1016/j.clim.2006.06.001
Odegaard, J., and Chawla, A. (2013). The immune system as a sensor of the metabolic state. Immunity 38, 644–654. doi:10.1016/j.immuni.2013.04.001
Ohnmacht, C., Pullner, A., King, S. B., Drexler, I., Meier, S., Brocker, T., et al. (2009). Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J. Exp. Med. 206, 549–559. doi:10.1084/jem.20082394
Park, J. S., Svetkauskaite, D., He, Q., Kim, J.-Y., Strassheim, D., Ishizaka, A., et al. (2004). Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279, 7370–7377. doi:10.1074/jbc.M306793200
Pearce, E., and Pearce, E. (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643. doi:10.1016/j.immuni.2013.04.005
Perl, A., Gergely, P. Jr, Nagy, G., Koncz, A., and Banki, K. (2004). Mitochondrial hyperpolarization: a checkpoint of T-cell life, death and autoimmunity. Trends Immunol. 25, 360–367. doi:10.1016/j.it.2004.05.001
Perl, A., Hanczko, R., and Doherty, E. (2012). Assessment of mitochondrial dysfunction in lymphocytes of patients with systemic lupus erythematosus. Methods Mol. Biol. 900, 61–89. doi:10.1007/978-1-60761-720-4_4
Peyssonnaux, C., Datta, V., Cramer, T., Doedens, A., Theodorakis, E. A., Gallo, R. L., et al. (2005). HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest. 115, 1806–1815. doi:10.1172/JCI23865
Pichlmair, A., and Reis e Sousa, C. (2007). Innate recognition of viruses. Immunity 27, 370–383. doi:10.1016/j.immuni.2007.08.012
Pisitkun, P., Deane, J. A., Difilippantonio, M. J., Tarasenko, T., Satterthwaite, A. B., and Bolland, S. (2006). Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312, 1669–1672. doi:10.1126/science.1124978
Pociot, F., Ronningen, K. S., and Nerup, J. (1993). Polymorphic analysis of the human MHC-linked heat shock protein 70 (HSP70-2) and HSP70-Hom genes in insulin-dependent diabetes mellitus (IDDM). Scand. J. Immunol. 38, 491–495. doi:10.1111/j.1365-3083.1993.tb02593.x
Poltorak, A., He, X., Smirnova, I., Liu, M. Y., Van Huffel, C., Du, X., et al. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088. doi:10.1126/science.282.5396.2085
Ramachandran, S., and Bell, R. B. (1995). Heat shock protein 70 gene polymorphisms and multiple sclerosis. Tissue Antigens 46, 140–141. doi:10.1111/j.1399-0039.1995.tb02492.x
Ratanachaiyavong, S., Demaine, A. G., Campbell, R. D., and Mcgregor, A. M. (1991). Heat shock protein 70 (HSP70) and complement C4 genotypes in patients with hyperthyroid Graves’ disease. Clin. Exp. Immunol. 84, 48–52.
Rath, E., and Haller, D. (2012). Mitochondria at the interface between danger signaling and metabolism: role of unfolded protein responses in chronic inflammation. Inflamm. Bowel Dis. 18, 1364–1377. doi:10.1002/ibd.21944
Reis e Sousa, C. (2006). Dendritic cells in a mature age. Nat. Rev. Immunol. 6, 476–483. doi:10.1038/nri1845
Rescigno, M. (2010). Intestinal dendritic cells. Adv. Immunol. 107, 109–138. doi:10.1016/B978-0-12-381300-8.00004-6
Ridge, J. P., Di Rosa, F., and Matzinger, P. (1998). A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393, 474–478. doi:10.1038/30989
Riteau, N., Baron, L., Villeret, B., Guillou, N., Savigny, F., Ryffel, B., et al. (2012). ATP release and purinergic signaling: a common pathway for particle-mediated inflammasome activation. Cell Death Dis. 3, e403. doi:10.1038/cddis.2012.144
Robinson, A. P., Caldis, M. W., Harp, C. T., Goings, G. E., and Miller, S. D. (2013). High-mobility group box 1 protein (HMGB1) neutralization ameliorates experimental autoimmune encephalomyelitis. J. Autoimmun. 43, 32–43. doi:10.1016/j.jaut.2013.02.005
Robson, S., Sévigny, J., and Zimmermann, H. (2006). The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance. Purinergic Signal. 2, 409–430. doi:10.1007/s11302-006-9003-5
Rock, K. L., Kataoka, H., and Lai, J. J. (2013). Uric acid as a danger signal in gout and its comorbidities. Nat Rev Rheumatol 9, 13–23. doi:10.1038/nrrheum.2012.143
Rock, K. L., and Kono, H. (2008). The inflammatory response to cell death. Annu. Rev. Pathol. 3, 99–126. doi:10.1146/annurev.pathmechdis.3.121806.151456
Rock, K. L., Latz, E., Ontiveros, F., and Kono, H. (2010). The sterile inflammatory response. Annu. Rev. Immunol. 28, 321–342. doi:10.1146/annurev-immunol-030409-101311
Rofstad, E. K., Mathiesen, B., Kindem, K., and Galappathi, K. (2006). Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 66, 6699–6707. doi:10.1158/0008-5472.CAN-06-0983
Rosmalen, J. G., Homo-Delarche, F., Durant, S., Kap, M., Leenen, P. J., and Drexhage, H. A. (2000). Islet abnormalities associated with an early influx of dendritic cells and macrophages in NOD and NODscid mice. Lab. Invest. 80, 769–777. doi:10.1038/labinvest.3780080
Sallusto, F., Cella, M., Danieli, C., and Lanzavecchia, A. (1995). Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J. Exp. Med. 182, 389–400. doi:10.1084/jem.182.2.389
Salvador-Morales, C., Flahaut, E., Sim, E., Sloan, J., Green, M. L. H., and Sim, R. B. (2006). Complement activation and protein adsorption by carbon nanotubes. Mol. Immunol. 43, 193–201. doi:10.1016/j.molimm.2005.02.006
Santiago-Raber, M. L., Baudino, L., and Izui, S. (2009). Emerging roles of TLR7 and TLR9 in murine SLE. J. Autoimmun. 33, 231–238. doi:10.1016/j.jaut.2009.10.001
Sauter, B., Albert, M. L., Francisco, L., Larsson, M., Somersan, S., and Bhardwaj, N. (2000). Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells [see comments]. J. Exp. Med. 191, 423–434. doi:10.1084/jem.191.3.423
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi:10.1038/nature00858
Schiraldi, M., and Monestier, M. (2009). How can a chemical element elicit complex immunopathology? Lessons from mercury-induced autoimmunity. Trends Immunol. 30, 502–509. doi:10.1016/j.it.2009.07.005
Schiraldi, M., Raucci, A., Munoz, L. M., Livoti, E., Celona, B., Venereau, E., et al. (2012). HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 209, 551–563. doi:10.1084/jem.20111739
Schorn, C., Strysio, M., Janko, C., Munoz, L. E., Schett, G., and Herrmann, M. (2010). The uptake by blood-borne phagocytes of monosodium urate is dependent on heat-labile serum factor(s) and divalent cations. Autoimmunity 43, 236–238. doi:10.3109/08916930903510948
Semenza, G. L. (2009). Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 24, 97–106. doi:10.1152/physiol.00045.2008
Seong, S.-Y., and Matzinger, P. (2004). Opinion: hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 4, 469–478. doi:10.1038/nri1372
Sethi, V., Rubinstein, I., Kuzmis, A., Kastrissios, H., Artwohl, J., and Onyuksel, H. (2013). Novel, biocompatible, and disease modifying VIP nanomedicine for rheumatoid arthritis. Mol. Pharm. 10, 728–738. doi:10.1021/mp300539f
Seuwen, K., Ludwig, M.-G., and Wolf, R. M. (2006). Receptors for protons or lipid messengers or both? J. Recept. Signal Transduct. Res. 26, 599–610. doi:10.1080/10799890600932220
Shapiro, L., and Dinarello, C. (1995). Osmotic regulation of cytokine synthesis in vitro. Proc. Natl. Acad. Sci. U.S.A. 92, 12230–12234. doi:10.1073/pnas.92.26.12230
Sharp, A. J., Polak, P. E., Simonini, V., Lin, S. X., Richardson, J. C., Bongarzone, E. R., et al. (2008). P2×7 deficiency suppresses development of experimental autoimmune encephalomyelitis. J. Neuroinflammation 5:33. doi:10.1186/1742-2094-5-33
Shaw, P. J., Lukens, J. R., Burns, S., Chi, H., Mcgargill, M. A., and Kanneganti, T. D. (2010). Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J. Immunol. 184, 4610–4614. doi:10.4049/jimmunol.1000217
Shi, Y., Evans, J. E., and Rock, K. L. (2003). Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521. doi:10.1038/nature01991
Shi, Y., Zheng, W., and Rock, K. L. (2000). Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc. Natl. Acad. Sci. U.S.A. 97, 14590–14595. doi:10.1073/pnas.260497597
Shime, H., Yabu, M., Akazawa, T., Kodama, K., Matsumoto, M., Seya, T., et al. (2008). Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J. Immunol. 180, 7175–7183.
Shirali, A. C., and Goldstein, D. R. (2008). Activation of the innate immune system by the endogenous ligand hyaluronan. Curr. Opin. Organ Transplant 13, 20–25. doi:10.1097/MOT.0b013e3282f3df04
Sica, A., Melillo, G., and Varesio, L. (2011). Hypoxia: a double-edged sword of immunity. J. Mol. Med. 89, 657–665. doi:10.1007/s00109-011-0724-8
Simmen, H. P., and Blaser, J. (1993). Analysis of pH and pO2 in abscesses, peritoneal fluid, and drainage fluid in the presence or absence of bacterial infection during and after abdominal surgery. Am. J. Surg. 166, 24–27. doi:10.1016/S0002-9610(05)80576-8
Sims, G. P., Rowe, D. C., Rietdijk, S. T., Herbst, R., and Coyle, A. J. (2010). HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 28, 367–388. doi:10.1146/annurev.immunol.021908.132603
Spirig, R., Djafarzadeh, S., Regueira, T., Shaw, S. G., Von Garnier, C., Takala, J., et al. (2010). Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1alpha and dendritic cell maturation under normoxic conditions. PLoS ONE 5:e10983. doi:10.1371/journal.pone.0010983
Sriram, U., Varghese, L., Bennett, H. L., Jog, N. R., Shivers, D. K., Ning, Y., et al. (2012). Myeloid dendritic cells from B6.NZM Sle1/Sle2/Sle3 lupus-prone mice express an IFN signature that precedes disease onset. J. Immunol. 189, 80–91. doi:10.4049/jimmunol.1101686
Srivastava, P. (2002). Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2, 185–194. doi:10.1038/nri749
Steinman, R. M., Hawiger, D., and Nussenzweig, M. C. (2003). Tolerogenic dendritic cells. Annu. Rev. Immunol. 21, 685–711. doi:10.1146/annurev.immunol.21.120601.141040
Stern, S. T., and McNeil, S. E. (2008). Nanotechnology safety concerns revisited. Toxicol. Sci. 101, 4–21. doi:10.1093/toxsci/kfm169
Stetson, D. B., Ko, J. S., Heidmann, T., and Medzhitov, R. (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. doi:10.1016/j.cell.2008.06.032
Stocki, P., and Dickinson, A. M. (2012). The immunosuppressive activity of heat shock protein 70. Autoimmune Dis 2012, 617213. doi:10.1155/2012/617213
Stoecklein, V. M., Osuka, A., and Lederer, J. A. (2012). Trauma equals danger – damage control by the immune system. J. Leukoc. Biol. 92, 539–551. doi:10.1189/jlb.0212072
Stoye, J. P. (2012). Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 10, 395–406. doi:10.1038/nrmicro2783
Stranger, B. E., and De Jager, P. L. (2012). Coordinating GWAS results with gene expression in a systems immunologic paradigm in autoimmunity. Curr. Opin. Immunol. 24, 544–551. doi:10.1016/j.coi.2012.09.002
Tang, D., Billiar, T., and Lotze, M. (2012). A Janus tale of two active high mobility group box 1 (HMGB1) redox states. Mol. Med. 18, 1360–1362. doi:10.2119/molmed.2012.00314
Taniguchi, T., and Takaoka, A. (2002). The interferon-alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr. Opin. Immunol. 14, 111–116. doi:10.1016/S0952-7915(01)00305-3
Termeer, C., Benedix, F., Sleeman, J., Fieber, C., Voith, U., Ahrens, T., et al. (2002). Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 195, 99–111. doi:10.1084/jem.20001858
Theofilopoulos, A. N., Baccala, R., Beutler, B., and Kono, D. H. (2005). Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 23, 307–336. doi:10.1146/annurev.immunol.23.021704.115843
Thompson, M. R., Kaminski, J. J., Kurt-Jones, E. A., and Fitzgerald, K. A. (2011). Pattern recognition receptors and the innate immune response to viral infection. Viruses 3, 920–940. doi:10.3390/v3060920
Tomura, H., Mogi, C., Sato, K., and Okajima, F. (2005). Proton-sensing and lysolipid-sensitive G-protein-coupled receptors: a novel type of multi-functional receptors. Cell. Signal. 17, 1466–1476. doi:10.1016/j.cellsig.2005.06.002
Tong, J., Wu, W.-N., Kong, X., Wu, P.-F., Tian, L., Du, W., et al. (2011). Acid-sensing ion channels contribute to the effect of acidosis on the function of dendritic cells. J. Immunol. 186, 3686–3692. doi:10.4049/jimmunol.1001346
Toscano, M. G., Delgado, M., Kong, W., Martin, F., Skarica, M., and Ganea, D. (2010). Dendritic cells transduced with lentiviral vectors expressing VIP differentiate into VIP-secreting tolerogenic-like DCs. Mol. Ther. 18, 1035–1045. doi:10.1038/mt.2009.293
Treuhaft, P. S., and McCarty, D. J. (1971). Synovial fluid pH, lactate, oxygen and carbon dioxide partial pressure in various joint diseases. Arthritis Rheumat. 14, 475–484.
Trevani, A. S., Andonegui, G., Giordano, M., López, D. H., Gamberale, R., Minucci, F., et al. (1999). Extracellular acidification induces human neutrophil activation. J. Immunol. 162, 4849–4857.
Ulbrich, W., and Lamprecht, A. (2010). Targeted drug-delivery approaches by nanoparticulate carriers in the therapy of inflammatory diseases. J. R. Soc. Interface 7(Suppl. 1), S55–66. doi:10.1098/rsif.2009.0285.focus
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2010). Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313. doi:10.1016/j.chom.2010.03.006
Urbonaviciute, V., Furnrohr, B. G., Meister, S., Munoz, L., Heyder, P., De Marchis, F., et al. (2008). Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J. Exp. Med. 205, 3007–3018. doi:10.1084/jem.20081165
Urbonaviciute, V., and Voll, R. E. (2011). High-mobility group box 1 represents a potential marker of disease activity and novel therapeutic target in systemic lupus erythematosus. J. Intern. Med. 270, 309–318. doi:10.1111/j.1365-2796.2011.02432.x
Vabulas, R. M., Wagner, H., and Schild, H. (2002). Heat shock proteins as ligands of toll-like receptors. Curr. Top. Microbiol. Immunol. 270, 169–184. doi:10.1007/978-3-642-59430-4_11
Valko, M., Leibfritz, D., Moncol, J., Cronin, M., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84. doi:10.1016/j.biocel.2006.07.001
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi:10.1038/nrm2970
Vermeulen, M. N., Giordano, M., Trevani, A. S., Sedlik, C., Gamberale, R., Fernández-Calotti, P., et al. (2004). Acidosis improves uptake of antigens and MHC class I-restricted presentation by dendritic cells. J. Immunol. 172, 3196–3204.
Walmsley, S. R., Print, C., Farahi, N., Peyssonnaux, C., Johnson, R. S., Cramer, T., et al. (2005). Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 201, 105–115. doi:10.1084/jem.20040624
Wang, H., Bloom, O., Zhang, M., Vishnubhakat, J. M., Ombrellino, M., Che, J., et al. (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251. doi:10.1126/science.285.5425.248
Wang, Q., Liu, C., Zhu, F., Liu, F., Zhang, P., Guo, C., et al. (2010). Reoxygenation of hypoxia-differentiated dendritic cells induces Th1 and Th17 cell differentiation. Mol. Immunol. 47, 922–931. doi:10.1016/j.molimm.2009.09.038
Witte, M. E., Geurts, J. J. G., De Vries, H. E., Van Der Valk, P., and Van Horssen, J. (2010). Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration? Mitochondrion 10, 411–418. doi:10.1016/j.mito.2010.05.014
Wu, C., Yosef, N., Thalhamer, T., Zhu, C., Xiao, S., Kishi, Y., et al. (2013). Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–517. doi:10.1038/nature11984
Wu, T., Xie, C., Han, J., Ye, Y., Weiel, J., Li, Q., et al. (2012). Metabolic disturbances associated with systemic lupus erythematosus. PLoS ONE 7:e37210. doi:10.1371/journal.pone.0037210
Yang, D., De La Rosa, G., Tewary, P., and Oppenheim, J. J. (2009). Alarmins link neutrophils and dendritic cells. Trends Immunol. 30, 531–537. doi:10.1016/j.it.2009.07.004
Yang, H., Antoine, D. J., Andersson, U., and Tracey, K. J. (2013). The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 93, 865–873. doi:10.1189/jlb.1212662
Yang, H., Wang, H., Czura, C. J., and Tracey, K. J. (2005). The cytokine activity of HMGB1. J. Leukoc. Biol. 78, 1–8. doi:10.1189/jlb.1104648
Yang, Y. G., Lindahl, T., and Barnes, D. E. (2007). Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886. doi:10.1016/j.cell.2007.10.017
Yao, D., and Brownlee, M. (2010). Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 59, 249–255. doi:10.2337/db09-0801
Yeste, A., Nadeau, M., Burns, E. J., Weiner, H. L., and Quintana, F. J. (2012). Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 109, 11270–11275. doi:10.1073/pnas.1120611109
Zeiser, R., Penack, O., Holler, E., and Idzko, M. (2011). Danger signals activating innate immunity in graft-versus-host disease. J. Mol. Med. 89, 833–845. doi:10.1007/s00109-011-0767-x
Zetterstrom, C. K., Jiang, W., Wahamaa, H., Ostberg, T., Aveberger, A. C., Schierbeck, H., et al. (2008). Pivotal advance: inhibition of HMGB1 nuclear translocation as a mechanism for the anti-rheumatic effects of gold sodium thiomalate. J. Leukoc. Biol. 83, 31–38. doi:10.1189/jlb.0507323
Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal, T., Junger, W., et al. (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. doi:10.1038/nature08780
Zhang, S., Zhong, J., Yang, P., Gong, F., and Wang, C. Y. (2009). HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. Int. J. Clin. Exp. Pathol. 3, 24–38.
Zitvogel, L., and Kroemer, G. (2010). The multifaceted granulysin. Blood 116, 3379–3380. doi:10.1182/blood-2010-08-299214
Keywords: dendritic cells, autoimmunity, DAMPs, mitochondria, nanomaterial, acidosis, hypoxia, osmolarity
Citation: Gallo PM and Gallucci S (2013) The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front. Immunol. 4:138. doi: 10.3389/fimmu.2013.00138
Received: 28 February 2013; Paper pending published: 12 March 2013;
Accepted: 23 May 2013; Published online: 10 June 2013.
Edited by:
Francesca Granucci, University of Milano-Bicocca, ItalyReviewed by:
Francesca Granucci, University of Milano-Bicocca, ItalyCopyright: © 2013 Gallo and Gallucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Stefania Gallucci, Laboratory of Dendritic Cell Biology, Department of Microbiology and Immunology, Temple Autoimmunity Center, Temple University School of Medicine, Philadelphia, PA 19140, USA e-mail:Z2FsbHVjY2lAdGVtcGxlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.