





94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 January 2013
Sec. Microbial Immunology
Volume 3 - 2012 | https://doi.org/10.3389/fimmu.2012.00396
This article is part of the Research Topic Interaction of Trypanosoma cruzi with Host Cells View all 10 articles
Chronic chagasic myocarditis (CCM) depends on Trypanosoma cruzi persistence in the myocardium. Studies of the proteolytic mechanisms governing host/parasite balance in peripheral sites of T. cruzi infection revealed that tissue culture trypomastigotes (TCTs) elicit inflammatory edema and stimulate protective type-1 effector T cells through the activation of the kallikrein-kinin system. Molecular studies linked the proinflammatory phenotype of Dm28c TCTs to the synergistic activities of tGPI, a lipid anchor that functions as a Toll-like receptor 2 (TLR2) ligand, and cruzipain, a kinin-releasing cysteine protease. Analysis of the dynamics of inflammation revealed that TCTs activate innate sentinel cells via TLR2, releasing CXC chemokines, which in turn evoke neutrophil/CXCR2-dependent extravasation of plasma proteins, including high molecular weight kininogen (HK), in parasite-laden tissues. Further downstream, TCTs process surface bound HK, liberating lysyl-BK (LBK), which then propagates inflammatory edema via signaling of endothelial G-protein-coupled bradykinin B2 receptors (BK2R). Dm28 TCTs take advantage of the transient availability of infection-promoting peptides (e.g., bradykinin and endothelins) in inflamed tissues to invade cardiovascular cells via interdependent signaling of BKRs and endothelin receptors (ETRs). Herein we present a space-filling model whereby ceramide-enriched endocytic vesicles generated by the sphingomyelinase pathway might incorporate BK2R and ETRs, which then trigger Ca2+-driven responses that optimize the housekeeping mechanism of plasma membrane repair from cell wounding. The hypothesis predicts that the NF-κB-inducible BKR (BK1R) may integrate the multimolecular signaling platforms forged by ceramide rafts, as the chronic myocarditis progresses. Exploited as gateways for parasite invasion, BK2R, BK1R, ETAR, ETBR, and other G protein-coupled receptor partners may enable persistent myocardial parasitism in the edematous tissues at expense of adverse cardiac remodeling.
Afflicting nearly 10 million people in Latin America (Coura and Dias, 2009), Chagas disease is a pleiomorphic clinical entity caused by Trypanosoma cruzi, a parasitic protozoan that undergoes obligate intracellular development in the mammalian host. Extremely polymorphic (Macedo and Pena, 1998), the natural populations of T. cruzi have been recently subdivided into six discrete taxonomic units (DTUs) named T. cruzi I to T. cruzi VI (Zingales et al., 2009), of which at least four are known to be involved with human pathology (Miles et al., 2009). Whether transmitted to humans via mucosal wounds inflicted by hematophagous vectors of the reduviid family or, indirectly, by oral ingestion of contaminated juices (Coura and Dias, 2009; Cortez et al., 2012), the insect-derived infective forms (metacyclic trypomastigotes) induce an acute phase that may be asymptomatic, or life-threatening. Characterized by high blood parasitemia, the sequels of severe acute disease may include hepatosplenic pathology, myocarditis, and more rarely, encephalitis. Lasting a few months, the acute symptoms subside with the onset of immunity, but the effector response is not capable of eradicating the intracellular parasites, leading to a chronic infection, characterized by low-grade tissue parasitism and positive serology. Several years later, about 30% of the patients develop a full-blown chronic chagasic myocardiopathy (CCM), characterized by the presence of inflammatory T cell infiltrates, myocardial fibrosis, complex arrhythmias, thromboembolism, and ventricular aneurysms (Marin-Neto et al., 2007). Patients with severe forms of CCM may have heart failure and sudden death, while the remaining chagasic patients (indeterminate stage) remain asymptomatic for decades. In the south cone of America, chagasic patients may also develop digestive system abnormalities (megacolon and/or megaesophagus), albeit in lower frequency than CCM.
Nearly a century after the discovery of Chagas disease, we have come to realize that the mechanisms responsible for the variable clinical manifestations during the chronic phase are still elusive. Cardiac parasympathetic depopulation, microvascular derangement, and low-grade myocardial inflammation directly induced by parasites and T cell-dependent immunopathology seem to converge in the genesis of CCM. After decades of debate, there are persuasive arguments supporting the notion that the primary cause of CCM is a low-grade, persistent parasitism of the myocardium (Tarleton, 2001). A large body of studies in mice and humans indicated that chronic myocarditis is critically dependent on the recruitment of parasite-specific (type 1) effector CD8 T cells to the infected cardiac tissues (Padilla et al., 2009; Silverio et al., 2012).
While not dismissing the relevance of intracardiac infiltrates in the progression of CCM, vascular pathologists argued that low-grade infection could lead to the accumulation of microvascular lesions in the chagasic heart, ultimately resulting in myocardial hypoxia, which in turn may aggravate collateral injury inflicted by pathogenic T cells infiltrating the heart (Morris et al., 1990; Rossi, 1990; Higuchi et al., 1999, 2003). Subsequent studies in experimentally infected animals shed light on the mechanisms by which T. cruzi induces microvasculopathy (Andrade et al., 1994; Tanowitz et al., 1999). Initial observations ascribed the formation of vasospasm to the pathogenic activity of endothelins (ETs), a potent class of vasoconstrictor polypeptides (Tanowitz et al., 1999). Of further interest, these workers reported that endothelin-1 (ET-1) expression is up-regulated in parasitized cardiovascular cells (Petkova et al., 2000). Follow-up studies in chronically infected mice demonstrated that cardiac remodeling significantly ameliorated in transgenic lines in which the ET gene was specifically removed from cardiomyocytes, while ablation of this gene in endothelial cells has not significantly reduced heart fibrosis (Tanowitz et al., 2005). Of further interest, the plasma levels of ETs are elevated both in chagasic patients and infected mice (Petkova et al., 2000; Salomone et al., 2001).
While the research linking infection-associated vasculopathy to the function of ETs progressed, our group reported that trypomastigotes generate proinflammatory kinin peptides extravascularly. Follow-up studies indicated that the local activation of the kallikrein-kinin system (KKS) translates into mutual benefits to the host/parasite relationship during the course of chagasic infection (Scharfstein and Andrade, 2011; Scharfstein and Svensjö, 2012). In the present article, we will review the results of these studies and advance the proposition that T. cruzi may take advantage of interstitial edema in the inflamed heart to potentiate their infectivity via cooperative signaling of multiple G protein-coupled receptors (GPCRs). The rationale of this hypothesis lies on two fundamental premises: (i) due to the low-grade parasitism observed in chronic infection, there are intermittent "flares" of plasma leakage in the inflamed myocardium (ii) the microvascular edema is temporally linked to the release of parasites from ruptured pseudocysts (Scharfstein and Morrot, 1999).
Studies in various experimental models indicated that tissue culture-derived trypomastigotes (Dm28c) swiftly activate microvascular beds through the activation of the KKS (Todorov et al., 2003; Monteiro et al., 2006; Schmitz et al., 2009; Scharfstein and Andrade, 2011; Andrade et al., 2012). Based on these initial observations, we predicted that the sudden diffusion of plasma-borne constituents (antibodies, complement components, kininogens, ETs) through parasite-laden tissues may affect the delicate host/parasite balance established in the chronically infected myocardium. Although the flagellated trypomastigotes released from pseudocysts may rapidly move away from the primary foci of infection, hence seeking for safer targets elsewhere in the myocardium, we proposed that the transient rise of plasma proteins in the edematous interstitial spaces might favor generation of infection-promoting peptides, such as bradykinin (BK), in the peripheral tissues (Scharfstein et al., 2000; Todorov et al., 2003; Andrade et al., 2012). Before outlining the arguments supporting this working hypothesis, we will present readers with an overview of the essential structural and functional features of the KKS.
Recently implicated in thrombo-inflammatory processes (Müller et al., 2009; von Brühl et al., 2012), the KKS is a hub-like network of proteolytic enzymes which, among other biological functions, release the proinflammatory “kinin” peptides from an internal segment of their plasma-borne precursors, the kininogens. Generation of kinins may involve multiple processing enzymes: in the bloodstream, plasma kallikrein (PK) releases the nonapeptide BK from high molecular weight kininogen (HK) upon activation of the contact system by negatively charged surfaces, such as platelet-derived polyphosphates (Figure 1A). In the extravascular spaces, lysyl-BK (LBK) is excised from low molecular weight kininogen (LK) or HK by tissue kallikrein, a serine protease that is constitutively expressed in multiple tissues. It is also known that kinins can be generated by alternative proteases. For example, in chronic inflammation kininogens may be processed by the concerted action of neutrophil elastase and mast cell tryptase, leading to the release of a slightly larger kinin, Met-LBK (Kozik et al., 1998). In the context of infections, kinins can be directly liberated from the kininogens by the action of microbial cysteine proteases, such as gingipain from Porphyromonas gingivalis (Imamura et al., 1994), staphopain A from Staphylococcus aureus (Imamura et al., 2005), streptopain from Streptococcus pyogenes (Herwald et al., 1996), and cruzipain (Del Nery et al., 1997; Scharfstein et al., 2000; Monteiro et al., 2006).
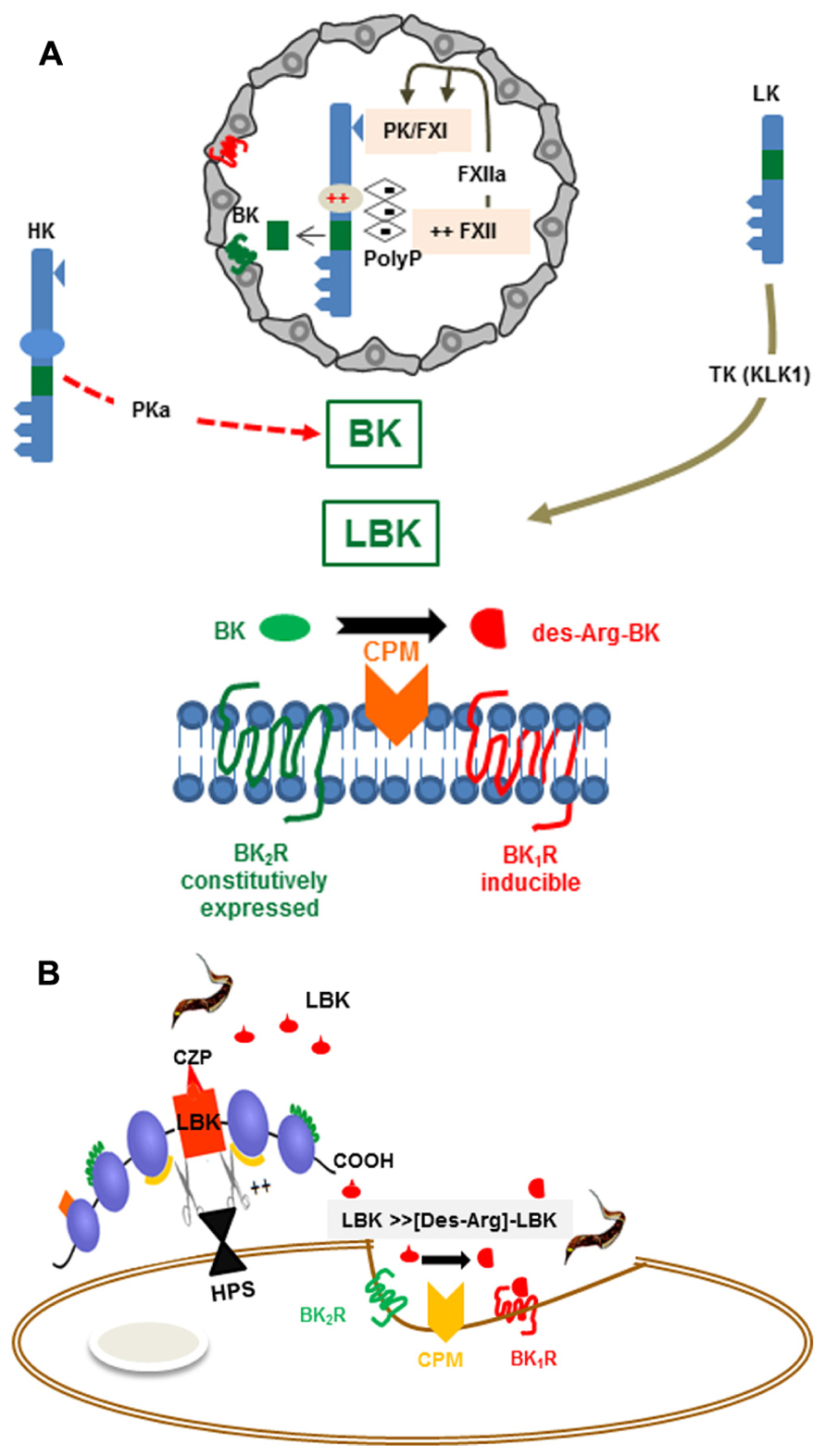
FIGURE 1. (A) Activation of the kallikrein-kinin system Top: The scheme depicts platelet-derived polyphosphates (PolyP) acting (intravascularly) as endogenous activators of the KKS (contact phase of coagulation). The proteolytic cascade is initiated as the negatively charged PolyP (long chains) bind to (i) FXII and (ii) HK, via the histidine-rich (D5) domain. PolyP-induced conformational changes in FXII lead to its autocatalytic cleavage. FXIIa then cleaves the plasma kallikrein (PK) zymogen, generating PKa. After several cycles of reciprocal activation between PKa/FXIIa, FXIIa activates FXI, the downstream effector of the intrinsic coagulation pathway, whereas PKa cleaves HK, releasing the internal nonapeptide bradykinin (BK, green rectangle). In inflammatory conditions, the plasma-borne HK/LK diffuse through post-capillary venules. As the extravascular levels of kininogens rise, these kinin-precursor molecules undergo cleavage by tissue kallikrein (TK), which liberates the decapeptide lysyl-BK. Acting by the paracrine mode, the intact form of kinins (BK or Lysyl-BK) binds at high-affinity to BK2R, a subtype of BKR that is constitutively expressed in a broad range of cell types. Bottom: The scheme depicts the activation requirements of BK1R, a GPCR subtype whose expression is strongly upregulated in inflamed/damaged tissues. Generation of high-affinity ligands for the inducible BK1R depends on metabolic processing of the intact kinins (BK or lysyl-BK) by the metallopeptidase kininase I (carboxypeptidase N or membrane-bound carboxypeptidase M). (B) BK1R is an inducible gateway for T. cruzi entry in cardiovascular cells. T. cruzi relies on the enzymatic versatility of the major lysosomal cysteine protease (cruzipain) to release LBK from HK molecules docked to the host cell surfaces via binding to heparan sulfate proteoglycans. Once released, the short-lived lysyl-BK is rapidly processed by membrane-bound carboxypeptidase M, generating [des-Arg]-lysyl-BK, which then enhances parasite infectivity through the signaling of the inducible BK1R.
The biological responses mediated by BK and LBK are mediated by bradykinin B2 receptor (BK2R), a subtype of heterotrimeric G-protein-coupled BKR (Figure 1A) which is constitutively expressed by several cell types, including cardiomyocytes, pain-sensitive neurons, vascular endothelial cells, smooth muscle cells (Leeb-Lundberg et al., 2005). As explained later on in this text, kinins (BK/LBK) also activate BK2R expressed by sentinel cells of the innate immune system, such as conventional dendritic cells (DCs; Aliberti et al., 2003; Bertram et al., 2007; Kaman et al., 2009).
Once released from HK/LK, the intact kinins (BK/LBK) have a half-life of <15 s in the plasma, hence must swiftly activate BK2R via the paracrine mode (Figure 1A). Since excessive activation of the kinin system may have adverse effects to the vascular system, the kinin/BK2R pathway is fined-tunned by overlapping regulatory mechanisms, such as (i) down-regulation of surface BK2R (ii) degradation of intact kinins by various metallopeptidases, including angiotensin converting enzyme (ACE, kininase II) – a transmembrane di-peptidyl carboxypeptidase (Skidgel and Erdos, 2004) highly expressed in the endothelium lining and, to a less extent, in other cell types, including innate sentinel cells such as monocytes and DCs (Danilov et al., 2003). Besides degrading intact kinins, thus reducing hypotension, ACE increases blood pressure through the formation of angiotensin II, a potent vasopressor octapeptide. Noteworthy, the transmembrane (somatic) enzyme undergoes cleavage by disintegrin and metalloproteinase (ADAM)-type “sheddase” (Parkin et al., 2004), leading to the accumulation of soluble forms of ACE in the blood and other body fluids.
In contrast to the constitutive BK2R, the expression of BK1R is strongly – but transiently – up-regulated during inflammation (Marceau and Bachvarov, 1998; Figure 1A). For example, ET-1 and angiotensin II induce BK1R expression through the signaling of ETAR and AT1 (angiotensin 1 receptor) respectively, leading to activation of phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinases (MAPK) cascade (Medeiros et al., 2004). BK1R is also induced by IL-1β, TNF-α, and IFN-γ via the NF-κB transcription factor (Marceau and Bachvarov, 1998; Medeiros et al., 2004; Moreau et al., 2007). Differently from BK2R, the inducible BK1R is not triggered by “intact” kinins (BK/LBK). Instead, BK1R is activated by the kinin metabolites [des-Arg]-BK or [des-Arg]-LBK, both of which are generated by carboxypeptidase N (CPN)/carboxypeptidase M (CPM) (kininase I)-mediated cleavage of the C-terminal arginine residue of BK/LBK (Figure 1A).
In the cardiovascular system, kinins/BK2R control the blood flow through nitric oxide (NO)-dependent vasodilation. In contrast to the beneficial role of kinins in cardiovascular homeostasis, there is evidence that dysregulated BK1R signaling drives myocardial fibrosis and impairs heart function in different experimental models (Spillmann and Tschöpe, 2012).
Beyond inducing detrimental cardiac responses, it is well-established that BK1R plays a major role in hyperalgesia (Calixto et al., 2004; Gabra et al., 2005; Cunha et al., 2007). Studies in mice subjected to traumatic brain injury revealed that blood-brain leakage and recruitment of inflammatory leukocytes to the CNS is blocked by a specific BK1R antagonist (Raslan et al., 2010). Although inflammation is usually initiated through the signaling of the constitutive BK2R, the sustenance of the inflammatory response depends on the signaling of endothelial BK1R. McLean et al. (2000) were the first to report that the trans-endothelial leukocyte migration is enhanced as result of up-regulated expression/signaling of endothelial BK1R. Recent progress in studies of experimental autoimmune encephalitis (EAE) demonstrated that BK1R increases the recruitment of pathogenic effector T cells into the CNS (Dutra et al., 2011; Göbel et al., 2011; Table 1).
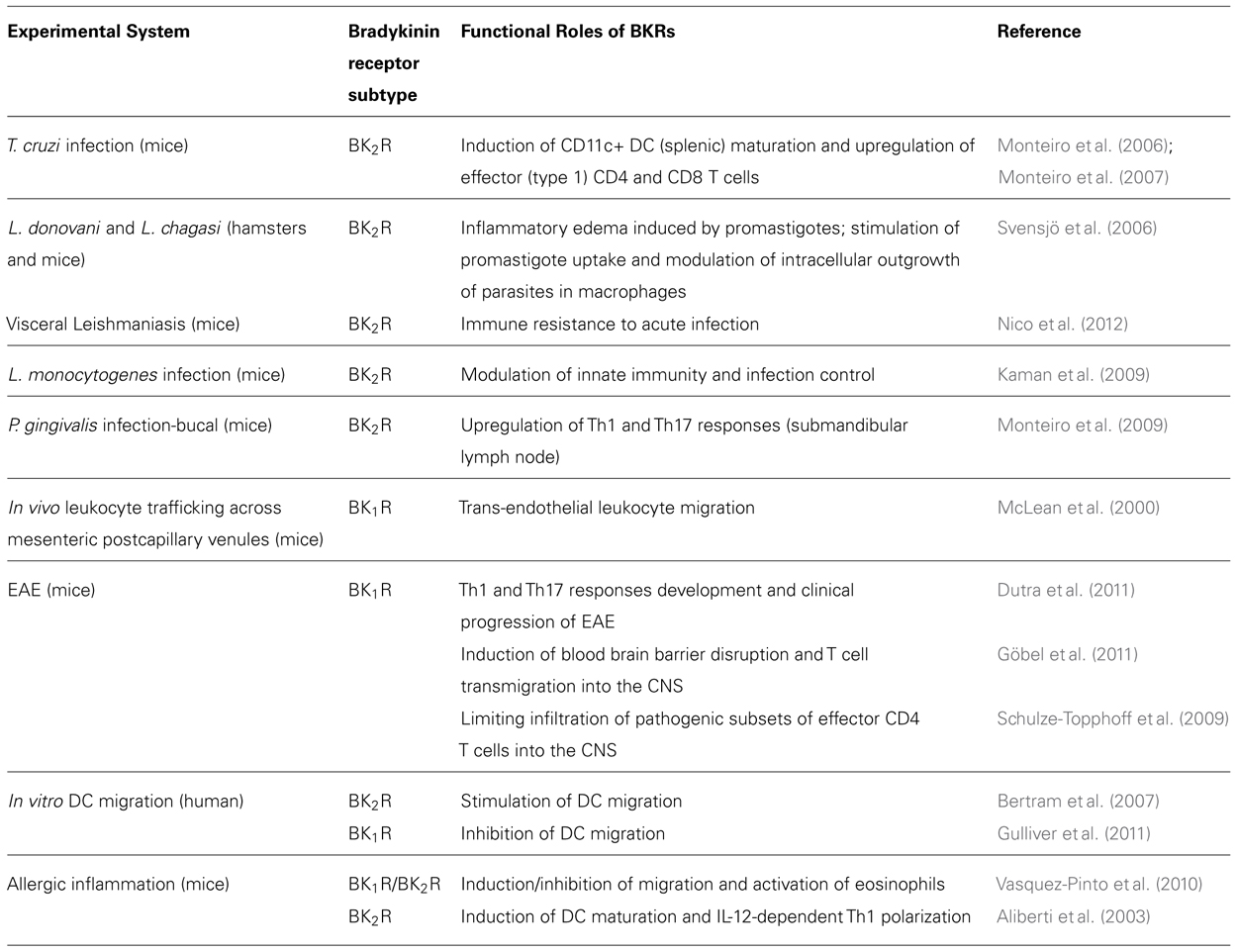
TABLE 1. Role of BKRs in immunity: literature update.
More recently, a growing number of studies indicated that immune resistance against infection by parasitic protozoan (Monteiro et al., 2006, 2007; Svensjö et al., 2006; Nico et al., 2012; Scharfstein and Svensjö, 2012) and bacterial pathogens (Kaman et al., 2009) is critically dependent on activation of innate sentinel cells via the BK2R pathway (Table 1).
Stably expressed at the cell surface, the constitutively expressed BK2R is rapidly desensitized/internalized via GRK4α-mediated receptor phosphorylation upon ligand binding, thus yielding a transient signaling response (Blaukat et al., 2001). Using BK2R-fusion constructs, Enquist et al. (2007) revealed that upon BK binding, the internalized receptors colocalize with transferring in endosomes, prior to entry in the arrestin-dependent, clathrin-mediated recycling pathway. A second trafficking pathway, described in smooth muscle cells and fibroblasts, indicated that BK also promotes the redistribution of BK2R and their coupling G proteins to caveolar rafts (de Weerd and Leeb-Lundberg, 1997).
Although the cellular systems vary from one report to another, it is well-established that BK binding to BK2R promotes the formation of BK2R homodimers and oligomer complexes of higher order (Leeb-Lundberg et al., 2005). Kang et al. (2004) reported that BK2R and BK1R form heterodimers in transfected HEK293 cells. Notably, BK2R also forms heterodimeric complexes with-type-1 angiotensin receptors (AT1) in vascular smooth muscle cells and transfected HEK293 (AbdAlla et al., 2001).
As to the signal transduction pathways, it is well established that BK frequently induces BK2R-dependent [Ca2+]i transients and PLC-β-dependent hydrolysis of phosphoinositides, both of which commonly coupled to the activation of PKC isoenzymes α, ε, and ξ. Although BK2R often signals through Gαq and Gαi, in some cellular systems BK-driven activation of the constitutively expressed BKR is coupled to Gαs and Gα12/13 (Leeb-Lundberg et al., 2005). Similar to the effects induced by other GPCR agonists, BK sequesters BK2R along with Gαq and Gαi in caveolar compartments of smooth muscle cells (de Weerd and Leeb-Lundberg, 1997).
In fibroblasts, a cell-type often used in models studies of T. cruzi interaction with non-professional phagocytic cells, BK2R activation is associated with transient tyrosine phosphorylation and activation of focal adhesion kinase as well as other focal-adhesion substrates (Leeb-Lundberg et al., 1994). In another study with fibroblasts, BK stimulated peripheral actin microspikes and membrane ruffling via activation of Cdc42 and Rac-1 (Kozma et al., 1995). Studies with endothelial cells showed that BK, acting as a potent vasodilator, transiently stimulates endothelial cell production of NO via endothelial nitric oxide synthase (eNOS) through mechanisms involving both calcium-dependent and phosphorylation cascades mediated by PI3K/Akt (Kuhr et al., 2010).
All the components of the KKS are present in the heart. In addition to contributing to vascular tone and inflammation in the cardiac tissues, the KKS influences extent of extracellular matrix (ECM) remodeling, angiogenesis, and stem cell mobilization (Spillmann et al., 2006). Of possible relevance to the understanding of KKS function in CCM, experimental studies in mouse models of diabetic cardiomyopathy and myocardial ischemia indicate that specific alterations in kinin homeostasis may either ameliorate or worsen cardiac pathology after cardiac pathology (Spillmann and Tschöpe, 2012).
As mentioned earlier, the beneficial effects of ACE inhibitors are due to their dual biological functions: these potent drugs block ACE-driven conversion of angiotensin I into the vasopressor angiotensin II and inhibit the enzymatic breakdown of kinins (BK2R agonists), thus increasing the half-life of “intact” kinins in the circulation. Noteworthy, ACE inhibitors reduce myocardial damage (e.g., reduction of infarction size) yet these effects were nullified upon treatment with BK2R antagonists or in BK2R-deficient mice (Yang et al., 2001), thus indicating that signaling of BK2R has a major function in the remarkable protective effects of ACE inhibitors.
Studies in streptozotocin (STZ)-induced models of diabetic cardiomyopathy have shown that early left ventricle (LV) and systolic dysfunction is improved in mice overexpressing tissue kallikrein (KLK1), whereas wild-type heart develop massive fibrosis (interstitial and perivascular) due to focal accumulation of collagen and fibrillar fragmentation. Although KLK1 has a pro-collagenase activity by itself and may activate matrix metalloproteinases (MMPs), the host-protective effects of KLK1 overexpression in diabetic cardiomyopathy were abolished upon treatment with HOE-140 (BK2R antagonist), thus implying that KLK1-mediated release of kinins improves cardiac function. Additional studies suggested that activation of the KLK1/BK2R pathway reduces cardiac fibrosis through the activation of the plasminogen activator/MMP2-dependent fibrinolytic cascade (Spillmann et al., 2006). Additional studies revealed that BK2R signaling that BK2R signaling improves cardiac function by (i) reducing apoptosis and chamber dilatation in the myocardium (Chao et al., 2008), (ii) optimizing angiotensin II-induced (NO-dependent) neovascularization (Munk et al., 2007), (iii) restoring S2a-mediated sarcoplasmatic Ca2+ uptake (Tschöpe et al., 2005), and/or (iv) promoting homing of endothelial progenitor cells to ischemic muscles (Kränkel et al., 2008).
The first clues suggesting that T. cruzi is equipped with a kininogenase came from enzymatic analysis of the substrate specificity of cruzipain (Del Nery et al., 1997), a lysosomal-like cysteine protease classified as member of clan A of the C1 peptidase family (Cazzulo et al., 2001; Alvarez et al., 2012). At first sight, the discovery that cruzipain acted as a “kininogenase” seemed paradoxical because kininogens are members of the cystatin family of cysteine protease inhibitors, hence rely on cystatin-like domains to potently inactivate papain-like enzymes, including cruzipain itself (Stoka et al., 1995). Consistent with this, in vitro data showed that cruzipain hydrolyzes soluble forms of HK at slower rates as compared to tissue kallikrein. This conundrum was settled by awareness that HK binds to negatively charged sulfated proteoglycans, such as heparan or chondroitin sulfates via the histidine-rich positively charged motif (D5H) localized at the C-terminal end of the BK (D4) sequence (Renné et al., 2000; Renné and Muller-Esterl, 2001). Based on this information, Lima et al. (2002) hypothesized that the spatial orientation of cell-bound HK docked to heparan sulfate proteoglycans was not suitable for cruzipain binding and inactivation by the cystatin-like inhibitory domain (Figure 1B). Indeed, model studies performed with cruzipain and HK in the test tube offered circumstantial support to this hypothesis (Lima et al., 2002): the addition of heparan sulfate (at optimal concentrations) drastically reduced the cysteine inhibitory activity of soluble HK on cruzipain while reciprocally increasing the catalytic efficiency of the parasite protease, measured with synthetic peptides flanking the BK sites. Consistent with these results, heparan sulfate potentiated HK processing by cruzipain, generating multiple HK breakdown products and promoting accelerated kinin release (Figure 1B). Combined, these biochemical studies suggested that the substrate specificity of the parasite protease was re-directed as result of reciprocal interactions between sulfated proteoglycans with the substrate (HK) and protease (cruzipain) molecules (Lima et al., 2002), hence increasing the efficiency of the kinin-release reaction in peripheral sites of infection.
Previously characterized as a therapeutic target of Chagas disease (McKerrow et al., 2009), cruzipain is a remarkably versatile virulence factor of T. cruzi. Beyond activation of the KKS (see below), the proteolytic activity of cruzipain was implicated in mechanisms of parasite virulence/pathogenicity, such as lence/pathogenicity, such as (i) enhancement of trypomastigote invasion and intracellular replication of amastigotes in cardiomyocytes (Meirelles et al., 1992; Scharfstein et al., 2000), (ii) degradation of proinflammatory chemokines (Benítez-Hernández et al., 2010), and (iii) subversion of innate microbicidal responses in parasitized macrophages through interference with the activation of NF-κB (Doyle et al., 2011).
Vertebrate hosts may control infection by rapidly mobilizing soluble and cellular-based microbicidal systems that destroy pathogen invaders at the cost of limited self-tissue destruction. Perturbations of steady-stated tissue homeostasis are sensed by sentinel cells of the innate immune system through specialized pattern-recognition receptors (PRRs; Medzhitov and Janeway, 1997). In many infections, the activation of PRRs lead to rapid secretion of pre-formed vasoactive mediators by innate sentinel cells (e.g., eicosanoids, leukotrienes, chemokines, TNF-α), which then activate the endothelium lining, rendering them sticky for circulating neutrophils. Further downstream, vasoactive mediators generated/released at the neutrophil/endothelial interface impair the integrity of the endothelial barrier, hence opening the “flood gates” (DiStasi and Ley, 2009).
Studies in a mouse model of subcutaneous (footpad) infection provided the first evidence that Dm28c TCTs (tissue culture trypomastigotes) induce inflammatory edema through the activation of the kinin system (Todorov et al., 2003; Figure 2A). A footpad edema potentiated by the ACE inhibitor captopril was observed 2 h p.i. in wild-type B6 mice, but not in BK2R-deficient mutants. Using pharmacological tools, we showed that the constitutively expressed BK2R orchestrated the early phase (2 h) edema, whereas BK1R (acting in “cross-talk” with BK2R) sustained the inflammatory response (24 h; Todorov et al., 2003). Intriguingly, Dm28c epimastigotes failed to induce a conspicuous inflammatory edema via the kinin pathway despite the fact that these vector-borne non-infective forms express abundant levels of cruzipain. In a subsequent study, it became clear, for reasons that will be explained further below, that cruzipain was necessary but not sufficient to generate kinins in hamster cheek pouch (HCP) topically exposed to Dm28c epimastigotes.
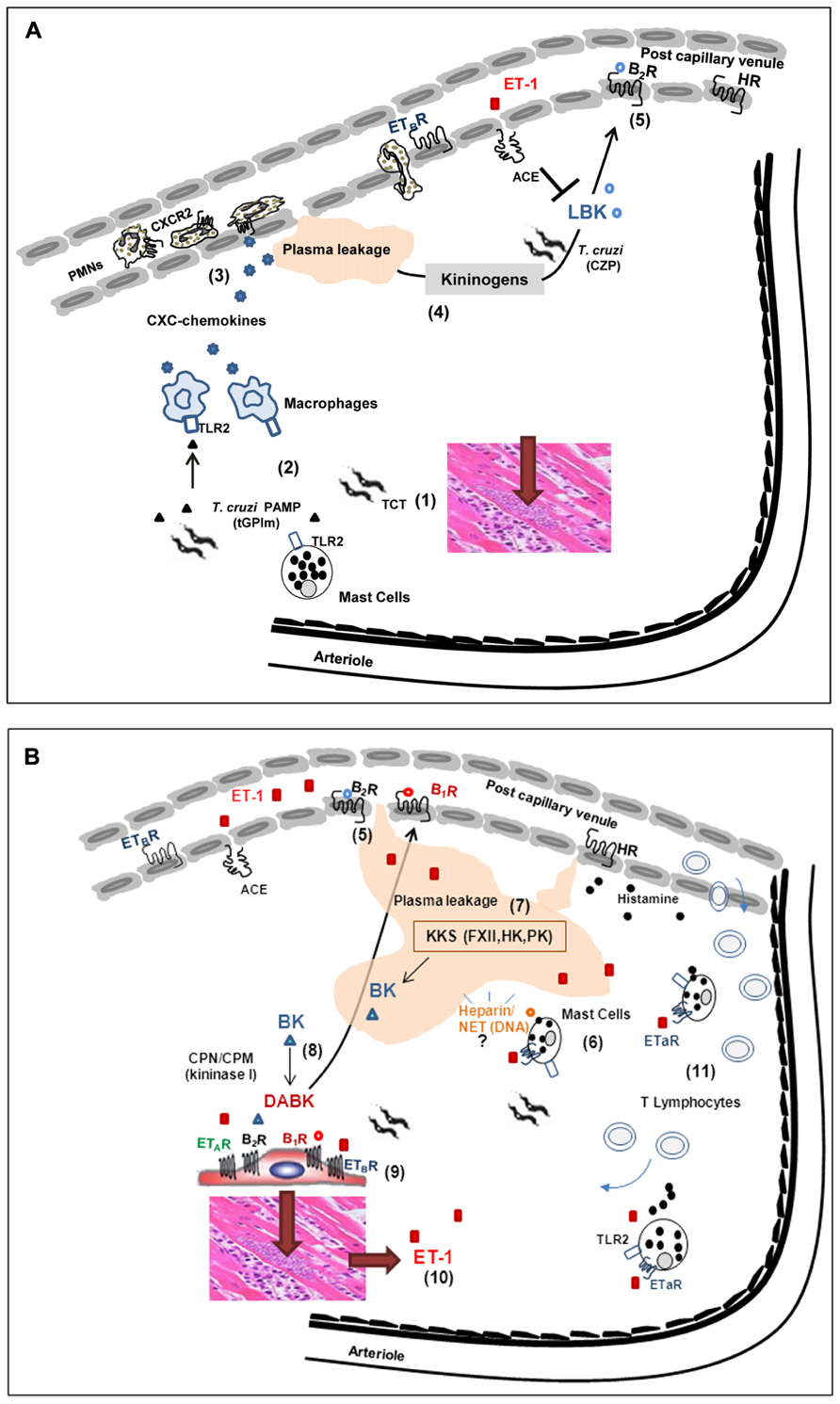
FIGURE 2. Influence of interstitial edema on host/parasite balance in the myocardium: lessons from studies with the Dm28c strain of T. cruzi. (A) (1) Despite the scanty parasitism in the myocardium of chronic chagasic patients, parasite-containing pseudocysts occasionally burst, releasing high numbers of pro-inflammatory trypomastigotes into the surrounding interstitial spaces. (2) Innate sentinel cells, such as macrophages and/or mast cells, initiate inflammation through the sensing of tGPI, a TLR2 ligands shed by trypomastigotes. (3) In response to TLR2 engagement, tissue macrophages release CXC chemokines, which in turn activate neutrophils/endothelium via CXCR2. (4) Vascular permeability is increased, leading to the diffusion/accumulation of plasma proteins (including kininogens) through parasite-laden interstitial spaces. Once bound to heparan sulfate proteoglycans, HK suffers proteolytic attack by cruzipain (CZP) secreted by trypomastigotes. (5) The released kinins (LBK) amplify plasma leakage through the activation of endothelium BK2R. The levels of intact kinins keep rising, despite the counter-regulatory role of ACE. (B) (6) Whether originating from plasma (following diffusion to sites of infection) or secreted by parasitized cardiomyocytes, ET-1 activates perivascular mast cells via ETAR. Upon degranulation, the mast cell release histamine (which further increases plasma leakage) and heparin, in the peripheral tissues. (7) The plasma “contact” phase system may be activated by mast cell-derived heparin and/or other negatively charged molecules of endogenous origin, such as DNA present in extracellular traps. Once activated by FXIIa, PKa releases BK from HK, thus propagating the proteolytic cascade. (8) Intact kinins are metabolized by kininase I (CPM and/or CPN), generating [des-Arg]-BK, which in turn activate BK1R (whose expression is up-regulated in a broad range of heart cells, including cardiomyocytes-shown at the center of the illustration). (9) The parasites take advantage of the local availability of infection-promoting peptides (ET1, BK and/or [des-Arg]-kinins) to persistently invade cardiovascular cells through the signaling (“cross-talk”) between BK2R/BK1R/ETAR/ETBR. (10) Intracellular parasite outgrowth leads to increased expression of pro-oxidative/pro-fibrotic ET-1 and proinflammatory chemokines. (11) Adverse cardiac remodeling ensues, as result of vicious cycles of ET-1-driven triggering of cardiac mast cells, endothelial activation and BK1R-driven recruitment of pathogenic subsets of effector CD8+ T cells into the heart.
Insight of the functional interplay between Toll-like receptor (TLR)-driven innate responses and the KKS emerged from analysis of the mechanisms underlying TCT-evoked microvascular leakage responses induced by topically applying the pathogen to the HCP (Monteiro et al., 2006). This unconventional system proved advantageous because it dispensed the use of needle to inject parasites, hence avoiding activation of the KKS by bleeding and traumatic injury. Using intravital microscopy, we found that Dm28c TCTs induced discrete plasma leakage responses in the HCP within a few minutes of parasite application. Noteworthy, the microvascular reactions were attenuated by HOE-140 (BK2R antagonist) or by pretreating the TCTs with an irreversible cruzipain inhibitor N-methyl-piperazine-Phe-homoPhe-vinyl sulfone (K11777), an anti-parasite drug that will be soon tested in clinical trials (Engel et al., 1998; McKerrow et al., 2009).
Monteiro et al. (2006) highlighted the fact that purified cruzipain (activated) topically applied to the HCP failed to induce significant plasma leakage even in the presence of ACE inhibitors. However, purified (activated) cruzipain induced a strong BK2R-driven leakage response when it was applied in combination to purified HK. The dependence on a supply of exogenous HK suggested that, in resting state conditions (i.e., in the absence of inflammation), the levels of endogenous kinin-precursor proteins in the interstitial spaces are insufficient to allow for significant activation of the kinin system. Based on these observations, Monteiro et al. (2006) predicted that Dm28c TCTs might bear developmentally regulated proinflammatory molecules (i.e., absent in Dm28c epimastigotes) which control the rate-limiting step of KKS activation in parasite-laden tissues: the influx of the plasma-borne kininogen (cruzipain substrate) into peripheral sites of T. cruzi infection (Figure 2A). Seeking to identify these factors, Monteiro et al. (2006) then turned their attention to tGPI, a mucin-linked lipid anchor previously characterized by Almeida and Gazzinelli (2001) as a TLR2 ligand that was expressed at high-levels exclusively in TCTs. Consistent with the working hypothesis, Dm28 TCTs failed to evoke inflammatory edema in TLR2-/- mice or in BK2R-/- mice (Monteiro et al., 2006). In contrast, these infective parasites evoked a prominent edema both in wild-type mice and TLR4 mutant (C3H/HeJ). These results argued against a role for GIPL, a lipid anchor of epimastigotes previously characterized as TLR4 ligand (Oliveira et al., 2004), in the activation pathway that generates vasoactive kinins in peripheral sites of infection.
Next, Monteiro et al. (2006) studied the functional interplay between TLR2 and the KKS by injecting TLR2-deficient mice with a parasite suspension supplemented (or not) with purified HK. Strikingly, the injection of HK in infected TLR2-/- mice rescued the edema response. Moreover, the edema responses which HK induced in TLR2-/- infected mice were abolished by HOE-140, or alternatively, by pre-incubating the TCTs with the irreversible cruzipain inhibitor K11777. Combined, these results supported the hypothesis that TCTs induce the initial leakage/accumulation of the HK (cruzipain substrate) in peripheral tissues via TLR2, whereas cruzipain amplifies this inflammatory response through the release of BK2R agonist, at the downstream end of the inflammatory cascade (Figure 2A).
In order to further confirm this hypothesis, Monteiro et al. (2006) injected (s.c.) purified tGPI, combined or not to activated cruzipain, in wild-type naïve mice or in TLR2-/- or BK2R-/- mutants. Edema measurements showed that tGPI/cruzipain potently induced microvascular responses via the TLR2/BK2R-pathway and these proinflammatory effects were further potentiated by ACE inhibitors. Beyond their effects on the microcirculation, the released kinins link TLR2-driven inflammation to innate immunity by triggering BK2R expressed on resident/migrating DCs, converting them into TH1-directing antigen-presenting cells (APCs; Monteiro et al., 2006). Conversely, ACE counter-modulates TH1-polarization via the trans-cellular the TLR2/BK2R pathway by degrading BK/LBK, an endogenous signal that drives DC maturation (Aliberti et al., 2003; Monteiro et al., 2006, 2007; Scharfstein et al., 2007).
A key event at early stage of infection, the interaction of circulating neutrophils with the endothelium has profound effects on the outcome of the inflammatory process. One of the most common consequences of the interactions that take place on the luminal side of post-capillary vessels is the increased vascular permeability, a biological response that leads to the accumulation of protein-rich edema fluid in interstitial spaces. Although the list of endogenous soluble factors that increase vascular permeability is extensive, they usually impair the integrity of the endothelial barrier through the triggering of [Ca2+]I – the via signaling of heterotrimeric G-proteins, i.e., a pathway required to induce myosin-dependent contraction and junctional disruption of the endothelial cells.
In the HCP studies described in the previous section, Monteiro et al. (2006) observed that circulating leukocytes rapidly adhered to the luminal face of post-capillary venules. Complementary studies performed in neutrophil-depleted wild-type mice revealed that Dm28 TCTs failed to evoke a conspicuous edema in these animals. Similar to the pharmacological maneuvers employed with TLR2-/- mice, already described, the deficient phenotype of neutrophil-depleted mice was rescued upon injection of TCT supplemented with HK (Monteiro et al., 2006). Collectively, these results supported the concept that neutrophils, acting at the early stages of the inflammatory response, link innate immunity (TLR2-driven) to the KKS by driving the diffusion/accumulation of plasma-borne kininogens into parasite-laden tissues (Figure 2A).
In a subsequent study, Schmitz et al. (2009) investigated whether macrophages could also play a role in the trans-cellular “cross-talk” mediated by TLR2/BK2R. First, they asked whether macrophages incubated with TCTs or tGPI in vitro stimulated the secretion of CXC chemokines (KC/MIP-2). The results revealed that wild-type macrophages robustly responded to Dm28c TCTs (but not to epimastigotes), whereas TLR2-/- macrophages were unresponsive to both stimuli. Turning to the HCP model, intravital microscopy studies showed that the drug repertaxin (CXCR2 antagonist) blocked leukocyte accumulation in the microvascular beds of HCP topically exposed to TCTs and inhibited the paw edema in T. cruzi-infected mice (Schmitz et al., 2009). Collectively, these findings supported the concept that Dm28c TCTs evoke inflammatory edema via the TLR2/CXCR2/BK2R axis, a sequential pathway of activation forged by the trans-cellular “cross-talk” of tissue resident macrophages, neutrophils and the endothelium (Figure 2A).
Encoded by distinct genes, ET-1, 2, and 3 are closely related peptides expressed by endothelial cells, cardiac myocytes, and cardiac fibroblasts (Goto, 2001; Kedzierski and Yanagisawa, 2001). Synthesized as pre-pro-endothelin, these precursor proteins are cleaved by ET-converting enzymes (ECE) forming big-endothelin, which upon further processing yields 21 amino acids ET peptides that activate cardiovascular cells via GPCRs subtypes, ETAR and ETBR (Dhaun et al., 2007).
Apart from its powerful vasoconstrictor effects, the pleiotropic ET-1 induces plasma exudation (Filep et al., 1993; Sampaio et al., 2000). Hemodynamic changes such as those provoked by shear stress are sensed by the endothelium, which responds by activating eNOS and early up-regulating ET-1 mRNA. Once released, NO and ET-1 modulate tissue homeostasis extravascularly through the activation of other cell types, such as mast cells (Maurer et al., 2004).
Recently, Andrade et al. (2012) investigated the functional interplay between the kinin and ETs pathways at the early stages of infection by Dm28c TCTs. Remarkably, the application of subtype-specific antagonists of ETAR or ETBR, or the BK2R antagonist (HOE-140) on the HCP prior to TCTs markedly reduced (~70%) leukocyte accumulation in microvascular beds. In addition, these endothelin receptor (ETR) antagonists blunted plasma leakage in the hamster cheek pouch and blocked the inflammatory edema in T. cruzi-infected mice. Collectively, these results indicated that ETRs (both subtypes) and BKRs, may propagate the inflammatory wave intiated via TLR2/CXCR2 through the proteolytic activation of the KKS.
Levick et al. (2010) have recently drawn attention to the importance of cardiac mast cells in adverse myocardial remodeling. Given the precedent that mast cells sense Mycobacterium tuberculosis via TLR2 (Carlos et al., 2009), Andrade et al. (2012) have speculated that Dm28c TCTs may directly activate ET-positive mast cells (Ehrenreich et al., 1992) via TLR2, perhaps upregulating the extravascular levels of ETs in the chagasic myocardium. Although plausible, the pathogenic role of TLR2-positive cardiac mast cells in CCM was not object of systematic investigations so far. Focusing on other aspects of chagasic pathology, Meuser-Batista et al. (2011) have recently reported that cardiac mast cells die by apoptosis in CBA-infected mice, presumably reflecting reduced production of stem cell factor in chagasic heart.
Studies in other disease settings have shown that ET-1 activates ETAR-positive mast cells in autocrine manner, inducing secretion of potent vasoactive mediators, such as histamine, leukotriene C4 (Yamamura et al., 1994), and TNF-α (Coulombe et al., 2002). Of further interest, there is evidence that ET-1 induces metalloproteinase-driven ventricular remodeling in models of chronic heart pressure/volume overload (Murray et al., 2004; Janicki et al., 2006) and modulate cardiac contractility through the induction of mast cell degranulation via ETAR (Eszlári et al., 2008). Although the role of the ET-1/mast cell axis was not directly investigated in CCM, there is evidence that plasma levels of ET-1 are elevated both in chagasic patients and experimentally infected mice (Petkova et al., 2000; Salomone et al., 2001), presumably reflecting increased shear stress or other infection-associated hemodynamic alterations in these individuals. Furthermore, as highlighted earlier in this text, the expression of ETs is up-regulated in parasitized cardiomyocytes (Tanowitz et al., 2005). Thus, irrespective of the source of ET-1, it is conceivable that the sudden rise in the extravascular levels of this potent pro-oxidative mediator may lead to the activation of perivascular ETAR-positive mast cells in the chagasic heart. According to this hypothetical scenario, mast cell degranulation via ETAR may release histamine, chemokines, along with a myriad of vasoactive mediators, in the parasite-ladden cardiac tissues (Figure 2B). More recently, Nascimento et al. (2012) have started to investigate this hypothesis by interfering with mast cell function in HCP topically exposed to Dm28 TCTs. Using mast cell stabilizers, we found that plasma leakage (BK2R-driven) was indeed inhibited, thus suggesting that mast cell degranulation is required for overt activation of the KKS in peripheral sites of chagasic infection (Figure 2B).
Thus far, the mechanisms by which mast cells may propagate the KKS cascade in peripheral sites of T. cruzi infection remain unknown. In an interesting precedent coming from mouse models of allergic inflammation, Oschatz et al. (2011) have recently proposed that heparin (a mast cell storage product) acts as a typical endogenous “contact” activator, i.e., it activates PKa in FXIIa/HK-dependent manner, thereby releasing BK. Alternatively, parasite-induced activation of cardiac mast cells may activate the KKS through the formation of DNA-containing extracellular traps (von Köckritz-Blickwede et al., 2008). Further studies may determine if the ET/mast cell/KKS pathway plays an important role in the pathogenesis of CCM.
Comprising a heterogeneous population of professional APCs, DCs are widely but sparsely distributed in peripheral tissues and lymphoid organs (Shortman and Naik, 2007). Strategically positioned in T cell-rich areas of secondary lymphoid tissues, the resident DCs are specialized in antigen-presentation to CD4+ and CD8+ T cells. In steady-state conditions, immature DCs present MHC-restricted antigen peptides to virgin T cells in the absence of co-stimulatory molecules, hence contributing to the maintenance of peripheral tolerance (Sela et al., 2011). However, during infection, the immature DCs sense “danger” motifs expressed by pathogens through distinct families of PRRs, such as TLRs or intracellular NOD2-like receptors (NLR; Akira, 2009). In addition, conventional DCs may sense the threat to tissue integrity via receptors for endogenous proinflammatory mediators, such as ATP, uric acid (Sansonetti, 2006), and BK (Aliberti et al., 2003; Monteiro et al., 2007). Stabilized by cognate interactions with co-stimulatory molecules (CD80/86, CD40, and MHC), the prolonged encounters between antigen-bearing DCs and naïve T cells are essential for TCR activation. During the course of DC/T cell interaction, the “mature” APCs deliver TH polarizing cytokines, such as IL-12p-70, which is critically required for TH1 development.
Efforts to characterize the activation pathways controlling DC maturation in the context of T. cruzi infection have initially converged to nucleic acid-sensing TLRs (3, 7, and 9). In a key study, Caetano et al. (2011) showed that transgenic mice strains which lack functional UNC93B1 as well as functional endosomal TLRs (TLR3, 7, and 9), were as susceptible to T. cruzi infection as mice deficient in TLR3/7/9 (Caetano et al., 2011). In the same study, it was documented that T. cruzi-infected macrophages and DCs from 3 day mice displayed low IL-12p40 and INF-γ responses. Based on these results, it was inferred that recognition of intracellular parasites require UNC93B1-driven translocation of the nuclei acid-sensing TLRs from the endoplasmic reticulum to the endolysosomes (Caetano et al., 2011). In spite of this conceptual advance, other studies suggest that DCs might sense T. cruzi through TLR-independent pathways. For example, Kayama et al. (2009) showed that T. cruzi induces maturation (up-regulation of MHC class II, CD40, and CD86) of MyD88-/-TRIF-/- mice bone-marrow-derived DCs as efficiently as wild-type bone-marrow-derived DCs. Using fetal liver DCs as target cells, these authors linked the T. cruzi-induced responses (IFN-γ production and DC maturation) to the activation of the NFATc1 pathway. These results implied that innate immunity is not exclusively controlled by nucleic acid-sensing TLRs.
Several years ago, we reported that that BK, acting as a typical endogenous danger signal (i.e., TH1-directed endogenous adjuvant) induced DC maturation (IL-12 and up-regulated expression of co-stimulatory molecules) via BK2R (Aliberti et al., 2003). In a key finding, we subsequently reported that BK2R-deficient mice succumbed to acute challenge by Dm28c TCTs (i.p. route). Analysis of the immune dysfunctions underlying the susceptible phenotype of BK2R-/- mice at early stages of infection showed a modest, but significant drop in the frequency of intracardiac type-1 effector T cells. Intriguingly, however, as the acute infection progressed in BK2R-/- mice, the immune deficiency was intensified and generalized, involving both the extra-lymphoid and lymphoid compartment. Of note, the decayed TH1 response of BK2R-/- was accompanied by a corresponding rise in IL-17-producing T cells (TH17). The premise that the deficient adaptive response of BK2R-/- mice was a secondary manifestation resulting from impaired BK2R-/- DC maturation was confirmed by systemically injecting wild-type BK2R+/+ DCs into the susceptible BK2R-/- mice, prior to pathogen injection. Remarkably, this DC transfer maneuver rendered the recipient BK2R-/- mice resistant to acute T. cruzi challenge, and restored their capability to generate protective IFN-γ-producing CD4+ CD44+ and CD8+ CD44+ effector T cells, while conversely suppressing the potentially detrimental TH17 (CD4+ subset) anti-parasite responses.
In the same study, Monteiro et al. (2007) further demonstrated that Dm28c TCTs potently activated BK2R+/+ CD11c+ DCs (splenic origin) but not BK2R-/- DCs, using IL-12 secretion and expression of co-stimulatory molecules (CD86, CD80, CD40) as read-out for DC maturation in vitro. Of further interest, K11777-treated trypomastigote failed to robustly activate wild-type DCs, thus linking generation of the BK2R agonist (DC maturation signal) to the proteolytic activity of cruzipain. Noteworthy, Dm28c TCTs also induced the maturation of (splenic) TLR2-/- CD11c+ DCs and TLR4 mutant (C3H/HeJ) via BK2R, thus precluding cooperative signaling between this GPCR and either one of these surface PRRs. Admittedly, these results do not exclude the possibility that enhanced parasite uptake via the BK2R pathway might have indirectly facilitated nucleic acid-sensing by TLRs residing in endolysosomes of immature splenic DCs. In any case, whether acting as a classical sensor receptor, and/or as a upstream pathway that potentiates TLR-signaling by parasite DNA or ssRNA, these results are in line with the concept that kinin “danger” signals proteolytically released by TCTs convert splenic BK2R+/+ DCs into inducers of type 1 immunity (Monteiro et al., 2007). Considering that the splenic parenchyma is continuously exposed to plasma proteins, it is conceivable that flagellated trypomastigotes navigating through the splenic stroma might be faced with abundant levels of blood-borne kininogens, most likely associated to ECM or cell-surface sulfated proteoglycans. Accordingly, we may predict that antigen-bearing CD11c+ DCs (bearing T. cruzi antigens) residing in the spleen (and/or liver) stroma are converted into TH1 inducers following exposure to high-levels of kinin “danger” signals. To this date, studies of BK2R function in human DCs were limited to lineages derived from monocytes exposed to granulocyte-macrophage colony-stimulating factor (GM-CSF)/IL-4. Using this experimental model, Bertram et al. (2007) reported that BK2R signaling promotes DC migration although BK, per se, did not induce the maturation of human DCs. However, in subsequent studies Kaman et al. (2009) suggested that BK2R signaling potentiates the maturation of DCs previously “primed” via TLR4. In view of the marked phenotypic heterogeneity of DCs (Shortman and Naik, 2007), studies with a more representative range of human DCs are required to determine whether BK2R is a key sensor of T. cruzi.
In conclusion, the analysis of BK2R function in different models of acute T. cruzi infection strongly suggests that activation of the kinin system fuels anti-parasite immunity. In the next section we will review evidences indicating that generation of kinins in sites of infection may also benefit T. cruzi, hence translating into mutual benefits to the host–parasite equilibrium.
As mentioned earlier, studies of T. cruzi interaction with mouse cardiomyocytes showed that membrane-permeable irreversible inhibitors of cruzipain impaired trypomastigote invasion and halted intracellular growth of amastigotes (Meirelles et al., 1992). While the studies on cruzipain-mediated pathways of invasion were in progress, Tardieux et al. (1992) reported that T. cruzi invades non-phagocytic cells through the induction of calcium-regulated pathway of lysosomal exocytosis. Subsequent studies revealed that the oligopeptidase B-mediated processing of a polypeptide precursor accumulating in the T. cruzi cytoplasm generated the calcium-inducing signal (Caler et al., 1998; Morty et al., 1999) that propelled parasite internalization. In a parallel development, Leite et al. (1998) reported evidences that GPCRs were the signal transducers of the [Ca2+]i-inducing signals generated by the oligopeptidase B-dependent peptide. Consistent with their working hypothesis, parasites genetically deficient in oligopeptidase B showed impaired infectivity, suggesting that proteolytic generation of the cytoplasmic Ca2+-inducing signal was indeed required for the development of infective phenotype. In another interesting study, Santana et al. (1997) described the biochemical properties of Tc80, a serine protease displaying collagenase activity. Subsequently characterized as a prolyl oligopeptidase, Tc80 localizes in a vesicular compartment close to the flagellar pocket of trypomastigotes. Synthetic inhibitors of Tc80 potently inhibited T. cruzi (Y strain) invasion of non-phagocytic cells without interfering with the [Ca2+]i-inducing activity of parasite extracts (Grellier et al., 2001).
Further exploring the role of cruzipain as a virulent factor, Scharfstein et al. (2000) reported that activation of BK2R potentiated parasite uptake by non-phagocytic cells. Whether using CHO transfected cells, human umbilical vein endothelial cells (HUVECs), primary cultures of mouse (neonatal) cardiomyocytes (Todorov et al., 2003), or primary culture of human smooth muscle cells (HSMCs; Andrade et al., 2012), these studies indicated that the parasites relied on cruzipain activity to generate a [Ca2+]i-inducing signal (BK2R agonist) from HK displayed on host cell surfaces. Although the genetic ablation of the multicopy cruzipain genes remained a technical obstacle, experiments performed with membrane-permeable irreversible inhibitors of cruzipain and parasites overexpressing the cruzipain gene strongly suggested that the trypomastigotes critically depended on the enzymatic activity of cruzipain to stimulate parasite uptake by BK2R-positive target cells (Scharfstein et al., 2000). The authors hypothesized that upon trypomastigote attachment (posterior end) to host cell surfaces, the enzymatically active forms of cruzipain – localized in the flagellar pocket (Murta et al., 1990; Souto-Padrón et al., 1990) – rapidly diffuse into the secluded spaces formed by the juxtaposed host/parasite membranes (Scharfstein et al., 2000). Once confined to “synapses” (Tyler et al., 2005; Butler and Tyler, 2012), the active protease should be protected from targeting by natural inhibitors, such as cystatins, soluble forms of kininogens (Stoka et al., 1998), and α2-macroglobulin (Araújo-Jorge et al., 1994; Morrot et al., 1997) present in interstitial fluids. Considering that BK2R is sequestered to membrane rafts/caveolae (de Weerd and Leeb-Lundberg, 1997; Haasemann et al., 1998), we hypothesized that surface-bound HK (cruzipain substrate) may translocate in the plasma membrane before being delivered to the site of synapse (Figure 3), thus ensuring efficient kinin release/BK2R-mediated signal transduction (Scharfstein et al., 2000; Santos et al., 2006; Scharfstein and Andrade, 2011; Andrade et al., 2012).
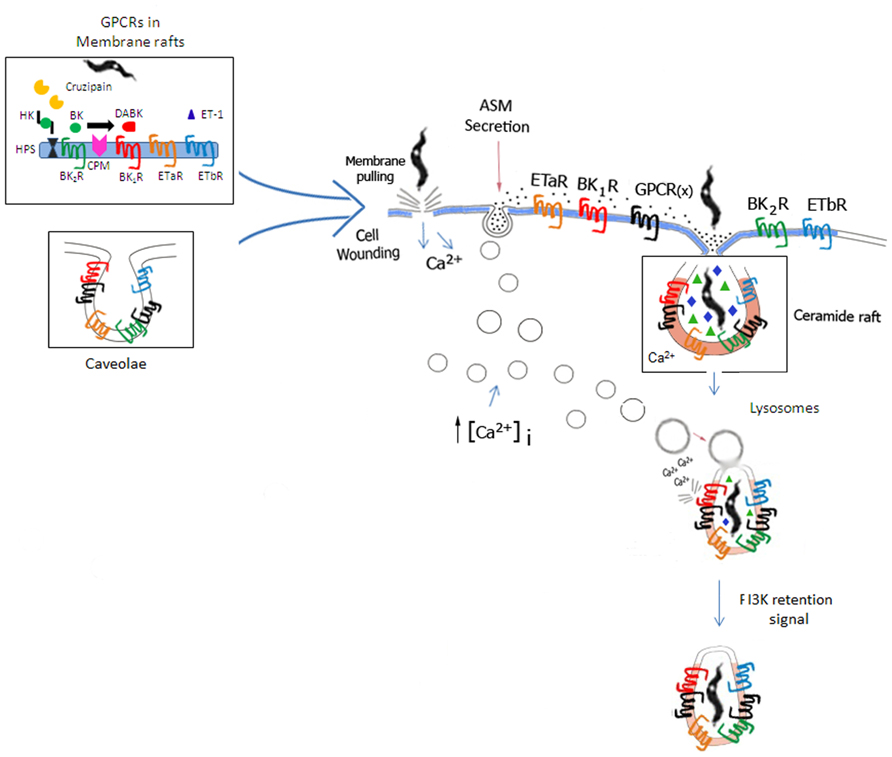
FIGURE 3. Space filling model of GPCR-mediated invasion of cardiovascular cells. Based on the unified model of T. cruzi invasion proposed by Fernandes and Andrews (2012), the scheme illustrates how GPCRs (including BKRs/ETRs) originally confined to lipid rafts or caveolae (left side of panel) might be incorporated into the molecular platforms that coalesce around ceramide-enriched vesicles generated by the housekeeping mechanisms of plasma membrane repair from cellular wounds. According to Fernandes and Andrews, extracellular calcium influx resulting from cell wounds (center of panel) trigger Ca2+-regulated lysosomal exocytosis, which in turn leads to the secretion of acid sphingomyelinase (ASM). Acting pericellularly, ASM generates ceramide-enriched endocytic vesicles. Next, BKRs, ETRs, along with several other GPCRs partners and several accessory signaling molecules translocate from lipid rafts/caveolae into the ceramide-platforms. In the resting state, signal transduction via BK2R/ETAR/ETBR (cross-talk) may likely depend on formation of homo or hetero-oligomers in multimolecular platforms coalescing in ceramide-enriched vesicles. During inflammation, the repertoire of GPCRs in the ceramide-rich endocytic vesicles is modified by the incorporation of inducible molecules, such as BK1R and CPM. ET-1 (host cell origin/autocrine route) and des-Arg-kinins (released from surface bound HK by cruzipain, then subsequently metabolized by CPM) bind to their respective GPCRs (ETAR/ETBR and BK1Rs). Feedback cycles might enhance the efficiency of the housekeeping plasma-membrane repair mechanisms via BKR/ETR-driven (i) induction of the canonical pathway of Ca2+-regulated lysosomal exocytosis and/or (ii) induction of the calcium-dependent lysosomal fusion with the nascent parasitophorous vacuole (Andrade and Andrews, 2004) and/or PIK3-dependent retention signals (Woolsey and Burleigh, 2004).
While analyzing the outcome of infection in cultures of HUVECs (resting state), we found that BK2R did not promote parasite uptake in the absence of ACE inhibitors. This was not unexpected, because expression of ACE, a transmembrane metallopeptidase that potently degrades intact kinins, is strongly upregulated by endothelial cells. As predicted, BK2R-dependent parasite uptake by HUVECs is potentiated by ACE inhibitors (Scharfstein et al., 2000; Todorov et al., 2003; Andrade et al., 2012).
Noteworthy, ACE inhibitors were not essential for BK2R-driven parasite invasion of cardiomyocytes or smooth muscle cells (Todorov et al., 2003; Andrade et al., 2012). Although the levels of ACE were not measured, it is likely that these muscle cells express lower levels of ACE as compared to HUVECs. Along similar lines, TCTs induced IL-12 responses on splenic CD11c+ DCs via BK2R irrespective of presence/absence of ACE inhibitors (Monteiro et al., 2007). In summary, the outcome of Dm28c TCT interaction with cells that constitutively express BK2R is controlled by ACE in cell-specific manner.
A major challenge in Chagas disease research is to predict the clinical outcome of the chronic cardiomyopathy. About 30% of these patients develop a progressive form of cardiomyopathy characterized by the presence of diffused inflammation/fibrosis. As mentioned in the introduction, pathologists noticed the presence of microvascular abnormalities and altered ECM patterns in the heart of CCM patients (Higuchi et al., 1999). To our knowledge, BK1R expression was not systematically investigated in heart autopsy studies of chagasic patients despite the indications that dysregulated BK1R function might be detrimental to the heart in other disease models. For example, in animal models of STZ-induced diabetic cardiomyopathy (Westermann et al., 2009), BK1R-deficient mice showed attenuated cardiomyopathy as compared to wild-type mice, as evidenced by the decrease of cardiac inflammation, fibrosis, oxidative stress, and significant improvement of left ventricular function. Pertinent to studies linking heart remodeling to the ET up-regulation by parasitized cardiomyocytes (Tanowitz et al., 2005), there is indication that oxidative stress induced by ET-1 (ETAR-driven) and angiotensin I (AT1-driven) up-regulates BK1R, leading to activation of PI3K and MAPK in smooth muscle (Morand-Contant et al., 2010). Although not directly examined, it is conceivable that NF-κB-induction by proinflammatory cytokines secreted by antigen-specific intracardiac CD8+ T cells (Padilla et al., 2009; Silverio et al., 2012) might also up-regulate BK1R in the chagasic heart.
Todorov et al. (2003) were the first to demonstrate that Dm28 TCTs evoke interstitial edema via the sequential activation of BK2R/BK1R. In the same study, they highlighted the dichotomic nature of kinin signaling pathways: the parasites were able to invade activated target cells via the BK1R/CPM pathway (Figure 1B). Differently from results observed in resting HUVECs (which critically depend on ACE blockade to efficiently internalize TCTs via BK2R), the trypomastigotes invade lipopolysaccharide (LPS)-treated HUVECs through interdependent signaling (“cross-talk”) between BK1R and BK2R (Todorov et al., 2003). Notably, the “cross-talk” between BK2R and BK1R was also observed in invasion assays performed with primary mouse cardiomyocytes (Todorov et al., 2003), which are cell types that spontaneously express BK1R in culture systems.
Although studies with non-specific kininase I inhibitors have tentatively linked BK1R-dependent parasite uptake to the processing activity of these carboxypeptidases, the role of the transmembrane carboxypeptidase CPM was not directly addressed in the above studies. As mentioned in the introduction, BK2R agonists (BK/LBK) are converted into the BK1R agonists (i.e., [des-Arg]-BK/LBK) either by surface CPM or by a soluble (plasma-borne) CPM (Figure 1A and Figure 2B). Since the interaction medium used in our invasion assays was free of serum, it seemed unlikely that CPN was the processing enzyme critically involved in BK1R-driven parasite invasion of LPS-HUVECs or cardiomyocytes. Instead, we inferred that CPM is likely the enzyme generating the BK1R agonist that propels parasite invasion in our in vitro assays. Interestingly, Sangsree et al. (2003) studied the functional interplay between CPM and BK1R in great details in human lung microvascular cells activated by IL-1β and IFN-γ. First, they showed that BK1R signaling is responsible for the sustained NO response induced by intact BK (BK2R agonist), implying that CPM converted the intact BK into [des-Arg]-kinins. Using cells transfected with genes conding for CPM and BK1R, the authors disrupted lipid rafts with methyl-beta-cyclodextrin (MβCD). As predicted, this maneuver reduced the BK1R-dependent increase in [Ca2+]i in response to stimulation with intact BK2R agonists, whereas addition of cholesterol rescued this BK1R-driven response. After showing that CPM and BK1R co-localized in lipid raft/caveolin-enriched membrane fractions (Zhang et al., 2008), they found that CPM/BK1R physically interact on the cell membrane, based on co-immunoprecipitation, cross-linking, and fluorescence resonance energy transfer analysis. In an elegant experiment using a novel fusion protein containing CPM at the N-terminus of the BK1R, Zhang et al. (2008) showed that these transfected cells were [Ca2+]i responsive upon stimulation with intact kinins, but (as predicted) this response was no longer impaired by MβCD or by CPM antibody.
As already mentioned, in invasion assays performed with LPS-HUVECs or cardiomyocytes, we found that ACE inhibitors were not required to promote parasite uptake via BK1R, as opposed to the ACE inhibitor-dependent phenotype displayed by resting HUVECs (Todorov et al., 2003; Andrade et al., 2012). This implies that CPM-dependent generation of the BK1R is prioritized over the ACE-dependent pathway of BK/LBK degradation, hence consistent with the findings reported by Zhang et al. (2008). Since both CPM/BK1R and BK1R/BK2R are sequestered into lipid rafts and/or caveolae, it is conceivable that these GPCRs segregate together with HK (bound to heparan sulfate proteoglycans) into specialized plasma membrane microdomains (Figure 3). Of further interest, although BK2R and BK1R signal cells through fairly similar intracellular pathways, the regulation of inducible BK1R differs from BK2R in that the former is desensitized upon agonist binding only to a limited degree (Enquist et al., 2007).
In a previous section, we reviewed results of studies showing that Dm28c TCTs induce inflammatory edema through mechanisms involving cooperation between BKRs and ETRs (Andrade et al., 2012). To evaluate whether the parasites could exploit the ET pathway for invasion purposes, Andrade et al. (2012) studied the interaction of Dm28c TCTs with three types of host cells: HUVECs (which only express ETBR), primary cultures of HSMCs (which express ETAR and ETBR), and mouse neonatal cardiomyocytes (expressing both subtypes of ETRs). Using multiple pharmacological tools (subtype-specific ETR antagonists; neutralizing antibodies for each GPCR) and, in addition, interference RNA, Andrade et al. (2012) demonstrated that TCTs invade “resting” HUVECs via ETBR, while invasion of the muscle cells involved activation of both subtypes of ETRs. Notably, the combined treatment of the muscle cell cultures with ETAR and ETBR subtype-specific antagonists failed to decrease parasite infectivity of HSMC or cardiomyocytes over values induced by the individual drugs, thus recapitulating the “cross-talk” observed between BK2R/BK1R in cardiomyocytes or LPS-HUVECs (Todorov et al., 2003). Importantly, studies with CHO-ETAR or CHO-ETBR-transfected cells demonstrated that parasite invasion was efficiently blocked in subtype receptor specific manner, implying that formation of ETAR/ETBR heterodimers, “per se,” is not an absolute requirement for parasite entry. Since G-protein-coupled B2R and ETR compartmentalize in lipid rafts/caveolae (de Weerd and Leeb-Lundberg, 1997; Bremnes et al., 2000; Okamoto et al., 2000; Harada et al., 2002; Ostrom, 2002), we reasoned that in transfected mammalian cells the density levels of any particular GPCR (in lipid rafts/caveolae) might be well above those found in target host cell types that naturally overexpress these receptors (Andrade et al., 2012). If true, in natural muscle cells, the reduced proportion of any given subtype of GPCR may be compensated by cooperative interactions involving physical association with alternative GPCR “partners.” Assuming that ligand generation is not a limiting factor, this “space-filling model” predicts that GPCRs that might be present at higher density in such microdomains should have a better chance to coordinate signaling responses upon ligand binding, as seems to be the case for BK2R, ETAR, and ETBR (Andrade et al., 2012; Figure 3).
Although we did not prove that BK2R, ETAR, and ETBR form homo or hetero-oligomers of higher order in host cell attachment sites, our pharmacological studies support this possibility. Whether using subtype-specific GPCR antagonists, neutralizing antibodies, or iRNA interference (ETAR), our results showed that parasite invasion was inhibited roughly to the same extent. Furthermore, we did not observe additive effects by combining receptor subtype antagonists or iRNA approaches. These results suggested that pharmacological blockade of one GPCR partner is sufficient to dismantle the signaling function of entire unit, consequently reducing parasite internalization in 40–60% (Andrade et al., 2012).
Several years ago, it was demonstrated that cholesterol-depleting drugs reduce host cell susceptibility to T. cruzi infection (Fernandes et al., 2007). In a subsequent study, we explored the possibility that BKRs and ETRs compartmentalize in lipid rafts/caveolae, by treating HSMCs with MβCD (Andrade et al., 2012). As predicted, the cholesterol-depleting drug drastically reduced parasite (Dm28c) entry in HSMCs whereas addition of exogenous cholesterol to MβCD-HSMCs restored ETAR/ETBR/BK2R-dependent pathways of T. cruzi invasion. Although the physical association of these GPCRs was not demonstrated at the molecular level, these results suggest that the cross-talk between ETRs and BK2R may critically depend on the integrity of lipid rafts/caveolae (Figure 3). Interestingly, confocal microscopy studies performed with antibodies to anti-ETAR or anti-ETBR showed that parasite–cell interaction sites contained increased clusters of these GPCRs. These results are consistent with the concept that ET-1 and “kinins” activate their cognate GPCRs in lipid rafts/caveolae, perhaps translocated to synaptic sites (Scharfstein et al., 2000). As explained below, it is also possible that ET-1, ET precursors and HK (bound to heparan sulfate) exert their functional roles as agonists and/or precursors of GPCR ligands after their internalization in ceramide-enriched vesicles (Figure 3).
In a breakthrough, Fernandes et al. (2011) and Fernandes and Andrews (2012) elaborated a unified mechanistic concept for parasite invasion of non-phagocytic host cells which nicely embraced two seemingly distinct internalization routes (Andrade and Andrews, 2004; Woolsey and Burleigh, 2004), the first one driven by calcium-regulated lysosomal exocytosis, leading to plasma membrane fusion and a lysosomal-independent pathway involving parasite entry though nascent parasitophorous vacuoles. In this unified mechanism of infection, trypomastigotes invade host cells by subverting a housekeeping mechanism of repair from cell wounds which crucially depends on acid sphingomyelinase (ASM)-driven formation of ceramide-enriched endocytic vesicles. Triggered by the formation of stable lesions in the host plasma membrane, the repair response is initiated by the influx of extracellular calcium. This event in turn leads to calcium-regulated lysosomal exocytosis and ASM secretion to the extracellular environment. Further downstream, ASM cleaves the polar head group of sphingomyelin, generating ceramide. The repair process is terminated by the formation of endocytic vesicles, which then reseal the original plasma membrane lesions as they internalize. T. cruzi trypomastigotes are thought to inflict plasma membrane injuries upon adherence of the vibrant flagellum/cell body to the cell surface. Invasion “per se” occurs after the formation of ceramide-rich endocytic vesicles (Fernandes et al., 2011).
Although we have not provided direct evidence that BK2R, BK1R, ETAR, ETBR are translocated from lipid and/or caveolar microdomains into ceramide rafts of muscle cells (HSMC or cardiomyocytes) or HUVECs, this possibility deserves to be explored in light of evidence that ceramide-enriched microdomains spontaneously fuse to generate large macrodomains containing receptor clusters in multimolecular platforms (Jin et al., 2008). Using endothelial cells derived from coronary arteries, these authors reported that Fas-ligand driven activation of death receptor mediates the formation of redox signaling platforms in lipid rafts via ceramide production by ASM-driven hydrolysis of sphingomyelin. They also showed that formation of ceramide-enriched signaling platforms was canceled in endothelial cells treated with inhibitors of lysosomal function. In other words, the requirements for the formation of multimolecular signaling platforms in ceramide rafts of endothelial cells seem to recapitulate the requirements for parasite invasion via the house-keeping mechanisms of repair from cell wounds described by Fernandes and Andrews (2012). According our “space-filling model” (Figure 3), the signal transduction responses coordinated by these various GPCRs (e.g,., BK2R/ETAR/ETBR) may either occur (i) within the synaptic sites formed soon after adhesion and/or (ii) after translocation of these GPCRs into ceramide rafts, hence integrating the above mentioned multimolecular signaling platforms. Sufficiently flexible, this GPCR-dependent model of parasite internalization is compatible with linear or converging activation pathways. For example, the linear activation mode predicts that specific GPCR subtypes are differentially targeted to lipid rafts/caveolae or to ceramide-enriched vesicles and/or to nascent parasitophorous vacuoles (Figure 3). Once translocated to nascent parasitophorous vacuoles, some of these GPCR subtypes may generate parasite retention signals through PI3K-dependent remodelling of the actin cytoskeleton (Woolsey and Burleigh, 2004), and/or drive calcium-dependent lysosomal fusion to parasitophorous vacuole (Andrade and Andrews, 2004). Multiple contacts with the pathogen and signals transmitted by soluble agonists available at high levels in the edematous extracellular environment, such as ETs, BK, DABK, may then fuel parasite uptake in vivo through converging signaling pathways.
The possibility that these GPCRs might serve as gateways for T. cruzi infection of heart cells is worth exploring. According to Fernandes and Andrews (2012), T. cruzi propensity to invade muscle cells is dictated by the need of muscle cells to constantly repair sarcolemma injuries via the Ca2+-regulated pathway of lysosomal exocytosis and ASM-driven ceramide generation (McNeil and Steinhardt, 2003). Future studies may clarify whether the signals that promote parasite uptake by cardiomyocytes via interdependent signaling of (BK2R/ETAR/ETBR) may increase the efficiency of the house-keeping mechanisms of muscle cell repair from sarcolemma wounds. If confirmed, the convergence of house-keeping and GPCR-mediated signaling pathways may translate into mutual benefits to the host/parasite relationship, at least so during the indeterminate stage of chronic infection. Interestingly, the concept that BK2R signaling is protective to the heart tissues is supported by evidences that this pathway restores S2a-mediated sacroplasmatic Ca2+ uptake (Tschöpe et al., 2005; Spillmann et al., 2006). As chronic pathology evolves, perhaps the harmonious trade-off between parasite and cardiac muscle is gradually compromised owing to the ubiquitous up-regulation of BK1R by heart cells, including cardiomyocytes, cardiac fibroblasts, and inflammatory and/or TGF-β (regulatory/pro-fibrotic) macrophages. Flares of plasma leakage elicited by trypomastigotes occasionally released by from ruptured pseudocysts may propitiate the rise of extravascular levels of [des-Arg]-BK/LBK (BK1R agonists) and ET-1. Furthermore, the extravascular levels of ET-1 are further elevated as result of up-regulated expression of this the pro-oxidative mediator by parasitized cardiomyocytes (Tanowitz et al., 2005). A vicious cycle may be installed, favoring parasite entry in heart cells via the ETAR/ETBR/BK1R pathway at expense of increased inflammation and adverse cardiac remodeling (Figure 2B).
The characterization of molecular determinants of pathogenic outcome is a major goal of basic and clinical research on CCM. Although the phenotype of the Dm28c may not be faithfully reproduced by every isolate of the highly polymorphic T. cruzi species, currently segregated into six DTUs (I–VI; Zingales et al., 2009), we may anticipate that comparative studies with other isolates should offer clues to understand the molecular determinant of pathogenicity of T. cruzi. For example, it will be interesting to know whether parasite strains belonging to particular DTUs share (or not) the ability to induce inflammatory edema through the cooperative activation of the TLR2/CXCR2/BKR2R/BK1R/ETAR/ETBR pathway. Although the expansion/contraction of this proteolytic cascade must certainly depend on additional parasite and host factors, the few players so far identified may help to investigate the phenotypic variability of natural T. cruzi populations. For example, strain-dependent variability on the expression levels of tGPI or, alternatively, in the extent of obscure mechanisms controlling shedding of lipid vesicles enriched with tGPI-linked mucins (Trocoli-Torrecilhas et al., 2009) might influence the trypomastigote ability to activate the KKS (Scharfstein and Andrade, 2011)
Alternative T. cruzi ligands are also known to activate innate sentinel cells through TLR4 (Oliveira et al., 2010), TLR9 (Bafica et al., 2006), or NOD1 (Silva et al., 2010), but it is presently unknown whether these pathways may interconnect to the KKS. Interestingly, Petersen et al. (2005) implicated TLR2 as the main upstream regulator of hypertrophy which T. cruzi trigger in isolated cardiomyocytes. On the other hand, T. cruzi strains that might express/shed higher levels of tGPI might somewhat blunt this potentially adverse phenotype by favoring low-grade release of cardioprotective kinins via cruzipain as described by Monteiro et al., 2006. In the chronic settings, the benefits of anti-apoptotic effects attributed to purified cruzipain (Aoki Mdel et al., 2006) and BK2R signaling (Chao et al., 2008) may be offset by the up-regulation of BK1R, a pathway that may synergize with TLR2 and ETAR, hence fueling inflammatory edema (Figure 2) and cardiac hypertrophy in chronically infected patients.
Another mechanism that may underlie the variable phenotype of T. cruzi strains is the variable expression profiles of cruzipain isoforms (Lima et al., 2001). For example, it is well established that cruzipain 2 (Dm28c strain) has narrow substrate specificity as compared to the major cruzipain isoform, i.e., the parasite kininogenase (Scharfstein, 2010). Predictably, strain-dependent variability in the ratio of expression between these two cruzipain isoforms may have impact on T. cruzi ability to invade host cells expressing BKRs (influence on tissue tropism) as well on its capacity to induce interstitial edema and TH1 responses via the kinin pathway. For similar reasons, we may predict that variations in the expression levels of chagasin, a tight-binding endogenous inhibitor of papain-like cysteine proteases – originally described in T. cruzi (Monteiro et al., 2001), may also influence the phenotype of T. cruzi strains. This possibility is supported by evidences (Aparício et al., 2004) indicating that TCTs of the G strain, which are poorly infective, display increased chagasin/cruzipain ratios as compared to Dm28c. Importantly, the infectivity of the G strain was enhanced upon addition of cruzipain-rich culture supernatants from Dm28 TCTs. In the same study, the authors pointed out that that vesicles shed by TCTs might serve as cruzipain substrates, presumably generating hitherto uncharacterized infection-promoting signals (Scharfstein and Lima, 2008). Hence, strain-dependent differences in the expression levels of tGPI and cruzipain isoforms may have impact on host/parasite relationship, either because kinins influence parasite infectivity, DC function and their ability to steer TH1-type effector development.
Host defense to infections depend on the mobilization of two distinct strategies in order to minimize infection-associated pathology: the first, innate immunity, is mobilized at the onset of infection with the purpose to eliminate or at least limit the spread of pathogens to the tissues (Medzhitov, 2009). The second strategy, involving products generated by proteolytic cascades and multiple endogenous “danger” signals emanating from injured tissues, has evolved to limit and repair the tissue damage inflicted by the microbial pathogen. The distinction between the mechanism underlying these overlapping processes in chronic parasitic infections, such as Chagas disease, is important because they profoundly affect the evolutionary dynamics of the of host–pathogen interactions (Rausher, 2001). Here we reviewed data showing that infective forms of T. cruzi can evoke interstitial edema through the activation of an inflammatory pathway initiated by one PRR (TLR2) and amplified through the “sequential” activation of the following GPCRs: CXCR2/BK2R/BK1R/ETAR/ETBR axis. Although presented as a linear cascade process for didactic reasons, this paradigm provides a useful framework to investigate the activation pathways that interconnect innate immunity to the KKS, a hub-like proteolytic network that generates proinflammatory kinin peptides while simultaneously activating three other proteolytic cascades, i.e., complement/coagulation/fibrinolytic systems – none of which were thus far explored in the context of CCM.
The evidence that activation of the kinin/BK2R pathway benefits the mammalian host emerged from our immunological studies (Monteiro et al., 2007). The analysis of the susceptible phenotype of BK2R-deficient mice (acute chagasic infection) demonstrated that DC sensing of the kinin “danger” signal via BK2R was essentially required for optimal generation of type 1 (host protective) effector T cells (Monteiro et al., 2007).
The dual role of the KKS in experimental Chagas disease is so far based on evidences that Dm28c trypomastigotes exploit BKRs/ETRs as (non-exclusive) gateways for cellular invasion of a broad range of non-phagocytic cells, including cardiomyocytes. We have proposed that trypomastigotes sharing the Dm28c phenotype might be able to transiently generate infection-promoting peptidergic ligands for GPCRs, such as BK and ET-1, in inflamed tissues through the induction of intersticial edema.
Interestingly, studies in other models of heart disease have associated KKS/BK2R signaling with protective cardiac functions, such as reducing apoptosis and chamber dilatation in the myocardium (Chao et al., 2008) and promoting restoration of S2a-mediated sarcoplasmatic Ca2+ uptake by cardiac cells (Tschöpe et al., 2005). Thus, it is tempting to speculate that BK2R signaling may bring about mutual benefits to the host/parasite equilibrium in the chagasic heart, perhaps during the quiescent phase (indeterminate) of chronic disease. Interestingly, there are a few reports describing beneficial effects of continuous ACE inhibitor treatment in animal models of Chagas disease (Leon et al., 2003; de Paula Costa et al., 2010). Although the clinical/pathological benefits of ACE inhibitors in CCM might be partially or totally ascribed to blockade in generation of pro-fibrotic angiotensin II, these results argue against an earlier proposition that ACE inhibitor-dependent up-regulation of the BK2R pathway might aggravate infection-associated pathology (Scharfstein et al., 2000).
In several models of sterile inflammation, the initial tissue damage is sufficient to initiate self-propagating biochemical and immunological processes with the aim to protect and rescue tissue integrity. Depending on the magnitude of the trauma, metabolic status, and host genetics, the collateral tissue damage associated with the activation of endogenous pathways of inflammation in the parasitized heart (Machado et al., 2000) may be extreme, causing irrevesible leison on cardiomyocytes. Studies in vitro have implicated TLR2 signaling in T. cruzi-induced pathways leading to cardiac hypertrophy (Petersen et al., 2005). It will be interesting to know whether the infection-associated damage of cardiomyocytes can be attenuated upon kinin release and BK2R signaling, perhaps reminiscent of some of the BK2R-dependent protective responses observed in ischemia reperfusion and diabetic cardiomyopathy (Messadi-Laribi et al., 2008; Tschöpe and Westermann, 2008).
Concerning BK1R, recent work in animal models of focal brain injury highlights the importance of therapeutic intervention on the KKS as a way to limit secondary damage caused by brain-barrier leakage (Raslan et al., 2010). As shown by these authors, pharmacological blockade of the inducible BK1R (but not BK2R) had remarkable protective effects on traumatic brain injury. Of interest in this context, we have previously reported that BK1R antagonist blunts the edematogenic response elicited by Dm28c TCTs in mice pretreated with LPS (Todorov et al., 2003).
Since chagasic patients display higher levels of ET-1 in the bloodstream (Petkova et al., 2000; Salomone et al., 2001), it is plausible that the edema resulting from pseudocyst rupture may promote the diffusion of blood-borne ET-1 through perivascular cardiac tissues. If true, these events may in turn trigger cardiac mast cell degranulation via the ET-1/ETAR axis, thus linking the ET pathway to the KKS (Figure 2B). Furthermore, considering that the expression of BK1R is up-regulated by the pro-oxidative ETs (Morand-Contant et al., 2010), and that ET-1 expression is robustly increased in parasitized cardiomyocytes (Tanowitz et al., 2005), it is conceivable that T. cruzi has evolved strategies to improve its infectivity at expense of increased inflammation, as recently shown in mice infected with the Y strain of T. cruzi (Paiva et al., 2012). Following a similar line of reasoning, we may predict that ET-1 and [des-Arg]-kinins, acting cooperatively, may propel parasite entry in cardiovascular cells through the signaling of BK1R, a subtype of GPCR that is ubiquitously up-regulated in inflamed tissues.
The sporadic formation of an intramyocardial edema in the proximity of an inflammatory lesion generated by pseudocyst rupture may alter tissue homeostasis, consequently modifying the dynamics of host/parasite interaction in this microenvironment. For example, the trypomastigotes navigating through intercellular cardiac spaces might be targeted by high-titered complement fixing/lytic antibodies (Krettli et al., 1982; Almeida et al., 1994) and/or by antibodies that inhibit host cell invasion (Schenkman et al., 1994; Tonelli et al., 2011). Induction of interstitial edema via activation of KKS might favor the rapid diffusion of these host protective antibody specificities into peripheral sites of infection. In addition, the inflammatory edema may allow the diffusion of antibodies against the C-terminal domain of cruzipain (GP25; Scharfstein et al., 1983). Antibody targeting of the immunodominant sulfated epitopes (Duschak and Couto, 2009) displayed on the C-terminal domain of cruzipain may either limit trypomastigote ability to efficiently invade cardiovascular cells (Meirelles et al., 1992; Scharfstein et al., 2000) and/or prevent anti-apoptotic responses which cruzipain otherwise induces in cardiomyocytes (Aoki Mdel et al., 2006).
For similar reasons, the heart conduction system might be targeted by auto-antibodies that are either specific for M2 muscarinic receptors (Medei et al., 2007) and/or β1-adrenergic receptors (Labovsky et al., 2007). Depending on their titers and fine-specificity, these auto-antibodies may evoke arrhythmia as a secondary consequence of plasma leakage elicited by trypomastigotes. If confirmed, this hypothesis may legitimate attempts to treat chagasic patients with drugs that target the ET/mast cell/KKS axis: by limiting leakage of auto-antibodies into the myocardium, these drugs may also mitigate cardiac arrhythmia.
Considering the long span of chronic infection, the myocardium of patients exhibiting deficient IL-10 production by macrophages or regulatory T cells (Costa et al., 2009) might be particularly prone to up-regulate the BK1R pathway in response to excessive collateral damage inflicted by pathogenic subsets of T cells. In this hypothetical scenario, the infiltrating type 1 effector CD8+ T cells may relentless fuel the pro-fibrotic pathways coordinated by BK1R/ETR. Of further interest, recent studies of sterile inflammation in transgenic mice subjected to cardiac pressure overload revealed that mitochondrial DNA (i.e., simulating symbiotic bacteria) leaks into autophagic vesicles of cardiomyocytes (Oka et al., 2012). Strikingly, the authors reported that the down-regulation of DNAse II, a lysosomal enzyme, fuels sterile inflammation in TLR9-dependent manner, provided that the mice are subjected to pressure overload (Oka et al., 2012). Although not explored in the context of CCM, it may not be surprising if chagasic heart debilitated by microvascular lesions, hypoxia, and immunopathology may be hyper-responsive to TLR9 signaling induced by T. cruzi mitochondrial DNA and/or self-DNA (from non-infected cardiomyocytes). If confirmed, it will be intriguing to know if BK1R and ETRs, acting cooperatively in the pro-oxidative environment that prevails in the chagasic heart (Dhiman and Garg, 2011) may up-regulate TLR9 and/or STING, another recently characterized sensor of self-DNA (Ahn et al., 2012). Ongoing studies in chagasic mice treated with BK1R blockers may clarify whether this therapeutic strategy may reduce parasite tissue load as well as myocardial fibrosis. Moreover, it is conceivable that BK1R antagonists may reduce chronic inflammation by preventing microvascular leakage and/or blocking parasite-induced recruitment of pathogenic subsets of anti-parasite CD8 T effector cells into the myocardium (Silverio et al., 2012), as reported in EAE (Dutra et al., 2011; Göbel et al., 2011).
In summary, our studies on the KKS have provided a general framework to investigate the intertwined proteolytic circuits that reciprocally couple anti-parasite immunity to inflammation and fibrosis in experimental CCM. Although limited to a single T. cruzi strain, Dm28c, the knowledge that emerged from 20 years of studies on this emerging field suggest that plasma leakage into the myocardium may shift the dynamics of host/parasite interaction in the inflamed heart through the activation of BKRs/ETRs. Hopefully, some of the lessons taken from this experimental work may help to elucidate the mechanisms underlying susceptibility to CCM, perhaps assisting efforts to improve the therapeutic management of chagasic patients.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by funds from the Instituto Nacional de Biologia Estrutural e Bio-Imagem do CNPq; PRONEX (26/110.562/2010), FAPERJ; CNPq; The authors acknowledge the help of Rafaela Serra in the preparation of one of the illustrations.
AbdAlla, S., Lother, H., Abdel-tawab, A. M., and Quitterer, U. (2001). The angiotensin II AT2 receptor is an AT1 receptor antagonist. Biol. Chem. 276, 39721–39726.
Ahn, J., Gutman, D., Saijo, S., and Barber, G. N. (2012). STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. U.S.A. 109, 19386–19391.
Akira, S. (2009). Pathogen recognition by innate immunity and its signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 85, 143–156.
Aliberti, J., Viola, J. P., Vieira-de-Abreu, A., Bozza, P. T., Sher, A., and Scharfstein, J. (2003). Cutting edge: bradykinin induces IL-12 production by dendritic cells: a danger signal that drives Th1 polarization. J. Immunol. 170, 5349–5353.
Almeida, I. C., Ferguson, M. A., Schenkman, S., and Travassos, L.R. (1994). Lytic anti-alpha-galactosyl antibodies from patients with chronic Chagas’ disease recognize novel O-linked oligosaccharides on mucin-like glycosyl-phosphatidylinositol-anchored glycoproteins of Trypanosoma cruzi. Biochem. J. 15, 793–802.
Almeida, I. C., and Gazzinelli, R. T. (2001). Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J. Leukoc. Biol. 70, 467–477.
Alvarez, V. E., Niemirowicz, G. T., and Cazzulo, J. J. (2012). The peptidases of Trypanosoma cruzi: digestive enzymes, virulence factors, and mediators of autophagy and programmed cell death. Biochim. Biophys. Acta 1824, 195–206.
Andrade, D., Serra, R., Svensjö, E., Lima, A. P., Ramos, E. S. Jr., Fortes, F. S., et al. (2012). Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: a converging pathway leading to chagasic vasculopathy. Br. J. Pharmacol. 165, 1333–1347.
Andrade, L. O., and Andrews, N. W. (2004). Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J. Exp. Med. 200, 1135–1143.
Andrade, Z. A., Andrade, S. G., Correa, R., Sadigursky, M., and Ferrans, V. J. (1994). Myocardial changes in acute Trypanosoma cruzi infection. Ultrastructural evidence of immune damage and the role of microangiopathy. Am. J. Pathol. 144, 1403–1411.
Aoki Mdel, P., Cano, R. C., Pellegrini, A. V., Tanos, T., Guiñazú, N. L., Coso, O. A., et al. (2006). Different signaling pathways are involved in cardiomyocyte survival induced by a Trypanosoma cruzi glycoprotein. Microbes Infect. 8, 1723–1731.
Aparício, I. M., Scharfstein, J., and Lima, A. P. (2004). A new cruzipain-mediated pathway of human cell invasion by Trypanosoma cruzi requires trypomastigote membranes. Infect. Immun. 72, 5892–5902.
Araújo-Jorge, T. C., Luz M. R., Coutinho, C. M., Medrano, N., Soeiro, M. N., Meirelles, M. N., et al. (1994). Alpha 2-macroglobulin in experimental and human Chagas’ disease. Ann. N. Y. Acad. Sci. 737, 453–455.
Bafica, A., Santiago, H. C., Goldszmid, R., Ropert, C., Gazzinelli, R. T., and Sher, A. (2006). Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J. Immunol. 177, 3515–3519.
Benítez-Hernández, I., Méndez-Enríquez, E., Ostoa, P., Fortoul, T., Ramírez, J. A., Stempin, C., et al. (2010). Proteolytic cleavage of chemokines by Trypanosoma cruzi’s cruzipain inhibits chemokine functions by promoting the generation of antagonists. Immunobiology 215, 413–426.
Bertram, C. M., Baltic, S., Misso, N. L., Bhoola, K. D., Foster, P. S., Thompson, P. J., et al. (2007). Expression of kinin B1 and B2 receptors in immature, monocyte-derived dendritic cells and bradykinin-mediated increase in intracellular Ca2+ and cell migration. J. Leukoc. Biol. 1, 1445–1454.
Blaukat, A., Pizard, A., Breit, A., Wernstedt, C., Alhenc-Gelas, F., Muller-Esterl, W., et al. (2001). Determination of bradykinin B2 receptor in vivo phosphorylation sites and their role in receptor function. J. Biol. Chem. 276, 40431–40440.
Bremnes, T., Paasche, J. D., Mehlum, A., Sandberg, C., Bremnes, B., and Attramadal, H. (2000). Regulation and intracellular trafficking pathways of the endothelin receptors. J. Biol. Chem. 275, 17596–17604.
Butler, C. E., and Tyler, K. M. (2012). Membrane traffic and synaptic cross-talk during host cell entry by Trypanosoma cruzi. Cell. Microbiol. 14, 1345–1353.
Caetano, B. C., Carmo, B. B., Melo, M. B., Cerny, A., dos Santos, S. L., Bartholomeu, D. C., et al. (2011). Requirement of UNC93B1 reveals a critical role for TLR7 in host resistance to primary infection with Trypanosoma cruzi. J. Immunol. 187, 1903–1911.
Caler, E. V., Vaena de Avalos, S., Haynes, P. A., Andrews, N. W., and Burleigh, B. A. (1998). Oligopeptidase B-dependent signaling mediates host cell invasion by Trypanosoma cruzi. EMBO J. 17, 4975–4986.
Calixto, J. B., Medeiros, R., Fernandes, E. S., Ferreira, J., Cabrini, D. A., and Campos, M. M. (2004). Kinin B1 receptors: key G-protein-coupled receptors and their role in inflammatory and painful processes. Br. J. Pharmacol. 143, 803–818.
Carlos, D., Frantz, F. G., Souza-Júnior, D. A., Jamur, M. C., Oliver, C., Ramos, S. G., et al. (2009). TLR2-dependent mast cell activation contributes to the control of Mycobacterium tuberculosis infection. Microbes Infect. 11, 770–778.
Cazzulo, J. J., Stoka, V., and Turk, V. (2001). The major cysteine proteinase of Trypanosoma cruzi: a valid target for chemotherapy of Chagas disease. Curr. Pharm. Des. 7, 1143–1156.
Chao, J., Yin H., Gao, L., Hagiwara, M., Shen, B., Yang, Z. R., et al. (2008). Tissue kallikrein elicits cardioprotection by direct kinin b2 receptor activation independent of kinin formation. Hypertension 52, 715–720.
Cortez, C., Yoshida, N., Bahia, D., and Sobreira, T. J. (2012). Structural basis of the interaction of a Trypanosoma cruzi surface molecule implicated in oral infection with host cells and gastric mucin. PLoS ONE 7:e42153. doi: 10.1371/journal.pone.0042153
Costa, G. C., da Costa Rocha, M. O., Moreira, P. R., Menezes, C. A., Silva, M. R., Gollob, K. J., et al. (2009). Functional IL-10 gene polymorphism is associated with Chagas disease cardiomyopathy. J. Infect. Dis. 199, 451–454.
Coulombe, M., Battistini, B., Stankova, J., Pouliot, P., and Bissonnette, E. Y. (2002). Endothelins regulate mediator production of rat tissue-cultured mucosal mast cells. Up-regulation of Th1 and inhibition of Th2 cytokines. J. Leukoc. Biol. 71, 829–836.
Coura, J. R., and Dias, J. C. (2009). Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Mem. Inst. Oswaldo Cruz 104, 31–40.
Cunha, T. M., Verri, W. A. Jr., Fukada, S. Y., Guerrero, A. T., Santodomingo-Garzón, T., Poole, S., et al. (2007). TNF-alpha and IL-1beta mediate inflammatory hypernociception in mice triggered by B1 but not B2 kinin receptor. Eur. J. Pharmacol. 573, 221–229.
Danilov, S. M., Sadovnikova, E., Scharenborg, N., Balyasnikova, I. V., Svinareva, D. A., Semikina, E. L., et al. (2003). Angiotensin-converting enzyme (CD143) is abundantly expressed by dendritic cells and discriminates human monocyte-derived dendritic cells from acute myeloid leukemia-derived dendritic cells. Exp. Hematol. 31, 1301–1309.
de Paula Costa, G., Silva R. R., Pedrosa, M. C., Pinho, V., de Lima, W. G., Teixeira, M. M., et al. (2010). Enalapril prevents cardiac immune-mediated damage and exerts anti-Trypanosoma cruzi activity during acute phase of experimental Chagas disease. Parasite Immunol. 32, 202–208.
de Weerd, W. F., and Leeb-Lundberg, L. M. (1997). Bradykinin sequesters B2 bradykinin receptors and the receptor-coupled Galpha subunits Galphaq and Galphai in caveolae in DDT1 MF-2 smooth muscle cells. J. Biol. Chem. 272, 17858–17866.
Del Nery, E. D., Juliano, M. A., Meldal, M., Svendsen, I., Scharfstein, J., Walmsley, A., et al. (1997). Characterization of the substrate specificity of the major cysteine protease (cruzipain) from Trypanosoma cruzi using a portion-mixing combinatorial library and fluorogenic peptides. Biochem. J. 323, 427–433.
Dhaun, N., Pollock, D. M., Goddard, J., and Webb, D. J. (2007). Selective and mixed endothelin receptor antagonism in cardiovascular disease. Trends Pharmacol. Sci. 28, 573–579.
Dhiman, M., and Garg, N. J. (2011). NADPH oxidase inhibition ameliorates Trypanosoma cruzi-induced myocarditis during Chagas disease. J. Pathol. 225, 583–596.
DiStasi, M. R., and Ley, K. (2009). Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol. 30, 547–556.
Doyle, P. S., Zhou, Y. M., Hsieh, I., Greenbaum, D. C., McKerrow, J. H., and Engel, J. C. (2011). The Trypanosoma cruzi protease cruzain mediates immune evasion. PLoS Pathog. 7:e1002139. doi: 10.1371/journal.ppat.1002139
Duschak, V. G., and Couto, A. S. (2009). Cruzipain, the major cysteine protease of Trypanosoma cruzi: a sulfated glycoprotein antigen as relevant candidate for vaccine development and drug target. Curr. Med. Chem. 16, 3174–3202.
Dutra, R. C., Leite, D. F., Bento, A. F., Manjavachi, M. N., Patrício, E. S., Figueiredo, C. P., et al. (2011). The role of kinin receptors in preventing neuroinflammation and its clinical severity during experimental autoimmune encephalomyelitis in mice. PLoS ONE 6:e27875. doi: 10.1371/journal.pone.0027875
Ehrenreich, H., Burd, P. R., Rottem, M., Hültner, L., Hylton, J. B., Garfield, M., et al. (1992). Endothelins belong to the assortment of mast cell-derived and mast cell-bound cytokines. New Biol. 4, 147–156.
Engel, J. C., Doyle, P. S., Hsieh, I., and McKerrow, J. H. (1998). Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med. 188, 725–734.
Enquist, J., Skröder, C., Whistler, J. L., and Leeb-Lundberg, L. M. (2007). Kinins promote B2 receptor endocytosis and delay constitutive B1 receptor endocytosis. Mol. Pharmacol. 71, 494–507.
Eszlári, E., Czóbel, M., Molnár, G., Bogáts, G., Kaszaki, J., Nagy, S., et al. (2008). Modulation of cardiac contractility through endothelin-1 release and myocardial mast cell degranulation. Acta Physiol. Hung. 95, 267–285.
Fernandes, M. C., Cortez, M., Geraldo Yoneyama, K. A., Straus, A. H., Yoshida, N., and Mortara, R. A. (2007). Novel strategy in Trypanosoma cruzi cell invasion: implication of cholesterol and host cell microdomains. Int. J. Parasitol. 37, 1431–1441.
Fernandes, M. C., Cortez, M., Flannery, A. R., Tam, C., Mortara, R. A., and Andrews, N. W. (2011). Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J. Exp. Med. 208, 909–921.
Fernandes, M. C., and Andrews, N. W. (2012). Host cell invasion by Trypanosoma cruzi: a unique strategy that promotes persistence. FEMS Microbiol. Rev. 36, 734–747.
Filep, J. G., Sirois, M. G., Földes-Filep, E., Rousseau, A., Plante, G. E., Fournier, A., et al. (1993). Enhancement by endothelin-1 of microvascular permeability via the activation of ETA receptors. Br. J. Pharmacol. 109, 880–886.
Gabra, B. H., Merino V. F., Bader M., Pesquero J. B., and Sirois P. (2005). Absence of diabetic hyperalgesia in bradykinin B1 receptor-knockout mice. Regul. Pept. 127, 245–248.
Göbel, K., Pankratz, S., Schneider-Hohendorf, T., Bittner, S., Schuhmann, M. K., Langer, H. F., et al. (2011). Blockade of the kinin receptor B1 protects from autoimmune CNS disease by reducing leukocyte trafficking. J. Autoimmun. 36, 106–114.
Goto, K. (2001). Basic and therapeutic relevance of endothelin-mediated regulation. Biol. Pharm. Bull. 24, 1219–1230.
Grellier, P., Vendeville, S., Joyeau, R., Bastos, I. M., Drobecq, H., Frappier, F., et al. (2001). Trypanosoma cruzi prolyl oligopeptidase Tc80 is involved in nonphagocytic mammalian cell invasion by trypomastigotes. J. Biol. Chem. 276, 47078–47086.
Gulliver, R., Baltic, S., Misso, N. L., Bertram, C. M., Thompson, P. J., and Fogel-Petrovic, M. (2011). Lys-des[Arg9]-bradykinin alters migration and production of interleukin-12 in monocyte-derived dendritic cells. Am. J. Respir. Cell Mol. Biol. 45, 542–549.
Haasemann, M., Cartaud, J., Muller-Esterl, W., and Dunia, I. (1998). Agonist-induced redistribution of bradykinin B2 receptor in caveolae. J. Cell Sci. 111, 917–928.
Harada, N., Himeno, A., Shigematsu, K., Sumikawa, K., and Niwa, M. (2002). Endothelin-1 binding to endothelin receptors in the rat anterior pituitary gland: possible formation of an ETA-ETB receptor heterodimer. Cell. Mol. Neurobiol. 22, 207–226.
Herwald, H., Collin, M., Müller-Esterl, W., and Björck, L. (1996). Streptococcal cysteine proteinase releases kinins: a virulence mechanism. J. Exp. Med. 184, 665–673.
Higuchi, M. L., Fukasawa, S., De Brito, T., Parzianello, L. C., Bellotti, G., and Ramires, J. A. (1999). Different microcirculatory and intersticial matrix patterns in idiopathic dilated cardiomyopathy and Chagas’ disease: a three dimensional confocal microscopy study. Heart 82, 279–285.
Higuchi, M. L., Benvenuti, L. A., Martins Reis, M., and Metzger, M. (2003). Pathophysiology of the heart in Chagas’ disease: current status and new developments. Cardiovasc. Res. 60, 96–107.
Imamura, T., Pike, R. N., Potempa, J., and Travis, J. (1994). Pathogenesis of periodontitis: a major arginine-specific cysteine proteinase from Porphyromonas gingivalis induces vascular permeability enhancement through activation of the kallikrein/kinin pathway. J. Clin. Invest. 94, 361–367.
Imamura, T., Tanase, S., Szmyd, G., Kozik, A., Travis, J., and Potempa, J. (2005). Induction of vascular leakage through release of bradykinin and a novel kinin by cysteine proteinases from Staphylococcus aureus. J. Exp. Med. 201, 1669–1676.
Janicki, J. S., Brower, G. L., Gardner, J. D., Forman, M. F., Stewart, J. A. Jr., Murray, D. B., et al. (2006). Cardiac mast cell regulation of matrix metalloproteinase-related ventricular remodeling in chronic pressure or volume overload. Cardiovasc. Res. 69, 657–665.
Jin, S., Zhang, F., Poklis, J. L., and Li, P. L. (2008). Lysosomal targeting and trafficking of acid sphingomyelinase to lipid raft platforms in coronary endothelial cells. Arterioscler. Thromb. Vasc. Biol. 28, 2056–2062.
Kaman, W. E., Wolterink, A. F., Bader, M., Boele, L. C., and van der Kleij, D. (2009). The bradykinin B2 receptor in the early immune response against Listeria infection. Med. Microbiol. Immunol. 198, 39–46.
Kang, D. S., Ryberg, K., Mörgelin, M., and Leeb-Lundberg, L. M. (2004). Spontaneous formation of a proteolytic B1 and B2 bradykinin receptor complex with enhanced signaling capacity. J. Biol. Chem. 279, 22102–22107.
Kayama, H., Koga, R., Atarashi, K., Okuyama, M., Kimura, T., Mak, T. W., et al. (2009). NFATc1 mediates Toll-like receptor-independent innate immune responses during Trypanosoma cruzi infection. PLoS Pathog. 5:e1000514. doi: 10.1371/journal.ppat.1000514
Kedzierski, R. M., and Yanagisawa, M. (2001). Endothelin system: the double-edged sword in health and disease. Annu. Rev. Pharmacol. Toxicol. 41, 851–876.
Kozik, A., Moore, R. B., Potempa, J., Imamura, T., Rapala-Kozik, M., and Travis, J. (1998). A novel mechanism for bradykinin production at inflammatory sites. Diverse effects of a mixture of neutrophil elastase and mast cell tryptase versus tissue and plasma kallikreins on native and oxidized kininogens. J. Biol. Chem. 273, 33224–33229.
Kozma, R., Ahmed, S., Best, A., and Lim, L. (1995). The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 15, 1942–1952.
Kränkel, N., Katare, R. G., Siragusa, M., Barcelos, L. S., Campagnolo, P., Mangialardi, G., et al. (2008). Role of kinin B2 receptor signaling in the recruitment of circulating progenitor cells with neovascularization potential. Circ. Res. 103, 1335–1343.
Krettli, A. U., Cançado, J. R., and Brener, Z. (1982). Effect of specific chemotherapy on the levels of lytic antibodies in Chagas’s disease. Trans. R. Soc. Trop. Med. Hyg. 76, 334–340.
Kuhr, F., Lowry, J., Zhang, Y., Brovkovych, V., and Skidgel, R. A. (2010). Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 44, 145–154.
Labovsky, V., Smulski, C. R., Gómez, K., Levy, G., and Levin, M. J. (2007). Anti-beta1-adrenergic receptor autoantibodies in patients with chronic Chagas heart disease. Clin. Exp. Immunol. 148, 440–449.
Leeb-Lundberg, L. M., Song, X. H., and Mathis, S. A. (1994). Focal adhesion-associated proteins p125FAK and paxillin are substrates for bradykinin-stimulated tyrosine phosphorylation in Swiss 3T3 cells. J. Biol. Chem. 269, 24328–24334.
Leeb-Lundberg, L. M., Marceau, F., Müller-Esterl, W., Pettibone, D. J., and Zuraw, B. L. (2005). International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 57, 27–77.
Leite, M. F., Moyer, M. S., and Andrews, N. W. (1998). Expression of the mammalian calcium signaling response to Trypanosoma cruzi in Xenopus laevis oocytes. Mol. Biochem. Parasitol. 92, 1–13.
Leon, J. S., Wang, K., and Engman, D. M. (2003). Captopril ameliorates myocarditis in acute experimental Chagas disease. Circulation 107, 2264–2269.
Levick, S. P., Meléndez, G. C., Plante, E., McLarty, J. L., Brower, G. L., and Janicki, J. S. (2010). Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovasc. Res. 89, 12–19.
Lima, A. P., dos Reis, F. C., Serveau, C., Lalmanach, G., Juliano, L., Ménard, R., et al. (2001). Cysteine protease isoforms from Trypanosoma cruzi, cruzipain 2 and cruzain, present different substrate preference and susceptibility to inhibitors. Mol. Biochem. Parasitol. 114, 41–52.
Lima, A. P., Almeida, P. C., Tersariol, I. L., Schmitz, V., Schmaier, A. H., Juliano, L., et al. (2002). Heparan sulfate modulates kinin release by Trypanosoma cruzi through the activity of cruzipain. J. Biol. Chem. 277, 5875–5881.
Macedo, A. M., and Pena, S. D. (1998). Genetic variability of Trypanosoma cruzi: implications for the pathogenesis of Chagas disease. Parasitol. Today 14, 119–124.
Machado, F. S., Martins, G. A., Aliberti, J. C., Mestriner, F. L., Cunha, F. Q., and Silva, J. S. (2000). Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation 102, 3003–3008.
Marin-Neto, J. A., Cunha-Neto, E., Maciel, B. C., and Simẽes, M. V. (2007). Pathogenesis of chronic Chagas heart disease. Circulation 115, 1109–1123.
Maurer, M., Wedemeyer, J., Metz, M., Piliponsky, A. M., Weller, K., and Chatterjea, D., et al. (2004). Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature 432, 512–516.
McKerrow, J. H., Doyle, P. S., Engel, J. C., Podust, L. M., Robertson, S. A., Ferreira, R., et al. (2009). Two approaches to discovering and developing new drugs for Chagas disease. Mem. Inst. Oswaldo Cruz 104(Suppl. 1), 263–269.
McLean, P. G., Ahluwalia, A., and Perretti, M. (2000). Association between kinin B1 receptor expression and leukocyte trafficking across mesenteric postcapillary venules. J. Exp. Med. 192, 367–380.
McNeil, P. L., and Steinhardt, R. A. (2003). Plasma membrane disruption: repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 19, 697–731.
Medei, E., Pedrosa, R. C., Benchimol Barbosa, P. R., Costa, P. C., Hernández, C. C., Chaves, E. A., et al. (2007). Human antibodies with muscarinic activity modulate ventricular repolarization: basis for electrical disturbance. Int. J. Cardiol. 115, 373–380.
Medeiros, R., Cabrini, D. A., Ferreira, J., Fernandes, E. S., Mori, M. A., Pesquero, J. B., et al. (2004). Bradykinin B1 receptor expression induced by tissue damage in the rat portal vein: a critical role for mitogen-activated protein kinase and nuclear factor-kappaB signaling pathways. Circ. Res. 94, 1375–1382.
Medzhitov, R., and Janeway, C. A. Jr. (1997). Innate immunity: impact on the adaptive immune response. Curr. Opin. Immunol. 9, 4–9.
Medzhitov, R. (2009). Damage control in host-pathogen interactions. Proc. Natl. Acad. Sci. U.S.A. 106, 15525–15526.
Meirelles, M. N., Juliano, L., Carmona, E., Silva, S. G., Costa, E. M., Murta, A. C., et al. (1992). Inhibitors of the major cysteinyl proteinase (GP57/51) impair host cell invasion and arrest the intracellular development of Trypanosoma cruzi in vitro. Mol. Biochem. Parasitol. 52, 175–184.
Messadi-Laribi, E., Griol-Charhbili, V., Gaies, E., Vincent, M. P., Heudes, D., Meneton, P., et al. (2008). Cardioprotection and kallikrein-kinin system in acute myocardial ischaemia in mice. Clin. Exp. Pharmacol. Physiol. 35, 489–493.
Meuser-Batista, M., Corrêa, J. R., Carvalho, V. F., de Carvalho Britto, C. F., Moreira, O. C., Batista M. M., et al. (2011). Mast cell function and death in Trypanosoma cruzi infection. Am. J. Pathol. 179, 1894–1904.
Miles, M. A., Llewellyn, M. S., Lewis, M. D., Yeo, M., Baleela, R., Fitzpatrick, S., et al. (2009). The molecular epidemiology and phylogeography of Trypanosoma cruzi and parallel research on leishmania: looking back and to the future. Parasitology 136, 1509–1528.
Monteiro, A. C., Abrahamson, M., Lima, A. P., Vannier-Santos, M. A., and Scharfstein, J. (2001). Identification, characterization and localization of chagasin, a tight-binding cysteine protease inhibitor in Trypanosoma cruzi. J. Cell Sci. 114, 3933–3942.
Monteiro, A. C., Schmitz, V., Svensjo, E., Gazzinelli, R. T., Almeida, I. C., Todorov, A., et al. (2006). Cooperative activation of TLR2 and bradykinin B2 receptor is required for induction of type 1 immunity in a mouse model of subcutaneous infection by Trypanosoma cruzi. J. Immunol. 177, 6325–6335.
Monteiro, A. C., Schmitz, V., Morrot, A., de Arruda, L. B., Nagajyothi, F., Granato, A., et al. (2007). Bradykinin B2 Receptors of dendritic cells, acting as sensors of kinins proteolytically released by Trypanosoma cruzi, are critical for the development of protective type-1 responses. PLoS Pathog. 3:e185. doi: 10.1371/journal.ppat.0030185
Monteiro, A. C., Scovino, A., Raposo, S., Gaze, V. M., Cruz, C., Svensjö, E., et al. (2009). Kinin danger signals proteolytically released by gingipain induce Fimbriae-specific IFN-gamma- and IL-17-producing T cells in mice infected intramucosally with Porphyromonas gingivalis. J. Immunol. 183, 3700–3711.
Morand-Contant, M., Anand-Srivastava, M. B., and Couture, R. (2010). Kinin B1 receptor upregulation by angiotensin II and endothelin-1 in rat vascular smooth muscle cells: receptors and mechanisms. Am. J. Physiol. Heart Circ. Physiol. 299, H1625–H1632.
Moreau, M. E., Bawolak, M. T., Morissette, G., Adam, A., and Marceau, F. (2007). Role of nuclear factor-kappaB and protein kinase C signaling in the expression of the kinin B1 receptor in human vascular smooth muscle cells. Mol. Pharmacol. 71, 949–956.
Morris, S. A., Tanowitz, H. B., Wittner, M., and Bilezikian, J. P. (1990). Pathophysiological insights into the cardiomyopathy of Chagas’ disease. Circulation 82, 1900–1909.
Morrot, A., Strickland, D. K., Higuchi, M. de L., Reis, M., Pedrosa, R., and Scharfstein, J. (1997). Human T cell responses against the major cysteine proteinase (cruzipain) of Trypanosoma cruzi: role of the multifunctional alpha 2-macroglobulin receptor in antigen presentation by monocytes. Int. Immunol. 9, 825–834.
Morty, R. E., Lonsdale-Eccles, J. D., Morehead, J., Caler, E. V., Mentele, R., Auerswald, E. A., et al. (1999). Oligopeptidase B from Trypanosoma brucei, a new member of an emerging subgroup of serine oligopeptidases. J. Biol. Chem. 274, 26149–26156.
Müller, F., Mutch, N. J., Schenk, W. A., Smith, S. A., Esterl, L., Spronk, H. M., et al. (2009). Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 139, 1143–1156.
Munk, V. C., Sanchez de Miguel, L., Petrimpol, M., Butz, N., Banfi, A., Eriksson, U., et al. (2007). Angiotensin II induces angiogenesis in the hypoxic adult mouse heart in vitro through an AT2-B2 receptor pathway. Hypertension 49, 1178–1185.
Murray, D. B., Gardner, J. D., Brower, G. L., and Janicki, J. S. (2004). Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am. J. Physiol. Heart Circ. Physiol. 287, H2295–H2299.
Murta, A. C., Persechini, P. M., Padron Tde, S., de Souza, W., Guimarães, J. A., and Scharfstein, J. (1990). Structural and functional identification of GP57/51 antigen of Trypanosoma cruzi as a cysteine proteinase. Mol. Biochem. Parasitol. 43, 27–38.
Nascimento, C. R., Andrade, D., Cordovil, T., Andrade, M. V., Juliano, M. A., Juliano, L., et al. (2012). “Mast cells propagate inflammation in peripheral sites of Trypanosoma cruzi infection through the activation of the kallikrein-kinin cascade in” XXVIII Reunião Anual da Sociedade Brasileira de Protozoologia, Caxambú.
Nico, D., Feijó, D. F., Maran, N., Morrot, A., Scharfstein, J., Palatnik, M., et al. (2012). Resistance to visceral leishmaniasis is severely compromised in mice deficient of bradykinin B2-receptors. Parasit. Vectors 5:261. doi: 10.1186/1756-3305-5-261
Oka, T., Hikoso, S., Yamaguchi, O., Taneike, M., Takeda, T., Tamai, T., et al. (2012). Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255.
Okamoto, Y., Ninomiya, H., Miwa, S., and Masaki, T. (2000). Cholesterol oxidation switches the internalization pathway of endothelin receptor type A from caveolae to clathrin-coated pits in Chinese hamster ovary cells. J. Biol. Chem. 275, 6439–6446.
Oliveira, A. C., Peixoto, J. R., de Arruda, L. B., Campos, M. A., Gazzinelli, R. T., Golenbock, D. T., et al. (2004). Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J. Immunol. 173, 5688–5696.
Oliveira, A. C., de Alencar, B. C., Tzelepis, F., Klezewsky, W., da Silva, R. N., Neves, F. S., et al. (2010). Impaired innate immunity in Tlr4(-/-) mice but preserved CD8 T cell responses against Trypanosoma cruzi in Tlr4-, Tlr2-, Tlr9- or Myd88-deficient mice. PLoS Pathog. 6:e1000870. doi: 10.1371/journal.ppat.1000870
Oschatz, C., Maas, C., Lecher, B., Jansen, T., Björkqvist, J., Tradler, T., et al. (2011). Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 34, 258–268.
Ostrom, R. S. (2002). New determinants of receptor-effector coupling: trafficking and compartmentation in membrane microdomains. Mol. Pharmacol. 61, 473–476.
Padilla, A. M., Bustamante, J. M., and Tarleton, R. L. (2009). CD8+ T cells in Trypanosoma cruzi infection. Curr. Opin. Immunol. 21, 385–390.
Paiva, C. N., Feijó, D. F., Dutra, F. F., Carneiro, V. C., Freitas, G. B., Alves, L. S., et al. (2012). Oxidative stress fuels Trypanosoma cruzi infection in mice. J. Clin. Invest. 122, 2531–2542.
Parkin, E. T., Turner, A. J., and Hooper, N. M. (2004). Secretase-mediated cell surface shedding of the angiotensin-converting enzyme. Protein Pept. Lett. 11, 423–432.
Petersen, C. A., Krumholz, K. A., and Burleigh, B. A. (2005). Toll-like receptor 2 regulates interleukin-1beta-dependent cardiomyocyte hypertrophy triggered by Trypanosoma cruzi. Infect. Immun. 73, 6974–6980.
Petkova, S. B., Tanowitz, H. B., Magazine, H. I., Factor, S. M., Chan, J., Pestell, R. G., et al. (2000). Myocardial expression of endothelin-1 in murine Trypanosoma cruzi infection. Cardiovasc. Pathol. 9, 257–265.
Raslan, F., Schwarz, T., Meuth, S. G., Austinat, M., Bader, M., Renné, T., et al. (2010). Inhibition of bradykinin receptor B1 protects mice from focal brain injury by reducing blood-brain barrier leakage and inflammation. J. Cereb. Blood Flow Metab. 30, 1477–1486.
Renné, T., Dedio, J., David, G., and Müller-Esterl, W. (2000). High molecular weight kininogen utilizes heparan sulfate proteoglycans for accumulation on endothelial cells. J. Biol. Chem. 275, 33688–33696.
Renné, T., and Muller-Esterl, W. (2001). Cell surface-associated chondroitin sulfate proteoglycans bind contact phase factor H-kininogen. FEBS Lett. 500, 36–40.
Rossi, M. A. (1990). Microvascular changes as a cause of chronic cardiomyopathy in Chagas’ disease. Am. Heart J. 120, 233–236.
Salomone, O. A., Caeiro, T. F., Madoery, R. J., Amuchástegui, M., Omelinauk, M., Juri, D., et al. (2001). High plasma immunoreactive endothelin levels in patients with Chagas’ cardiomyopathy. Am. J. Cardiol. 87, 1217–1220.
Sampaio, A. L., Rae, G. A., and Henriques, M. G. (2000). Participation of endogenous endothelins in delayed eosinophil and neutrophil recruitment in mouse pleurisy. Inflamm. Res. 49, 170–176.
Sangsree, S., Brovkovych, V., Minshall, R. D., and Skidgel, R. A. (2003). Kininase I-type carboxypeptidases enhance nitric oxide production in endothelial cells by generating bradykinin B1 receptor agonists. Am. J. Physiol. Heart Circ. Physiol. 284, H1959–H1968.
Sansonetti, P. J. (2006). The innate signaling of dangers and the dangers of innate signaling. Nat. Immunol. 7, 1237–1242.
Santana, J. M., Grellier, P., Schrével, J., and Teixeira, A. R. (1997). A Trypanosoma cruzi-secreted 80 kDa proteinase with specificity for human collagen types I and IV. Biochem. J. 325, 129–137.
Santos, C. C., Scharfstein, J., and Lima, A. P. (2006). Role of chagasin-like inhibitors as endogenous regulators of cysteine proteases in parasitic protozoa. Parasitol. Res. 99, 323–324.
Scharfstein, J., Rodrigues, M. M., Alves, C. A., de Souza, W., Previato, J. O., and Mendonça-Previato, L. (1983). Trypanosoma cruzi: description of a highly purified surface antigen defined by human antibodies. J. Immunol. 131, 972–976.
Scharfstein, J., and Morrot, A. (1999). A role for extracellular amastigotes in the immunopathology of Chagas disease. Mem. Inst. Oswaldo Cruz 1, 51–63.
Scharfstein, J., Schmitz, V., Morandi, V., Capella, M. M., Lima, A. P., Morrot, A., et al. (2000). Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B2 receptors. J. Exp. Med. 192, 1289–1300.
Scharfstein, J., Schmitz, V., Svensjö, E., Granato, A., and Monteiro, A. C. (2007). Kininogens coordinate adaptive immunity through the proteolytic release of bradykinin, an endogenous danger signal driving dendritic cell maturation. Scand. J. Immunol. 66, 128–136.
Scharfstein, J., and Lima, A. P. (2008). Roles of naturally occurring protease inhibitors in the modulation of host cell signaling and cellular invasion by Trypanosoma cruzi. Subcell. Biochem. 47, 140–154.
Scharfstein, J. (2010). Trypanosoma cruzi cysteine proteases, acting in the interface between the vascular and immune systems, influence pathogenic outcome in experimental Chagas disease. Open Parasitol. 4, 60–71.
Scharfstein, J., and Andrade, D. (2011) Infection-associated vasculopathy in experimental Chagas disease pathogenic roles of endothelin and kinin pathways. Adv. Parasitol. 76, 101–127.
Scharfstein, J., and Svensjö, E. (2012). “The kallikrein-kinin system in parasitic infection,” in Kinins, ed. M. Bader (Berlin: Walter de Gryter GmbH & Co.), 321–330.
Schenkman, S., Eichinger, D., Pereira, M. E., and Nussenzweig, V. (1994). Structural and functional properties of Trypanosoma trans-sialidase. Annu. Rev. Microbiol. 48, 499–523.
Schmitz, V., Svensjö, E., Serra, R. R., Teixeira, M. M., and Scharfstein, J. (2009). Proteolytic generation of kinins in tissues infected by Trypanosoma cruzi depends on CXC chemokine secretion by macrophages activated via Toll-like 2 receptors. J. Leukoc. Biol. 85, 1005–1014.
Schulze-Topphoff, U., Prat, A., Prozorovski, T., Siffrin, V., Paterka, M., Herz, J., et al. (2009). Activation of kinin receptor B1 limits encephalitogenic T lymphocyte recruitment to the central nervous system. Nat. Med. 15, 788–793.
Sela, U., Olds, P., Park, A., Schlesinger, S. J., and Steinman, R. M. (2011). Dendritic cells induce antigen-specific regulatory T cells that prevent graft versus host disease and persist in mice. J. Exp. Med. 208, 2489–2496.
Shortman, K., and Naik, S. H. (2007). Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7, 19–30.
Silva, G. K., Gutierrez, F. R., Guedes, P. M., Horta, C. V., Cunha, L. D., Mineo, T. W., et al. (2010). Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against Trypanosoma cruzi infection. J. Immunol. 184, 1148–1152.
Silverio, J. C., Pereira, I. R., Cipitelli Mda, C., Vinagre, N. F., Rodrigues, M. M., Gazzinelli, R. T., et al. (2012). CD8+ T-cells expressing interferon gamma or perforin play antagonistic roles in heart injury in experimental Trypanosoma cruzi-elicited cardiomyopathy. PLoS Pathog. 8:e1002645. doi: 10.1371/journal.ppat.1002645
Skidgel, R. A., and Erdos, E. G. (2004). Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow-ups to early studies. Peptides 25, 521–525.
Souto-Padrón, T., Campetella, O. E., Cazzulo, J. J., and de Souza, W. (1990). Cysteine proteinase in Trypanosoma cruzi: immunocytochemical localization and involvement in parasite-host cell interaction. J. Cell Sci. 96, 485–490.
Spillmann, F., Van Linthout, S., Schultheiss, H. P., and Tschöpe, C. (2006). Cardioprotective mechanisms of the kallikrein-kinin system in diabetic cardiopathy. Curr. Opin. Nephrol. Hypertens. 15, 22–29.
Spillmann, F., and Tschöpe, C. (2012). “Kallikrein-Kinin System in the heart,” in Kinins, ed. M. Bader (Walter de Gryter GmbH & Co.: Berlin), 117–127.
Stoka, V., Nycander, M., Lenarcic, B., Labriola, C., Cazzulo, J. J., Björk, I., et al. (1995). Inhibition of cruzipain, the major cysteine proteinase of the protozoan parasite, Trypanosoma cruzi, by proteinase inhibitors of the cystatin superfamily. FEBS Lett. 370,101–104.
Stoka, V., McKerrow, J. H., Cazzulo, J. J., and Turk, V. (1998). Substrate inhibition of cruzipain is not affected by the C-terminal domain. FEBS Lett. 429, 129–133.
Svensjö, E., Batista, P. R., Brodskyn, C. I., Silva, R., Lima, A. P., Schmitz, V., et al. (2006). Interplay between parasite cysteine proteases and the host kinin system modulates microvascular leakage and macrophage infection by promastigotes of the Leishmania donovani complex. Microbes Infect. 8, 206–220.
Tanowitz, H. B., Wittner, M., Morris, S. A., Zhao, W., Weiss, L. M., Hatcher, V. B., et al. (1999). The putative mechanistic basis for the modulatory role of endothelin-1 in the altered vascular tone induced by Trypanosoma cruzi. Endothelium 6, 217–230.
Tanowitz, H. B., Huang, H., Jelicks, L. A., Chandra, M., Loredo, M. L., Weiss, L. M., et al. (2005). Role of endothelin 1 in the pathogenesis of chronic chagasic heart disease. Infect. Immun. 73, 2496–2503.
Tardieux, I., Webster, P., Ravesloot, J., Boron, W., Lunn, J. A., Heuser, J. E., et al. (1992). Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 71, 1117–1130.
Tarleton, R. L. (2001). Parasite persistence in the aetiology of Chagas disease. Int. J. Parasitol. 31, 550–554.
Todorov, A. G., Andrade, D., Pesquero, J. B., Araujo Rde, C., Bader, M., Stewart, J., et al. (2003). Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor (B1/B2) subtypes. FASEB J. 17, 73–75.
Tonelli, R. R., Torrecilhas, A. C., Jacysyn, J. F., Juliano, M. A., Colli, W., and Alves, M. J. (2011). In vivo infection by Trypanosoma cruzi: the conserved FLY domain of the gp85/trans-sialidase family potentiates host infection. Parasitology 138, 481–492.
Trocoli-Torrecilhas, A. C., Tonelli, R. R., Pavanelli, W. R., da Silva, J. S., Schumacher, R. I., and de Souza, W., et al. (2009). Trypanosoma cruzi: parasite shed vesicles increase heart parasitism and generate an intense inflammatory response. Microbes Infect. 11, 29–39.
Tschöpe, C., Walther, T., Escher, F., Spillmann, F., Du, J., Altmann, C., et al. (2005). Transgenic activation of the kallikrein-kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. FASEB J. 19, 2057–2059.
Tschöpe, C., and Westermann, D. (2008). Development of diabetic cardiomyopathy and the kallikrein-kinin system – new insights from B1 and B2 receptor signaling. Biol. Chem. 389, 707–711.
Tyler, K. M., Luxton, G. W., Applewhite, D. A., Murphy, S. C., and Engman, D. M. (2005). Responsive microtubule dynamics promote cell invasion by Trypanosoma cruzi. Cell. Microbiol. 11, 1579–1591.
Vasquez-Pinto, L. M., Nantel, F., Sirois, P., and Jancar, S. (2010). Bradykinin B(1) receptor antagonist R954 inhibits eosinophil activation/proliferation/migration and increases TGF-beta and VEGF in a murine model of asthma. Neuropeptides 44, 107–113.
von Brühl, M. L., Stark, K., Steinhart, A., Chandraratne, S., Konrad, I., Lorenz, M., et al. (2012). Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209, 819–835.
von Köckritz-Blickwede, M., Goldmann, O., Thulin, P., Heinemann, K., Norrby-Teglund, A., Rohde, M., et al. (2008). Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111, 3070–3080.
Wang, P. H., Cenedeze, M. A., Pesquero, J. B., Pacheco-Silva, A., and Câmara, N. O. (2006). Influence of bradykinin B1 and B2 receptors in the immune response triggered by renal ischemia-reperfusion injury. Int. Immunopharmacol. 6, 1960–1965.
Westermann, D., Walther, T., Savvatis, K., Escher, F., Sobirey, M., Riad, A., et al. (2009). Gene deletion of the kinin receptor B1 attenuates cardiac inflammation and fibrosis during the development of experimental diabetic cardiomyopathy. Diabetes Metab. Res. Rev. 58, 1373–1381.
Woolsey, A. M., and Burleigh, B. A. (2004). Host cell actin polymerization is required for cellular retention of Trypanosoma cruzi and early association with endosomal/lysosomal compartments. Cell. Microbiol. 6, 829–838.
Yamamura, H., Nabe, T., Kohno, S., and Ohata, K. (1994). Endothelin-1 induces release of histamine and leukotriene C4 from mouse bone marrow-derived mast cells. Eur. J. Pharmacol. 257, 235–242.
Yang, X. P., Liu, Y. H., Mehta, D., Cavasin, M. A., Shesely, E., and Xu, J., et al. (2001). Diminished cardioprotective response to inhibition of angiotensin-converting enzyme and angiotensin II type 1 receptor in B(2) kinin receptor gene knockout mice. Circ. Res. 88, 1072–1079.
Zhang, X., Tan, F., Zhang, Y., and Skidgel, R. A. (2008). Carboxypeptidase M and kinin B1 receptors interact to facilitate efficient b1 signaling from B2 agonists. J. Biol. Chem. 283, 7994–8004.
Keywords: bradykinin, cardiomyopathy, cruzipain, endothelins, GPCRs, proteases, kallikrein, Trypanosoma cruzi
Citation: Scharfstein J, Andrade D, Svensjö E, Oliveira AC and Nascimento CR (2013) The kallikrein-kinin system in experimental Chagas disease: a paradigm to investigate the impact of inflammatory edema on GPCR-mediated pathways of host cell invasion by Trypanosoma cruzi. Front. Immun. 3:396. doi: 10.3389/fimmu.2012.00396
Received: 16 September 2012; Paper pending published: 17 October 2012;
Accepted: 07 December 2012; Published online: 25 January 2013.
Edited by:
Wanderley De Souza, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Ravi Durvasula, University of New Mexico School of Medicine, USACopyright: © 2013 Scharfstein, Andrade, Svensjö, Oliveira and Nascimento. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Julio Scharfstein, Laboratório de Imunologia Molecular, Instituto de Biofísica Carlos Chagas Filho, Centro de Ciências da Saúde, Universidade Federal do Rio de Janeiro, Cidade Universitária Rio de Janeiro, Rio de Janeiro, Brazil. e-mail:anNjaGFyZjJAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.