


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Immunol. , 26 June 2012
Sec. Inflammation
Volume 3 - 2012 | https://doi.org/10.3389/fimmu.2012.00173
This article is part of the Research Topic The Molecular Mechanisms of Chronic Inflammation Development View all 13 articles
Metabolic disorders of the cardiovascular system, including atherosclerosis, obesity, diabetes, and hypertension, are the leading cause of morbidity and mortality in the western world. Metabolic syndrome is associated with a combination of risk factors, including nutrient excess, hyperglycemia, hyperlipidemia, insulin-resistance, and obesity that, when occurring together, increase the risk of developing cardiovascular metabolic disorders (CVMDs). In individuals with this syndrome, different CVMDs often develop simultaneously, so that obese patients present with an increased risk of suffering also from type II diabetes and insulin-resistance, these being frequently associated with atherosclerosis and hypertension, suggesting the sharing of similar pathogenic mechanisms.
Chronic low-grade inflammation is a key feature of CVMDs. Macrophages and innate immunity have been classically implicated in the pathogenesis of atherosclerosis, and more recently of hypertension, as well as other metabolic disorders associated with cardiovascular disease such as obesity and type II diabetes (Hotamisligil, 2006; Rocha and Libby, 2009; Harrison et al., 2010; Matarese et al., 2010; Schiffrin, 2010; Baker et al., 2011; Donath and Shoelson, 2011; Hansson and Hermansson, 2011; Lahoute et al., 2011).
Evidence for the contribution of T cell-mediated immunity to CVMDs has only recently emerged. T cells infiltrate atherosclerotic plaques, obese adipose tissue, pancreatic islets, and the perivascular fat and kidneys in hypertensive individuals. In these sites, proinflammatory immune responses mediated by CD4+ T-helper type 1 (TH1) and CD8+ cytotoxic T lymphocytes are preponderant over immunomodulatory (TH2) and immunosuppressive (Treg) immunity, and drive disease progression (Rocha and Libby, 2009; Baker et al., 2011). Interferon (IFN)-γ, the signature TH1 cytokine, is found in human atherosclerotic plaques and obese adipose tissue, where it exerts pathogenic effects by promoting the recruitment and activation of macrophages and the release of proinflammatory cytokines such as Tumor Necrosis Factor (TNF), all of which perpetuate the local inflammation (Gupta et al., 1997; Ranjbaran et al., 2007; Rocha and Libby, 2009; Hansson and Hermansson, 2011). In contrast, Interleukin (IL)-4, the prototypical anti-inflammatory cytokine of the TH2 lineage, is not frequently observed in human plaques (Rocha and Libby, 2009; Hansson and Hermansson, 2011). TH1-mediated proinflammatory mechanisms have also been involved in the pathogenesis of hypertension (Mahmoud et al., 2003; Shao et al., 2003; Seaberg et al., 2005; Guzik et al., 2007; Harrison et al., 2010; Schiffrin, 2010). RAG1−/− mice, which lack both T and B cells, develop neither angiotensin II- nor deoxycorticosterone acetate (DOCA) salt-induced hypertension, and adoptive transfer of T cells, but not B cells, restores the hypertensive phenotype induced by these stimuli (Guzik et al., 2007). Immunosuppressive Treg lymphocytes appear to be reduced in number in human atherosclerotic plaques (de Boer et al., 2007; Lahoute et al., 2011) and in the adipose tissue of insulin-resistant obese mice (Feuerer et al., 2009; Matarese et al., 2010). Blockade of IL-10 and Transforming Growth Factor (TGF)-β, the two cytokines responsible for most of the immunosuppressive effects mediated by Treg cells, accelerates lesion development (Lahoute et al., 2011). Adoptive transfer of Treg cells reduces atherosclerosis in Apo−/− mice (Ait-Oufella et al., 2006; Lahoute et al., 2011) and insulin-resistance in obese mice (Feuerer et al., 2009; Matarese et al., 2010). How proinflammatory T cells accumulate in the inflamed sites is an open question.
The regulation of energy metabolism is crucial to T cell-mediated immunity, including activation, proliferation, and differentiation toward effector versus regulatory T cells, and, as we discuss here, migration. Naïve T cells rely upon a catabolic type of metabolism whereby ATP is mainly generated via oxidative phosphorylation (OXPHOS). This slow metabolism is sufficient to support their requirements for survival, maintain housekeeping functions, such as ion transport and membrane integrity, and keep them away from engaging into cell proliferation (Rathmell et al., 2000; Frauwirth and Thompson, 2004). Upon TCR engagement by cognate antigen, T cells switch from catabolism to anabolism, with phosphatidylinositol 3′-kinase (PI3K) leading to the activation of the serine-threonine kinase AKT. This step promotes glucose metabolism by stimulating the localization of the glucose transporter Glut1 to the plasma membrane and the activity of hexokinase and phosphofructokinase, two rate limiting enzymes of the glycolytic pathway. Increased glycolytic flux enables activated T cells to generate ATP and, at the same time, efficiently utilize carbon sources in the form of amino acids and lipids for the biosynthesis of proteins and membranes necessary for the expansion phase that characterizes the immune response. Downstream of TCR, AKT also controls the activation state of the mammalian target of rapamycin (mTOR), a sensor of nutritional and energetic status in cells that promotes protein synthesis. mTOR is a key regulator of T cell differentiation toward proinflammatory subsets, and its inhibition with rapamycin promotes immunosuppression via the induction of anergic and Treg cells (Jones and Thompson, 2007; Zheng et al., 2009; Peter et al., 2010; Powell and Delgoffe, 2010; Marelli-Berg et al., 2012; Mauro et al., 2012).
Indirect evidence suggests that metabolic status can influence T cell homing patterns. Expression of the adhesion molecule CD62L (also known as L-selectin) and the chemokine receptors CC-chemokine receptor 7 (CCR7) and sphingosine-1-phosphate receptor 1 (S1P1) on the surface of naïve T cells facilitates their trafficking to secondary lymphoid organs (SLOs). Upon TCR engagement, the PI3K-AKT-mTOR axis promotes the downregulation of CD62L, CCR7, and S1P1, and prompts effector T cells expressing adhesion molecules [such as VLA4 (very late antigen 4) and ligands for P-selectin and E-selectin] and chemokine receptors (such as CXCR3 and CCR5) to traffic to the sites of inflammation (Sinclair et al., 2008).
Upon resolution of the immune response, the number of antigen specific T cells contracts, as effector cells die and only the memory subset survives. This step is characterized by a metabolic transition from anabolism to catabolism, from high mTOR activity to low mTOR activity, and from effector to memory cells (Sinclair et al., 2008; Finlay and Cantrell, 2011). This transition is associated with the re-expression of CD62L and CCR7, thus allowing the newly formed memory cells to continue surveillance by trafficking in and out of SLOs. Consistently, rapamycin-mediated inhibition of mTOR causes effector T cells to re-express CD62L and CCR7, and home to SLOs where they are trapped away from the target sites in the periphery (Sinclair et al., 2008; Finlay and Cantrell, 2011). Therefore, rapamycin can promote immunosuppression by redirecting effector T cells from peripheral tissues to SLOs.
Nuclear Factor (NF)-κB is a family of transcription factors that promote immunity by controlling the expression of genes involved in inflammation (Baker et al., 2011). A number of recent studies have demonstrated a key role for NF-κB signaling pathways in the development of inflammation-driven metabolic disorders in adipose tissue, pancreatic β-cells, arterial walls, and the central nervous system (Yuan et al., 2001; Gareus et al., 2008; Baker et al., 2011; Purkayastha et al., 2011).
An important role for NF-κB in the organization of energy metabolism networks in the cell through the control of the balance between the utilization of glycolysis and OXPHOS has recently been demonstrated. NF-κB acts as a physiological regulator of mitochondrial respiration and via this function suppresses the metabolic reprogramming to aerobic glycolysis in cells and prevents necrosis upon nutrient starvation. This metabolic function of NF-κB involves the p53-dependent upregulation of mitochondrial Synthesis of Cytochrome c Oxidase 2 (SCO2), a crucial component of the electron transfer chain Complex IV or Cytochrome c Oxidase, which increases OXPHOS and reduces glycolytic flux in cells (Mauro et al., 2011).
Due to its ability to control both inflammatory and metabolic responses, NF-κB represents a major target, in addition to the mTOR pathway, for the development of novel therapeutic strategies in CVMDs (Figure 1).
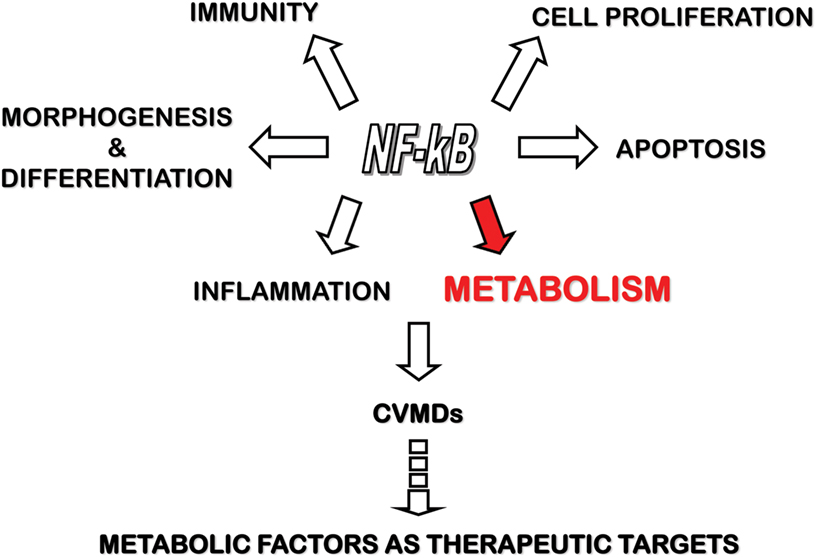
Figure 1. The control of inflammation and metabolism by NF-κB and its relevance to CVMDs. NF-κB transcription factors control several physiologic functions, including morphogenesis and differentiation, cell proliferation and apoptosis, immunity and inflammation. Recent work has shed light in the control of cell energy metabolism by NF-κB. By regulating both inflammatory and metabolic pathways, NF-κB has been involved in the pathogenesis of CVMDs, thereby opening the way to the identification of metabolic factors under NF-κB control as target for therapy.
The control of T cell migration by TCR engagement and co-stimuli (Mirenda et al., 2007; Jarmin et al., 2008) implies that metabolic changes downstream of these receptors can influence both efficiency and topography of T cell trafficking. The metabolic machinery is also likely to directly affect and be affected by T cell migratory events, as T cells continuously recirculate between different microenvironments (e.g., blood, lymphoid tissues, and peripheral tissues) in which they are exposed to different nutrient availability and oxygen (O2) tension, and must adapt their metabolic pathways to effectively mediate immune responses. The direct effect of metabolism on the trafficking ability of T cells, however, has not been investigated.
Memory lymphocyte trafficking is regulated by a number of complex mechanisms, which involve both pro-migratory and pro-static events. Following priming in the lymph nodes, T cells migrate to antigen-rich sites to exert their effector function. Effector T cells are retained in the tissue for a time sufficient to achieve effective immunity. Homeostatic mechanisms prescribe that long-lived memory T cells leave the tissue once the immune response is complete, and inflammation then subsides. A failure of T cells to be released from the tissue leads to persistent inflammation and progressive tissue damage (Burman et al., 2005). T cells are exposed to different metabolic environments during these migratory events, which are likely to affect their motility and ability to respond to migratory cues from the surrounding microenvironment. Thus, it is conceivable that altered migration might result from an altered metabolic microenvironment (blood, tissues, or the inflammatory milieu itself) and that this might in turn initiate or sustain chronic inflammation, as it happens in CVMDs. Specifically, metabolic alterations that characterize the metabolic syndrome, including nutrient excess, hyperglycemia and hyperlipidemia, might promote T cells infiltration of inflammatory sites by modifying their intracellular metabolism. Once inside the inflamed tissue, T cells could become immobile due to a drop in O2 tension, nutrient availability, or pH that affects their metabolic status. The inability of differentiated T cells to exit inflamed tissues would favor the persistence of chronic inflammation.
Immunosuppressive therapies have been proposed in CVMDs. TNF blockade reduced the incidence of cardiovascular events in patients with rheumatoid arthritis (RA), an autoimmune disease associated with a strong inflammatory component and with accelerated atherosclerosis (Jacobsson et al., 2005; Greenberg et al., 2011). Sirolimus (rapamycin), an inhibitor of the mTOR approved for use in the prevention of transplant rejection, is being tested in pre-clinical studies of cardiovascular disease in animal models and has been used with some success in the clinic for the local treatment of restenosis (Adelman, 2010).
In addition to conventional immunosuppression, compounds targeting metabolic pathways have been shown to exert anti-inflammatory properties. For instance, essential amino acid depletion has been shown to contribute to tolerance induction in experimental heart transplantation (Sucher et al., 2012). Similarly, n−3 polyunsaturated fatty acids (PUFAs) from fish oils, EPA, and DHA, popularly referred to as omega-3 fatty acids, with hypotriglyceridemic, hypotensive, and antithrombotic properties, have been shown to inhibit inflammatory responses associated with alteration of lipid metabolism (Cottin et al., 2011). The inhibition of immune-inflammation by correcting altered metabolism is an attractive therapeutic strategy which would specifically target CVMDs without the severe side-effects of conventional immunosuppression.
Investigation of the mechanisms for metabolic control of T cell trafficking represents a fascinating area – mostly unexplored – of research for the forthcoming years. The observations arising from such studies will provide key insights into both the physiology of T cell trafficking and the physiopathologic mechanisms of T cell infiltration in inflammatory sites, where T cells are retained and perpetuate the chronic inflammation that drives the disease progression. Importantly, while the antigen specificities promoting the activation of the adaptive immunity in humans are controversial, these diseases have been associated to non-antigen specific mechanisms involving altered T cell migration patterns (Burman et al., 2005; Full and Monaco, 2011). We propose that altered metabolism can fuel chronic inflammation in an antigen-non-specific manner, i.e., that otherwise harmless immune responses might fail to resolve leading to chronic inflammation and bystander damage when they occur in a metabolically altered environment. Similar mechanisms are likely to be important in other cardiovascular diseases (e.g., hypertension, stroke, heart transplantation, etc.) with a T cell-mediated inflammatory component. Additional associations are being drawn between CVMDs, such as obesity, and diseases that are less obviously linked to metabolic alterations, including asthma, arthritis, lupus, Alzheimer, and several forms of cancer (Mathis and Shoelson, 2011; Gerriets and Rathmell, 2012). Inflammation has been etiologically associated to the pathogenesis of each of these conditions, and the binomium metabolism-inflammation might provide new effective therapeutic protocols for a wide range of diseases that share similar pathogenic mechanisms.
We thank E. Simpson for critical comments on the manuscript. Claudio Mauro is supported by the British Heart Foundation. Federica M. Marelli-Berg is supported by the British Heart Foundation, the Medical Research Council of the UK, and the Gates Foundation. This work was facilitated by National Institute for Health Research Cardiovascular Biomedical Research Unit at Barts and The London School of Medicine and Dentistry.
Adelman, S. J. (2010). Sirolimus and its analogs and its effects on vascular diseases. Curr. Pharm. Des. 16, 4002–4011.
Ait-Oufella, H., Salomon, B. L., Potteaux, S., Robertson, A. K., Gourdy, P., Zoll, J., Merval, R., Esposito, B., Cohen, J. L., Fisson, S., Flavell, R. A., Hansson, G. K., Klatzmann, D., Tedgui, A., and Mallat, Z. (2006). Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12, 178–180.
Baker, R. G., Hayden, M. S., and Ghosh, S. (2011). NF-kappaB, inflammation, and metabolic disease. Cell Metab. 13, 11–22.
Burman, A., Haworth, O., Hardie, D. L., Amft, E. N., Siewert, C., Jackson, D. G., Salmon, M., and Buckley, C. D. (2005). A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J. Immunol. 174, 1693–1700.
Cottin, S. C., Sanders, T. A., and Hall, W. L. (2011). The differential effects of EPA and DHA on cardiovascular risk factors. Proc. Nutr. Soc. 70, 215–231.
de Boer, O. J., van der Meer, J. J., Teeling, P., van der Loos, C. M., and van der Wal, A. C. (2007). Low numbers of FOXP3 positive regulatory T cells are present in all developmental stages of human atherosclerotic lesions. PLoS ONE 2, e779. doi: 10.1371/journal.pone.0000779
Donath, M. Y., and Shoelson, S. E. (2011). Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11, 98–107.
Feuerer, M., Herrero, L., Cipolletta, D., Naaz, A., Wong, J., Nayer, A., Lee, J., Goldfine, A. B., Benoist, C., Shoelson, S., and Mathis, D. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939.
Finlay, D., and Cantrell, D. A. (2011). Metabolism, migration and memory in cytotoxic T cells. Nat. Rev. Immunol. 11, 109–117.
Frauwirth, K. A., and Thompson, C. B. (2004). Regulation of T lymphocyte metabolism. J. Immunol. 172, 4661–4665.
Full, L. E., and Monaco, C. (2011). Targeting inflammation as a therapeutic strategy in accelerated atherosclerosis in rheumatoid arthritis. Cardiovasc. Ther. 29, 231–242.
Gareus, R., Kotsaki, E., Xanthoulea, S., van der Made, I., Gijbels, M. J., Kardakaris, R., Polykratis, A., Kollias, G., de Winther, M. P., and Pasparakis, M. (2008). Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 8, 372–383.
Gerriets, V. A., and Rathmell, J. C. (2012). Metabolic pathways in T cell fate and function. Trends Immunol. 33, 168–173.
Greenberg, J. D., Furer, V., and Farkouh, M. E. (2011). Cardiovascular safety of biologic therapies for the treatment of RA. Nat. Rev. Rheumatol. 15, 13–21.
Gupta, S., Pablo, A. M., Jiang, X., Wang, N., Tall, A. R., and Schindler, C. (1997). IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Invest. 99, 2752–2761.
Guzik, T. J., Hoch, N. E., Brown, K. A., McCann, L. A., Rahman, A., Dikalov, S., Goronzy, J., Weyand, C., and Harrison, D. G. (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 204, 2449–2460.
Hansson, G. K., and Hermansson, A. (2011). The immune system in atherosclerosis. Nat. Immunol. 12, 204–212.
Harrison, D. G., Vinh, A., Lob, H., and Madhur, M. S. (2010). Role of the adaptive immune system in hypertension. Curr. Opin. Pharmacol. 10, 203–207.
Jacobsson, L. T., Turesson, C., Gulfe, A., Kapetanovic, M. C., Petersson, I. F., Saxne, T., and Geborek, P. (2005). Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J. Rheumatol. 32, 1213–1218.
Jarmin, S. J., David, R., Ma, L., Chai, J. G., Dewchand, H., Takesono, A., Ridley, A. J., Okkenhaug, K., and Marelli-Berg, F. M. (2008). T cell receptor-induced phosphoinositide-3-kinase p110 delta activity is required for T cell localization to antigenic tissue in mice. J. Clin. Invest. 118, 1154–1164.
Jones, R. G., and Thompson, C. B. (2007). Revving the engine: signal transduction fuels T cell activation. Immunity 27, 173–178.
Lahoute, C., Herbin, O., Mallat, Z., and Tedgui, A. (2011). Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat. Rev. Cardiol. 8, 348–358.
Mahmoud, F., Omu, A., Abul, H., El-Rayes, S., and Haines, D. (2003). Lymphocyte subpopulations in pregnancy complicated by hypertension. J. Obstet. Gynaecol. 23, 20–26.
Marelli-Berg, F. M., Fu, H., and Mauro, C. (2012). Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T cell-mediated immunity. Immunology. doi: 10.1111/j.1365- 2567.2012.03583.x. [Epub ahead of print].
Matarese, G., Procaccini, C., De Rosa, V., Horvath, T. L., and La Cava, A. (2010). Regulatory T cells in obesity: the leptin connection. Trends Mol. Med. 16, 247–256.
Mathis, D., and Shoelson, S. E. (2011). Immunometabolism: an emerging frontier. Nat. Rev. Immunol. 11, 81.
Mauro, C., Fu, H., and Marelli-Berg, F. M. (2012). T cell trafficking and metabolism: novel mechanisms and targets for immunomodulation. Curr. Opin. Pharmacol. doi: 10.1016/j.coph.2012.02.018. [Epub ahead of print].
Mauro, C., Leow, S. C., Anso, E., Rocha, S., Thotakura, A. K., Tornatore, L., Moretti, M., De Smaele, E., Beg, A. A., Tergaonkar, V., Chandel, N. S., and Franzos, G. (2011). NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 13, 1272–1279.
Mirenda, V., Jarmin, S. J., David, R., Dyson, J., Scott, D., Gu, Y., Lechler, R. I., Okkenhaug, K., and Marelli-Berg, F. M. (2007). Physiologic and aberrant regulation of memory T-cell trafficking by the costimulatory molecule CD28. Blood 109, 2968–2977.
Peter, C., Waldmann, H., and Cobbold, S. P. (2010). mTOR signalling and metabolic regulation of T cell differentiation. Curr. Opin. Immunol. 22, 655–661.
Powell, J. D., and Delgoffe, G. M. (2010). The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 33, 301–311.
Purkayastha, S., Zhang, G., and Cai, D. (2011). Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat. Med. 17, 883–887.
Ranjbaran, H., Sokol, S. I., Gallo, A., Eid, R. E., Iakimov, A. O., D’Alessio, A., Kapoor, J. R., Akhtar, S., Howes, C. J., Aslan, M., Pfau, S., Pober, J. S., and Tellides, G. (2007). An inflammatory pathway of IFN-gamma production in coronary atherosclerosis. J. Immunol. 178, 592–604.
Rathmell, J. C., Vander Heiden, M. G., Harris, M. H., Frauwirth, K. A., and Thompson, C. B. (2000). In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol. Cell 6, 683–692.
Rocha, V. Z., and Libby, P. (2009). Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 6, 399–409.
Schiffrin, E. L. (2010). T lymphocytes: a role in hypertension? Curr. Opin. Nephrol. Hypertens. 19, 181–186.
Seaberg, E. C., Munoz, A., Lu, M., Detels, R., Margolick, J. B., Riddler, S. A., Williams, C. M., and Phair, J. P. (2005). Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to 2003. AIDS 19, 953–960.
Shao, J., Nangaku, M., Miyata, T., Inagi, R., Yamada, K., Kurokawa, K., and Fujita, T. (2003). Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension 42, 31–38.
Sinclair, L. V., Finlay, D., Feijoo, C., Cornish, G. H., Gray, A., Ager, A., Okkenhaug, K., Hagenbeek, T. J., Spits, H., and Cantrell, D. A. (2008). Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 9, 513–521.
Sucher, R., Fischler, K., Oberhuber, R., Kronberger, I., Margreiter, C., Ollinger, R., Schneeberger, S., Fuchs, D., Werner, E. R., Watschinger, K., Zelger, B., Tellides, G., Pilat, N., Pratschke, J., Margreiter, R., Wekerle, T., and Brandacher, G. (2012). IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated Ag-4 Ig-mediated long-term solid organ allograft survival. J. Immunol. 188, 37–46.
Yuan, M., Konstantopoulos, N., Lee, J., Hansen, L., Li, Z. W., Karin, M., and Shoelson, S. E. (2001). Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293, 1673–1677.
Citation: Mauro C and Marelli-Berg FM (2012) T cell immunity and cardiovascular metabolic disorders: does metabolism fuel inflammation? Front. Immun. 3:173. doi: 10.3389/fimmu.2012.00173
Received: 08 May 2012; Accepted: 08 June 2012;
Published online: 26 June 2012.
Edited by:
Masaaki Murakami, Osaka University, JapanReviewed by:
Daisuke Kamimura, Osaka University, JapanCopyright: © 2012 Mauro and Marelli-Berg. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence:Yy5tYXVyb0BxbXVsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.