
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 June 2012
Sec. T Cell Biology
Volume 3 - 2012 | https://doi.org/10.3389/fimmu.2012.00159
This article is part of the Research Topic How does T cell antigen receptor signaling begin? View all 14 articles
 Michael S. Kuhns1,2* and Mark M. Davis3,4,5*
Michael S. Kuhns1,2* and Mark M. Davis3,4,5*“How does T cell receptor signaling begin?” Answering this question requires an understanding of how the parts of the molecular machinery that mediates this process fit and work together. Ultimately this molecular architecture must (i) trigger the relay of information from the TCR-pMHC interface to the signaling substrates of the CD3 molecules and (ii) bring the kinases that modify these substrates in close proximity to interact, initiate, and sustain signaling. In this contribution we will discuss advances of the last decade that have increased our understanding of the complex machinery and interactions that underlie this type of signaling.
In our experience, immunologists who have not directly worked on this problem are often shocked to learn that, 25+ years after its initial discovery and characterization, we consider the question of “How does T cell receptor signaling begin?” to remain unanswered. No doubt, a great deal of progress has been made. For example, it is well established that TCR signaling is initiated when the TCR binds composite surfaces of peptides embedded in major histocompatibility complex (pMHC) molecules. It is also accepted that the Src kinase Lck, which is associated with CD4 or CD8, phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMs) of the TCR-associated CD3γε, δε, and ζζ signaling dimmers when both the TCR and CD4 or CD8 bind class II or class I pMHC, respectively. Modification of the ITAMs then connects the TCR-CD3 complex to the intracellular signaling apparatus. Additionally, a great deal is now known about the ensuing signaling cascade to the nucleus. Thus, for most, this extensive understanding of the “business ends” of the TCR-CD3 complex represents a satisfactory explanation for how TCR signaling begins. It also highlights the significant advances made by the field in defining the rules that govern TCR-pMHC interactions and identifying the major players and sequence of events that transmit information from the ITAMs to the nucleus (Rudolph et al., 2006; Garcia et al., 2009; Smith-Garvin et al., 2009). Yet, there remains a lack of clarity about the mechanism(s) that trigger the initial transmission of information from the TCR-pMHC interface to the ITAMS. Our limited understanding of these processes reflects the complexity of the molecular machinery under discussion as well as the challenges researchers have faced when attempting to deconstruct and understand the processes of how TCR signaling begins. Nevertheless, the significance of the problem invites and requires persistence.
TCR triggering remains one of the most important basic research problems facing the field of immunology. The significance of this question is rooted in the fact that properly functioning T cells are essential for vertebrate immunity–without them you cannot survive for very long, even in the relatively hygienic developed world (Haase, 1999; Morgan et al., 2011). But, merely having T cells is insufficient to ensure survival in an environment full of pathogens. If they cannot properly relay information about the quantity and quality of pMHC on the surfaces of antigen-presenting cells (APCs) to their intracellular signaling apparatus, and then on to the nucleus, they will not be able to make the appropriate cell fate decisions regarding: if a precursor T cell develops to join the mature T cell repertoire (Starr et al., 2003); if a T cell becomes activated upon antigen encounter (Corse et al., 2010, 2011); the number of daughter cells made by any single T cell upon clonal expansion (Malherbe et al., 2008; Zehn et al., 2009; Corse et al., 2010, 2011); the phenotype to which those daughter cells differentiate upon activation; and the execution of effector functions upon repeated antigen encounter (Constant and Bottomly, 1997; Fazilleau et al., 2009; McHeyzer-Williams et al., 2009; Gottschalk et al., 2010). Thus, the TCR-CD3 complex represents the chief molecular checkpoint for T cell development, activation, and function. Defects in these fate decisions can result in immune-compromised individuals who are likely to suffer from complex diseases associated with immune-disregulation (Ohashi, 2002; Holst et al., 2008; Vercelli, 2008).
The central role of the TCR-CD3 complex in immunity and human health is a prime motivator for understanding the molecular mechanisms by which it functions. Such knowledge will inform practical efforts aimed at promoting T cell responses to vaccines, tumor, or pathogen. It will also guide efforts to attenuate responses to auto-antigens or beneficial commensal organisms. Indeed, the TCR-CD3 complex has been and continues to be targeted for the development of immune-modulating reagents. Modified anti-CD3ε antibodies are used to attenuate graft rejection, and such reagents hold potential for treating autoimmunity (Chatenoud, 2005; Chatenoud and Bluestone, 2007; You and Chatenoud, 2010). But, the mechanism of action of these antibodies is poorly understood. The complex is also the primary focus of gene therapy strategies aimed at reprograming the specificity of T cells (e.g., to target tumor cells; Rosenberg et al., 2008). However, the nature of TCR-CD3 complex assembly does not favor the incorporation of exogenous TCRs into complexes on cells expressing endogenous receptors. A better understanding of the assembly and function of the complex is important for guiding gene therapy (Cohen et al., 2006). Undoubtedly, more insights into how the parts of this molecular machine fit and work together will inform the next generation of a variety of immunotherapeutic strategies.
Increased insights into the TCR-CD3 complex are also likely to have a broader impact on our basic and practical knowledge. This complex represents one of the most complicated and sensitive molecular machines identified to date (Kuhns et al., 2006; Davis et al., 2007). Nature often employs similar strategies to accomplish related tasks; thus, elucidating the mechanisms by which the TCR-CD3 complex functions is likely to provide valuable insights into the functions of related activating immune receptor complexes (e.g., NK or Fc receptors) as well as other multi-molecular signaling complexes (Call and Wucherpfennig, 2007).
We will discuss the question of how TCR signaling begins from the standpoint of what we consider to be the minimal essential molecular components needed for robust TCR signaling: the TCR-CD3 complex subunits, the CD4, and CD8 co-receptors, and agonist pMHC.
There are two fundamental requirements that must be met in order for modular biosensors to transduce a signal across a membrane. The first is that signaling substrates inside the cell must be made accessible to modification by signaling enzymes upon ligand engagement if they are not accessible by default. The second is that these signaling substrates must be brought into the appropriate spatial relationship with the enzymes that modify them in order to initiate signaling upon ligand engagement. At present, we do not fully understand (i) how TCR engagement triggers the relay of information from the TCR-pMHC interface to the CD3 ITAMs to make them accessible for phosphorylation by the Src kinase Lck, or (ii) how the ITAMs and Lck are brought into a high local concentration to interact and initiate signaling.
For technical reasons, and an incomplete understanding of the processes outlined above, few studies have reported measuring these events. Therefore, much of what we discuss below will be based on calcium mobilization data. This is a commonly used measure of TCR-proximal signaling events that has been reviewed extensively elsewhere (Hogan et al., 2010). This event is downstream of the initial ITAM phosphorylation events outlined above and occurs rapidly after TCR engagement (Huse et al., 2007). In addition, it can occur in response to as few as one agonist pMHC (Irvine et al., 2002; Purbhoo et al., 2004). Thus, it is a sensitive readout of TCR engagement and signaling even though it not a direct measurement of the events outlined above.
It is of note, however, that the molecular mechanisms that initiate transient signaling in response to low ligand densities may be less complex than those that drive T cells to secrete cytokines and proliferate. TCR signaling must be sustained for several hours in order for T cells to reach this more classically defined state of T cell activation (Huppa et al., 2003). Understanding the molecular mechanisms by which sustained TCR-CD3 signaling informs T cell fate decisions is, for us, a natural extension of asking how TCR signaling begins. As with any complex mechanical or molecular machinery, understanding how the parts of this machinery fit and work together is key to unraveling the mystery of how they function. We think it is likely that “full activation” results from a multi-step process for assembly of, and signaling through, these modular biosensors.
Determining the structure of an intact TCR-CD3 complex at atomic resolution will represent a major step forward in our understanding of the form and function of this macromolecular machine. However, there are severe technical challenges associated with working on a complex composed of eight polypeptides that associate through a continuum of transmembrane and extracellular interactions (Call and Wucherpfennig, 2005; Kuhns et al., 2006; Kuhns and Davis, 2007). In addition, the mechanistic basis for TCR triggering may involve a change of the ECD of the subunits with respect to each other and/or the outer leaf of the membrane (Kuhns et al., 2006). This could occur as a consequence of some sort of force being induced by pushing or pulling upon pMHC engagement that ultimately changes the orientation of the intracellular domains of the CD3 ITAMs with respect to the inner leaf of the membrane. If so, then static structures of unliganded and liganded complexes removed from their native environment, and in the absence of a source of counter posing force, may not reveal the mechanistic basis for triggering. Indeed, it is generally accepted that monomers of pMHC do not induce TCR signaling. Clearly, there is a long road ahead regarding an atomic level picture of this complex in action. Nevertheless, progress from a number of groups is allowing us to piece together a sketch of the complex architecture.
A great deal of structural information has been acquired regarding the individual parts of this molecular machine (Kuhns et al., 2006). The first crystal structure of the TCR ECD revealed that the TCR is remarkably similar to a Fab fragment of an antibody (Garcia et al., 1996). The clonotypic TCRα and TCRβ variable (Vα and Vβ) domains adopt classic Ig-folds that associate via their bottom beta-sheets. The TCRβ constant domain (Cβ) also adopts an Ig-fold and associates with the bottom beta-sheet of the TCRα constant domain (Cα). Interestingly, the Cα domain has a clear Ig-fold ancestry but deviates structurally from the Vα, Vβ, and Cβ domains due to its lack of a top beta-sheet. Instead, it has two loosely associated top strands that expose an apolar pocket, reminiscent of a protein interaction hot spot (DeLano et al., 2000). The evolutionary pressures that caused this structural divergence are unknown. But, our recent data suggests that this unusual structure plays a role in mediating homotypic TCR interactions (Kuhns et al., 2010). The connecting peptides that span from the Cα and Cβ domains to the membrane are also quite interesting due to their unusually length (∼20 amino acids). No structural information is available for these segments of the TCR extracellular regions, but they have been implicated in complex assembly, stability, and signaling (Backstrom et al., 1996, 1997; Werlen et al., 2000; Naeher et al., 2002; Mallaun et al., 2008). The solution and crystal structures of the ECD of CD3δ, γ, and ε also reveal Ig domains (Sun et al., 2001, 2004; Arnett et al., 2004; Kjer-Nielsen et al., 2004). Interestingly, the subunits of each heterodimer associate via the intra-strand loops in a side-by-side manner. In contrast to the TCR heterodimer, CD3γε and CD3δε have short, rigid connecting peptides with RxCxxCxE motifs that facilitate pairing of these heterodimers and play a role in signaling (Sun et al., 2001; Martinez-Martin et al., 2009; Wang et al., 2009b). The transmembrane domains of each complex subunit are predicted to form alpha helices that are known to associate via a complex network of electrostatic interactions. The solution structure of the transmembrane domains of CD3ζζ in SDS micelles does reveal helices that run parallel through the membrane with a small crossing angle (Call et al., 2006). Likewise, by analogy, the solution structure of a NKG2C transmembrane helix interacting with two DAP12 helices suggests that the associating TCRα-CD3δε and TCRβ-CD3γε transmembrane helices are likely to form a tight trimer of parallel non-crossing helices through the membrane (Call et al., 2010). Finally, as first proposed by Stern and colleagues (Aivazian and Stern, 2000), the intracellular domains of CD3ε and CD3ζ, but not CD3γ or CD3δ, contain stretches of basic residues that are thought to facilitate interactions with the acidic inner leaf of the membrane. These interactions are thought to protect the critical tyrosines of the ITAMs from Src kinase phosphorylation when the TCR is unliganded (Aivazian and Stern, 2000; Sigalov et al., 2006; Xu et al., 2008; Deford-Watts et al., 2009; DeFord-Watts et al., 2011; Zhang et al., 2011); however, paradoxically, these polybasic stretches are also important to facilitate signaling (Deford-Watts et al., 2009; DeFord-Watts et al., 2011; Zhang et al., 2011). Altogether, these individual pieces of information provide important insights into the structural characteristics of the key components of the TCR-CD3 complex. But, with the exception of the transmembrane domain data, they provide very few insights into how the TCR and CD3 subunits fit together to form a molecular machine that can transfer pMHC-specific information from the outside to the inside of a cell.
When we last reviewed this topic we highlighted all of the potential extracellular TCR-CD3 interaction sites that had been proposed at that point in time. The state of the field had lead us to conclude that nearly any conceivable TCR-CD3 complex architecture could have been possible (Kuhns et al., 2006). Experiments performed by the Wucherpfennig and colleagues had clearly shown the stoichiometry of a nascent complex to be 1TCR:1CD3γε:1CD3δε:1CD3ζζ (Call et al., 2002). Furthermore, due to the periodicity of the transmembrane helices that play an important role in mediating complex assembly, the TCRα-CD3δε and TCRα-CD3ζζ transmembrane interactions would be spaced roughly 140° from each other. But, there was no information regarding the relationship between the turns of the transmembrane helices and the ECD that could provide insights into the spatial relationship between the ECD of the TCR and CD3 subunits. In addition, while antibody-mapping data suggested that one of the CD3ε subunits resides in close proximity to the distinctive FG loop of TCRβ (Ghendler et al., 1998), and crosslinking data also placed the ECD domain of CD3γ in close associate with TCRβ (Brenner et al., 1985), no experimental data existed for the relationship between TCRα and CD3δε or CD3δε and CD3γε. Thus, we undertook a reverse engineering project to determine the spatial relationship between the ECD of the TCR and CD3 heterodimers (Kuhns and Davis, 2007).
In order to deconstruct and deduce how the ECD domains of the TCR and CD3 heterodimers fit together, we generated TCRs mutated on distinct solvent-accessible surfaces of the Cα and Cβ domains (Kuhns and Davis, 2007). We found that mutating the highly conserved CαDE loop destabilizes TCR-CD3δε interactions in co-immunoprecipitation experiments, suggesting that the Cα DE loop composes a portion of the extracellular interaction site with TCRα. We also observed that mutations in the highly conserved Cβ CC′ loop, which is adjacent to the Cα DE loop on one side of the TCR, weakened the association between the TCR and both CD3 heterodimers. This phenotype was similar to that observed by the Reinherz group when they deleted the FG loop (Touma et al., 2006), suggesting that both the Cβ FG and CC′ loops contribute to TCR-CD3 complex stability. By extension, this indicates that the face of the Cβ domain bordered by these loops is the likely site for extracellular TCR-CD3γε interactions. Importantly, the close proximity of the Cα DE and Cβ CC′ loops, the similar phenotypes of our Cβ CC′ loop mutant and the FG loop deletion, and data from Call et al. (2002) showing that TCR-CD3δε interactions occur normally in the absence of CD3γε but that TCR-CD3γε interactions require TCR-CD3δε interactions, lead us to postulate that relatively strong TCR-CD3δε interactions form a composite binding surface for TCR-CD3γε interactions. One important prediction of this model was that mutating both the Cα DE and Cβ CC′ loops should completely destabilize the TCR-CD3 complex. This is in fact what we observed in our immunopreciptiation experiments. These data were consistent with our hypothesis that the CD3 heterodimers are clustered on one side of the TCR. And, of note, very recent mutagenesis experiments independently confirm this location (Fernandes et al., 2012).
We next used a kinase-based dimerization assay to independently verify that the two CD3 heterodimers are clustered on one side of the TCR. This was important for two reasons: (i) our model was controversial because the dominant model at the time docked the CD3 heterodimers on opposite sides of the TCR (Sun et al., 2004); (ii) our mutagenesis data did not provide any information about the specific spatial arrangement of these CD3 subunits with respect to each other (Kuhns and Davis, 2007).
This assay operates on two key principles/components (Yamasaki et al., 2006). The first takes advantage of the signaling mechanism of the erythropoietin receptor (EPOR). In the unliganded state, EPOR exists as a homodimer with the intracellular signaling domains held ∼70 Å apart (Livnah et al., 1999). Upon ligand engagement, these domains are brought within ∼40 Å of each other. This conformational change brings the two signaling domains and their constitutively associated Jak2 molecules into juxtaposition. In turn, the two Jak2 molecules are positioned to directly interact and transautophosphorylate. BaF3 cells are the second key component of this system. These cells are dependent upon Jak-Stat signaling for growth. Thus, chimeric proteins containing the intracellular signaling domains of EPOR will drive BaF3 cell growth if two EPOR domains are positioned within ∼40 Å of one another. For our experiments, we generated a polycistronic construct encoding truncated CD3 subunits with the picornavirus 2A cleavage system, similarly to that pioneered by Vignali and colleagues (Szymczak et al., 2004). The subunits lacked all of the ITAMs in order to eliminate the potential for any signaling through these domains. Variants of this construct were then generated encoding chimeras in which the EPOR intracellular signaling domain was fused directly to the last transmembrane residue of CD3δ, γ, or ε. We also generated constructs encoding both CD3δ and CD3γ fused to EPOR. Each CD3 construct was then expressed in BaF3 cells in the presence or absence of TCRα and TCRβ. The results of these experiments were consistent with the CD3 heterodimers being clustered on one side of the TCR. Specifically, they showed that the two CD3ε domains are juxtaposed upon assembly of the TCR-CD3 complex because the CD3e-EPOR chimera drove BaF3 growth in a TCR dependent manner (Kuhns et al., 2010). The CD3δ- and CD3γ-EPOR chimeras did not drive BaF3 growth in a TCR dependent manner and therefore are unlikely to be positioned next to each other as has been recently proposed (Fernandes et al., 2012). The TCR-CD3 complex architecture that is most consistent with the sum of the existing experimental data is one in which CD3δε and CD3γε associate with the TCR in an open face orientation. They dock on the side of the TCR that includes the Cα DE and Cβ CC′ loops in the order δ:ε:ε:γ (Figure 1; Kuhns et al., 2010). The periodicity of the TCRα transmembrane helix and the spacing (∼140°) of the key charged residues that mediate interactions with CD3δε and CD3ζζ indicates that CD3ζζ resides on the opposite side of the TCR.
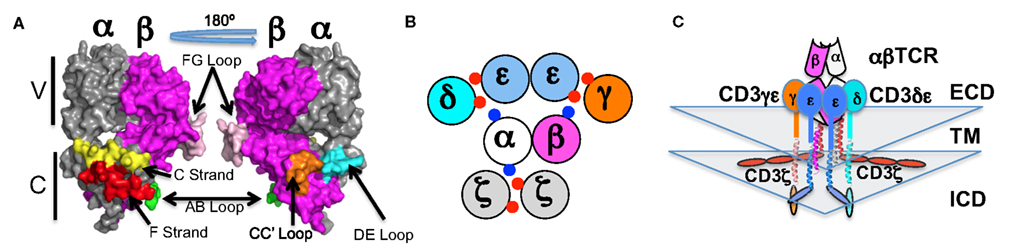
Figure 1. The architecture of the TCR-CD3 complex. (A) The αβTCR is shown (PDB: 1TCR) as a surface rendered structure with the variable (V) regions on top and the constant (C) regions on the bottom. The constant region surfaces discussed herein are colored: Cα AB loop (green), Cα C strand (yellow), Cα DE loop (cyan) Cα F strand (red), Cβ CC′ loop (orange), and Cβ FG loop (pink). (B) A top down view of the transmembrane domains, with red and blue dots indicating the acidic and basic transmembrane charge residues (respectively) that mediate assembly of the complex (Call et al., 2002; Kuhns and Davis, 2007; Kuhns et al., 2010). (C) A front-on view of the extracellular (ECD), transmembrane helices (TM), and intracellular (ICD) domains according to our current interpretation of the existing data. The subunits are color coded as labeled: TCRα (white), TCRβ (magenta), CD3γ (orange), CD3δ (cyan), CD3ε (marine).
Our understanding of the architecture of a complex in isolation is rounded out by data indicating that the intracellular domains of CD3ε and CD3ζ, but not CD3δ and CD3γ, associate with the acidic inner leaf of the membrane (Aivazian and Stern, 2000; Sigalov et al., 2006; Kuhns and Davis, 2008; Xu et al., 2008; Deford-Watts et al., 2009; DeFord-Watts et al., 2011; Zhang et al., 2011). Several features are note worthy when all of these data are considered in aggregate. The picture that is taking shape is one in which the two CD3ε intracellular domains emerge from the membrane side-by-side and interact with the acidic inner leaf of the membrane (Figure 1). The CD3γ and CD3δ intracellular domains would emerge from the membrane to flank the CD3ε intracellular domains on one side of the TCR. CD3ζζ would emerge from and associate with the acidic inner leaf of the membrane below the unique surface of the Cα domain on the other side of the TCR (Kuhns et al., 2010). Thus, the four ITAMs (one each for CD3γ, δ, and ε) of the CD3γε/CD3δε signaling module and the six ITAMs of the CD3ζζ signaling module (three per chain) reside on opposite sides of the TCR.
A discussion of how the function of this complex is influenced by its architecture requires an introduction to the idea that TCR-CD3 multimerization plays a role in signaling. Dimerization, higher-order multimerization, and/or general clustering of the TCR-CD3 complex have long been thought to be a key aspect of TCR signaling. Consequently, it is a concept that will be discussed at various points herein.
The connection between TCR-CD3 complex clustering and signaling has its origins in crosslinking studies performed with anti-TCR or anti-CD3 monoclonal antibodies (mAbs) that induce cytokine secretion or T cell proliferation (Rojo and Janeway, 1988; Yoon et al., 1994; Janeway, 1995). These data suggest that there are geometric constraints placed on signaling induced by clustering, meaning that random clustering is insufficient for signaling. More recent studies using inventive approaches to crosslink TCR-CD3 complexes with antibodies or soluble pMHC dimers, trimers, or tetramers also support the notion that forcing TCR-CD3 complexes together can lead to signaling (Boniface et al., 1998; Cochran et al., 2000; Krogsgaard et al., 2005; Minguet et al., 2007). These reagents and experimental approaches have been extremely helpful in establishing what parameters might play a role in signaling; they have also been very useful in studying the intracellular signaling cascades that connect the complex to the nucleus (Smith-Garvin et al., 2009). However, they have not addressed the issue of geometric constraints. Also, as discussed later, the fact that mitogenic reagents such as mAbs or more physiological reagents such as multimerized pMHCs can force TCR-CD3 complexes together and induce signaling does not prove that higher-order TCR-CD3 complexes plays a role in signaling when the TCR engages cognate pMHC. The evidence for direct homotypic interactions between soluble TCRs upon binding pMHC has been decidedly mixed (Reich et al., 1997; Alam et al., 1999; Baker and Wiley, 2001). Thus, the role of TCR-CD3 complex dimerization or higher-order multimerization in recognition of pMHC on APCs remains a question of great significant in need of further inquiry and clarification.
We have used the kinase-based assay outlined above to address this problem. The data represent the first evidence for TCR–TCR interactions in the cell membrane, and thus the first insights into the architecture of higher-order TCR-CD3 complex assemblies (Kuhns et al., 2010). In addition, using mutagenesis we mapped the regions mediating these interactions to the surface of the unusual Cα domain composed of the C and F strands as well as the AB loop (Figure 1). Interestingly, the sum of the data discussed to this point position the CD3ζζ subunits immediately below this surface in a single TCR-CD3 complex. Furthermore, it would place two CD3ζζ homodimers at the interface between two TCR-CD3 complexes. These data are in-line with an increasing body of evidence that suggest a link between the geometric organization of TCR-CD3 complexes, and their function, upon ligand engagement.
We coupled functional studies with our structural analysis in order to contribute to our understanding of this link. Importantly, we observed that mutations that destabilize extracellular TCR-CD3 interactions on one side of the TCR and those that impair TCR dimerization on the other each have distinct impacts on TCR signaling (Kuhns and Davis, 2007; Kuhns et al., 2010). More specifically, mutations in the Cα DE loop that destabilize TCR-CD3 complex stability lead to a significant lag in the time between T cell contact with a peptide-pulsed APC and intracellular calcium mobilization, suggesting that complex stability is important for how TCR signaling begins. But, these mutations did not impact T cell polarization to the site of antigen contact or immunological synapse (IS) formation. In contrast, impairing dimerization with mutations in either the Cα, C, or F strands had minor impacts on the initial magnitude, but not the kinetics of calcium flux; however, these mutations significantly impaired IS formation and T cell polarization toward the site of antigen engagement. We concluded from these studies that there is a functional sidedness to the TCR at the nucleus of each TCR-CD3 complex and that the interactions mediated by its constant domains play important roles in TCR signaling (Kuhns et al., 2010). A better understanding to how this functional sidedness contributes to signaling requires a more in-depth understanding of how monomers and dimers of the complex fit within the higher-order macromolecular machinery responsible for full TCR signaling.
The TCR-CD3 complex is the primary component of the molecular machinery that transfers pMHC-specific information from the outside of a T cell to the intracellular signaling apparatus, but it cannot efficiently complete this task in isolation. The complex fails to activate T cells when confronted with monomeric pMHC in solution. Yet, it is not known if this is because the complex is unable to make the ITAMs accessible to Lck when bound to a soluble monomer, if a monomer is unable to successfully recruit Lck to the ITAMs, or both. A single agonist pMHC on an APC can elicit a transient, co-receptor-dependent calcium flux in CD4+ and CD8+ T cells (Irvine et al., 2002; Purbhoo et al., 2004). Sustained calcium mobilization requires 10 pMHC on an APC as well as co-receptors, whereas more than 20 pMHC complexes must be present on an APC before the TCR-CD3 complex can sustain calcium mobilization in the absence of a co-receptors. Similarly, on a planar bilayer a single agonist pMHC only elicits a transient calcium flux (Ma et al., 2008b). More recently Manz et al. (2011) used partitioned planar bilayers, where the total number of agonist pMHC encountered by cells are the same, to show that an average concentration of one pMHC per partition cannot induce half-maximal calcium flux whereas two pMHC per partition can. Importantly, the authors calculated that at an average concentration of one pMHC per partition there was a high probability of TCR clusters encountering two pMHC. At an average concentration of two pMHC per partition the probability of a triggering TCR cluster encountering more than two pMHC is sufficiently high to conclude that the minimal triggering unit per partition in this experimental system to induce half-maximal calcium flux was four to six agonist pMHC. In other systems it has been shown that soluble pMHC dimers, trimers, and tetramers can also induce TCR signaling in a co-receptor dependent manner (Boniface et al., 1998; Cochran et al., 2000; Krogsgaard et al., 2005). Each of these experimental systems have strengths and weaknesses and there are finer points to be argued. Nevertheless, the existing data indicate that sustained TCR signaling requires the assembly of higher-order TCR-CD3 complexes, co-receptors, and multi-valent arrays of 2+ agonist pMHC. These macromolecular assemblies are not likely to come together randomly. Rather, the structural and mutagenesis data accumulated to date point to geometrically constrained macromolecular complexes mediating sustained TCR signaling (Figure 2).
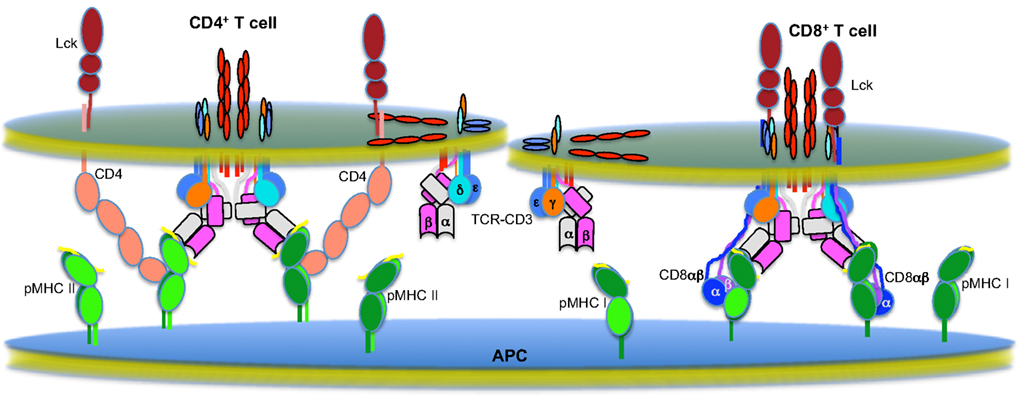
Figure 2. The multi-molecular T cell signaling machinery. Shown is the state of the field as we understand it regarding the multi-molecular higher-order assemblies that mediate sustained TCR signaling on CD4+ (left) and CD8+ (right) T cells (Garcia et al., 1996, 2009; Sun et al., 2001, 2004; Wang et al., 2001, 2009a; Call et al., 2002; Arnett et al., 2004; Kjer-Nielsen et al., 2004; Kuhns and Davis, 2007; Kuhns et al., 2010).
TCRs bound to pMHC are the cornerstones of these assemblies. One of the most interesting observations to come out of the comparison of a number of TCR-pMHC crystal structures is that the assembly of this basic building block is not random (Rudolph et al., 2006; Garcia et al., 2009). Instead, the structures of productive TCR-pMHC interactions indicate that there is a canonical docking polarity driven by germline-encoded interactions that Garcia et al. (2009) have termed “interaction codons.” Interestingly, in spite of these apparent restrictions, highly conserved germline contacts still allow for great diversity in specificity and affinity of TCR-pMHC interactions due to significant diversity and flexibility in the CDR3 loops (Newell et al., 2011).
As we have previously noted, this docking polarity will fix the relationship between the CD4 or CD8 co-receptors and the CD3 subunits within a TCR-CD3 complex (Kuhns and Davis, 2007); specifically, the CD3 heterodimers will always be on the same face of a TCR-pMHC unit as the docking sites for the CD4 or CD8 co-receptors on class I or class II pMHC, respectively, that have been identified by crystal structures and/or mutagenesis (Konig et al., 1992; Wang et al., 2001, 2009a; Yin et al., 2012). Dimerization of TCR-CD3 complexes would extend onto this base unit via the opposite face of the TCR, which is composed of the unusual Cα surface (Kuhns et al., 2010). Recent data also suggests that intercomplex transmembrane interactions between CD3ζζ homodimers play a role in stabilizing oligomers of TCR-CD3 complexes (Kumar et al., 2011). Thus, the core of this stable signaling unit would consist of at least two mirroring TCR-CD3-pMHC units. For class II MHC specific T cells, the CD4 co-receptor is thought to assume a V-like relationship with respect to the TCR-CD3-pMHC unit. This is based on a crystal structure of the D1 and D2 domains of CD4 in association with class II MHC (Wang et al., 2001) as well as a recent report of the ternary complex of a pMHC bound to a TCR and an affinity maturated CD4 (Yin et al., 2012). It should be noted however that CD4 has also been proposed to interact directly with the TCR-CD3 complex. This may appear in conflict with a V-like organization. But, the side-chains of the residues identified by mutagenesis that impaired CD4 function were either completely or partially buried in the hydrophobic core of the D3 Ig-fold; thus, decreased CD4 function could have reflected destabilized CD4 molecules rather than TCR-CD4 interactions (Vignali et al., 1996; Vignali and Vignali, 1999). Alternatively, CD4 may directly interact with a TCR that is not engaged to the same pMHC through a “pseudodimer” type orientation (Irvine et al., 2002). In such a scenario, the base unit illustrated in Figure 2 could serve as a nucleus from which higher-order macromolecular assemblies branch out (not shown). Likewise, CD4 has been observed to crystallize as a homodimers in the D4 domain and mutagenesis data suggest that this is biologically relevant for CD4 function (Wu et al., 1997; Moldovan et al., 2006). This may also provide a mechanism for an outgrowth of higher-order assemblies from this core TCR-CD3-pMHC-CD4 unit. The CD8 heterodimer, with its long tethers, is thought to hang down in close proximity to the TCR-CD3 complex, and some studies indicate that it interacts with CD3δ or TCRα (Naeher et al., 2002; Doucey et al., 2003; Mallaun et al., 2008).
A variety of functional studies also support the macromolecular models shown in Figure 2. TCR signaling clearly begins when TCRs bind cognate pMHCs, and these interactions are required to initiate and sustain signaling. The central role of the canonical docking polarity to the proper assembly of macromolecular signaling units predicts that any TCRs found to deviate from this polarity should signal poorly or not at all. Two autoreactive TCRs, specific for myelin basic protein (MBP)-derived peptides presented in the class II MHC HLA-DR2, that are presumed to have escaped negative selection by signaling poorly do deviate from the norm in their docking configurations (Hahn et al., 2005; Li et al., 2005). Both are unusual in that they are docked over the N-terminal half of the peptide, rather than the central portion of the peptide. This could place the CD3 ITAMs, and particularly those for CD3ζζ, at a greater distance from the CD4 associated Lck and may lead to incomplete phosphorylation during pMHC engagement. In addition, one of these TCRs docks at an angle that the authors proposed might impair the proper spatial relationship with CD4 and thus impair CD4 function (Hahn et al., 2005). More recent structural data supports this view (Yin et al., 2012). Very recently, Garcia and colleagues have used an elegant yeast display approach to circumvent the selective pressures of the thymus, including any co-receptor constraints, and identified a peptide (p3A1) that is recognized by the 42F3 TCR when presented by H2-Ld via a docking configuration that significantly deviates from the norm (Adams et al., 2011). Importantly, this pMHC complex can bind the 42F3 TCR, as measured by surface plasmon resonance, with an affinity that should be activating when compared with other known agonist TCR-pMHC interactions. But presentation on an APC fails to induce signaling through the 42F3 TCR. This finding represents a major advance in our understanding of how TCR signaling is initiated because it lends critical experimental support to the idea that signaling not only begins with TCR-pMHC engagement, but that it can only occur if a specific docking polarity is achieved. Whether the unusual docking of 42F3 on the p3A1-Ld complex prevents the transfer of information from the binding interface to the ITAMs, prevents dimerization of the TCR-CD3 complex, prevents recruitment of CD8-associated Lck to the ITAMs, or all of the above remains to be determined.
CD4 or CD8 are also critical for the initiation of TCR signaling, particularly to low density and low affinity ligands. As clearly demonstrated by Singer and colleagues, the co-receptors provide binding and signaling specificity to MHC by sequestering Lck away from the TCR until the TCR engages cognate pMHC; consequently, they have also been proposed to play a key role in modulating TCR signaling (Van Laethem et al., 2007). The existing structural and mutagenesis data, which largely overlap in their determination of the general location where the co-receptors bind MHC, indicate that there are specific surfaces on class I and class II MHC bound by either CD8 or CD4, respectively, to recruit Lck to the TCR-CD3 complex (Potter et al., 1989; Salter et al., 1990; Konig et al., 1992; Wang et al., 2001, 2009a; Yin et al., 2012). Mutagenesis of the CD4 or CD8 binding sites on their respective MHC ligands abrogates signaling, presumably because this prevents recruitment of Lck to the ITAMs and favors sequestration of Lck away from the complex by the co-receptor (Potter et al., 1989; Salter et al., 1990; Konig et al., 1992). Given that a significant fraction of the Lck within T cells has been determined to be in an active state (Nika et al., 2010), sequestering Lck away from the ITAMs is likely to be of critical importance to keeping the complex off when agonist pMHC is not present (Van Laethem et al., 2007). This would also allow for a hair trigger response when antigen is encountered. It is interesting, when looking at Figure 2, to wonder whether CD4 and CD8 contribute in the same manner to TCR signaling. It is difficult to understand how CD4 associated Lck is capable of reaching and phosphorylating all of the CD3ζζ ITAMs. But, Förster resonance energy (FRET) pairs at the C-terminus of CD4 and the C-terminus of CD3ζζ do transfer energy, suggesting that these domains can come within close proximity of each other (Zal et al., 2002). In addition, particularly if CD8 does directly interact with the complex via the TCRα connecting peptide (Mallaun et al., 2008), it is interesting to consider if the co-receptors play some role in making the ITAMs accessible to Lck in addition to recruiting the kinase to its substrate. In any event, the co-receptors are clearly key components of the macromolecular machinery important for the initiation of TCR signaling and without them we may be addressing a very different question, or approaching this issue in a very different way.
Finally, as discussed above, we have shown that mutations in the unusual top strands of the Cα domain that disrupt TCR dimerization also impair TCR signaling, and in particular those signals that lead to cytoskeletal polarization and IS formation (Kuhns et al., 2010). Schamel and colleagues have also implicated TCR dimerization as an important component of TCR signaling (Minguet et al., 2007) and, more recently, it has been reported that mutations in transmembrane domain residue of CD3ζ that appear to stabilize dimerization or oligomerization of TCR-CD3 complexes also impair TCR signaling (Kumar et al., 2011). Together, these data build a strong case for a role of TCR dimerization in signaling. But, as discussed in greater detail below, it is unclear if dimerization is necessary to initiate signaling or if it is more important for sustained signaling.
Overall, the state of the field both structurally and functionally can be illustrated as shown in Figure 2. This represents our best approximation of the nature of the basic unit of higher-order macromolecular assemblies that initiate and/or sustain robust TCR signaling on CD4+ and CD8+ T cells. Intriguingly, it suggests that slightly different mechanisms might be employed for CD4 versus CD8 assisted signaling due to the potential spatial differential between the co-receptors-associated Lck recruited to the complex and the CD3 ITAMs.
Is the assembly of all of the parts, as illustrated in Figure 2, important to initiate signaling, required for sustained signaling, or both? These are important distinctions to consider because the requirements for initiating a transient stop signal upon contact to scan the pMHC on the surface of an APC may not be as complex as the requirements for the sustained signaling that is necessary for cytokine production and proliferation. As we have previously discussed (Kuhns et al., 2006), the reigning models for how signaling begins involve some sort of conformational change of the TCR-CD3 complex subunits with respect to each other and/or the membrane; TCR dimerization is also commonly proposed. But, little attention has been paid to whether or not the molecular assemblies required for transient signaling versus sustained signaling may be different. Nevertheless, a variety of recent functional studies have provided us with some important clues upon which to evolve our thinking regarding how TCR signaling is initiated versus how it may be sustained.
First, when the stability of the TCR-CD3 complex or the rigidity of the CD3ε stalks have been compromised signaling has been compromised (Backstrom et al., 1996, 1998; Sun et al., 2001; Sasada et al., 2002; Touma et al., 2006; Kuhns and Davis, 2007; Wang et al., 2009b). These data do not support models in which the ECD of the complex subunits undergo a conformational change with respect to each other to transmit information across the cell membrane. But, they make a strong case for the idea that the subunits of the complex move as a unit to undergo a conformational change with respect to the membrane under the influence of a mechanical force presumably exerted by the cytoskeleton. This force would push and/or pull the complex upon engagement of pMHC on the surface of an APC (Sun et al., 2001; van der Merwe, 2001; Kuhns et al., 2006; Kuhns and Davis, 2007; Ma et al., 2008a).
As we have speculated before, the architecture of the complex is interesting with respect to mechanical force models because the TCR may be thought to resemble a lever with the CD3 heterodimers serving as a wedge at its base (Kuhns and Davis, 2007). The unusual stalks of the CD3 heterodimers, with their RxCxxCxE motifs, have been proposed to stiffen to transfer force through the membrane (Sun et al., 2001; Martinez-Martin et al., 2009). So, we can envision a potential mechanism for TCR-CD3 complex signaling similar to that by which a classic-style water cooler normally retains water and then transfers it from one reservoir to another in response to force (Figure 3). In short, pushing down on a water cooler lever causes the lever to pivot over the top of its protruding base as one unit. This causes the valve in the back to be pulled upward and open to allow water to flow. Likewise, TCR engagement of pMHC on an APC might cause the TCR to pivot over the top of the CD3 heterodimers, moving as a unit, to push the CD3γε and CD3δε down while also pulling up on the CD3ζζ situated on the opposite side of the complex. This opposing action would allow pMHC-specific information to flow across the membrane. Such a mechanism would cause the transmembrane domains to pivot with respect to each other, as has previously been proposed (Engelman, 2003). Importantly, given that a 1-Å change of transmembrane domains with respect to each other can induce signaling in the aspartate receptor (Ottemann et al., 1999), any type of lever-like pivot may not have to be particularly extreme to relay pMHC-specific information across the cell membrane. The co-receptors may contribute force in this model by holding the pMHC in a particular orientation with respect to the TCR and/or the membrane. This “water cooler” model is one of several ways in which a mechanical force may be transmitted across the membrane and testing such models will likely provide insights into how the critical tyrosines of the CD3ε and CD3ζζ ITAMs are made accessible for Src kinase phosphorylation.
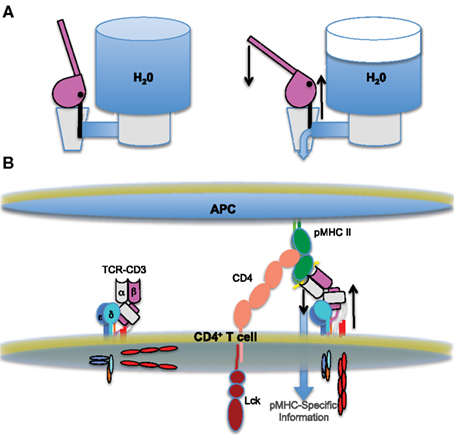
Figure 3. The water cooler model of TCR triggering. (A) Cartoons of a “classic-style” water cooler in action. The valve is closed on the left and water is retained in the tank. Once the lever is pushed down it pivots over its rigid base (right), causing an upward pull on the attached valve. This opposing action opens the valve and allows water to flow from the tank. (B) An analogous mechanism may trigger the flow of pMHC-specific information from the outside to the inside of a T cell. A cartoon of the basic triggering components is shown, similarly to Figures 1 and 2. The unengaged TCR-CD3 complex on the left is relatively upright. Upon ligand engagement (right), the TCR pivots over the rigid CD3 heterodimers at its base, possibly pushing them into the membrane while pulling up on the CD3ζζ on the opposite side of the complex.
Several lines of evidence suggest that a single pMHC can elicit a signaling event (Irvine et al., 2002; Purbhoo et al., 2004; Ma et al., 2008b); however, as introduced above, dimerization or oligomerization of the TCR-CD3 complex has long been considered a possible mechanism for signaling (Symer et al., 1992; Reich et al., 1997; Boniface et al., 1998; Alam et al., 1999; Cochran et al., 2000). It is not immediately obvious why dimerization would be necessary. The complex has 10 ITAMs that are already pre-clustered within a single complex and it only lacks an intrinsic Src kinase activity with which to modify the ITAMs. So, it is not clear why there would be a need to bring more ITAMs together to initiate signaling given the high concentration of signaling substrates in one complex.
The failure of soluble pMHC monomers to activate T cells may be taken as evidence that multi-valent arrays are important to facilitate TCR dimerization and signaling. Yet, this would also be consistent with a requirement for force to be transmitted across a membrane. A force requirement is more difficult to reconcile with the fact that soluble pMHC dimers can activate T cells (Cochran et al., 2001). The increased avidity of the dimer and potential constraints imposed by the linker may result in some sort of torque being transmitted through the TCR-CD3 complex upon pMHC engagement. This could be facilitated and/or enhanced by the co-engagement of the pMHC dimers by TCRs and co-receptors, which are required for activation with dimers (Krogsgaard et al., 2005), such that the constraints of adopting a higher-order macromolecular structure by the TCR-CD3 complex, co-receptor, and pMHC could cause a force to be transmitted across the membrane. Alternatively, triggering may not involve a change in the conformation of the complex with respect to the membrane under force. Rather, the relative stability and/or rigidity of the complex as a whole, as well as a canonical TCR-pMHC docking motif, may be important for the natural process of TCR dimerization; soluble dimers may simply force two TCRs together to induce signaling in a non-physiological manner. Clearly, any discussion of TCR signaling warrants further consideration of the role of TCR dimerization in how TCR signaling begins or is sustained.
As is often the case when the number of models for a process outstrip the data, key aspects such as geometric constraints, force, and dimerization or higher-order oligomerization are all likely to be at least partially correct. Indeed, there are data to support each of these mechanisms. While it is known that TCR-CD3 complexes are co-associated spatially in protein islands in the T cell membrane (Schamel et al., 2005; Lillemeier et al., 2006, 2010; Kumar et al., 2011) it is not known to what extent unliganded TCRs are physically engaged in homotypic protein: protein interactions in the absence of ligand engagement. As reviewed by Dustin and Depoil (2011), this is an entirely different scale, at the molecular level, rather than the much larger units of protein islands, micro clusters, or macro clusters described by various imaging techniques.
We interpret the existing TCR dimerization data as suggesting that TCR: TCR interactions via the Cα top strands are of low affinity and stabilized upon ligand engagement (Kuhns et al., 2010); thus, homotypic TCR dimers may normally be stabilized, particularly at high ligand densities, within higher-order macromolecular signaling units after TCR and co-receptor engage pMHC and transmit a force across the membrane. Soluble dimers might be able to induce TCR signaling at high concentrations by skipping the step that normally requires mechanical force when dispersed, and low density, pMHC is encountered on an APC. Dimers would simply force two TCR-CD3 complexes together. We suggest this possibility because neither our mutant TCRs that destabilize the complex, and presumably impair the transfer of force through the complex, nor those that impair TCR dimerization, completely abrogate signaling (Kuhns and Davis, 2007; Kuhns et al., 2010). Rather, as noted above, when we measured calcium flux we observed that destabilizing the complex leads to a significant delay in both the onset and initial magnitude of intracellular calcium mobilization after first T cell contact with an APC pulsed with high concentrations of agonist peptide. In contrast, impairing TCR dimerization only slightly reduces the magnitude of the initial influx of calcium mobilization in the same experimental system. This suggests that a stable complex, and presumably a transfer of force, is more important for the initiation of signaling than dimerization when a T cell encounters cognate ligand on an APC. Thus, the use of soluble dimers as a stimulant may limit the mechanisms by which the cells can signal. In contrast, when the cells are confronted with agonist pMHC on an APC, particularly at low or dispersed ligand densities they may preferentially employ other mechanisms, such as force, to initiate calcium mobilization.
Such a discrimination of force and dimerization steps might explain why endogenous co-agonist peptides appear to have a greater observed impact on the frequency of T cells that flux calcium in response to soluble dimers of agonist and co-agonist peptides than when the co-agonist peptides are presented on a planar bilayer with agonist pMHC (Krogsgaard et al., 2005; Ma et al., 2008b). Interestingly, under conditions where the agonist and co-agonist peptides are immobilized on planar bilayers the magnitude of the response seems to be most greatly affected by a co-agonist peptide, suggesting that endogenous peptides may play modulatory roles (Manz et al., 2011). This may provide the T cell with important environmental cues about the self-peptide repertoire on an APC. Or, transient engagement of endogenous ligands might help to orient the parts of this macromolecular machinery to facilitate assembly of the higher-order units composed of two agonist pMHC, much in the way that certain peptides bound to class II MHC can facilitate loading of higher affinity binders (Rabinowitz et al., 1998).
The discussion to this point has been leading to the idea of multi-step signaling. What might a multi-step signaling process look like? Very low ligand densities, including monomers on an APC or mobile bilayer, can induce a transient co-receptor-dependent calcium flux for both CD4+ and CD8+ T cells (Irvine et al., 2002; Purbhoo et al., 2004; Manz et al., 2011). This suggests that TCR and co-receptor bind to pMHC in relative isolation and may initiate signaling by transiently relaying information from the TCR-pMHC interface to some or all of the ITAMs such that they can be phosphorylated by Lck. Presumably this is where a force component would be relevant to induce a change in the ITAMs that alters the positional relationship of the key tyrosines with respect to the inner leaf of the membrane to make them accessible for Lck phosphorylation. Based on the geometric constraints imposed by the canonical docking polarity, the co-receptor-associated Lck would be in closest proximity to the ITAMs of the CD3γε/CD3δε signaling module (particularly CD3δε for CD8, Figure 2) and thus these, and not the ITAMs of the CD3ζζ signaling module, may be the most likely to initially get phosphorylated at low or dispersed ligand densities when a T cell first scans the surface of an APC for agonist pMHCs that can deliver a stop signal. We find this intriguing for three reasons. First, recent data from Alarcon and colleagues showed that mutants of the rigid CD3ε stalk have a dominant negative impact on signaling, which indicates that the CD3ε subunits act cooperatively in TCR signaling (Martinez-Martin et al., 2009). Second, even though association of the CD3ε and intracellular domains might mask the critical tyrosines of the ITAMs from Src kinase phosphorylation under certain conditions (Xu et al., 2008), the polybasic regions that mediate such interactions with the acidic inner leaf of the membrane have paradoxically been shown to be important for optimal TCR signaling (Deford-Watts et al., 2009); perhaps an association with the membrane is important to allow these ITAMs to radiate out toward an approaching co-receptor-associated Lck. Thirdly, Weiss and colleagues have proposed that two ZAP-70 molecules bound to paired ITAMs might trans-autophosphorylate to amplify signaling (Brdicka et al., 2005). Our experiments using the EPOR intracellular signaling domain as a surrogate for the native CD3ε intracellular domain show that within a full complex the two Jak2 molecules associated with the two EPOR domains do trans-autophosphorylate and signal through the Jak/Stat pathway (Kuhns et al., 2010). This suggests that the two side-by-side CD3ε ITAMs could potentially serve as a substrate for signal amplification by ZAP-70 and, by extension, all of the ITAMs of the CD3γε and CD3δε may present a bank of four ITAMs for signal amplification that could increase the sensitivity of signaling. Thus, when a T cell is scanning the surface of an APC for low or dispersed densities of agonist pMHC, the first step in a multi-step signaling process may involve TCR and co-receptor binding pMHC to: (i) modify the orientation of the CD3γε and CD3δε ITAMs such that they are accessible for Lck phosphorylation; (ii) recruit co-receptor-associated Lck to the ITAMs of the CD3δε/CD3γε signaling module.
Functionally, the initial signaling events that rely on force may be transient when agonist pMHC densities are low, either due to a low ligand density on an APC or as a consequence of the pMHC being dispersed across the surface of an APC. Yet, they are likely to deliver the stop signals that ultimately cause a T cell to focus its full attention toward the pMHC composition on the surface of an APC. If the T cell detects higher ligand densities on the surface of the APC then this could allow for TCR dimerization, which we have shown is important for T cells to polarize to the point of antigen contact and form a mature IS that effectively collects the agonist pMHC into a high local concentration and is a hallmark of sustained TCR signaling (Kuhns et al., 2010). Higher ligand densities may more efficiently allow the formation of the higher-order units shown in Figure 2. This would bring the CD3ζζ homodimers from two complexes into close proximity, which as Stern and colleagues have proposed might cause a release of the critical ITAM tyrosines from the membrane for phosphorylation by Src kinases (Aivazian and Stern, 2000). Likewise, a more stable macromolecular assembly may facilitate phosphorylation of some or all of the CD3ζζ ITAMs by co-receptor-associated Lck by holding the substrates and kinase in close proximity for a greater amount of time. This could then increase the total number of ITAMs, particularly for CD3ζζ, which get phosphorylated within a complex based on the quantity and/or quality of the TCR-pMHC interactions. In such a scenario, dimerization may not be essential to initiate signaling, but may play a role in modulating or enhancing sustained signaling as well as delivering information about the quantity and quality of the pMHC encountered on the surface of an APC. The formation of these higher-order complexes may also be important for the sustained signaling that has been reported upon TCR-CD3 internalization (Yudushkin and Vale, 2010). In short, returning to the idea of a functional sidedness to the TCR, we hypothesize that there are multiple steps involved in initiating and sustaining TCR signaling that control the quantity and quality of ITAMs that get phosphorylated within a complex upon ligand engagement. This in turn would impact the outcome of ligand engagement with regards to T cell responses such as cytokine secretion or proliferation.
“How does T cell receptor signaling begin?” We have approached this Research Topic as a forum to reflect on what we know, or think we know, about TCR signaling and to speculate about what it might mean. The system is complicated, and there is much we still do not know. Nevertheless, it is clear that engagement of agonist pMHC by TCRs and co-receptors are the key initiating events for TCR signaling. From there, the existing data indicate that the extent to which signaling is sustained depends upon the assembly of a complex multi-molecular machine, and that signals emerge from the sum of these parts.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Evan Newell, Neha Deshpande, Mark Lee, and Hemant Badgandi for critically reading the manuscript and thoughtful discussions. This work was funded by the Pew Charitable Trusts (Michael S. Kuhns), the NIH (Mark M. Davis), and the HHMI (Mark M. Davis).
ECD, extracellular domain; ICD, intracellular domain; ITAM, immune receptor tyrosine-based activation motifs; pMHC, peptide-major histocompatibility complex; TCR, T cell receptor; TM, transmembrane domain.
Adams, J. J., Narayanan, S., Liu, B., Birnbaum, M. E., Kruse, A. C., Bowerman, N. A., Chen, W., Levin, A. M., Connolly, J. M., Zhu, C., Kranz, D. M., and Garcia, K. C. (2011). T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity 35, 681–693.
Aivazian, D., and Stern, L. J. (2000). Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat. Struct. Biol. 7, 1023–1026.
Alam, S. M., Davies, G. M., Lin, C. M., Zal, T., Nasholds, W., Jameson, S. C., Hogquist, K. A., Gascoigne, N. R., and Travers, P. J. (1999). Qualitative and quantitative differences in T cell receptor binding of agonist and antagonist ligands. Immunity 10, 227–237.
Arnett, K. L., Harrison, S. C., and Wiley, D. C. (2004). Crystal structure of a human CD3-epsilon/delta dimer in complex with a UCHT1 single-chain antibody fragment. Proc. Natl. Acad. Sci. U.S.A. 101, 16268–16273.
Backstrom, B. T., Hausmann, B. T., and Palmer, E. (1997). Signaling efficiency of the T cell receptor controlled by a single amino acid in the beta chain constant region. J. Exp. Med. 186, 1933–1938.
Backstrom, B. T., Milia, E., Peter, A., Jaureguiberry, B., Baldari, C. T., and Palmer, E. (1996). A motif within the T cell receptor alpha chain constant region connecting peptide domain controls antigen responsiveness. Immunity 5, 437–447.
Backstrom, B. T., Muller, U., Hausmann, B., and Palmer, E. (1998). Positive selection through a motif in the alphabeta T cell receptor. Science 281, 835–838.
Baker, B. M., and Wiley, D. C. (2001). Alpha beta T cell receptor ligand-specific oligomerization revisited. Immunity 14, 681–692.
Boniface, J. J., Rabinowitz, J. D., Wüfing, C., Hampl, J., Reich, Z., Altman, J. D., Kantor, R. M., Beeson, C., McConnell, H. M., and Davis, M. M. (1998). Initiation of signal transduction through the T cell receptor requires the multivalent engagement of peptide/MHC ligands. Immunity 9, 459–466. [corrected].
Brdicka, T., Kadlecek, T. A., Roose, J. P., Pastuszak, A. W., and Weiss, A. (2005). Intramolecular regulatory switch in ZAP-70: analogy with receptor tyrosine kinases. Mol. Cell. Biol. 25, 4924–4933.
Brenner, M. B., Trowbridge, I. S., and Strominger, J. L. (1985). Cross-linking of human T cell receptor proteins: association between the T cell idiotype beta subunit and the T3 glycoprotein heavy subunit. Cell 40, 183–190.
Call, M. E., Pyrdol, J., Wiedmann, M., and Wucherpfennig, K. W. (2002). The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 111, 967–979.
Call, M. E., Schnell, J. R., Xu, C., Lutz, R. A., Chou, J. J., and Wucherpfennig, K. W. (2006). The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 127, 355–368.
Call, M. E., and Wucherpfennig, K. W. (2005). The T cell receptor: critical role of the membrane environment in receptor assembly and function. Annu. Rev. Immunol. 23, 101–125.
Call, M. E., and Wucherpfennig, K. W. (2007). Common themes in the assembly and architecture of activating immune receptors. Nat. Rev. Immunol. 7, 841–850.
Call, M. E., Wucherpfennig, K. W., and Chou, J. J. (2010). The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat. Immunol. 11, 1023–1029.
Chatenoud, L. (2005). CD3-specific antibodies restore self-tolerance: mechanisms and clinical applications. Curr. Opin. Immunol. 17, 632–637.
Chatenoud, L., and Bluestone, J. A. (2007). CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat. Rev. Immunol. 7, 622–632.
Cochran, J. R., Cameron, T. O., and Stern, L. J. (2000). The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity 12, 241–250.
Cochran, J. R., Cameron, T. O., Stone, J. D., Lubetsky, J. B., and Stern, L. J. (2001). Receptor proximity, not intermolecular orientation, is critical for triggering T-cell activation. J. Biol. Chem. 276, 28068–28074.
Cohen, C. J., Zhao, Y., Zheng, Z., Rosenberg, S. A., and Morgan, R. A. (2006). Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 66, 8878–8886.
Constant, S. L., and Bottomly, K. (1997). Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu. Rev. Immunol. 15, 297–322.
Corse, E., Gottschalk, R. A., and Allison, J. P. (2011). Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J. Immunol. 186, 5039–5045.
Corse, E., Gottschalk, R. A., Krogsgaard, M., and Allison, J. P. (2010). Attenuated T cell responses to a high-potency ligand in vivo. PLoS Biol. 8, e1000481. doi:10.1371/journal.pbio.1000481
Davis, M. M., Krogsgaard, M., Huse, M., Huppa, J., Lillemeier, B. F., and Li, Q. J. (2007). T cells as a self-referential, sensory organ. Annu. Rev. Immunol. 25, 681–695.
DeFord-Watts, L. M., Dougall, D. S., Belkaya, S., Johnson, B. A., Eitson, J. L., Roybal, K. T., Barylko, B., Albanesi, J. P., Wulfing, C., and Van Oers, N. S. (2011). The CD3 zeta subunit contains a phosphoinositide-binding motif that is required for the stable accumulation of TCR-CD3 complex at the immunological synapse. J. Immunol. 186, 6839–6847.
Deford-Watts, L. M., Tassin, T. C., Becker, A. M., Medeiros, J. J., Albanesi, J. P., Love, P. E., Wulfing, C., and Van Oers, N. S. (2009). The cytoplasmic tail of the T cell receptor CD3 epsilon subunit contains a phospholipid-binding motif that regulates T cell functions. J. Immunol. 183, 1055–1064.
DeLano, W. L., Ultsch, M. H., De Vos, A. M., and Wells, J. A. (2000). Convergent solutions to binding at a protein-protein interface. Science 287, 1279–1283.
Doucey, M. A., Goffin, L., Naeher, D., Michielin, O., Baumgartner, P., Guillaume, P., Palmer, E., and Luescher, I. F. (2003). CD3 delta establishes a functional link between the T cell receptor and CD8. J. Biol. Chem. 278, 3257–3264.
Dustin, M. L., and Depoil, D. (2011). New insights into the T cell synapse from single molecule techniques. Nat. Rev. Immunol. 11, 672–684.
Engelman, D. M. (2003). Electrostatic fasteners hold the T cell receptor-CD3 complex together. Mol. Cell 11, 5–6.
Fazilleau, N., McHeyzer-Williams, L. J., Rosen, H., and McHeyzer-Williams, M. G. (2009). The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 10, 375–384.
Fernandes, R. A., Shore, D. A., Vuong, M. T., Yu, C., Zhu, X., Pereira-Lopes, S., Brouwer, H., Fennelly, J. A., Jessup, C. M., Evans, E. J., Wilson, I. A., and Davis, S. J. (2012). T-cell receptors are structures capable of initiating signaling in the absence of large conformational rearrangements. J. Biol. Chem. 287, 13324–13335.
Garcia, K. C., Adams, J. J., Feng, D., and Ely, L. K. (2009). The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat. Immunol. 10, 143–147.
Garcia, K. C., Degano, M., Stanfield, R. L., Brunmark, A., Jackson, M. R., Peterson, P. A., Teyton, L., and Wilson, I. A. (1996). An alphabeta T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science 274, 209–219.
Ghendler, Y., Smolyar, A., Chang, H. C., and Reinherz, E. L. (1998). One of the CD3epsilon subunits within a T cell receptor complex lies in close proximity to the Cbeta FG loop. J. Exp. Med. 187, 1529–1536.
Gottschalk, R. A., Corse, E., and Allison, J. P. (2010). TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J. Exp. Med. 207, 1701–1711.
Haase, A. T. (1999). Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 17, 625–656.
Hahn, M., Nicholson, M. J., Pyrdol, J., and Wucherpfennig, K. W. (2005). Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat. Immunol. 6, 490–496.
Hogan, P. G., Lewis, R. S., and Rao, A. (2010). Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28, 491–533.
Holst, J., Wang, H., Eder, K. D., Workman, C. J., Boyd, K. L., Baquet, Z., Singh, H., Forbes, K., Chruscinski, A., Smeyne, R., Van Oers, N. S., Utz, P. J., and Vignali, D. A. (2008). Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat. Immunol. 9, 658–666.
Huppa, J. B., Gleimer, M., Sumen, C., and Davis, M. M. (2003). Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat. Immunol. 4, 749–755.
Huse, M., Klein, L. O., Girvin, A. T., Faraj, J. M., Li, Q. J., Kuhns, M. S., and Davis, M. M. (2007). Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity 27, 76–88.
Irvine, D. J., Purbhoo, M. A., Krogsgaard, M., and Davis, M. M. (2002). Direct observation of ligand recognition by T cells. Nature 419, 845–849.
Janeway, C. A. Jr. (1995). Ligands for the T-cell receptor: hard times for avidity models. Immunol. Today 16, 223–225.
Kjer-Nielsen, L., Dunstone, M. A., Kostenko, L., Ely, L. K., Beddoe, T., Mifsud, N. A., Purcell, A. W., Brooks, A. G., McCluskey, J., and Rossjohn, J. (2004). Crystal structure of the human T cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mAb OKT3. Proc. Natl. Acad. Sci. U.S.A. 101, 7675–7680.
Konig, R., Huang, L. Y., and Germain, R. N. (1992). MHC class II interaction with CD4 mediated by a region analogous to the MHC class I binding site for CD8. Nature 356, 796–798.
Krogsgaard, M., Li, Q. J., Sumen, C., Huppa, J. B., Huse, M., and Davis, M. M. (2005). Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature 434, 238–243.
Kuhns, M. S., and Davis, M. M. (2007). Disruption of extracellular interactions impairs T cell receptor-CD3 complex stability and signaling. Immunity 26, 357–369.
Kuhns, M. S., Davis, M. M., and Garcia, K. C. (2006). Deconstructing the form and function of the TCR/CD3 complex. Immunity 24, 133–139.
Kuhns, M. S., Girvin, A. T., Klein, L. O., Chen, R., Jensen, K. D., Newell, E. W., Huppa, J. B., Lillemeier, B. F., Huse, M., Chien, Y. H., Garcia, K. C., and Davis, M. M. (2010). Evidence for a functional sidedness to the alphabetaTCR. Proc. Natl. Acad. Sci. U.S.A. 107, 5094–5099.
Kumar, R., Ferez, M., Swamy, M., Arechaga, I., Rejas, M. T., Valpuesta, J. M., Schamel, W. W., Alarcon, B., and Van Santen, H. M. (2011). Increased sensitivity of antigen-experienced T cells through the enrichment of oligomeric T cell receptor complexes. Immunity 35, 375–387.
Li, Y., Huang, Y., Lue, J., Quandt, J. A., Martin, R., and Mariuzza, R. A. (2005). Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J. 24, 2968–2979.
Lillemeier, B. F., Mortelmaier, M. A., Forstner, M. B., Huppa, J. B., Groves, J. T., and Davis, M. M. (2010). TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 11, 90–96.
Lillemeier, B. F., Pfeiffer, J. R., Surviladze, Z., Wilson, B. S., and Davis, M. M. (2006). Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc. Natl. Acad. Sci. U.S.A. 103, 18992–18997.
Livnah, O., Stura, E. A., Middleton, S. A., Johnson, D. L., Jolliffe, L. K., and Wilson, I. A. (1999). Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 283, 987–990.
Ma, Z., Janmey, P. A., and Finkel, T. H. (2008a). The receptor deformation model of TCR triggering. FASEB J. 22, 1002–1008.
Ma, Z., Sharp, K. A., Janmey, P. A., and Finkel, T. H. (2008b). Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLoS Biol. 6, e43. doi:10.1371/journal.pbio.0060043
Malherbe, L., Mark, L., Fazilleau, N., McHeyzer-Williams, L. J., and McHeyzer-Williams, M. G. (2008). Vaccine adjuvants alter TCR-based selection thresholds. Immunity 28, 698–709.
Mallaun, M., Naeher, D., Daniels, M. A., Yachi, P. P., Hausmann, B., Luescher, I. F., Gascoigne, N. R., and Palmer, E. (2008). The T cell receptor’s alpha-chain connecting peptide motif promotes close approximation of the CD8 coreceptor allowing efficient signal initiation. J. Immunol. 180, 8211–8221.
Manz, B. N., Jackson, B. L., Petit, R. S., Dustin, M. L., and Groves, J. (2011). T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc. Natl. Acad. Sci. U.S.A. 108, 9089–9094.
Martinez-Martin, N., Risueno, R. M., Morreale, A., Zaldivar, I., Fernandez-Arenas, E., Herranz, F., Ortiz, A. R., and Alarcon, B. (2009). Cooperativity between T cell receptor complexes revealed by conformational mutants of CD3epsilon. Sci. Signal. 2, ra43.
McHeyzer-Williams, L. J., Pelletier, N., Mark, L., Fazilleau, N., and McHeyzer-Williams, M. G. (2009). Follicular helper T cells as cognate regulators of B cell immunity. Curr. Opin. Immunol. 21, 266–273.
Minguet, S., Swamy, M., Alarcon, B., Luescher, I. F., and Schamel, W. W. (2007). Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity 26, 43–54.
Moldovan, M. C., Sabbagh, L., Breton, G., Sekaly, R. P., and Krummel, M. F. (2006). Triggering of T cell activation via CD4 dimers. J. Immunol. 176, 5438–5445.
Morgan, N. V., Goddard, S., Cardno, T. S., McDonald, D., Rahman, F., Barge, D., Ciupek, A., Straatman-Iwanowska, A., Pasha, S., Guckian, M., Anderson, G., Huissoon, A., Cant, A., Tate, W. P., Hambleton, S., and Maher, E. R. (2011). Mutation in the TCRalpha subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRalphabeta+ T cells. J. Clin. Invest. 121, 695–702.
Naeher, D., Luescher, I. F., and Palmer, E. (2002). A role for the alpha-chain connecting peptide motif in mediating TCR-CD8 cooperation. J. Immunol. 169, 2964–2970.
Newell, E. W., Ely, L. K., Kruse, A. C., Reay, P. A., Rodriguez, S. N., Lin, A. E., Kuhns, M. S., Garcia, K. C., and Davis, M. M. (2011). Structural basis of specificity and cross-reactivity in T cell receptors specific for cytochrome c-I-E(k). J. Immunol. 186, 5823–5832.
Nika, K., Soldani, C., Salek, M., Paster, W., Gray, A., Etzensperger, R., Fugger, L., Polzella, P., Cerundolo, V., Dushek, O., Hofer, T., Viola, A., and Acuto, O. (2010). Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity 32, 766–777.
Ohashi, P. S. (2002). T-cell signalling and autoimmunity: molecular mechanisms of disease. Nat. Rev. Immunol. 2, 427–438.
Ottemann, K. M., Xiao, W., Shin, Y. K., and Koshland, D. E. Jr. (1999). A piston model for transmembrane signaling of the aspartate receptor. Science 285, 1751–1754.
Potter, T. A., Rajan, T. V., Dick, R. F. II, and Bluestone, J. A. (1989). Substitution at residue 227 of H-2 class I molecules abrogates recognition by CD8-dependent, but not CD8-independent, cytotoxic T lymphocytes. Nature 337, 73–75.
Purbhoo, M. A., Irvine, D. J., Huppa, J. B., and Davis, M. M. (2004). T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 5, 524–530.
Rabinowitz, J. D., Vrljic, M., Kasson, P. M., Liang, M. N., Busch, R., Boniface, J. J., Davis, M. M., and McConnell, H. M. (1998). Formation of a highly peptide-receptive state of class II MHC. Immunity 9, 699–709.
Reich, Z., Boniface, J. J., Lyons, D. S., Borochov, N., Wachtel, E. J., and Davis, M. M. (1997). Ligand-specific oligomerization of T-cell receptor molecules. Nature 387, 617–620.
Rojo, J. M., and Janeway, C. A. Jr. (1988). The biologic activity of anti-T cell receptor V region monoclonal antibodies is determined by the epitope recognized. J. Immunol. 140, 1081–1088.
Rosenberg, S. A., Restifo, N. P., Yang, J. C., Morgan, R. A., and Dudley, M. E. (2008). Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 8, 299–308.
Rudolph, M. G., Stanfield, R. L., and Wilson, I. A. (2006). How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24, 419–466.
Salter, R. D., Benjamin, R. J., Wesley, P. K., Buxton, S. E., Garrett, T. P., Clayberger, C., Krensky, A. M., Norment, A. M., Littman, D. R., and Parham, P. (1990). A binding site for the T-cell co-receptor CD8 on the alpha 3 domain of HLA-A2. Nature 345, 41–46.
Sasada, T., Touma, M., Chang, H. C., Clayton, L. K., Wang, J. H., and Reinherz, E. L. (2002). Involvement of the TCR Cbeta FG loop in thymic selection and T cell function. J. Exp. Med. 195, 1419–1431.
Schamel, W. W., Arechaga, I., Risueno, R. M., Van Santen, H. M., Cabezas, P., Risco, C., Valpuesta, J. M., and Alarcon, B. (2005). Coexistence of multivalent and monovalent TCRs explains high sensitivity and wide range of response. J. Exp. Med. 202, 493–503.
Sigalov, A. B., Aivazian, D. A., Uversky, V. N., and Stern, L. J. (2006). Lipid-binding activity of intrinsically unstructured cytoplasmic domains of multichain immune recognition receptor signaling subunits. Biochemistry 45, 15731–15739.
Smith-Garvin, J. E., Koretzky, G. A., and Jordan, M. S. (2009). T cell activation. Annu. Rev. Immunol. 27, 591–619.
Starr, T. K., Jameson, S. C., and Hogquist, K. A. (2003). Positive and negative selection of T cells. Annu. Rev. Immunol. 21, 139–176.
Sun, Z. J., Kim, K. S., Wagner, G., and Reinherz, E. L. (2001). Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3 epsilon gamma heterodimer. Cell 105, 913–923.
Sun, Z. Y., Kim, S. T., Kim, I. C., Fahmy, A., Reinherz, E. L., and Wagner, G. (2004). Solution structure of the CD3epsilondelta ectodomain and comparison with CD3epsilongamma as a basis for modeling T cell receptor topology and signaling. Proc. Natl. Acad. Sci. U.S.A. 101, 16867–16872.
Symer, D. E., Dintzis, R. Z., Diamond, D. J., and Dintzis, H. M. (1992). Inhibition or activation of human T cell receptor transfectants is controlled by defined, soluble antigen arrays. J. Exp. Med. 176, 1421–1430.
Szymczak, A. L., Workman, C. J., Wang, Y., Vignali, K. M., Dilioglou, S., Vanin, E. F., and Vignali, D. A. (2004). Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat. Biotechnol. 22, 589–594.
Touma, M., Chang, H. C., Sasada, T., Handley, M., Clayton, L. K., and Reinherz, E. L. (2006). The TCR Cbeta FG loop regulates {alpha}beta T cell development. J. Immunol. 176, 6812–6823.
Van Laethem, F., Sarafova, S. D., Park, J. H., Tai, X., Pobezinsky, L., Guinter, T. I., Adoro, S., Adams, A., Sharrow, S. O., Feigenbaum, L., and Singer, A. (2007). Deletion of CD4 and CD8 coreceptors permits generation of alphabetaT cells that recognize antigens independently of the MHC. Immunity 27, 735–750.
Vercelli, D. (2008). Discovering susceptibility genes for asthma and allergy. Nat. Rev. Immunol. 8, 169–182.
Vignali, D. A., Carson, R. T., Chang, B., Mittler, R. S., and Strominger, J. L. (1996). The two membrane proximal domains of CD4 interact with the T cell receptor. J. Exp. Med. 183, 2097–2107.
Vignali, D. A., and Vignali, K. M. (1999). Profound enhancement of T cell activation mediated by the interaction between the TCR and the D3 domain of CD4. J. Immunol. 162, 1431–1439.
Wang, J. H., Meijers, R., Xiong, Y., Liu, J. H., Sakihama, T., Zhang, R., Joachimiak, A., and Reinherz, E. L. (2001). Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc. Natl. Acad. Sci. U.S.A. 98, 10799–10804.
Wang, R., Natarajan, K., and Margulies, D. H. (2009a). Structural basis of the CD8 alpha beta/MHC class I interaction: focused recognition orients CD8 beta to a T cell proximal position. J. Immunol. 183, 2554–2564.
Wang, Y., Becker, D., Vass, T., White, J., Marrack, P., and Kappler, J. W. (2009b). A conserved CXXC motif in CD3epsilon is critical for T cell development and TCR signaling. PLoS Biol. 7, e1000253. doi:10.1371/journal.pbio.1000253
Werlen, G., Hausmann, B., and Palmer, E. (2000). A motif in the alphabeta T-cell receptor controls positive selection by modulating ERK activity. Nature 406, 422–426.
Wu, H., Kwong, P. D., and Hendrickson, W. A. (1997). Dimeric association and segmental variability in the structure of human CD4. Nature 387, 527–530.
Xu, C., Gagnon, E., Call, M. E., Schnell, J. R., Schwieters, C. D., Carman, C. V., Chou, J. J., and Wucherpfennig, K. W. (2008). Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 135, 702–713.
Yamasaki, S., Ishikawa, E., Sakuma, M., Ogata, K., Sakata-Sogawa, K., Hiroshima, M., Wiest, D. L., Tokunaga, M., and Saito, T. (2006). Mechanistic basis of pre-T cell receptor-mediated autonomous signaling critical for thymocyte development. Nat. Immunol. 7, 67–75.
Yin, Y., Wang, X. X., and Mariuzza, R. A. (2012). Crystal structure of a complete ternary complex of T-cell receptor, peptide-MHC, and CD4. Proc. Natl. Acad. Sci. U.S.A. 109, 5405–5410.
Yoon, S. T., Dianzani, U., Bottomly, K., and Janeway, C. A. Jr. (1994). Both high and low avidity antibodies to the T cell receptor can have agonist or antagonist activity. Immunity 1, 563–569.
You, S., and Chatenoud, L. (2010). New generation CD3 monoclonal antibodies: are we ready to have them back in clinical transplantation? Curr. Opin. Organ Transplant. 15, 720–724.
Yudushkin, I. A., and Vale, R. D. (2010). Imaging T-cell receptor activation reveals accumulation of tyrosine-phosphorylated CD3zeta in the endosomal compartment. Proc. Natl. Acad. Sci. U.S.A. 107, 22128–22133.
Zal, T., Zal, M. A., and Gascoigne, N. R. (2002). Inhibition of T cell receptor-coreceptor interactions by antagonist ligands visualized by live FRET imaging of the T-hybridoma immunological synapse. Immunity 16, 521–534.
Zehn, D., Lee, S. Y., and Bevan, M. J. (2009). Complete but curtailed T-cell response to very low-affinity antigen. Nature 458, 211–214.
Keywords: TCR, CD3, complex, co-receptor, multi-molecular machinery, triggering
Citation: Kuhns MS and Davis MM (2012) TCR signaling emerges from the sum of many parts. Front. Immun. 3:159. doi: 10.3389/fimmu.2012.00159
Received: 25 February 2012; Accepted: 27 May 2012;
Published online: 25 June 2012.
Edited by:
Oreste Acuto, University of Oxford, UKReviewed by:
Tomasz Zal, University of Texas MD Anderson Cancer Center, USACopyright: © 2012 Kuhns and Davis. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Michael S. Kuhns, Department of Immunobiology, The University of Arizona College of Medicine, PO Box 245221, Tucson, AZ 85724, USA. e-mail:bWt1aG5zQHUuYXJpem9uYS5lZHU=; Mark M. Davis, Department of Microbiology and Immunology, Beckman Center, Stanford University, B221, 279 Campus Drive, Stanford, CA 94305, USA. e-mail:bW1kYXZpc0BzdGFuZm9yZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.