 Agostinho Carvalho1,2,3
Agostinho Carvalho1,2,3 Cristina Cunha1
Cristina Cunha1 Silvia Bozza1
Silvia Bozza1 Silvia Moretti1
Silvia Moretti1 Cristina Massi-Benedetti1 Francesco Bistoni1 Franco Aversa4
Cristina Massi-Benedetti1 Francesco Bistoni1 Franco Aversa4 Luigina Romani1*
Luigina Romani1*- 1 Department of Experimental Medicine and Biochemical Sciences, University of Perugia, Perugia, Italy
- 2 Life and Health Sciences Research Institute, School of Health Sciences, University of Minho, Braga, Portugal
- 3 ICVS/3B’s – PT Government Associate Laboratory, Braga/Guimarães, Portugal
- 4 Hematology and Bone Marrow Transplant Unit, Department of Internal Medicine and Biomedical Sciences, University of Parma, Parma, Italy
Resistance and tolerance are two complementary host defense mechanisms that increase fitness in response to low-virulence fungi. Resistance is meant to reduce pathogen burden during infection through innate and adaptive immune mechanisms, whereas tolerance mitigates the substantial cost of resistance to host fitness through a multitude of anti-inflammatory mechanisms, including immunological tolerance. In experimental fungal infections, both defense mechanisms are activated through the delicate equilibrium between Th1/Th17 cells, which provide antifungal resistance, and regulatory T cells limiting the consequences of the ensuing inflammatory pathology. Indoleamine 2,3-dioxygenase (IDO), a rate-limiting enzyme in the tryptophan catabolism, plays a key role in induction of tolerance against fungi. Both hematopoietic and non-hematopoietic compartments contribute to the resistance/tolerance balance against Aspergillus fumigatus via the involvement of selected innate receptors converging on IDO. Several genetic polymorphisms in pattern recognition receptors influence resistance and tolerance to fungal infections in human hematopoietic transplantation. Thus, tolerance mechanisms may be exploited for novel diagnostics and therapeutics against fungal infections and diseases.
Introduction
Allogeneic hematopoietic stem cell transplantation (allogeneic HSCT) remains a curative treatment for hematological malignancies resistant to other treatment approaches. The unique post-transplantation milieu, which is characterized by lymphopenia, regulatory T (Treg) cell depletion, and the release of growth factors and cytokines (Matsuoka et al., 2010), provides a unique opportunity for the occurrence of severe infections. Fungal infections have historically been, and remain important causes of transplant-related morbidity in HSCT (Mulanovich and Kontoyiannis, 2011). The ability to combine antifungal agents may provide new treatment options, but prevention and resolution of these infections have been difficult to achieve. Several studies have reported the predominance of aspergillosis occurring in the post-engraftment rather than the neutropenic period in allogeneic HSCT recipients (Grow et al., 2002; Marr et al., 2002). Clinically, severe fungal infections occur in patients with immune reconstitution syndrome (IRS), an entity characterized by local and systemic reactions that have both beneficial and deleterious effects on infection (Singh and Perfect, 2007). Intriguingly, IRS responses are also found in immunocompetent individuals and after rapid resolution of immunosuppression, indicating that inflammatory responses can result in quiescent or latent infections manifesting as opportunistic mycoses. These considerations indicate that host immunity is crucial in eradicating infection, but immunological recovery can also be detrimental and may contribute toward worsening disease in opportunistic and non-opportunistic infections (Gupta and Singh, 2011). Ultimately, control of infection depends on the restoration of adequate antifungal immunity, and thus, strategies to augment immunity against fungal pathogens are complementary to those targeting the pathogens. As a matter of fact, part of the antifungal effect of antifungal agents may occur via immunomodulation (Simitsopoulou et al., 2011).
Current understanding of the pathophysiology underlying fungal infections and diseases highlights the multiple cell populations and cell-signaling pathways involved in these complex conditions beyond the dysregulated chaos in which fungal infection and disease are perceived. Because the immune response is a complex entity, a systems biology approach that integrates investigations of immunity at the systems-level is required to generate novel insights into this complexity (Santamaria et al., 2011). At the population level, single nucleotide polymorphisms (SNPs), by altering protein-protein interactions or transcriptional regulation, may add further complexity to the system. Applying systems biology approaches to these complex processes is required for a better appreciation of the intricate cross-talk provided by temporal changes in mediators, metabolites and cell phenotypes underlining the coordinated processes.
In this review we will discuss how mechanisms that regulate both resistance and tolerance to fungi could be successfully exploited to elicit antimicrobial immunity and concomitant tolerance via acquired local immune privilege in HSCT.
Insight into the Mechanisms Regulating Immune Homeostasis in Response to Fungi: The Resistance and Tolerance Paradigm
The immune system protects from infections primarily by detecting and eliminating the invading pathogens; however, the host organism can also protect itself from infectious diseases by reducing the negative impact of infections on host fitness (Figure 1). This ability to tolerate a pathogen’s presence is a distinct host defense strategy that, in many circumstances, favors the evolution of protective mechanisms that do not involve pathogen killing (Medzhitov et al., 2012). This strategy likely occurs in the host-microbial symbiosis, thus suggesting that the majority of host defense mechanisms that have arisen during evolution are perhaps tolerance mechanisms. Little attempt to formally decompose human or animal health into resistance and tolerance components has been done. Resistance is meant to reduce pathogen burden during infection through innate and adaptive immune mechanisms, whereas tolerance mitigates the substantial cost to host fitness of resistance (Read et al., 2008; Schneider and Ayres, 2008). Even in the absence of overt tissue damage, resistance mechanisms commonly occur at a cost to normal tissue function, thus causing immunopathology. This means that the optimal immune response is determined by the balance between efficient pathogen clearance and an acceptable level of immunopathology. A plethora of tolerance mechanisms, despite less known relative to resistance mechanisms, protect the host from immune- or pathogen-induced damage (Cobbold et al., 2010; Saraiva and O’Garra, 2010). Therefore, the term tolerance is semantically used here to refer to the multitude of anti-inflammatory mechanisms, including immunological tolerance, that is, unresponsiveness to self-antigens.
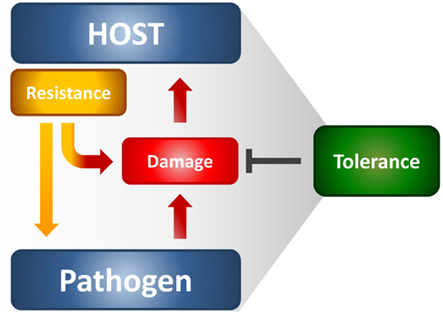
Figure 1. The resistance and tolerance defense strategies in infections. Resistance reduces microbial burden during infection through innate and adaptive immune mechanisms, whereas tolerance mitigates the damage to host tissues caused by both the resistance mechanisms and pathogen’s virulence. Both the host and the pathogen can reduce fitness costs through tolerance mechanisms that reduce both the direct tissue damage by pathogens as well as immunopathology.
It has been argued that a high rate of infection but low-virulence should select for host tolerance, whereas the opposite should favor resistance (Restif and Koella, 2004). Thus, it is not surprising that resistance and tolerance are two complementary host defense traits that increase fitness in response to low-virulence fungi (Romani, 2011). In experimental fungal infections, both defense mechanisms are activated through the delicate equilibrium between Th1/Th17 cells, which provide antifungal resistance mechanisms, and Treg cells limiting the consequences of the associated inflammatory pathology. Indeed, while some degree of inflammation is required for protection, particularly at mucosal tissues during the transitional response occurring between the rapid innate and slower adaptive response, progressive inflammation worsens disease, and ultimately prevents pathogen eradication. Recent observations highlight a truly bipolar nature of the inflammatory process against fungi (Romani and Puccetti, 2007; Romani et al., 2008a). The conceptual principle highlighting a truly bipolar nature of the inflammatory process in infection is best exemplified by the occurrence of severe fungal infections and diseases in patients with IRS (Gupta and Singh, 2011) and in the hyper-IgE syndrome in which increased levels of pro-inflammatory gene transcripts have recently been described (Holland et al., 2007). For A. fumigatus, the association of persistent inflammation with intractable infection is common in non-neutropenic patients after allogeneic HSCT (Ortega et al., 2006) as well as in allergic fungal diseases (Schubert, 2006). A main implication of these findings is that, at least in specific clinical settings, it is an exaggerated inflammatory response that likely compromises a patient’s ability to eradicate infection, and not an “intrinsic” susceptibility to infection that determines a state of chronic or intractable disease. The above findings may serve to accommodate fungi within the host immune system and at the same time explain why, despite the fact that human beings are constantly exposed to fungi, fungal diseases are relatively rare. Should a degree of coexistence had occurred between fungi and their mammalian hosts, this would implicate the possible, underestimated, contribution of fungi to the plasticity of the immune system. Evidence suggest that the continued integration of pro-inflammatory and anti-inflammatory stimuli in response to fungi is critical for a proper control of infection and T cell homeostasis (Romani, 2011).
Indoleamine 2,3-Dioxygenase is a Critical Regulator of Tolerance to Fungi
IDO is an interferon-gamma-inducible intracellular enzyme which catalyzes the catabolism of tryptophan (Puccetti and Grohmann, 2007; Mellor and Munn, 2008). Work has demonstrated a complex and crucial role for tryptophan catabolism in modulating inflammatory processes and T cell tolerance after HSCT (Hainz et al., 2007; Brandacher et al., 2008). The effects of IDO activity are tryptophan deficiency, excess tryptophan breakdown products (kynurenines) and consumption of reactive oxygen species. In transplantation, increased IDO activity in transplanted cells has been demonstrated to have anti-rejection properties both in vitro and in vivo (Hainz et al., 2007). Overall, the available data suggest a potential role of IDO in governing transplantation tolerance through mechanistic pathways possibly involving IDO induction by reverse signaling through costimulatory receptors (Puccetti and Grohmann, 2007) and IDO-mediated long-term tolerance (Pallotta et al., 2011).
IDO and kynurenines serve many roles in fungal infections; most relevant, the induction of CD4+CD25+ Treg cells via IDO+ dendritic cells (DCs; Montagnoli et al., 2006). In experimental aspergillosis, IDO blockade greatly exacerbated infections and allergy to the fungus, as a result of deregulated innate and adaptive immune responses caused by the impaired activation and functioning of suppressor CD4+CD25+ Tregs producing IL-10 (Montagnoli et al., 2006). A number of studies have established that the proper control of the infection and associated inflammatory reactions require IDO induction and consequent production of tryptophan metabolites with immune-regulatory activities, contributing to the maintenance of the Treg/Th17 balance (Romani et al., 2008b). As clearly shown in susceptible mice, Treg and Th17 cells mediate antagonizing roles in aspergillosis, where increasing levels of IL-17-driven inflammation occurred alongside decreased anti-inflammatory Treg responses, resulting in inflammatory overreactions (Romani et al., 2008b). A reciprocal antagonistic relationship was also found between IDO and the Th17 pathway, with IDO restraining Th17 responses and IL-17A inhibiting IDO (Zelante et al., 2007). Further adding to the complexity, a recent study has revealed the ability of IL-17A to increase survival and virulence of fungi (Zelante et al., 2012).
Evidence indicates that the non-hematopoietic compartment also contributes to tolerance to fungi (Cunha et al., 2010; de Luca et al., 2010). Epithelial cells (ECs) are known to determine the balance between a state of “mucosal homeostasis,” required for optimal organ function, and “mucosal injury,” leading to mucosal inflammation and barrier breakdown. However, recent evidence has also indicated ECs as key players in tolerance to respiratory pathogens via an IFN-γ/IDO axis culminating in the inhibition of Th17 cell responses (Desvignes and Ernst, 2009; de Luca et al., 2010). IDO over-expression in airway ECs was found to restrain CD4+ T cell activation to the fungus, an activity that was nevertheless dispensable in the presence of IDO-expressing tolerogenic DCs. However, IDO induction in ECs could compensate for the lack of IDO on hematopoietic cells (Paveglio et al., 2011). The expression of IDO on ECs occurred through the TLR3/TRIF-dependent pathway, a finding consistent with the abundant expression of TLR3 both intracellularly and on the cell surface of ECs. The failure to activate IDO likely accounted for the lack of tolerance to the fungus observed in experimental HSCT in condition in which either the recipient or the donor, or even more when both, were TRIF- or TLR3-deficient (de Luca et al., 2010). Overall, these data shed light on pathways of immune resistance and tolerance to the fungus that likely take place in a hematopoietic transplantation setting. It appears that protective tolerance to the fungus is achieved through a TLR3/TRIF-dependent pathway activating Th1/Treg cells via IDO expressed on both the hematopoietic/non-hematopoietic compartments. In contrast, the MyD88 pathway provided antifungal resistance, i.e., the ability to restrict the fungal growth through defensins and likely, other effector mechanisms (de Luca et al., 2010). However, the ability of mice to clear the fungus in the relative absence of the MyD88 pathway (Bretz et al., 2008) clearly indicates redundancies and hierarchy in antifungal mechanisms of resistance. Ultimately, the finding that both Candida albicans (De Luca et al., 2007) and A. fumigatus (de Luca et al., 2010), two major human fungal pathogens, exploit the TRIF-dependent pathway at the interface with the mammalian hosts, indicates that the exploitation of tolerance mechanisms is an advantageous option.
Metabolic Regulation of Tolerance to Fungi
The activation of distinct signaling pathways in DCs translates recognition of fungi into distinct inflammatory and adaptive immune responses (Bonifazi et al., 2009, 2010). The screening of signaling pathways in DCs through a systems biology approach was exploited for the development of therapeutics to attenuate inflammation in experimental fungal infections and diseases. In vivo targeting inflammatory [PI3K/Akt/mammalian target of rapamycin (mTOR)] or anti-inflammatory (STAT3/IDO) DC pathways by intranasally delivered small interfering RNA (siRNA) modified resistance and tolerance to infection. Thus, the screening of signaling pathways in DCs through a systems biology approach may be exploited for the development of siRNA therapeutics to attenuate inflammation in respiratory fungal infections and diseases (Bonifazi et al., 2010). It is of interest that the mTOR pathway has emerged as a key player in sensing nutritional/energetic signals and integrating signals controlling metabolism and cellular fate. The mTOR pathway has been shown to play an important role in determining the differentiation of CD4+ T cells into inflammatory and regulatory subsets, the induction of anergy, the development of CD8+ memory T cells, and the regulation of T cell trafficking (Araki et al., 2009; Delgoffe et al., 2009; Cobbold et al., 2010; Peter et al., 2010). The inhibition of mTOR promoted immune tolerance in mouse models of transplantation, by favoring the expansion of Treg cells over effector T cells (Zuber et al., 2011). However, recent data have shed light on the unexpected pro-inflammatory burst observed in some transplant recipients treated with mTOR inhibitors (Saemann et al., 2009). Therefore, the potential therapeutic utility of mTOR modulation in tolerance to fungi in HSCT awaits clarification.
Exploiting Pathogen-Induced Tolerance in Experimental HSCT through Dendritic Cells
The potential use of tolerogenic DCs as negative cellular vaccines to induce experimental transplantation tolerance has been suggested (Turnquist and Thomson, 2008). Plasmacytoid DCs may contribute to the T cell repertoire reconstitution, facilitate engraftment (Fugier-Vivier et al., 2005), and prevent graft-versus-host disease in HSCT (Arpinati et al., 2003). As DC function is impaired during the immediate post-transplantation period (Reddy et al., 2004), the administration of donor DCs may have beneficial effects in immune recovering in the early HSCT. Over recent years experimental models have shown that it is possible to exploit the mechanisms that normally maintain immune homeostasis and tolerance to self-antigens to induce tolerance to alloantigens (Waldmann and Cobbold, 2004; Martinic and von Herrath, 2006). Like natural tolerance, transplantation tolerance is achieved through control of T cell reactivity by central and peripheral mechanisms of tolerance. We have recently found that this goal is achievable by the adoptive cellular therapy of fungus-pulsed or RNA-transfected IDO+DCs that could induce antifungal resistance within a regulatory environment (Bozza et al., 2003; Romani et al., 2006). In experimental HSCT, a model in which allogeneic reconstitution of host stem cells is greatly reduced to the benefit of a long-term, donor type chimerism in more than 95% of the mice and low incidence of graft-versus-host disease (Bozza et al., 2003), protection was associated with myeloid and T cell recovery, the activation of CD4+ Th1 lymphocytes, and the concomitant IL-10-driven Treg cells. Distinct DC subsets activated specialized antifungal effector and regulatory functions upon adoptive transfer in experimental HSCT. FLT3-ligand derived DCs (mainly B220+IDO+pDCs) fulfilled the requirement for Th1/Treg antifungal priming. In contrast, conventional CD11c+DCs contributed to inflammatory pathology via the activation of Th1/Th17 responses (Romani et al., 2006; Bonifazi et al., 2010). Thus, IDO+DCs proved to be pivotal in the generation of some form of dominant regulation that ultimately controlled inflammation, pathogen immunity and tolerance in transplant recipients eventually leading to prevention of graft-versus-host reaction and reduction of aspergillosis incidence rates.
The ability of Aspergillus-induced Treg cells to inhibit alloreactivity while sparing responsiveness to pathogen, suggests that pathogen-induced Treg cells may be associated with minimal bystander suppression. From a mechanistic perspective, this implies that the function of Treg cells in transplantation can be controlled by the specificity of the T cell receptor expressed on Treg cells (Albert et al., 2006) and is in line with the observation of a positive effect on post-transplant immunity of antigen exposure at the time of transplantation (Mori et al., 2005). Several studies have addressed the effect that infections have on transplantation tolerance, and the overall view is that both prior and concurrent exposure to pathogens can prevent tolerance induction. However, much less attention has been paid to the effect that pathogen-directed tolerance based on active T cell regulation might have on tolerance to donor antigens. Because of the cross-reactivity in the T cell repertoire between antimicrobial, environmental, and transplantation antigens (Mason, 1998), our results raise the intriguing possibility that pathogen-conditioned DCs could be potential reagents to promote donor-specific transplantation tolerance through the induction of CD4+CD25+ Treg cells with indirect anti-donor allospecificity. Strategies to generate human CD4+CD25+ T cell lines with indirect allospecificity for therapeutic use for the induction of donor-specific transplantation tolerance have recently been described (Jiang and Lombardi, 2006). Thus, transplantation tolerance and concomitant pathogen clearance could be achieved through the therapeutic induction of antigen-specific Tregs via instructive immunotherapy with pathogen-conditioned donor DCs.
Exploiting Pathogen-Induced Tolerance in Experimental HSCT through PAMP/DAMP/PRR Signaling
Although allograft rejection is mainly a T cell-mediated process, the innate immune system can participate in the immune response to organ transplantation (Larosa et al., 2007). Toll-like receptors (TLRs) and others innate pattern recognition receptors (PRRs; Romani, 2011) are critical innate immune receptors expressed on a variety of cells that sense not only pathogen-associated molecular patterns (PAMPs) but also damaged host cell components, collectively known as damage-associated molecular patterns (DAMPs; Bianchi, 2007). This suggests that PRR signaling participates in inflammation that may occur in the absence of overt infection and promotes acute allograft rejection and prevention of transplantation tolerance (Alegre et al., 2008). Strategies to prevent innate immunity-mediated rejection have already been described (Land, 2007). However, although signaling through TLRs can prevent tolerance induction and promote graft rejection (Chen et al., 2006), TLR signaling also promotes the induction of Treg cells (Kabelitz et al., 2006). This implies that selected TLR ligands can be useful candidate adjuvants for Treg induction/maintenance in transplantation. This appears to be the case for thymosin α1 (Tα1), a naturally occurring thymic peptide (Goldstein and Badamchian, 2004), that promoted maturation of and cytokine production by human and murine DCs (Romani et al., 2006). By signaling through TLR9, Tα1 induced IDO expression and kynurenin production by murine DCs, promoted pDC-mediated generation of CD4+CD25+ Treg cells and created tolerance in the inflammatory milieu of HSCT (Romani et al., 2006). Tα1 acted as a fine regulator of peripheral inflammation via tolerance induction through Treg cell expansion. In addition, by taming inflammatory DCs, Tα1 successfully primed for antifungal Th1/Treg cells devoid of alloreactivity in hematopoietic transplantation. Thus, Tα1 is a unique immunoregulatory molecule capable of fine-tuning and controlling the quality of the immune response, which may result in the control of inflammation and restoration of protective antimicrobial immunity in the relative absence of immunopathology.
Despite the identification of specific signaling pathways that negatively regulate responses to PAMPs or DAMPs (Bianchi, 2007), the unexpected convergence of molecular pathways responsible for recognition of PAMPs and DAMPs raised the question of whether and how the host discriminates between the two molecular patterns and the relative contribution of either one to inflammation, immune homeostasis, and mechanisms of repair during infection. A mechanism that discriminates between pathogen- and danger-induced immune responses via the spatiotemporal integration of signals from TLRs and the receptor for advanced glycation end-products (RAGE) has recently been described in mice and a genetically determined hyperfunction of the DAMP signaling was associated with invasive aspergillosis in human HSCT (Cunha et al., 2011b; Sorci et al., 2011). The mechanism exploits a previously unrecognized role for the S100B/RAGE axis that, in sensing danger, plays a critical and unanticipated role as a fine modulator of inflammation in Aspergillus pneumonia. Thus, the cross-talk between RAGE and TLRs details an evolving braking circuit whereby an endogenous danger protects the host against pathogen-induced inflammation and a pathogen-sensing mechanism terminates danger-induced inflammation.
Exploiting Pathogen-Induced Tolerance in Human HSCT through Functional Genomics
It is now clear that genetic variants of molecules involved in innate recognition of fungi may account, in part, for the inherited differences in human susceptibility to fungal infections (Carvalho et al., 2009; Mezger et al., 2010). Although the dissection of the genetic traits modulating susceptibility to fungal infections is complex, the contribution of host genetics may hold the key to elucidate genetic markers for fungal diseases occurring in high-risk patients. Understanding which patients are at highest risk of developing a life-threatening infection is at present a major unmet need, and genetic markers will probably assist in risk assessment. Figure 2 summarizes known association of SNPs in human immune genes and susceptibility to fungal infections and diseases.
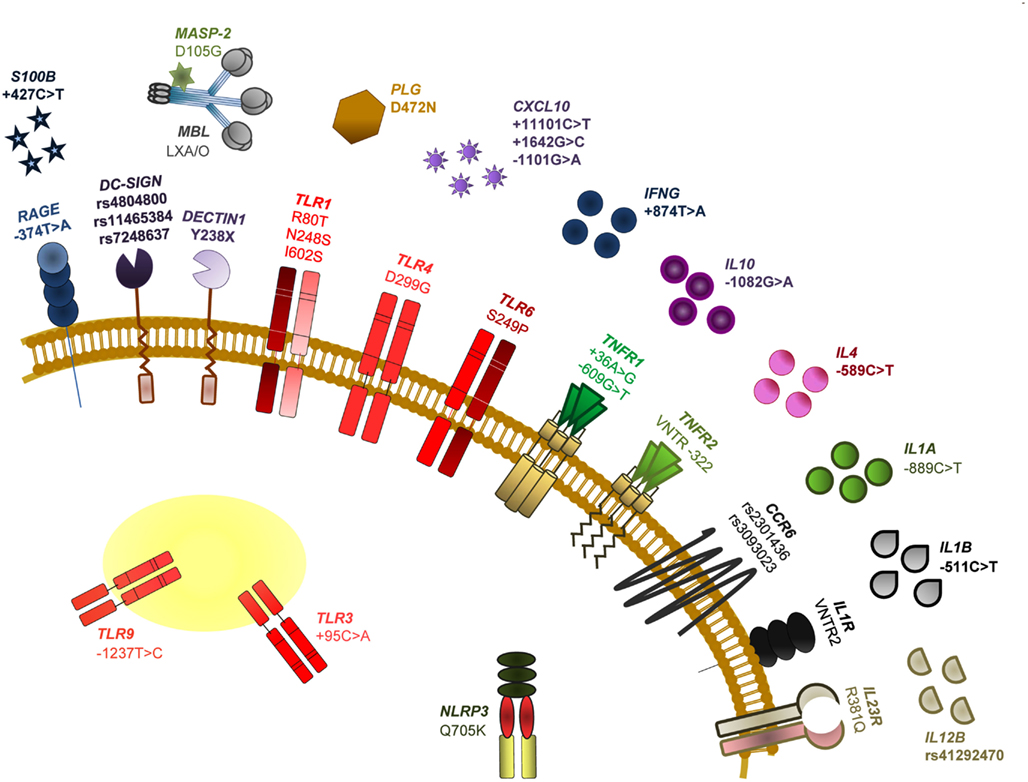
Figure 2. Single nucleotide polymorphisms of human immune genes associated with susceptibility to fungal infections and diseases. The SNPs specifically addressed in this review are among those most notoriously associated with impaired PAMP/DAMP/PRR signaling and consequent susceptibility to fungal disease in HSCT recipients. In particular, we discuss the relevance of TLR4 D299G (a non-synonymous mutation demonstrated to increase the risk for fungal colonization or disease, likely depending on the type of transplant and associated clinical variables), DECTIN1 Y238X (an early stop codon mutation affecting dectin-1 function and that has been revealed to modulate susceptibility to mucosal candidiasis, as well as Candida colonization and invasive aspergillosis) and RAGE −374T > A and S100B +427C > T (SNPs compromising their transcriptional regulation and demonstrated to underlie an hyperfunctional RAGE-mediated DAMP signaling and consequent susceptibility to aspergillosis). For further details on these and other SNPs and relevant references, see Romani (2011).
A number of polymorphisms in several PRRs, mostly TLRs and C-type lectin receptors (CLRs), has been shown to affect, through distinct intracellular signaling pathways, resistance and tolerance to fungi in HSCT (Cunha et al., 2011a). The association of the TLR4 polymorphism D299G with colonization by A. fumigatus, but not invasive disease, in a cohort of T cell depleted transplant recipients from related donors, was meant to signify impaired fungal recognition, but also protection from excessive inflammation leading to immunopathology (Carvalho et al., 2009). Thus, and although further validation studies are ultimately required, by limiting an exacerbated inflammatory response to the fungus, the D299G polymorphism could contribute to tolerance in aspergillosis. More recently, a polymorphism affecting the CLR dectin-1, Y238X, has also been shown to affect tolerance, besides resistance, to A. fumigatus (Cunha et al., 2010). Although dectin-1 has been regarded as one major innate receptor leading to Th17 activation in response to A. fumigatus (Werner et al., 2009), and the Y238X polymorphism was associated with impaired IL-17 production in response to C. albicans or β-glucan (Rosentul et al., 2011), IFN-γ, and IL-10 production by human mononuclear cells carrying the Y238X polymorphism were also defective upon β-glucan or conidia stimulation. Thus, these findings point to a previously unsuspected role for dectin-1 in modulating the resistance and tolerance balance in antifungal responses. The contribution of recipient dectin-1 deficiency to the high-risk of infection in these patients also highlights the distinct, yet complementary, mechanisms of immune resistance and tolerance that are dependent on the hematopoietic/non-hematopoietic compartmentalization (Carvalho et al., 2009).
Conclusion
The past decades have brought important progress in the development of more effective and safe antifungal agents. However, medical treatments that increase host resistance, such as antibiotics, place selective pressures on pathogens, ultimately resulting in the generation of a bewildering array of immuno-evasive or immuno-suppressive strategies (Read et al., 2008). Thus, targeting mechanisms of resistance only may not always work. As tolerance mechanisms are not expected to have the same selective pressure on pathogens, new drugs that target tolerance will provide therapies to which pathogens will not develop resistance. The distinction between failed resistance and failed tolerance is important because it can dictate the choice of therapeutic approaches. In the case of failed tolerance, boosting immunity, and reducing fungal burden with antifungals may be ineffective, whereas enhancing tolerance may have salutary effects. Targeting disease tolerance mechanisms may thus provide new approaches for patient stratification, donor selection, and therapeutic management in HSCT. In this regard, the immunogenetic approach will help design tailored therapies and immunotherapies in high-risk patients and to move beyond hand-me-down data.
Conflict of Interest Statement
This work is supported by Gilead Sciences srl.
Acknowledgments
This work is supported by the Specific Targeted Research Project FUNMETA (ERC-2011-AdG-293714) and the project entitled “Genetic polymorphisms in patients with invasive aspergillosis: a new frontier for diagnosis and therapy” funded by Gilead Sciences S.r.l. Agostinho Carvalho and Cristina Cunha were financially supported by fellowships from Fundação para a Ciência e Tecnologia, Portugal (contracts SFRH/BPD/46292/2008 and SFRH/BD/65962/2009, respectively).
References
Albert, M. H., Anasetti, C., and Yu, X. Z. (2006). T regulatory cells as an immunotherapy for transplantation. Expert Opin. Biol. Ther. 6, 315–324.
Alegre, M. L., Leemans, J., Le Moine, A., Florquin, S., De Wilde, V., Chong, A., and Goldman, M. (2008). The multiple facets of toll-like receptors in transplantation biology. Transplantation 86, 1–9.
Araki, K., Turner, A. P., Shaffer, V. O., Gangappa, S., Keller, S. A., Bachmann, M. F., Larsen, C. P., and Ahmed, R. (2009). mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112.
Arpinati, M., Chirumbolo, G., Urbini, B., Perrone, G., Rondelli, D., and Anasetti, C. (2003). Role of plasmacytoid dendritic cells in immunity and tolerance after allogeneic hematopoietic stem cell transplantation. Transpl. Immunol. 11, 345–356.
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5.
Bonifazi, P., D’Angelo, C., Zagarella, S., Zelante, T., Bozza, S., De Luca, A., Giovannini, G., Moretti, S., Iannitti, R. G., Fallarino, F., Carvalho, A., Cunha, C., Bistoni, F., and Romani, L. (2010). Intranasally delivered siRNA targeting PI3K/Akt/mTOR inflammatory pathways protects from aspergillosis. Mucosal Immunol. 3, 193–205.
Bonifazi, P., Zelante, T., D’Angelo, C., De Luca, A., Moretti, S., Bozza, S., Perruccio, K., Iannitti, R. G., Giovannini, G., Volpi, C., Fallarino, F., Puccetti, P., and Romani, L. (2009). Balancing inflammation and tolerance in vivo through dendritic cells by the commensal Candida albicans. Mucosal Immunol. 2, 362–374.
Bozza, S., Perruccio, K., Montagnoli, C., Gaziano, R., Bellocchio, S., Burchielli, E., Nkwanyuo, G., Pitzurra, L., Velardi, A., and Romani, L. (2003). A dendritic cell vaccine against invasive aspergillosis in allogeneic hematopoietic transplantation. Blood 102, 3807–3814.
Brandacher, G., Margreiter, R., and Fuchs, D. (2008). Clinical relevance of indoleamine 2,3-dioxygenase for alloimmunity and transplantation. Curr. Opin. Organ Transplant. 13, 10–15.
Bretz, C., Gersuk, G., Knoblaugh, S., Chaudhary, N., Randolph-Habecker, J., Hackman, R. C., Staab, J., and Marr, K. A. (2008). MyD88 signaling contributes to early pulmonary responses to Aspergillus fumigatus. Infect. Immun. 76, 952–958.
Carvalho, A., Cunha, C., Carotti, A., Aloisi, T., Guarrera, O., Di Ianni, M., Falzetti, F., Bistoni, F., Aversa, F., Pitzurra, L., Rodrigues, F., and Romani, L. (2009). Polymorphisms in Toll-like receptor genes and susceptibility to infections in allogeneic stem cell transplantation. Exp. Hematol. 37, 1022–1029.
Chen, L., Wang, T., Zhou, P., Ma, L., Yin, D., Shen, J., Molinero, L., Nozaki, T., Phillips, T., Uematsu, S., Akira, S., Wang, C. R., Fairchild, R. L., Alegre, M. L., and Chong, A. (2006). TLR engagement prevents transplantation tolerance. Am. J. Transplant. 6, 2282–2291.
Cobbold, S. P., Adams, E., Nolan, K. F., Regateiro, F. S., and Waldmann, H. (2010). Connecting the mechanisms of T-cell regulation: dendritic cells as the missing link. Immunol. Rev. 236, 203–218.
Cunha, C., Aversa, F., Bistoni, G., Casagrande, A., Rodrigues, F., Romani, L., and Carvalho, A. (2011a). Immunogenetic profiling to predict risk of invasive fungal diseases: where are we now? Immunol. Invest. 40, 723–734.
Cunha, C., Giovannini, G., Pierini, A., Bell, A. S., Sorci, G., Riuzzi, F., Donato, R., Rodrigues, F., Velardi, A., Aversa, F., Romani, L., and Carvalho, A. (2011b). Genetically-determined hyperfunction of the S100B/RAGE axis is a risk factor for aspergillosis in stem cell transplant recipients. PLoS ONE 6, e27962. doi:10.1371/journal.pone.0027962
Cunha, C., Di Ianni, M., Bozza, S., Giovannini, G., Zagarella, S., Zelante, T., D’Angelo, C., Pierini, A., Pitzurra, L., Falzetti, F., Carotti, A., Perruccio, K., Latge, J. P., Rodrigues, F., Velardi, A., Aversa, F., Romani, L., and Carvalho, A. (2010). Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 116, 5394–5402.
de Luca, A., Bozza, S., Zelante, T., Zagarella, S., D’Angelo, C., Perruccio, K., Vacca, C., Carvalho, A., Cunha, C., Aversa, F., and Romani, L. (2010). Non-hematopoietic cells contribute to protective tolerance to Aspergillus fumigatus via a TRIF pathway converging on IDO. Cell. Mol. Immunol. 7, 459–470.
De Luca, A., Montagnoli, C., Zelante, T., Bonifazi, P., Bozza, S., Moretti, S., D’Angelo, C., Vacca, C., Boon, L., Bistoni, F., Puccetti, P., Fallarino, F., and Romani, L. (2007). Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J. Immunol. 179, 5999–6008.
Delgoffe, G. M., Kole, T. P., Zheng, Y., Zarek, P. E., Matthews, K. L., Xiao, B., Worley, P. F., Kozma, S. C., and Powell, J. D. (2009). The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844.
Desvignes, L., and Ernst, J. D. (2009). Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31, 974–985.
Fugier-Vivier, I. J., Rezzoug, F., Huang, Y., Graul-Layman, A. J., Schanie, C. L., Xu, H., Chilton, P. M., and Ildstad, S. T. (2005). Plasmacytoid precursor dendritic cells facilitate allogeneic hematopoietic stem cell engraftment. J. Exp. Med. 201, 373–383.
Goldstein, A. L., and Badamchian, M. (2004). Thymosins: chemistry and biological properties in health and disease. Expert Opin. Biol. Ther. 4, 559–573.
Grow, W. B., Moreb, J. S., Roque, D., Manion, K., Leather, H., Reddy, V., Khan, S. A., Finiewicz, K. J., Nguyen, H., Clancy, C. J., Mehta, P. S., and Wingard, J. R. (2002). Late onset of invasive Aspergillus infection in bone marrow transplant patients at a university hospital. Bone Marrow Transplant. 29, 15–19.
Gupta, A. O., and Singh, N. (2011). Immune reconstitution syndrome and fungal infections. Curr. Opin. Infect. Dis. 24, 527–533.
Hainz, U., Jurgens, B., Wekerle, T., Seidel, M. G., and Heitger, A. (2007). Indoleamine 2,3-dioxygenase in hematopoietic stem cell transplantation. Curr. Drug Metab. 8, 267–272.
Holland, S. M., Deleo, F. R., Elloumi, H. Z., Hsu, A. P., Uzel, G., Brodsky, N., Freeman, A. F., Demidowich, A., Davis, J., Turner, M. L., Anderson, V. L., Darnell, D. N., Welch, P. A., Kuhns, D. B., Frucht, D. M., Malech, H. L., Gallin, J. I., Kobayashi, S. D., Whitney, A. R., Voyich, J. M., Musser, J. M., Woellner, C., Schaffer, A. A., Puck, J. M., and Grimbacher, B. (2007). STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 357, 1608–1619.
Jiang, S., and Lombardi, G. (2006). New trends in immunosuppression and immunotherapy. Int. Immunopharmacol. 6, 1874–1878.
Kabelitz, D., Wesch, D., and Oberg, H. H. (2006). Regulation of regulatory T cells: role of dendritic cells and toll-like receptors. Crit. Rev. Immunol. 26, 291–306.
Land, W. G. (2007). Innate immunity-mediated allograft rejection and strategies to prevent it. Transplant. Proc. 39, 667–672.
Larosa, D. F., Rahman, A. H., and Turka, L. A. (2007). The innate immune system in allograft rejection and tolerance. J. Immunol. 178, 7503–7509.
Marr, K. A., Carter, R. A., Boeckh, M., Martin, P., and Corey, L. (2002). Invasive aspergillosis in allogeneic stem cell transplant recipients: changes in epidemiology and risk factors. Blood 100, 4358–4366.
Martinic, M. M., and von Herrath, M. G. (2006). Control of graft-versus-host disease by regulatory T cells: which level of antigen specificity? Eur. J. Immunol. 36, 2299–2303.
Mason, D. (1998). A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 19, 395–404.
Matsuoka, K., Kim, H. T., Mcdonough, S., Bascug, G., Warshauer, B., Koreth, J., Cutler, C., Ho, V. T., Alyea, E. P., Antin, J. H., Soiffer, R. J., and Ritz, J. (2010). Altered regulatory T cell homeostasis in patients with CD4+ lymphopenia following allogeneic hematopoietic stem cell transplantation. J. Clin. Invest. 120, 1479–1493.
Medzhitov, R., Schneider, D. S., and Soares, M. P. (2012). Disease tolerance as a defense strategy. Science 335, 936–941.
Mellor, A. L., and Munn, D. H. (2008). Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat. Rev. Immunol. 8, 74–80.
Mezger, M., Einsele, H., and Loeffler, J. (2010). Genetic susceptibility to infections with Aspergillus fumigatus. Crit. Rev. Microbiol. 36, 168–177.
Montagnoli, C., Fallarino, F., Gaziano, R., Bozza, S., Bellocchio, S., Zelante, T., Kurup, W. P., Pitzurra, L., Puccetti, P., and Romani, L. (2006). Immunity and tolerance to Aspergillus involve functionally distinct regulatory T cells and tryptophan catabolism. J. Immunol. 176, 1712–1723.
Mori, S., Kocak, U., Shaw, J. L., and Mullen, C. A. (2005). Augmentation of post transplant immunity: antigen encounter at the time of hematopoietic stem cell transplantation enhances antigen-specific donor T-cell responses in the post transplant repertoire. Bone Marrow Transplant. 35, 793–801.
Mulanovich, V. E., and Kontoyiannis, D. P. (2011). Fungal pneumonia in patients with hematologic malignancies: current approach and management. Curr. Opin. Infect. Dis. 24, 323–332.
Ortega, M., Rovira, M., Filella, X., Martinez, J. A., Almela, M., Puig, J., Carreras, E., and Mensa, J. (2006). Prospective evaluation of procalcitonin in adults with non-neutropenic fever after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 37, 499–502.
Pallotta, M. T., Orabona, C., Volpi, C., Vacca, C., Belladonna, M. L., Bianchi, R., Servillo, G., Brunacci, C., Calvitti, M., Bicciato, S., Mazza, E. M., Boon, L., Grassi, F., Fioretti, M. C., Fallarino, F., Puccetti, P., and Grohmann, U. (2011). Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 12, 870–878.
Paveglio, S. A., Allard, J., Foster Hodgkins, S. R., Ather, J. L., Bevelander, M., Campbell, J. M., Whittaker Leclair, L. A., Mccarthy, S. M., Van Der Vliet, A., Suratt, B. T., Boyson, J. E., Uematsu, S., Akira, S., and Poynter, M. E. (2011). Airway epithelial indoleamine 2,3-dioxygenase inhibits CD4+ T cells during Aspergillus fumigatus antigen exposure. Am. J. Respir. Cell Mol. Biol. 44, 11–23.
Peter, C., Waldmann, H., and Cobbold, S. P. (2010). mTOR signalling and metabolic regulation of T cell differentiation. Curr. Opin. Immunol. 22, 655–661.
Puccetti, P., and Grohmann, U. (2007). IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat. Rev. Immunol. 7, 817–823.
Read, A. F., Graham, A. L., and Raberg, L. (2008). Animal defenses against infectious agents: is damage control more important than pathogen control. PLoS Biol. 6, e4. doi:10.1371/journal.pbio.1000004
Reddy, V., Iturraspe, J. A., Tzolas, A. C., Meier-Kriesche, H. U., Schold, J., and Wingard, J. R. (2004). Low dendritic cell count after allogeneic hematopoietic stem cell transplantation predicts relapse, death, and acute graft-versus-host disease. Blood 103, 4330–4335.
Restif, O., and Koella, J. C. (2004). Concurrent evolution of resistance and tolerance to pathogens. Am. Nat. 164, E90–E102.
Romani, L., Bistoni, F., Perruccio, K., Montagnoli, C., Gaziano, R., Bozza, S., Bonifazi, P., Bistoni, G., Rasi, G., Velardi, A., Fallarino, F., Garaci, E., and Puccetti, P. (2006). Thymosin alpha1 activates dendritic cell tryptophan catabolism and establishes a regulatory environment for balance of inflammation and tolerance. Blood 108, 2265–2274.
Romani, L., Fallarino, F., De Luca, A., Montagnoli, C., D’Angelo, C., Zelante, T., Vacca, C., Bistoni, F., Fioretti, M. C., Grohmann, U., Segal, B. H., and Puccetti, P. (2008a). Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 451, 211–215.
Romani, L., Zelante, T., De Luca, A., Fallarino, F., and Puccetti, P. (2008b). IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J. Immunol. 180, 5157–5162.
Romani, L., and Puccetti, P. (2007). Controlling pathogenic inflammation to fungi. Expert Rev. Anti. Infect. Ther. 5, 1007–1017.
Rosentul, D. C., Plantinga, T. S., Oosting, M., Scott, W. K., Velez Edwards, D. R., Smith, P. B., Alexander, B. D., Yang, J. C., Laird, G. M., Joosten, L. A., Van Der Meer, J. W., Perfect, J. R., Kullberg, B. J., Netea, M. G., and Johnson, M. D. (2011). Genetic variation in the dectin-1/CARD9 recognition pathway and susceptibility to candidemia. J. Infect. Dis. 204, 1138–1145.
Saemann, M. D., Haidinger, M., Hecking, M., Horl, W. H., and Weichhart, T. (2009). The multifunctional role of mTOR in innate immunity: implications for transplant immunity. Am. J. Transplant. 9, 2655–2661.
Santamaria, R., Rizzetto, L., Bromley, M., Zelante, T., Lee, W., Cavalieri, D., Romani, L., Miller, B., Gut, I., Santos, M., Pierre, P., Bowyer, P., and Kapushesky, M. (2011). Systems biology of infectious diseases: a focus on fungal infections. Immunobiology 216, 1212–1227.
Saraiva, M., and O’Garra, A. (2010). The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 10, 170–181.
Schneider, D. S., and Ayres, J. S. (2008). Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 8, 889–895.
Simitsopoulou, M., Roilides, E., and Walsh, T. J. (2011). Immunomodulatory properties of antifungal agents on phagocytic cells. Immunol. Invest. 40, 809–824.
Singh, N., and Perfect, J. R. (2007). Immune reconstitution syndrome associated with opportunistic mycoses. Lancet Infect. Dis. 7, 395–401.
Sorci, G., Giovannini, G., Riuzzi, F., Bonifazi, P., Zelante, T., Zagarella, S., Bistoni, F., Donato, R., and Romani, L. (2011). The danger signal S100B integrates pathogen- and danger-sensing pathways to restrain inflammation. PLoS Pathog. 7, e1001315. doi:10.1371/journal.ppat.1001315
Turnquist, H. R., and Thomson, A. W. (2008). Taming the lions: manipulating dendritic cells for use as negative cellular vaccines in organ transplantation. Curr. Opin. Organ Transplant. 13, 350–357.
Waldmann, H., and Cobbold, S. (2004). Exploiting tolerance processes in transplantation. Science 305, 209–212.
Werner, J. L., Metz, A. E., Horn, D., Schoeb, T. R., Hewitt, M. M., Schwiebert, L. M., Faro-Trindade, I., Brown, G. D., and Steele, C. (2009). Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J. Immunol. 182, 4938–4946.
Zelante, T., De Luca, A., Bonifazi, P., Montagnoli, C., Bozza, S., Moretti, S., Belladonna, M. L., Vacca, C., Conte, C., Mosci, P., Bistoni, F., Puccetti, P., Kastelein, R. A., Kopf, M., and Romani, L. (2007). IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur. J. Immunol. 37, 2695–2706.
Zelante, T., Iannitti, R. G., De Luca, A., Arroyo, J., Blanco, N., Servillo, G., Sanglard, D., Reichard, U., Palmer, G. E., Latge, J. P., Puccetti, P., and Romani, L. (2012). Sensing of mammalian IL-17A regulates fungal adaptation and virulence. Nat. Commun. 3, 683.
Keywords: fungal infections, resistance, tolerance, IDO, single nucleotide polymorphism
Citation: Carvalho A, Cunha C, Bozza S, Moretti S, Massi-Benedetti C, Bistoni F, Aversa F and Romani L (2012) Immunity and tolerance to fungi in hematopoietic transplantation: principles and perspectives. Front. Immun. 3:156. doi: 10.3389/fimmu.2012.00156
Received: 03 May 2012; Paper pending published: 23 May 2012;
Accepted: 25 May 2012; Published online: 13 June 2012.
Edited by:
Stephen Paul Cobbold, University of Oxford, UKReviewed by:
Yi-Chi Kong, Wayne State University School of Medicine, USABin Li, Chinese Academy of Sciences, China
Copyright: © 2012 Carvalho, Cunha, Bozza, Moretti, Massi-Benedetti, Bistoni, Aversa and Romani. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Luigina Romani, Department of Experimental Medicine and Biochemical Sciences, University of Perugia, Via del Giochetto, 06122 Perugia, Italy. e-mail:bHJvbWFuaUB1bmlwZy5pdA==