 Sophie Borel
Sophie Borel

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 07 May 2012
Sec. Antigen Presenting Cell Biology
Volume 3 - 2012 | https://doi.org/10.3389/fimmu.2012.00097
This article is part of the Research Topic Eat Thyself, Heal Thyself: Autophagy in pathogen recognition and antigen processing View all 13 articles
Autophagy is an intracellular mechanism whereby pathogens, particularly viruses, are destroyed in autolysosomes after their entry into targets cells. Therefore, to survive and replicate in host cells, viruses have developed multiple strategies to either counteract or exploit this process. The aim of this review is to outline the known relationships between HIV-1 and autophagy in CD4+ T lymphocytes and macrophages, two main HIV-1 cell targets. The differential regulation of autophagy in these two cell-types is highlighted and its potential consequences in terms of viral replication and physiopathology discussed.
Although progress has been made in the global fight against HIV/AIDS, more than 33 million people are still living with HIV worldwide (data from UNAIDS, 2009). HIV infection is characterized by an acute phase with very high levels of circulating viruses and a rapid decline in CD4+ T cells. Despite a strong immune response, the host cannot clear the infection and during the ensuing long, clinically latent phase, CD4+ T cells are progressively lost, eventually leading to AIDS in untreated patients. Long-lived, latent HIV reservoirs, established early during infection, prevent complete virus eradication although the anti-viral therapy effectively reduces the plasma HIV levels below detection limits (Chang and Altfeld, 2010; Mogensen et al., 2010). As a consequence, the fight against HIV is very difficult and a better understanding of the relationship between HIV and the human innate and adaptive immune systems is needed for the discovery of new drugs. Indeed, at present, drugs block the virus life cycle at different steps (entry, reverse transcription, integration, maturation) but do not act on the host response, which is responsible for the establishment of the reservoirs.
Macroautophagy (hereafter referred to as autophagy) is a physiologically controlled intracellular degradation mechanism that plays a role in both innate and adaptive immunity. Cytoplasmic pathogens can be degraded by autophagy, a process that has been termed xenophagy (Deretic and Levine, 2009). Moreover, autophagy participates in antigen processing for their presentation via the major histocompatibility complex (MHC) class I and II proteins (Schmid and Munz, 2007; English et al., 2009; Lunemann and Munz, 2009). As an evolutionary counterpoint, certain pathogens, including HIV-1, can inhibit or subvert autophagy to replicate more efficiently (Espert et al., 2007; Levine and Deretic, 2007).
HIV-1 can infect different types of immune cells that express CD4 and a coreceptor, mainly CCR5 or CXCR4 [i.e., CD4+ T lymphocytes, macrophages, monocytes, and dendritic cells (DCs)], but replicates efficiently only in CD4+ T cells and macrophages, indicating that several aspects of the virus–host relationship are different in these target cell populations. Although both CD4+ T cells and macrophages are infected by HIV-1 and produce large amounts of virions, many differences have been identified at each step of the HIV-1 life cycle in these target cells (Table 1). The ultimate difference of the complex relationship between host cell and HIV-1 is the death of CD4+ T cells by apoptosis and the resistance of macrophages to HIV-1 cytopathic effects (Carter and Ehrlich, 2008). This review summarizes the most recent information on the regulation and the role of autophagy during HIV-1 infection of CD4+ T cells and macrophages and proposes that the differential regulation of autophagy in these target cells might contribute to HIV-1 physiopathology.
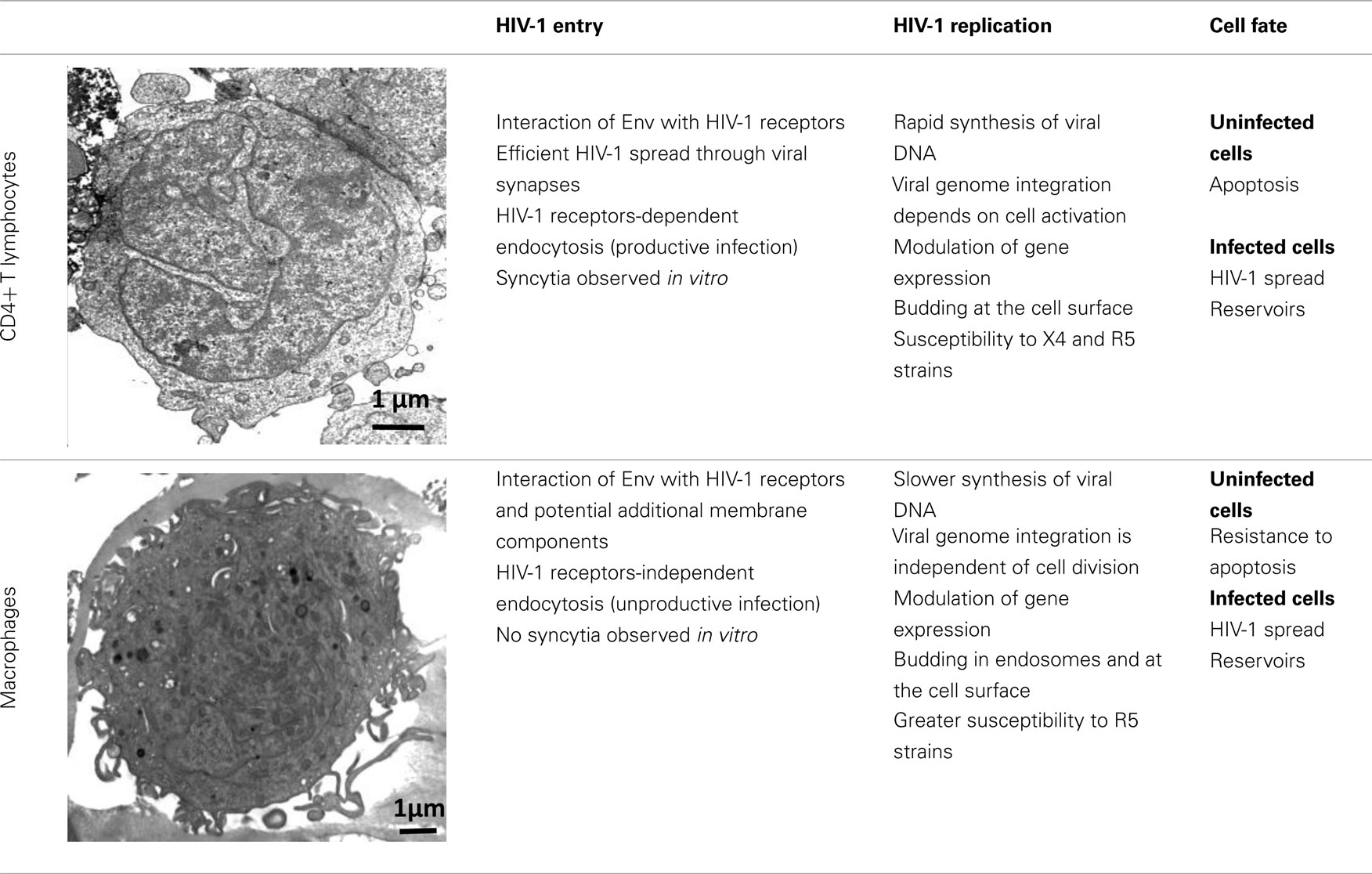
Table 1. Major differences between CD4+ T lymphocytes and macrophages during HIV-1 infection.
HIV-1 envelope (Env), composed of the glycoproteins gp120 and gp41, plays a crucial role in virus entry. In most instances, to enter a target cell, HIV-1 Env gp120, which is expressed at the surface of free virions and infected cells, must bind to CD4. The interaction between gp120 and CD4 triggers conformational changes leading to increased exposure of gp120 regions (including the V3 loop) that can bind to CCR5 or CXCR4 (Pierson and Doms, 2003). Finally, interaction of gp120 with the coreceptor induces a structural rearrangement of the transmembrane Env subunit gp41 and insertion of the fusion domain at the N-terminus of gp41 into the target cell membrane. At this stage, gp41 adopts a trimeric extended pre-hairpin intermediate conformation before the formation of a stable six-helix bundle structure and virus/cell-to-cell fusion.
Infection by cell-free viruses or through contact between infected and uninfected cells leads to HIV-1 spread (Sattentau, 2008), but the efficiency of HIV-1 infection is higher when the virus is delivered through cell-to-cell contacts. DCs and macrophages, which are among the first cells encountering HIV-1, efficiently transmit HIV-1 to CD4+ T cells through virological synapses (McDonald et al., 2003; Arrighi et al., 2004; Groot et al., 2008; Felts et al., 2010). This way of spread also occurs between HIV-1-infected and uninfected CD4+ T cells (Jolly et al., 2004; Rudnicka et al., 2009). In vitro, cell-to-cell fusion leads to the formation of giant, multinucleated cells called syncytia.
Coreceptor (CCR5 or CXCR4) use is correlated, at least in part, with the different phases of the disease. R5 viruses, which utilize CCR5, are predominantly isolated during the early stages of HIV-1 infection. The emergence in a patient of X4 variants, which use CXCR4, is almost invariably associated with faster decline of circulating CD4+ T cells, accelerated disease progression and poor prognosis for survival (Koot et al., 1993; Richman and Bozzette, 1994). However, the presence of X4 viruses is not an obligatory prerequisite for disease progression and a significant proportion of individuals who progress to AIDS harbor exclusively R5 variants. The selective transmission of R5 viruses is not fully understood, but it may depend on the superimposition of multiple imperfect gatekeepers that restrict HIV-1 X4 transmission at different steps of the infection process (Margolis and Shattock, 2006).
Only about 15–30% of CD4+ T lymphocytes express detectable levels of CCR5 at the cell surface, whereas CXCR4 is expressed on nearly all of these T cells (Bleul et al., 1997; Grivel and Margolis, 1999). High levels of CCR5 are observed in activated/memory CD4+ T cells, which are the CD4+ T cells that can be productively infected. In addition, the first CD4+ T cells to undergo intense HIV-1 replication after infection are the resting memory CD4+ T cells present in the gut-associated lymphoid tissue (GALT), which also express CCR5 (Li et al., 2005).
Macrophages are also infected by X4 and R5 strains through its receptors, CD4, CXCR4, and CCR5. However, they are more frequently infected by HIV-1 R5 strains. This phenomenon is not fully understood, but one explanation is that R5 strains can exploit low levels of CD4 and/or CCR5 to enter macrophages (Peters et al., 2004, 2006).
Besides this route of entry, endocytosis of HIV-1 has been described, especially in macrophages. Most virions are subsequently degraded, but productive infection may nevertheless occur through this CD4-independent mechanism (Marechal et al., 2001). Entry by endocytosis has also been described in CD4+ T cells, but in such cells it requires the presence of CD4 and leads to productive infection (Pauza and Price, 1988). Recently, Miyauchi et al. (2009) have demonstrated that HIV-1 likely enters lymphoid cells via an endocytic pathway, and that fusion between Env and its receptors occur in intracellular compartments.
Additional membrane components may support HIV-1 entry in macrophages, including syndecan, a heparan sulfate proteoglycan (Saphire et al., 2001; de Parseval et al., 2005); gp340, a cysteine-rich scavenger receptor (Wu et al., 2004); the macrophage mannose receptor (Larkin et al., 1989; Chehimi et al., 2003; Nguyen and Hildreth, 2003); elastase (Bristow et al., 2003); and α-v-integrin (Bosch et al., 2006). These constituents may facilitate virus attachment, binding, entry and/or fusion. Another potential membrane ligand of HIV-1 is annexin II, which is expressed on the membrane of macrophages, but not of T cells. Annexin II, which binds to phosphatidyl serine (PS), an anionic phospholipid captured during HIV-1 budding, contributes to the early events of macrophage HIV-1 infection (Ma et al., 2004). Other candidate host cell surface proteins that are incorporated in HIV-1 membranes and are potentially needed for HIV-1 entry might include CD28, CD44, and CD62L (Herzberg et al., 2006).
After reaching the cytoplasm of the infected cell, HIV-1 reverse transcription takes place in the reverse transcription complex (RTC) that is constituted by virion core proteins, cellular proteins, and the RNA genome (McDonald et al., 2002; Nermut and Fassati, 2003). Double-stranded DNA is synthesized within a few hours in CD4+ T cells, whereas this process is slower in macrophages (Collin and Gordon, 1994; O’Brien et al., 1994). The RTC moves along the microtubule network to reach the microtubule organizing center (MTOC) and the nucleus. The viral DNA is then imported into the nucleus and integrated in the cell genome through a process that requires mitosis in CD4+ T cells but not in macrophages. This suggests that cellular rather than viral components of the preintegration complex (PIC), in which the viral DNA is associated with many proteins, may play a major role in viral nuclear import and integration in macrophages (Yamashita and Emerman, 2005). After integration of the viral genome, most of the assembling HIV-1 particles localize at the plasma membrane in infected CD4+ T cells, whereas in macrophages viral particles are mostly concentrated in apparently internal compartments that possess the characteristics of late endosomes/multivesicular bodies (LEs/MVBs). However, a fraction of the intracellular virus-containing compartments in macrophages could be large and complex invaginations of the plasma membrane (Deneka et al., 2007; Jouve et al., 2007; Welsch et al., 2007).
Several microarray studies also indicate that HIV-1 infection leads to changes in host gene expression, depending on the target cell-type. Specifically, modulation of genes associated with the host defense, signal transduction, cell cycle transcription, and arrest occurs predominantly in HIV-1-infected macrophages (Coberley et al., 2004; Vazquez et al., 2005). This differential modulation of gene expression contributes to the greater survival of macrophages in comparison to CD4+ T cells following HIV-1 infection (Giri et al., 2006). Transcription of the HIV-1 DNA is also a highly regulated process that exploits the specific environment of the cell host and involves multiple interplays between cell and viral factors (Rohr et al., 2003).
Acute HIV-1 infection leads to a dramatic decrease in the number of CD4+ T cells, which then return to normal level in the majority of patients. Afterward, the asymptomatic phase is characterized by a progressive and continuous decline in the level of circulating CD4+ T cells. In contrast, macrophages are more resistant to HIV-1 cytopathic effects and their number is stable over years, although several macrophage functions are affected by HIV-1 infection (Biggs et al., 1995; Yoo et al., 1996; Polyak et al., 1997; Kumar et al., 1999). CD4+ T lymphocyte depletion is due to continued and accelerated apoptosis, probably triggered by multifactorial mechanisms (Espert et al., 2006; Cummins and Badley, 2010). HIV-1-induced apoptosis in bystander, uninfected immune cells is likely to be a key factor in the gradual depletion of T lymphocytes observed in HIV-1-infected patients, since the degree of cell loss largely exceeds the number of infected cells. Furthermore, the vast majority of CD4+ T cells undergoing apoptosis in peripheral blood and lymph nodes of HIV-1 patients are uninfected (Krammer et al., 1994; Finkel et al., 1995; Doitsh et al., 2010).
Among the HIV-1 proteins known to induce apoptosis of CD4+ T cells, cumulative data have demonstrated a major role of Env in apoptosis of uninfected, bystander lymphocytes (Laurent-Crawford et al., 1993; Heinkelein et al., 1995; Ohnimus et al., 1997; Blanco et al., 1999, 2000; Roggero et al., 2001; Roshal et al., 2001; Ahr et al., 2004). Indeed, binding of Env to its receptors constitutes the primary interface between HIV-1 and its target cells, and both HIV-1 X4 and R5 Env can induce CD4+ T cell death (Joshi et al., 2011). Although interaction of gp120 with CD4 and the coreceptor is required for apoptosis induction, Env-mediated apoptosis of target CD4+ T cells is induced through the fusogenic function of gp41. Interestingly, hemifusion, an intermediate step in the fusion process characterized by the merger of the outer membrane leaflets of two biological membranes without the formation of a fusion pore or mixing of the inner leaflets, is sufficient to trigger Env-mediated apoptosis of bystander CD4+ T cells (Garg and Blumenthal, 2006, 2008; Garg et al., 2007).
Many fundamental questions remain unsolved, such as how host factors can influence the susceptibility to HIV-1 infection and resistance to death and what are the mechanisms leading to the establishment and maintenance of HIV-1 latency. This latter point is of major importance because the latent viral reservoirs, which are constituted mainly of resting CD4+ T cells (Chun et al., 1995, 1997; Finzi et al., 1997; Wong et al., 1997; Siliciano et al., 2003) and cells of the monocyte–macrophage lineage (Folks et al., 1988; Biswas et al., 1992, 1994; Coleman and Wu, 2009), are unaffected by highly active anti-retroviral therapies (HAART; Finzi et al., 1999) and can reinitiate systemic infection upon interruption of HAART, or following the development of resistance (Marcello, 2006; Suyama et al., 2009).
Autophagy is a highly regulated mechanism that involves specific genes called Atg (autophagy-related genes) and has an essential role in cell homeostasis. Accordingly, autophagy has been implicated in several pathologies, including cancer, neurodegeneration, and myopathies (Marino and Lopez-Otin, 2004). Autophagy has also a specialized function in the innate immune response against intracellular pathogens through their degradation in autolysosomes (Deretic, 2006). Its implication in antigen presentation by MHC class I and II molecules extends its function to adaptive immunity as well (Nimmerjahn et al., 2003; Dengjel et al., 2005; Schmid et al., 2006). Besides its role as an intracellular host defense mechanism against viruses, autophagy can also be used by the virus for its own profit to replicate more efficiently in cells, or to control cell survival (Espert et al., 2007; Deretic and Levine, 2009; Espert and Biard-Piechaczyk, 2009; Lunemann and Munz, 2009; Lin et al., 2010; Sumpter and Levine, 2010). Data from a genome-wide RNAi screen (Brass et al., 2008) and silencing of 30 candidate cofactors (Eekels et al., 2011) indicate that HIV-1 replication in cells requires the presence of several Atg (Atg7, GABARAPL2, Atg12, and Atg16L). A very recent study underlines the role of several Atgs, in particular Atg5 and Atg16, in HIV-1 replication in CD4+ T cells (Eekels et al., 2012).
Induction of autophagy and its regulation during viral infection have different biological consequences on pathogen degradation and on the innate and adaptive immune responses, depending on both the pathogen and the host cell. The differential regulation of autophagy by HIV-1 in CD4+ T cells and macrophages (presented below) is only the tip of the iceberg, but demonstrates the importance of this process during HIV-1 infection, although many issues are not fully understood yet.
Autophagy of uninfected CD4+ T cells, when co-cultured with lymphocytes that express Env, was described for the first time in 2006 (Espert et al., 2006). It is triggered by binding of Env to its receptors CD4 and CXCR4, but is independent of CD4 or CXCR4 signaling pathways. Autophagy can be induced in single cells (hemifusion) and syncytia (complete fusion) and depends on the fusogenic function of gp41. These results indicate that the entire process leading to HIV-1 entry into target cells through binding of Env to its receptors CD4 and CXCR4 is responsible for autophagy in CD4+ T lymphocytes (Denizot et al., 2008). As autophagy is gp41-dependent, it can be induced whatever the coreceptor used for HIV-1 entry (CCR5 or CXCR4; Espert et al., 2009). The mechanisms by which gp41 induces autophagy are unknown, but it could be a consequence of the cellular stress induced after hemifusion of the membranes. A proteomics study has already demonstrated that rapid and sustained accumulation of ROS is induced in CD4+ T cells after contact with Env-expressing cells (Molina et al., 2007), and the production of ROS is known to be directly involved in autophagy (Scherz-Shouval et al., 2007).
Importantly, Env-mediated autophagy is required to trigger CD4+ T cell apoptosis. Indeed, blockade of autophagy at different steps by drugs (3-methyladenine or Bafilomycin A1) or short interfering RNAs against Beclin 1 and Atg7 fully inhibits Env-mediated apoptosis. At present, nothing is known about the mechanisms that link autophagy to apoptosis, but Beclin 1 might play a key role since this Atg also binds to anti-apoptotic Bcl-2 proteins (Bcl-2 and Bcl-xL). Interaction of Beclin 1 with Bcl-2 or Bcl-xL inhibits Beclin 1-dependent autophagy, demonstrating a direct crosstalk between the core machineries regulating autophagy and apoptosis (Pattingre et al., 2005).
Interestingly, Beclin 1 is accumulated during the early steps of the Env-induced signaling cascade, and this phenomenon precedes autophagic vacuolization. Accumulation of Beclin 1 has also been reported in response to drugs that trigger cell death with autophagic features (Scarlatti et al., 2004; Shimizu et al., 2004; Furuya et al., 2005). Deciphering the molecular mechanism of Beclin 1 accumulation will be a crucial step for understanding the role of autophagy in HIV-1 pathogenicity.
Conversely, when cells that express HIV-1 R5 or X4 Env are co-cultured with cells from the monocyte/macrophage lineage, autophagy is not observed in uninfected cells, although they are susceptible to autophagy induced by different agents. Env-mediated autophagy is thus a cell-type dependent process. The state of differentiation is not responsible for their intrinsic resistance to Env-mediated autophagy because cells at different stage of differentiation (from promonocytic cells to monocyte-differentiated macrophages) are all equally resistant. Macrophages also do not undergo Env-mediated apoptosis and they are resistant to cell depletion during HIV-1 infection.
These major differences between CD4+ T cells and macrophages raise unsolved questions: what are the mechanisms that lead to Env-mediated autophagy, how autophagy is regulated in the different cell-types, and how autophagy triggers apoptosis in CD4+ T lymphocytes.
The fact that the fusogenic function of gp41 induces Env-mediated autophagy only in CD4+ T cells is surprising since fusion also occurs in uninfected macrophages co-cultured with HIV-1-infected cells. An important and still unexplained difference between these two target cells is the ability to form syncytia. This has been observed since the discovery of the disease and HIV strains were first classified based on this observation. Indeed “syncytia-inducing” viruses correspond to HIV-1 strains capable of infecting CD4+ T cells through CXCR4, while “non-syncytia-inducing” viruses are HIV-1 strains that infect macrophages through CCR5 (Goodenow and Collman, 2006). One hypothesis to explain why Env-mediated autophagy is a cell-specific mechanism is that the gp41-induced perturbations, triggered at the membrane of both macrophages and CD4+ T cells, could be different in the two cell-types or differentially regulated. Indeed, HIV-1 entry in macrophages is supported by additional specific interactions with host membrane molecules following Env binding to the receptor/coreceptor (see HIV-1 Entry in CD4+ T Cells and Macrophages). HIV-1 can also enter macrophages by endocytosis. Furthermore, recent data have demonstrated that ceramides, which are known inducers of autophagy (Pattingre et al., 2009), play an important role in the reorganization of membrane proteins (Chiantia et al., 2008) that is necessary for HIV-1 entry into cells. Another hypothesis is that gp120 binding to CD4 and to the coreceptor, two steps that precede gp41 insertion into the target membrane, transduces signals that counteract Env-mediated autophagy in a cell-type dependent manner. We also cannot exclude the involvement of secreted chemokines and/or cytokines in the regulation of gp41-induced autophagy. Further investigation is needed to elucidate this point.
HIV-1 X4 or R5 Env expression at the cell surface (Env-transfected or HIV-1-infected cells) triggers autophagy in uninfected CD4+ T cells, leading to apoptosis. Surprisingly, when this contact leads to a productive infection, Env-mediated autophagy is inhibited in CD4+ T cells (Zhou and Spector, 2008; Espert et al., 2009). Moreover, the levels of the autophagy factor LC3-II (Zhou and Spector, 2008; Espert et al., 2009) and of Beclin 1 (Zhou and Spector, 2008) are dramatically decreased in these newly infected CD4+ T cells, suggesting that HIV-1 can actively down-regulate autophagy. Interestingly, when autophagy is triggered by different inducers in HIV-1-infected CD4+ T cells, production of HIV-1 is decreased, suggesting that autophagy can limit HIV-1 replication (unpublished data from our group).
Thus, HIV-1, as many viruses, can counteract the anti-viral function of autophagy, but the mechanism(s) by which HIV-1 can interfere with this essential cellular pathway is (are) still unknown. One or several viral proteins might block autophagy to avoid HIV-1 destruction.
Conversely, in cells from the monocyte/macrophage lineage, autophagy is induced following productive infection through contact with HIV-1 X4 or R5-infected effector cells (Espert et al., 2009). However, the observation that viruses are present only in moderately autophagic cells (Espert et al., 2009), suggests that autophagy is still controlled by HIV-1 in these cells to avoid degradation. Interestingly, early, non-degradative steps of autophagy promote HIV-1 production since blockade of this process dramatically decreases the quantity of p24 Gag (Espert et al., 2009; Kyei et al., 2009). In addition, the HIV-1 precursor Gag is found in complexes with LC3 and is present at LC3-II-enriched membranes, suggesting that autophagy could favor Gag processing and thus production of viral particles (Kyei et al., 2009). In contrast, the degradative step of autophagy has an anti-HIV-1 activity that must be controlled by the virus to prevent its degradation. Indeed, blockage of the degradative step of autophagy increases HIV-1 production (Espert et al., 2009; Kyei et al., 2009). Interestingly, the auxiliary HIV-1 protein Nef plays a major role in the inhibition of the degradative stage of autophagy by binding to Beclin 1 (Kyei et al., 2009). Nef also interacts with immunity-associated GTPase family M (IRGM), a protein known to play an autophagy-dependent anti-bacterial function (Deretic et al., 2006; McCarroll et al., 2008; Deretic, 2010a; Singh et al., 2010) and to bind to several key proteins of the autophagy process such as Atg5 and Atg10 (Pombo-Grégoire et al., 2011). Nef/IRGM interaction promotes autophagosome accumulation and improves HIV-1 replication (Pombo-Grégoire et al., 2011). In contrast, its absence is detrimental for the viral production. IRGM also triggers autophagy in cells infected by other RNA viruses, such as hepatitis C virus (HCV) and measles virus (MeV), suggesting that different RNA virus families use similar strategies, involving IRGM, to fine-tune autophagy to their own benefit.
Taken together, all these results (summarized in Table 2) suggest a complex, cell-type specific relationship between HIV-1 and the autophagic response and highlight the complexity of HIV-1 pathogenesis.
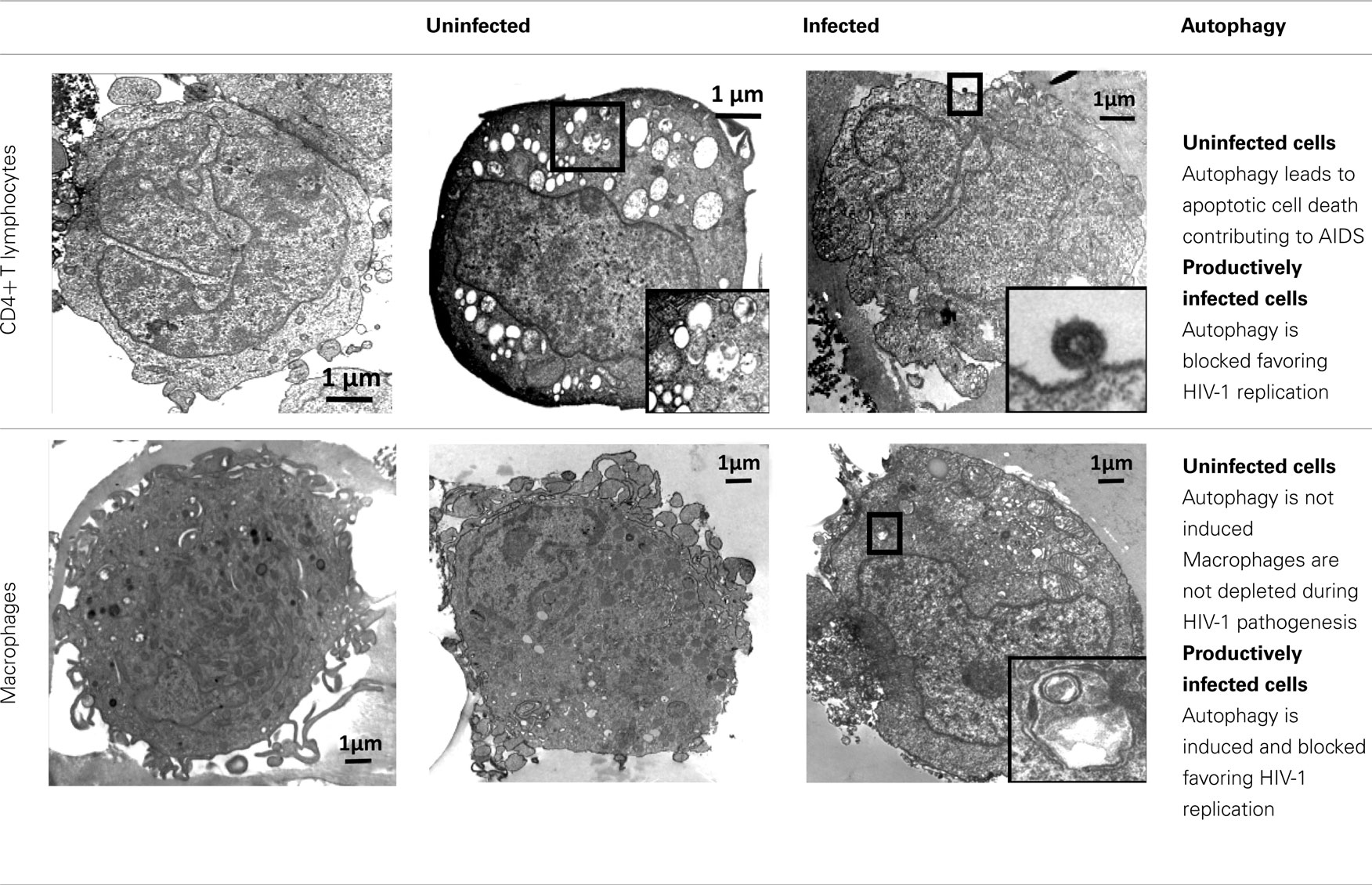
Table 2. Regulation of autophagy in CD4+ T lymphocytes and macrophages during HIV-1 infection.
Although autophagy is now acknowledged to have a major role in HIV-1 infection, the available data are still quite fragmentary and more research work is needed to clarify the contribution of autophagy to viral replication, host immune responses, and viral pathogenesis. The available data indicate that autophagy is an anti-viral process and as such is inhibited by HIV-1 to avoid degradation and to promote replication. However, autophagy is also used by HIV-1 for its replication, especially in macrophages where autophagy is present all along the productive infection period.
Based on the current knowledge on the role of autophagy in different viral infections, several hypotheses can be proposed on how HIV-1 uses autophagy for its own profit in macrophages: (i) autophagy could be required to transport the RTC to the MTOC using the microtubule network. Indeed, the RTC and autophagosomes that are formed in the cytoplasm use the microtubule network to traffic toward the MTOC to reach the nucleus and to fuse with late endosomes or lysosomes, respectively (Afonso et al., 2007; Jahreiss et al., 2008; Orsi et al., 2009; Geeraert et al., 2010); (ii) autophagy could be a mechanism for membrane remodeling to support viral replication and assembly, as already described for other viruses (Miller and Krijnse-Locker, 2008). The fact that HIV-1 Gag-derived proteins interact with LC3 suggests that autophagy plays a role in promoting certain steps of HIV biogenesis (Kyei et al., 2009); (iii) autophagy could be used to specifically degrade host cell proteins that are important for the defense against HIV-1, in association with the ubiquitin–proteasome system through adapter proteins, such as p62/SQSTM1 and NBR1 (Kirkin et al., 2009; Lamark et al., 2009; Deretic, 2010b); (iv) low levels of autophagy may promote the formation of latent cell reservoirs; and (v) autophagy could process antigen determinants for their presentation by the MHC, and could hide HIV-1 from recognition by the immune system. This point is discussed in other chapters of this issue.
In addition, autophagy is a cell fate-determining process that triggers apoptosis of bystander CD4+ T cells that cannot be productively infected by HIV-1. Inhibition of autophagy in productively infected CD4+ T cells may thus both prevent HIV-1 degradation and maintain cell viability long enough to allow efficient viral replication. Autophagy has been defined as a type II programmed cell death, based on the morphology of the dying cells. However, Env-mediated autophagy is not a cell death mechanism by itself as it leads to apoptosis by a yet unknown mechanism. It is important to note that autophagy deregulation may also play a role in a variety of diseases that are related directly and/or indirectly to HIV infection, including cancer, dementia, and premature aging.
To the best of our knowledge, HIV-1 infection of CD4+ T cells and macrophages is the first example of a viral infection in which autophagy governs both viral replication and the fate of uninfected cells. The data also strongly suggest that autophagy is responsible, at least in part, for HIV-1 pathogenesis, providing new insights into therapeutic strategies for the future.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work was supported by institutional funds from the Centre National de la Recherche Scientifique (CNRS) and the Montpellier I and II Universities, and grants from SIDACTION.
Afonso, P. V., Zamborlini, A., Saib, A., and Mahieux, R. (2007). Centrosome and retroviruses: the dangerous liaisons. Retrovirology 4, 27.
Ahr, B., Robert-Hebmann, V., Devaux, C., and Biard-Piechaczyk, M. (2004). Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology 1, 12.
Arrighi, J. F., Pion, M., Garcia, E., Escola, J. M., van Kooyk, Y., Geijtenbeek, T. B., and Piguet, V. (2004). DC-SIGN-mediated infectious synapse formation enhances X4 HIV-1 transmission from dendritic cells to T cells. J. Exp. Med. 200, 1279–1288.
Biggs, B. A., Hewish, M., Kent, S., Hayes, K., and Crowe, S. M. (1995). HIV-1 infection of human macrophages impairs phagocytosis and killing of Toxoplasma gondii. J. Immunol. 154, 6132–6139.
Biswas, P., Poli, G., Kinter, A. L., Justement, J. S., Stanley, S. K., Maury, W. J., Bressler, P., Orenstein, J. M., and Fauci, A. S. (1992). Interferon gamma induces the expression of human immunodeficiency virus in persistently infected promonocytic cells (U1) and redirects the production of virions to intracytoplasmic vacuoles in phorbol myristate acetate-differentiated U1 cells. J. Exp. Med. 176, 739–750.
Biswas, P., Poli, G., Orenstein, J. M., and Fauci, A. S. (1994). Cytokine-mediated induction of human immunodeficiency virus (HIV) expression and cell death in chronically infected U1 cells: do tumor necrosis factor alpha and gamma interferon selectively kill HIV-infected cells? J. Virol. 68, 2598–2604.
Blanco, J., Barretina, J., Henson, G., Bridger, G., De Clercq, E., Clotet, B., and Este, J. A. (2000). The CXCR4 antagonist AMD3100 efficiently inhibits cell-surface- expressed human immunodeficiency virus type 1 envelope-induced apoptosis. Antimicrob. Agents Chemother. 44, 51–56.
Blanco, J., Jacotot, E., Cabrera, C., Cardona, A., Clotet, B., DeClercq, E., and Esté, J. A. (1999). The implication of the chemokine receptor CXCR4 in HIV-1 envelope protein-induced apoptosis is independent of the G protein-mediated signalling. AIDS 13, 909–917.
Bleul, C., Wu, L. H., Hoxie, J. A., Springer, T. A., and MacKay, C. R. (1997). The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 94, 1925–1930.
Bosch, B., Clotet-Codina, I., Blanco, J., Pauls, E., Coma, G., Cedeno, S., Mitjans, F., Llano, A., Bofill, M., Clotet, B., Piulats, J., and Esté, J. A. (2006). Inhibition of human immunodeficiency virus type 1 infection in macrophages by an alpha-v integrin blocking antibody. Antiviral Res. 69, 173–180.
Brass, A. L., Dykxhoorn, D. M., Benita, Y., Yan, N., Engelman, A., Xavier, R. J., Lieberman, J., and Elledge, S. J. (2008). Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926.
Bristow, C. L., Mercatante, D. R., and Kole, R. (2003). HIV-1 preferentially binds receptors copatched with cell-surface elastase. Blood 102, 4479–4486.
Carter, C. A., and Ehrlich, L. S. (2008). Cell biology of HIV-1 infection of macrophages. Annu. Rev. Microbiol. 62, 425–443.
Chang, J. J., and Altfeld, M. (2010). Innate immune activation in primary HIV-1 infection. J. Infect. Dis. 202(Suppl. 2), S297–S301.
Chehimi, J., Luo, Q., Azzoni, L., Shawver, L., Ngoubilly, N., June, R., Jerandi, G., Farabaugh, M., and Montaner, L. J. (2003). HIV-1 transmission and cytokine-induced expression of DC-SIGN in human monocyte-derived macrophages. J. Leukoc. Biol. 74, 757–763.
Chiantia, S., Ries, J., Chwastek, G., Carrer, D., Li, Z., Bittman, R., and Schwille, P. (2008). Role of ceramide in membrane protein organization investigated by combined AFM and FCS. Biochim. Biophys. Acta 1778, 1356–1364.
Chun, T. W., Finzi, D., Margolick, J., Chadwick, K., Schwartz, D., and Siliciano, R. F. (1995). In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1, 1284–1290.
Chun, T. W., Stuyver, L., Mizell, S. B., Ehler, L. A., Mican, J. A., Baseler, M., Lloyd, A. L., Nowak, M. A., and Fauci, A. S. (1997). Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U.S.A. 94, 13193–13197.
Coberley, C. R., Kohler, J. J., Brown, J. N., Oshier, J. T., Baker, H. V., Popp, M. P., Sleasman, J. W., and Goodenow, M. M. (2004). Impact on genetic networks in human macrophages by a CCR5 strain of human immunodeficiency virus type 1. J. Virol. 78, 11477–11486.
Coleman, C. M., and Wu, L. (2009). HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology 6, 51.
Collin, M., and Gordon, S. (1994). The kinetics of human immunodeficiency virus reverse transcription are slower in primary human macrophages than in a lymphoid cell line. Virology 200, 114–120.
Cummins, N. W., and Badley, A. D. (2010). Mechanisms of HIV-associated lymphocyte apoptosis. Cell Death Dis. 1, e99.
de Parseval, A., Bobardt, M. D., Chatterji, A., Chatterji, U., Elder, J. H., David, G., Zolla-Pazner, S., Farzan, M., Lee, T. H., and Gallay, P. A. (2005). A highly conserved arginine in gp120 governs HIV-1 binding to both syndecans and CCR5 via sulfated motifs. J. Biol. Chem. 280, 39493–39504.
Deneka, M., Pelchen-Matthews, A., Byland, R., Ruiz-Mateos, E., and Marsh, M. (2007). In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J. Cell Biol. 177, 329–341.
Dengjel, J., Schoor, O., Fischer, R., Reich, M., Kraus, M., Muller, M., Kreymborg, K., Altenberend, F., Brandenburg, J., Kalbacher, H., Brock, R., Driessen, C., Rammensee, H. G., and Stevanovic, S. (2005). Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 7922–7927.
Denizot, M., Varbanov, M., Espert, L., Robert-Hebmann, V., Sagnier, S., Garcia, E., Curriu, M., Mamoun, R., Blanco, J., and Biard-Piechaczyk, M. (2008). HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 4, 998–1008.
Deretic, V. (2010b). A master conductor for aggregate clearance by autophagy. Dev. Cell 18, 694–696.
Deretic, V., and Levine, B. (2009). Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5, 527–549.
Deretic, V., Singh, S., Master, S., Harris, J., Roberts, E., Kyei, G., Davis, A., de Haro, S., Naylor, J., Lee, H. H., and Vergne, I. (2006). Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell. Microbiol. 8, 719–727.
Doitsh, G., Cavrois, M., Lassen, K. G., Zepeda, O., Yang, Z., Santiago, M. L., Hebbeler, A. M., and Greene, W. C. (2010). Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143, 789–801.
Eekels, J. J., Geerts, D., Jeeninga, R. E., and Berkhout, B. (2011). Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antiviral Res. 89, 43–53.
Eekels, J. M., Sagnier, S., Geerts, D., Jeeninga, R. E., Biard-Piechaczyk, M., and Berkhout, B. (2012). Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol. J. 9. (in press).
English, L., Chemali, M., Duron, J., Rondeau, C., Laplante, A., Gingras, D., Alexander, D., Leib, D., Norbury, C., Lippe, R., and Desjardins, M. (2009). Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487.
Espert, L., and Biard-Piechaczyk, M. (2009). Autophagy in HIV-induced T cell death. Curr. Top. Microbiol. Immunol. 335, 307–321.
Espert, L., Codogno, P., and Biard-Piechaczyk, M. (2007). Involvement of autophagy in viral infections: antiviral function and subversion by viruses. J. Mol. Med. 85, 811–823.
Espert, L., Denizot, M., Grimaldi, M., Robert-Hebmann, V., Gay, B., Varbanov, M., Codogno, P., and Biard-Piechaczyk, M. (2006). Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 116, 2161–2172.
Espert, L., Varbanov, M., Robert-Hebmann, V., Sagnier, S., Robbins, I., Sanchez, F., Lafont, V., and Biard-Piechaczyk, M. (2009). Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 4, e5787. doi:10.1371/journal.pone.0005787
Felts, R. L., Narayan, K., Estes, J. D., Shi, D., Trubey, C. M., Fu, J., Hartnell, L. M., Ruthel, G. T., Schneider, D. K., Nagashima, K., Bess, J. W., Bavari, S., Lowekamp, B. C., Bliss, D., Lifson, J. D., and Subramaniam, S. (2010). 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc. Natl. Acad. Sci. U.S.A. 107, 13336–13341.
Finkel, T. H., Tudor-Williams, G., Banda, N. K., Cotton, M. F., Curiel, T., Monks, C., Baba, T. W., Ruprecht, R. M., and Kupfer, A. (1995). Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1, 129–134.
Finzi, D., Blankson, J., Siliciano, J. D., Margolick, J. B., Chadwick, K., Pierson, T., Smith, K., Lisziewicz, J., Lori, F., Flexner, C., Quinn, T. C., Chaisson, R. E., Rosenberg, E., Walker, B., Gange, S., Gallant, J., and Siliciano, R. F. (1999). Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5, 512–517.
Finzi, D., Hermankova, M., Pierson, T., Carruth, L. M., Buck, C., Chaisson, R. E., Quinn, T. C., Chadwick, K., Margolick, J., Brookmeyer, R., Gallant, J., Markowitz, M., Ho, D. D., Richman, D. D., and Siliciano, R. F. (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300.
Folks, T. M., Justement, J., Kinter, A., Dinarello, C., and Fauci, A. S. (1988). Cytokine-induced expression of HIV-1 in a chronically infected promonocytic cell line. Science 238, 800–802.
Furuya, D., Tsuji, N., Yagihashi, A., and Watanabe, N. (2005). Beclin 1 augmented cis-diamminedichloroplatinum induced apoptosis via enhancing caspase-9 activity. Exp. Cell Res. 307, 26–40.
Garg, H., and Blumenthal, R. (2006). HIV gp41-induced apoptosis is mediated by caspase-3-dependent mitochondrial depolarization, which is inhibited by HIV protease inhibitor nelfinavir. J. Leukoc. Biol. 79, 351–362.
Garg, H., and Blumenthal, R. (2008). Role of HIV Gp41 mediated fusion/hemifusion in bystander apoptosis. Cell. Mol. Life Sci. 65, 3134–3144.
Garg, H., Joshi, A., Freed, E. O., and Blumenthal, R. (2007). Site-specific mutations in HIV-1 GP41 reveal a correlation between HIV-1-mediated bystander apoptosis and fusion/hemifusion. J. Biol. Chem. 282, 16899–16906.
Geeraert, C., Ratier, A., Pfisterer, S. G., Perdiz, D., Cantaloube, I., Rouault, A., Pattingre, S., Proikas-Cezanne, T., Codogno, P., and Pous, C. (2010). Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 285, 24184–24194.
Giri, M. S., Nebozhyn, M., Showe, L., and Montaner, L. J. (2006). Microarray data on gene modulation by HIV-1 in immune cells: 2000-2006. J. Leukoc. Biol. 80, 1031–1043.
Goodenow, M. M., and Collman, R. G. (2006). HIV-1 coreceptor preference is distinct from target cell tropism: a dual-parameter nomenclature to define viral phenotypes. J. Leukoc. Biol. 80, 965–972.
Grivel, J. C., and Margolis, L. B. (1999). CCR5- and CXCR4-tropic HIV-1 are equally cytopathic for their T-cell targets in human lymphoid tissue. Nat. Med. 5, 344–346.
Groot, F., Welsch, S., and Sattentau, Q. J. (2008). Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 111, 4660–4663.
Heinkelein, M., Sopper, S., and Jassoy, C. (1995). Contact of human immunodeficiency virus type 1-infected and uninfected CD4+ T lymphocytes is highly cytolytic for both cells. J. Virol. 69, 6925–6931.
Herzberg, M. C., Weinberg, A., and Wahl, S. M. (2006). (C3) The oral epithelial cell and first encounters with HIV-1. Adv. Dent. Res. 19, 158–166.
Jahreiss, L., Menzies, F. M., and Rubinsztein, D. C. (2008). The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 9, 574–587.
Jolly, C., Kashefi, K., Hollinshead, M., and Sattentau, Q. J. (2004). HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199, 283–293.
Joshi, A., Nyakeriga, A. M., Ravi, R., and Garg, H. (2011). HIV ENV glycoprotein-mediated bystander apoptosis depends on expression of the CCR5 co-receptor at the cell surface and ENV fusogenic activity. J. Biol. Chem. 286, 36404–36413.
Jouve, M., Sol-Foulon, N., Watson, S., Schwartz, O., and Benaroch, P. (2007). HIV-1 buds and accumulates in “nonacidic” endosomes of macrophages. Cell Host Microbe 2, 85–95.
Kirkin, V., Lamark, T., Sou, Y. S., Bjorkoy, G., Nunn, J. L., Bruun, J. A., Shvets, E., McEwan, D. G., Clausen, T. H., Wild, P., Bilusic, I., Theurillat, J. P., Øvervatn, A., Ishii, T., Elazar, Z., Komatsu, M., Dikic, I., and Johansen, T. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516.
Koot, M., Keet, I. P., Vos, A. H., de Goede, R. E., Roos, M. T., Coutinho, R. A., Miedema, F., Schellekens, P. T., and Tersmette, M. (1993). Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann. Intern. Med. 118, 681–688.
Krammer, P. H., Dhein, J., Walczak, H., Behrmann, I., Mariani, S., Matiba, B., Fath, M., Daniel, P. T., Knipping, E., Westendorp, M. O., Stricker, K., Bäumler, C., Hellbardt, S., Germer, M., Peter, M. E., and Debativ, K.-M. (1994). The role of APO-1-mediated apoptosis in the immune system. Immunol. Rev. 142, 175–191.
Kumar, A., Dhawan, S., Mukhopadhyay, A., and Aggarwal, B. B. (1999). Human immunodeficiency virus-1-tat induces matrix metalloproteinase-9 in monocytes through protein tyrosine phosphatase-mediated activation of nuclear transcription factor NF-kappaB. FEBS Lett. 462, 140–144.
Kyei, G. B., Dinkins, C., Davis, A. S., Roberts, E., Singh, S. B., Dong, C., Wu, L., Kominami, E., Ueno, T., Yamamoto, A., Federico, M., Panganiban, A., Vergne, I., and Deretic, V. (2009). Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186, 255–268.
Lamark, T., Kirkin, V., Dikic, I., and Johansen, T. (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8, 1986–1990.
Larkin, M., Childs, R. A., Matthews, T. J., Thiel, S., Mizuochi, T., Lawson, A. M., Savill, J. S., Haslett, C., Diaz, R., and Feizi, T. (1989). Oligosaccharide-mediated interactions of the envelope glycoprotein gp120 of HIV-1 that are independent of CD4 recognition. AIDS 3, 793–798.
Laurent-Crawford, A. G., Krust, B., Riviere, Y., Desgranges, C., Muller, S., Kieny, M. P., Dauguet, C., and Hovanessian, A. G. (1993). Membrane expression of HIV envelope glycoproteins triggers apoptosis in CD4 cells. AIDS Res. Hum. Retroviruses 9, 761–773.
Levine, B., and Deretic, V. (2007). Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7, 767–777.
Li, Q., Duan, L., Estes, J. D., Ma, Z. M., Rourke, T., Wang, Y., Reilly, C., Carlis, J., Miller, C. J., and Haase, A. T. (2005). Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 434, 1148–1152.
Lin, L. T., Dawson, P. W., and Richardson, C. D. (2010). Viral interactions with macroautophagy: a double-edged sword. Virology 402, 1–10.
Lunemann, J. D., and Munz, C. (2009). Autophagy in CD4+ T-cell immunity and tolerance. Cell Death Differ. 16, 79–86.
Ma, G., Greenwell-Wild, T., Lei, K., Jin, W., Swisher, J., Hardegen, N., Wild, C. T., and Wahl, S. M. (2004). Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J. Exp. Med. 200, 1337–1346.
Marechal, V., Prevost, M. C., Petit, C., Perret, E., Heard, J. M., and Schwartz, O. (2001). Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J. Virol. 75, 11166–11177.
Margolis, L., and Shattock, R. (2006). Selective transmission of CCR5-utilizing HIV-1: the “gatekeeper” problem resolved? Nat. Rev. Microbiol. 4, 312–317.
Marino, G., and Lopez-Otin, C. (2004). Autophagy: molecular mechanisms, physiological functions and relevance in human pathology. Cell. Mol. Life Sci. 61, 1439–1454.
McCarroll, S. A., Huett, A., Kuballa, P., Chilewski, S. D., Landry, A., Goyette, P., Zody, M. C., Hall, J. L., Brant, S. R., Cho, J. H., Duerr, R. H., Silverberg, M. S., Taylor, K. D., Rioux, J. D., Altshuler, D., Daly, M. J., and Xavier, R. J. (2008). Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 40, 1107–1112.
McDonald, D., Vodicka, M. A., Lucero, G., Svitkina, T. M., Borisy, G. G., Emerman, M., and Hope, T. J. (2002). Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159, 441–452.
McDonald, D., Wu, L., Bohks, S. M., KewalRamani, V. N., Unutmaz, D., and Hope, T. J. (2003). Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 300, 1295–1297.
Miller, S., and Krijnse-Locker, J. (2008). Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6, 363–374.
Miyauchi, K., Kim, Y., Latinovic, O., Morozov, V., and Melikyan, G. B. (2009). HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137, 433–444.
Mogensen, T. H., Melchjorsen, J., Larsen, C. S., and Paludan, S. R. (2010). Innate immune recognition and activation during HIV infection. Retrovirology 7, 54.
Molina, L., Grimaldi, M., Robert-Hebmann, V., Espert, L., Varbanov, M., Devaux, C., Granier, C., and Biard-Piechaczyk, M. (2007). Proteomic analysis of the cellular responses induced in uninfected immune cells by cell-expressed X4 HIV-1 envelope. Proteomics 7, 3116–3130.
Nermut, M. V., and Fassati, A. (2003). Structural analyses of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J. Virol. 77, 8196–8206.
Nguyen, D. G., and Hildreth, J. E. (2003). Involvement of macrophage mannose receptor in the binding and transmission of HIV by macrophages. Eur. J. Immunol. 33, 483–493.
Nimmerjahn, F., Milosevic, S., Behrends, U., Jaffee, E. M., Pardoll, D. M., Bornkamm, G. W., and Mautner, J. (2003). Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 33, 1250–1259.
O’Brien, W. A., Namazi, A., Kalhor, H., Mao, S. H., Zack, J. A., and Chen, I. S. (1994). Kinetics of human immunodeficiency virus type 1 reverse transcription in blood mononuclear phagocytes are slowed by limitations of nucleotide precursors. J. Virol. 68, 1258–1263.
Ohnimus, H., Heinkelein, M., and Jassoy, C. (1997). Apoptotic cell death upon contact of CD4+ T lymphocytes with HIV glycoprotein-expressing cells is mediated by caspases but bypasses CD95 (Fas/Apo-1) and TNF receptor 1. J. Immunol. 159, 5246–5252.
Orsi, A., Polson, H. E., and Tooze, S. A. (2009). Membrane trafficking events that partake in autophagy. Curr. Opin. Cell Biol. 22, 150–156.
Pattingre, S., Bauvy, C., Carpentier, S., Levade, T., Levine, B., and Codogno, P. (2009). Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 284, 2719–2728.
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., Packer, M., Schneider, M. D., and Levine, B. (2005). Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 122, 927–939.
Pauza, C. D., and Price, T. M. (1988). Human immunodeficiency virus infection of T cells and monocytes proceeds via receptor-mediated endocytosis. J. Cell Biol. 107, 959–968.
Peters, P. J., Bhattacharya, J., Hibbitts, S., Dittmar, M. T., Simmons, G., Bell, J., Simmonds, P., and Clapham, P. R. (2004). Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and lymph node tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusogenicity for macrophages. J. Virol. 78, 6915–6926.
Peters, P. J., Sullivan, W. M., Duenas-Decamp, M. J., Bhattacharya, J., Ankghuambom, C., Brown, R., Luzuriaga, K., Bell, J., Simmonds, P., Ball, J., and Clapham, P. R. (2006). Non-macrophage-tropic human immunodeficiency virus type 1 R5 envelopes predominate in blood, lymph nodes, and semen: implications for transmission and pathogenesis. J. Virol. 80, 6324–6332.
Pierson, T. C., and Doms, R. W. (2003). HIV-1 entry and its inhibition. Curr. Top. Microbiol. Immunol. 281, 1–27.
Polyak, S., Chen, H., Hirsch, D., George, I., Hershberg, R., and Sperber, K. (1997). Impaired class II expression and antigen uptake in monocytic cells after HIV-1 infection. J. Immunol. 159, 2177–2188.
Pombo-Grégoire, I., Richetta, C., Meyniel-Schicklin, L., Borel, S., Pradezynski, F., Diaz, O., Deloire, A., Le Breton, M., Azocar, O., Mangeot, P. E., Navratil, V., Joubert, P. E., Flacher, M., Vidalain, P. O., André, P., Lotteau, V., Biard-Piechaczyk, M., Rabourdin-Combe, C., and Faure, M. (2011). IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 7, e1002422. doi:10.1371/journal.ppat.1002422
Richman, D. D., and Bozzette, S. A. (1994). The impact of the syncytium-inducing phenotype of human immunodeficiency virus on disease progression. J. Infect. Dis. 169, 968–974.
Roggero, R., Robert-Hebmann, V., Harrington, S., Roland, J., Vergne, L., Jaleco, S., Devaux, C., and Biard-Piechaczyk, M. (2001). Binding of human immunodeficiency virus type 1 gp120 to CXCR4 induces mitochondrial transmembrane depolarization and cytochrome c-mediated apoptosis independently of Fas signaling. J. Virol. 75, 7637–7650.
Rohr, O., Marban, C., Aunis, D., and Schaeffer, E. (2003). Regulation of HIV-1 gene transcription: from lymphocytes to microglial cells. J. Leukoc. Biol. 74, 736–749.
Rudnicka, D., Feldmann, J., Porrot, F., Wietgrefe, S., Guadagnini, S., Prevost, M. C., Estaquier, J., Haase, A. T., Sol-Foulon, N., and Schwartz, O. (2009). Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J. Virol. 83, 6234–6246.
Saphire, A. C., Bobardt, M. D., Zhang, Z., David, G., and Gallay, P. A. (2001). Syndecans serve as attachment receptors for human immunodeficiency virus type 1 on macrophages. J. Virol. 75, 9187–9200.
Sattentau, Q. (2008). Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6, 815–826.
Scarlatti, F., Bauvy, C., Ventruti, A., Sala, G., Cluzeaud, F., Vandewalle, A., Ghidoni, R., and Codogno, P. (2004). Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 279, 18384–18391.
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760.
Schmid, D., Dengjel, J., Schoor, O., Stevanovic, S., and Munz, C. (2006). Autophagy in innate and adaptive immunity against intracellular pathogens. J. Mol. Med. 84, 194–202.
Schmid, D., and Munz, C. (2007). Innate and adaptive immunity through autophagy. Immunity 27, 11–21.
Shimizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thompson, C. B., and Tsujimoto, Y. (2004). Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 6, 1221–1228.
Siliciano, J. D., Kajdas, J., Finzi, D., Quinn, T. C., Chadwick, K., Margolick, J. B., Kovacs, C., Gange, S. J., and Siliciano, R. F. (2003). Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9, 727–728.
Singh, S. B., Ornatowski, W., Vergne, I., Naylor, J., Delgado, M., Roberts, E., Ponpuak, M., Master, S., Pilli, M., White, E., Komatsu, M., and Deretic, V. (2010). Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 12, 1154–1165.
Sumpter, R. Jr., and Levine, B. (2010). Autophagy and innate immunity: triggering, targeting and tuning. Semin. Cell Dev. Biol. 21, 699–711.
Suyama, M., Daikoku, E., Goto, T., Sano, K., and Morikawa, Y. (2009). Reactivation from latency displays HIV particle budding at plasma membrane, accompanying CD44 upregulation and recruitment. Retrovirology 6, 63.
UNAIDS. (2009). The Annual AIDS Epidemic Update Reports. “UNAIDS/09.36E/JC1700E”.1-100. Geneva: UNAIDS.
Vazquez, N., Greenwell-Wild, T., Marinos, N. J., Swaim, W. D., Nares, S., Ott, D. E., Schubert, U., Henklein, P., Orenstein, J. M., Sporn, M. B., and Wahl, S. M. (2005). Human immunodeficiency virus type 1-induced macrophage gene expression includes the p21 gene, a target for viral regulation. J. Virol. 79, 4479–4491.
Welsch, S., Keppler, O. T., Habermann, A., Allespach, I., Krijnse-Locker, J., and Krausslich, H. G. (2007). HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 3, e36. doi:10.1371/journal.ppat.0030036
Wong, J. K., Hezareh, M., Gunthard, H. F., Havlir, D. V., Ignacio, C. C., Spina, C. A., and Richman, D. D. (1997). Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278, 1291–1295.
Wu, Z., Golub, E., Abrams, W. R., and Malamud, D. (2004). gp340 (SAG) binds to the V3 sequence of gp120 important for chemokine receptor interaction. AIDS Res. Hum. Retroviruses 20, 600–607.
Yamashita, M., and Emerman, M. (2005). The cell cycle independence of HIV infections is not determined by known karyophilic viral elements. PLoS Pathog. 1, e18. doi:10.1371/journal.ppat.0010018
Yoo, J., Chen, H., Kraus, T., Hirsch, D., Polyak, S., George, I., and Sperber, K. (1996). Altered cytokine production and accessory cell function after HIV-1 infection. J. Immunol. 157, 1313–1320.
Keywords: autophagy, HIV-1, CD4+ T cells, macrophages, infection
Citation: Borel S, Espert L and Biard-Piechaczyk M (2012) Macroautophagy regulation during HIV-1 infection of CD4+ T cells and macrophages. Front. Immun. 3:97. doi: 10.3389/fimmu.2012.00097
Received: 05 December 2011; Accepted: 13 April 2012;
Published online: 07 May 2012.
Edited by:
Irina Caminschi, Burnet Institute, AustraliaReviewed by:
Marianne Boes, University Medical Centre Utrecht, NetherlandsCopyright: © 2012 Borel, Espert and Biard-Piechaczyk. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Martine Biard-Piechaczyk, Centre d’études d’agents Pathogènes et Biotechnologies pour la Santé, CNRS UMR5236, UM1/UM2, 1919 Route de Mende, 34293 Montpellier Cedex 5, France. e-mail:bWFydGluZS5iaWFyZEBjcGJzLmNucnMuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.