


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 April 2012
Sec. Antigen Presenting Cell Biology
Volume 3 - 2012 | https://doi.org/10.3389/fimmu.2012.00060
This article is part of the Research Topic Eat Thyself, Heal Thyself: Autophagy in pathogen recognition and antigen processing View all 13 articles
Macroautophagy (autophagy) is a cellular pathway facilitating several critical functions. First, autophagy is a major pathway of degradation. It enables elimination of microbes that have invaded intracellular compartments. In addition, it promotes degradation of damaged cellular content, thereby acting to limit inflammatory signals. Second, autophagy is a major trafficking pathway, shuttling content between the cytosol and the lysosomal compartment. Given these two key roles, autophagy can have significant and sometimes unexpected consequences on mechanisms that initiate robust immunity. Here, we will discuss the impact of autophagy on pathways of innate and adaptive immune responses including microbe elimination, inflammatory cytokine production, antigen processing and T and B lymphocyte immunity.
Macroautophagy (referred herein as autophagy) is a process that involves the formation of an autophagosome, a double membrane vesicle, which is trafficked to lysosomes where autophagosomal contents are degraded. Autophagy is considered both a major pathway of degradation, providing an alternative to the proteasome, and a significant trafficking pathway between the cytosol and lysosomal compartments. Activating autophagy mostly occurs via serine/threonine kinase, mammalian target of rapamycin (mTOR) that impedes autophagy by binding and inactivating the UNC-51-like kinase (ULK)1/2 kinase. Signaling via the ULK1/2 kinase complex is critical for recruiting autophagy-related gene (ATG) proteins to the site of autophagosome biogenesis (Jung et al., 2010; Weidberg et al., 2011). One ATG of particular importance is Atg8 (microtubule-associated light chain 3, LC3 in mammals), a widely used marker of autophagosomes. LC3 acts in autophagosome expansion and closure (Nakatogawa et al., 2007), in addition to selectively recruiting autophagosomal cargo (Noda et al., 2008). Prior to association with the autophagosome, LC3 is conjugated to phosphatidylethanolamine by a series of reactions that involves Atg7, Atg3 and an Atg5–Atg12–Atg16 complex (Tanida et al., 2004). Other critical contributors to autophagy are phosphatidylinositol kinase Vsp34 and Beclin 1, that promote autophagosomal membrane and fusion events (Liang et al., 2008; Matsunaga et al., 2009; Zhong et al., 2009). Here, we will discuss examples of the contribution of the autophagy pathway to innate (Figure 1) and adaptive (Figure 2) effector mechanisms that are critical for robust immunity.
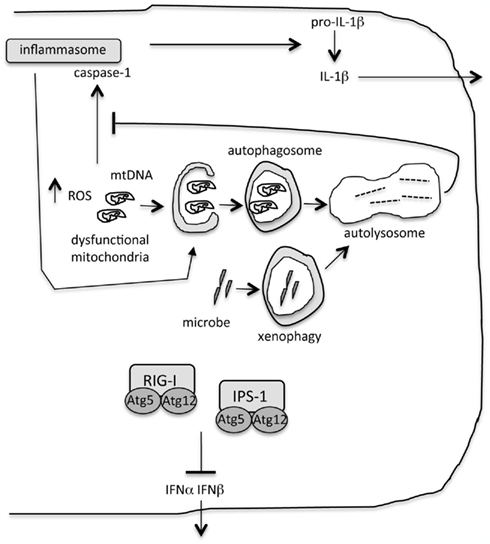
Figure 1. Role of autophagy during innate immunity. “Xenophagy” eliminates intracellular microbes. Autophagy limits inflammatory cytokine production by ensuring removal of dysfunctional organelles, including mitochondria. Accumulation of damaged mitochondria results in increased reactive oxygen species (ROS) and the escape of mitochondrial DNA into the cytosol. These are triggers of the inflammasome and interleukin-1β (IL-1β) production. Inflammasomes are targeted for degradation by autophagy. Proteins in the autophagy machinery bind to and inhibit signaling of key proteins in the type I interferon pathway, retinoic acid inducible gene-I (RIG-I) and the signaling molecule IFNβ promoter stimulator-1 (IPS-1).
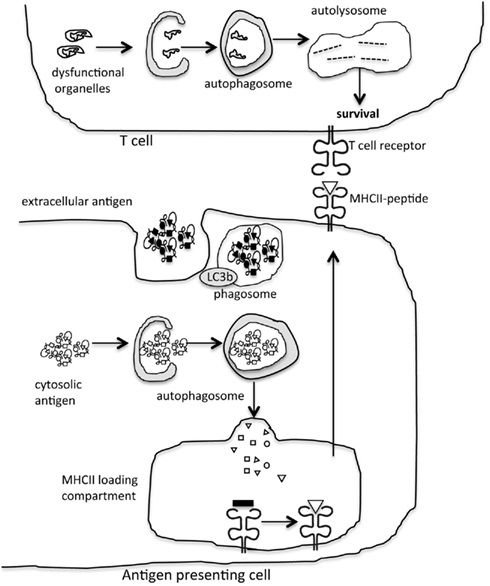
Figure 2. Autophagy modulation of adaptive immunity. Autophagy is a significant trafficking pathway for the delivery of cytosolic antigen to the MHCII loading compartment in antigen presenting cells. In addition, proteins in the autophagy machinery contribute to phagocytosis of extracellular antigen. In lymphocytes, the role of autophagy in eliminating damaged organelles promotes cell survival.
Autophagy is an innate effector mechanism that eliminates intracellular pathogens. Autophagy is induced downstream of signaling by “danger” receptors including Toll like receptors (TLR; Xu et al., 2007; Delgado et al., 2008), retinoic acid inducible gene I-like receptors (RLR; Yano et al., 2008; Cooney et al., 2010; Travassos et al., 2010), and alarmins (Tang et al., 2010). “Xenophagy” describes the process by which intracellular microbes are selectively degraded by autophagy (Levine, 2005). Xenophagy eliminates bacteria that access the cytosol including Streptococcus pyogenes (Nakagawa et al., 2004) and Mycobacterium tuberculosis (Gutierrez et al., 2004). Vacuolar bacteria are also targeted. Following RAW cell infection with M. tuberculosis var. bovis bacillus Calmette–Guerin (BCG), autophagy facilitates the fusion of mycobacteria containing vacuoles with late endosomes/lysosomes to promote mycobacteria degradation (Singh et al., 2006). Salmonella-containing vacuoles are targeted by LC3 (Birmingham et al., 2006) and can be engulfed by autophagosomes (Kageyama et al., 2011). Interestingly, recruitment of autophagy machinery to damaged bacterial vacuoles involves the exposure of host sugar residues that attract receptor galectin 8 (Thurston et al., 2012). Xenophagy also facilitates removal of viral pathogens. One example is the degradation of the Sindbis virus capsid by the autophagy pathway (Orvedahl et al., 2010). Autophagy can also impair viral replication by trafficking cytosolic viral replication products to the endolysosomal compartment where they are detected by TLR and a type I interferon (IFN) response is triggered. This is the case for vesicular stomatitis virus (VSV) infection of plasmacytoid dendritic cells (pDC; Lee et al., 2007). Given its role in anti-viral defense, many viruses encode proteins to disable autophagy. Gammaherpes virus 68 (Ku et al., 2008), influenza A virus (Gannage et al., 2009), herpes simplex virus-1 (HSV-1; Orvedahl et al., 2007), human cytomegalovirus (Chaumorcel et al., 2012), and human immunodeficiency virus (HIV;Kyei et al., 2009) are examples of viruses that encode proteins to block autophagy initiation by interfering with beclin 1 activity. Other interesting examples of viral interference with the autophagy pathway include Kaposi’s sarcoma-associated herpesvirus encoded Fadd-like interleukin-1 beta-converting enzyme (FLICE)-like inhibitor protein that blocks interaction between LC3 and Atg4 (Lee et al., 2009) and HIV nef that prevents autophagosomal maturation (Kyei et al., 2009). Like viruses, bacteria also subvert the autophagy pathway. L. monocytogenes, via the expression of ActA, recruits host proteins to its surface to disguise itself from autophagic recognition (Yoshikawa et al., 2009). Similarly, Shigella hides from the autophagy machinery by expressing IcsB, that inhibits Atg5 binding to the Shigella surface (Ogawa et al., 2005). In summary, autophagy is a major mode of innate immune defense against intracellular pathogens.
Autophagy participates in the control of pro-inflammatory cytokine cascades. Many examples illustrate a shutdown in autophagy results in excessive inflammatory cytokine production. This is particularly well described for interleukin (IL)-1β and IL-18, where the absence of autophagy exacerbates their secretion by murine macrophages when stimulated with pro-inflammatory compounds. Specifically, this has been described for beclin1+/− (Nakahira et al., 2011), LC3b−/− (Nakahira et al., 2011), Atg16L1−/− (Saitoh et al., 2008), and Atg7−/− (Saitoh et al., 2008) macrophages or following expression of inactive Atg4B or due to treatment with the chemical autophagy inhibitor 3-methyladenine (3-MA; Saitoh et al., 2008; Harris et al., 2011). Enhanced secretion of IL-1β also occurs when human peripheral blood mononuclear cells are stimulated in the presence of an impaired autophagy pathway due to treatment with 3-MA (Crisan et al., 2011). Conversely, stimulation of murine DC with rapamycin suppresses IL-1β production in response to inflammatory stimuli (Harris et al., 2011). Rapamycin administration in vivo, inhibits the detected increase in IL-1β but not IL-12, in the serum 4 h following lipopolysaccharide (LPS) treatment (Harris et al., 2011). Not all stimuli provoke excessive IL-1β secretion in the absence of autophagy. Stimuli that do include LPS, or treatment of LPS-primed macrophages with adenosine 5’-triphosphate (ATP) or monosodium urate (MSU). Salmonella typhimurium or muramyl dipeptide, a nucleotide-binding oligomerization domain-containing protein 2 (NOD2) ligand, do not provoke such a response (Saitoh et al., 2008). Ligands that engage TLR4 and to some extent those recognized by TLR3, TLR7, and TLR9, but not TLR2 or TLR5, elicit enhanced IL-1β in autophagy-deficient macrophages (Saitoh et al., 2008). In murine macrophages, excessive cytokine production following autophagy shutdown requires TIR-domain containing adapter inducing IFNβ (TRIF), an adapter molecule for TLR signaling (Saitoh et al., 2008), while in human cells the response is dependent on p38 mitogen activated protein kinase (MAPK) phosphorylation (Crisan et al., 2011). Excessive IL-1β secretion in the absence of autophagy is not associated with alterations in NFκB activation (Saitoh et al., 2008). Importantly, exaggerated inflammatory cytokine production in the absence of autophagy translates to the development of severe disease in vivo. Atg16L1-deficient mice display enhanced inflammatory infiltrates in the distal colon, elevated IL-1β, and IL-18 serum levels and reduced survival in a dextran sulfate sodium induced model of experimental colitis (Saitoh et al., 2008). LC3b-deficient mice exhibit enhanced susceptibility to LPS-induced lethality (Nakahira et al., 2011), while both LC3b-deficient and beclin1+/− mice have increased IL-1β and IL-18 serum levels in response to cecal ligation and puncture-induced sepsis (Nakahira et al., 2011). In addition to IL-1β/IL-18, the type I IFN pathway is also susceptible to modulation by autophagy. Again the absence of a functional autophagy pathway promotes amplified secretion of type I IFN in response to a given stimulus. VSV infection of Atg5-deficient murine embryonic fibroblasts (MEF) elicits increased production of IFNα, IFNβ, IL-6, and IP-10 and as such dramatically attenuates viral replication rendering cells resistant to infection (Jounai et al., 2007; Lee et al., 2010). Another example is Atg9a-deficient MEF that exhibit enhanced IFNβ production in response to stimulation with inflammatory double stranded DNA (Saitoh et al., 2009). Therefore, autophagy has the potential to exert a critical influence on the regulation of pro-inflammatory cytokine production and downstream effector mechanisms of innate immunity. The ability of autophagy to limit inflammatory cytokine production renders it a target pathway for pathogens to activate as a strategy to facilitate replication. Hepatitis C virus (HCV) is an example of a pathogen that is proposed to do this. If HCV infection occurs in the absence of a functional autophagy pathway, HCV viral replication is severely attenuated due to the corresponding activation of the type I IFN pathway. Therefore, HCV infection elicits active autophagy in the infected cell, limiting type I IFN production and enabling successful viral replication (Ke and Chen, 2011). Triggering autophagy is therefore a putative mode of immune escape.
How does autophagy act to regulate inflammatory cytokine activity? Secretion of IL-1β/IL-18 is a tightly regulated process. Pro-inflammatory stimuli induce the transcription of inactive precursors pro-IL-1β or pro-IL-18. Cleavage of the inactive proteins into their active secreted forms requires the assembly of an inflammasome, a large protein complex that, under inflammatory conditions, processes pro-caspase-1 to caspase-1. Active caspase-1 then cleaves pro-IL-1β and pro-IL-18 liberating the active cytokines for secretion by the cell. In mouse cells, the levels of precursor IL-1β are not altered following LPS stimulation in the absence of autophagy (Saitoh et al., 2008), rather there is an increase in the cleaved form of caspase-1 (Saitoh et al., 2008; Nakahira et al., 2011). In contrast, in human cells, transcription of IL-1β is elevated (Crisan et al., 2011). Autophagy exerts its influence on IL-1β and IL-18 secretion by several mechanisms. A major role for autophagy in inflammatory cytokine secretion is considered to be via its contribution to maintaining a healthy intracellular environment. Namely, autophagy has a pivotal role in the clearance and elimination of damaged and dysfunctional organelles. Studies have demonstrated autophagic degradation of depolarized mitochondria (Sandoval et al., 2008), expanded endoplasmic reticulum (Bernales et al., 2006), mature ribosomes (Kraft et al., 2008; Kundu et al., 2008), and excess peroxisomes (Dunn et al., 2005). The role of autophagy in the maintenance of mitochondrial integrity is a process known as “mitophagy.” In the absence of functional mitophagy, cells accumulate damaged mitochondria that produce high levels of reactive oxygen species (ROS), a side product of cellular respiration (Zhou et al., 2011). The accumulation of damaged mitochondria is further exacerbated following stimulation with inflammatory compounds as observed for LC3b or beclin 1-deficient macrophages treated with LPS plus ATP (Nakahira et al., 2011). Without the autophagy pathway to remove these dysfunctional organelles, ROS levels increase (Saitoh et al., 2008), a known trigger for the nucleotide binding domain, leucine rich-containing family, pyrin domain containing 3 (NLRP3) inflammasome (Zhou et al., 2011). Damaged mitochondria also display increased membrane permeability with mitochondrial DNA (mtDNA) then being able to translocate to the cytosol. mtDNA is detected in higher abundance in the cytosol of LC3b and beclin 1-deficient macrophages, most notably following stimulation with LPS plus ATP (Nakahira et al., 2011). As such, autophagy, by acting to maintain turnover of damaged organelles and preserving mitochondrial integrity, indirectly restricts excessive inflammatory cytokine production by removing endogenous triggers of the inflammasome.
The second mechanism by which autophagy controls inflammatory cytokine activity is through its direct interaction with proteins involved in inflammatory cascades. In this case, proteins are targeted by autophagosomes or the autophagy machinery. Interestingly, inflammasomes are engulfed and degraded by autophagy. Following inflammasome induction, components of the absent in melanoma 2 (AIM2) inflammasome associate with p62 and beclin1 and exhibit K63-linked ubiquitination (Shi et al., 2012). Other examples of the direct targeting of inflammatory proteins by autophagy include the co-localization of the autophagosomal protein LC3 and IL-1β in punctate autophagosomal structures following stimulation of bone marrow-derived macrophages with a variety of TLR ligands including LPS, Pam3Cys, poly (I:C), R837, or CpG (Harris et al., 2011). The authors suggest that autophagy limits IL-1β secretion by facilitating its degradation in the auto-lysosome. A direct role for autophagy in modulating components of the type I IFN response is illustrated by several examples where the autophagy machinery targets proteins involved in this signaling cascade. Atg5–Atg12 directly interacts with the cytosolic viral RNA sensor RIG-I and the signaling molecule IFNβ promoter stimulator-1 (IPS-1). Interaction with Atg5–Atg12 blocks caspase recruitment domain (CARD)-mediated signaling and the downstream production of type I IFN (Jounai et al., 2007). Another interesting example is stimulator of interferon genes (STING), a molecule critical for type I IFN signaling. Atg9a is critical for the localization of STING to cytoplasmic punctae following treatment of cells with dsDNA (Saitoh et al., 2009). Mislocalization of STING to the endoplasmic reticulum results in exacerbated IFN responses (Saitoh et al., 2009), although the specific details of why this is the case remain to be elucidated.
Not all inflammatory cytokine production is enhanced in the absence of functional autophagy. In contrast to IL-1β, IL-18 and type I IFN, mouse tumor necrosis factor (TNF)α and IL-16 (Harris et al., 2011), and human TNFα (Crisan et al., 2011) production is inhibited when autophagy is suppressed. This is the case for macrophages following stimulation with LPS or Pam3Cys in the presence of 3-MA. Another example is the reduced production of IL-8 by intestinal epithelial cells with silenced Atg7 (Li et al., 2011b). The mechanism of why these cytokine pathways require autophagy may involve autophagic inhibition of p38 phosphorylation (Crisan et al., 2011). Therefore the role of autophagy in modulating cytokine output is complex with its contribution varying for individual cytokines.
Another mechanism by which autophagy modulates inflammation is by restricting the expression of extracellular inflammatory signals. During programmed cell death, autophagy enables the generation of a cellular corpse that is efficiently phagocytized and less likely to elicit detrimental inflammatory outcomes. This is elegantly illustrated in a model of embryonic cavitation, where apoptotic embryonic stem cells lacking Atg5 or beclin1 fail to exhibit critical engulfment signals (Qu et al., 2007). These include failing to expose phosphatidylserine (PS) at the outer membrane and lack of secretion of the chemoattractant lysophosphatidylcholine. As a consequence, autophagy-deficient cellular corpses are not engulfed for clearance by immune cells. The failure to eliminate autophagy-deficient apoptotic corpses is implicated in the neonatal death of Atg5-deficient mice. Excess apoptotic corpses are detected in the lungs and retina, together with the presence of an abnormal inflammatory infiltrate in these tissues. Interestingly, Atg5−/− (Uhl et al., 2009), beclin1+/− (Li et al., 2008), or Atg12−/− (Li et al., 2008) cells loaded with antigen are less efficient at facilitating DC antigen presentation to CD8+ T cells (cross-presentation). This is postulated to be due to their poor engulfment. The requirement for autophagy in the display of engulfment signals is thought to be due to the pathway’s role in maintaining cellular bioenergetics. Restoration of ATP levels in autophagy-deficient cells by treatment with methylpyruvate restores corpse clearance (Qu et al., 2007). Induction of autophagy in dying cells also stimulates the release of immune modulator high mobility group B1 (HMGB1; Thorburn et al., 2009) that promotes immunogenicity and removal of apoptotic corpses (Scaffidi et al., 2002).
Proteolysis provides the peptide epitopes displayed by major histocompatibility complex (MHC) molecules to elicit T cell responses. All nucleated cells utilize the proteasome, a multiproteolytic complex located in the cytosol that generates ligands for MHCI presentation. In MHCII expressing cells, lysosomal proteases generate ligands for these molecules. Autophagy participates in generating the pool of peptides displayed by MHCII molecules due to its role in trafficking cytosolic proteins to the lysosomes, the same compartment where biosynthesis and loading of MHCII molecules occurs. Indeed, the autophagosomal protein LC3 co-localizes with MHCII loading compartments in DC (Schmid et al., 2007). Fusion of influenza matrix protein M1 (Schmid et al., 2007) or the hemagglutinin (HA) site 1 epitope (Comber et al., 2011) to LC3 enhances their presentation by MHCII to CD4+ T cells as a result of the intersection of autophagosomes and MHCII loading compartments. Autophagy contribution to MHCII antigen presentation is further supported by detailed analysis of the steady state MHCII repertoire where up to a third of peptides displayed by MHCII are shown to be derived from cytoplasmic or nuclear sources (Chicz et al., 1993; Dongre et al., 2001) and would potentially rely on autophagy trafficking to access the MHCII loading compartment. Induction of autophagy increases the relative display of these peptides compared to those derived from membrane proteins (Dengjel et al., 2005). Peptides derived from LC3b and gamma-aminobutyric acid receptor-associated protein (GABARAP), components of the autophagy machinery are eluted from MHCII molecules (Dengjel et al., 2005). There is now a growing list of antigens that are documented as being trafficked by autophagy for MHCII display. These include self proteins together with virus and bacterial-derived antigens (Munz, 2009). The role of autophagy in presenting self antigen by MHCII, means that autophagy contributes to CD4+ T cell selection in the thymus. Studies in which Atg5−/− thymi were grafted into normal recipients elegantly illustrate that autophagy trafficking in thymic epithelial cells participates in the display of self antigen by MHCII (Nedjic et al., 2008). Transplant of Atg5-deficient thymi into MHCII-restricted TCR transgenic hosts results in reduced positive selection of HA and SEP-specific CD4+ TCR T cells. Selection of AND, DO11.1, or DEP-specific CD4+ TCR transgenic T cells, on the other hand, proceeds efficiently in the absence of autophagy. Autophagy is therefore considered to facilitate the thymic MHCII presentation of some self proteins but not others. In doing so, autophagy contributes to the complex composition of the repertoire of MHCII epitopes that are displayed by the thymic epithelium. The role for autophagy in thymic selection is further evidenced by an increase in IEα52–68–IAb complexes at the surface of cortical thymic epithelial cells that lack Atg5 (Nedjic et al., 2008). In this case, it is postulated that the removal of autophagy dependent endogenous epitopes due to the absence of Atg5, reduces competition and increases access of the IEα peptide to MHCII. In addition to autophagy shaping the CD4+ T cell repertoire that emerges from the thymus, it also participates in effective CD4+ T cell immunity during infection. Autophagy dependent-MHCII presentation of cytosolic model antigens has been described for complement C5 (Brazil et al., 1997), tumor antigen mucin 1 (Dorfel et al., 2005) and neomycin phosphotransferase (Nimmerjahn et al., 2003) where presentation is inhibited following treatment with 3-MA. Nuclear antigen 1 of Epstein-Barr virus (EBNA-1) is a prominent example of a viral-derived antigen that requires autophagy for presentation by MHCII (Paludan et al., 2005). Treatment with 3-MA or silencing of Atg12 inhibits EBNA-1 specific CD4+ T cells from recognizing a lymphoblastoid cell line (Paludan et al., 2005). Antigen presenting cell types that utilize autophagy to deliver antigen for MHCII presentation include B cells (Brazil et al., 1997), macrophages (Brazil et al., 1997), and DC (Schmid et al., 2007; Jagannath et al., 2009; Lee et al., 2010). Autophagy-mediated delivery of antigen for MHCII presentation to CD4+ T cells means that triggering autophagy represents a promising immunotherapeutic strategy to promote robust CD4+ T cell responses. Indeed, immunization with rapamycin-treated DC, enhances MHCII presentation and CD4+ T cell immunity to the anti-tuberculosis BCG vaccine antigen (Jagannath et al., 2009). This is an exciting outcome, with major implications for improving vaccine efficacy in multiple settings.
Whether autophagy exhibits selectivity in the substrates targeted is currently not well understood. This is important if the autophagy pathway is to be manipulated for delivery of antigen to MHCII. While basal autophagy initiated under starvation conditions is considered to be a non-selective process, autophagy initiated under steady state conditions in immune cells is considered to be selective. A general observation is that long lived proteins are considered to be turned over by lysosomal proteolysis and hence are autophagy substrates, while the proteasome degrades short lived polypeptides (Henell et al., 1987). Aggregates of long lived poly-ubiquitinated proteins are known substrates of autophagy (Mizushima and Klionsky, 2007). Of interest, protein aggregates form in activated DC (Lelouard et al., 2002) and in macrophages following TLR4 stimulation (Fujita et al., 2011). Several “autophagy receptors” have been described that act to recruit ubiquitinated proteins to autophagosomes via an LC3-interacting motif (LIR). Known autophagy receptors identified to date include p62/sequestosome1 (Komatsu et al., 2007), neighbour of Brca1 (NBR1) (Kirkin et al., 2009), nuclear dot protein 52kDa (NDP52) (Thurston et al., 2009), and optineurin (Wild et al., 2011) with all of these proteins possessing binding domains for both ubiquitin (UBD) and LC3 (LIR). The interplay of these receptors in targeting autophagy substrates is beginning to begin explored. For example, p62, optineurin, and NDP52 all target ubiquitinated Salmonella enterica for destruction in lysosomes (Wild et al., 2011). While optineurin and NDP52 colocalize, p62 binds to a separate subdomain of the ubiquitinated bacteria. This implies selective recognition of ubiquitin chains by specific autophagy receptors. How upstream events regulate the activity of autophagy receptors is mostly unknown. Interestingly, optineurin is subject to phosphorylation by tank binding kinase (TBK)1, providing the molecular trigger to promote autophagic clearance of cytosolic bacteria (Wild et al., 2011). Identification of a growing number of autophagy receptors is exciting with the detail of the specific E3 ubiquitin ligases involved, the selective recognition of ubiquitin chains and their specific regulation by phosphorylation all remaining to be further studied.
Somewhat unexpectedly, in addition to assisting the display of cytosolic antigen by MHCII molecules, autophagy also contributes to the presentation of extracellular antigen and phagocytosis. Individual components of the autophagy machinery are recruited to phagosomes in the presence of TLR signaling. During a process termed “LC3-associated phagocytosis (LAP)” (Sanjuan and Green, 2008), LC3 is recruited to latex-bead phagosomes in macrophages, when beads are coupled with the TLR agonists LPS or Pam3Cys or in response to phagosomes containing killed yeast particles (zymosan; Sanjuan et al., 2007). LAP also contributes to the phagocytosis of apoptotic, necrotic, or receptor interacting kinase-3 (RIPK3)-necrotic cells with this response being triggered by the T cell immunoglobulin and mucin domain containing molecule (TIM)4 receptor (Martinez et al., 2011). Entosis, the engulfment of live cells, also involves localization of LC3 to the entotic vacuole (Florey et al., 2011). The recruitment of LC3 to phagosomes requires other components of the autophagy machinery given that it does not take place in the absence of Atg5 (Sanjuan et al., 2007; Martinez et al., 2011), Atg7 (Sanjuan et al., 2007; Martinez et al., 2011), or beclin-1 (Martinez et al., 2011). The role of autophagy in extracellular phagocytosis and MHCII antigen presentation is evident given that Atg5-deficient DC show impaired MHCII presentation of soluble and cell-associated antigen and elicit impaired CD4+ T cell responses in response to antigens derived from herpes simplex virus-2 (HSV-2; Lee et al., 2010). Infection of Atg5-deficient mice with HSV-2 results in a failure of Atg5-deficient mice to mount protective CD4+ T cell immunity and enhanced disease progression occurs (Lee et al., 2010). Autophagy also contributes to exogenous processing and MHCII presentation of HIV-1-derived protein (Blanchet et al., 2010). The autophagy machinery is considered to contribute to phagocytosis by facilitating phagosome to lysosome fusion and the delivery of lysosomal proteases to phagosomes, required for effective antigen degradation. Extracts of phagosomes isolated from Atg5-deficient DC displayed impaired cathepsin (Cat) S, CatB/L activity when assayed with fluorogenic substrates (Lee et al., 2010).
A contribution of autophagy to MHCI antigen presentation is less obvious, given that the major proteolytic source of peptide generation for MHCI is the proteasome and not lysosomal proteases. Indeed, there are several reports discarding a role for autophagy in MHCI presentation of antigen derived from extracellular sources (cross-presentation; Blanchet et al., 2010; Lee et al., 2010). Its role in MHCI presentation of cytosolic antigen is less well studied. One report demonstrates the requirement for autophagy in macrophage MHCI presentation of glycoprotein B (gB), a HSV-1-derived protein (English et al., 2009). In this case, treatment with 3-MA or silencing of Atg5 reduced presentation of gB by macrophages late after infection. HSV-1 gB is proposed to be trafficked from the nuclear envelope to the autophagosome from where its escapes for degradation by the proteasome and MHCI display. Whether this applies only to HSV-1 infection, macrophages, or is relevant to other antigens or cell types, remains to be elucidated. Another example is the cross-presentation of antigen loaded αAl2O3 nanoparticles (Li et al., 2011a). MHCI presentation of αAl2O3 nanoparticles loaded with OVA is inhibited following 3-MA treatment or silencing of Atg12 or beclin1. The mechanism is unknown, however internalized nanoparticles are shown to access autophagosomes that are considered to be immunogenic (Li et al., 2008). Autophagy may also impact MHCI presentation by degrading MHCI molecules themselves (Li et al., 2010).
An important contribution of autophagy to adaptive immunity is its cell intrinsic role in lymphocyte survival. An active autophagy pathway operates in T cells, with autophagosomes detected, particularly following activation via the T cell receptor (TCR) (Espert et al., 2006a; Li et al., 2006; Pua et al., 2007). Analysis of T cell compartments in Atg5-deficient mice illustrates a role for autophagy in mature T cell survival (Pua et al., 2007). While thymus cellularity is reduced in the absence of Atg5, thymocyte development is unperturbed. In the periphery, reduced numbers of CD4+ and CD8+ T cells are present in the spleen and lymph node. The CD8+ T cell compartment is more affected, with a large proportion of Atg5-deficient CD8+ T cells staining with Annexin V, indicative of cells undergoing early apoptosis. Both CD4+ and CD8+ T cells that survive in the absence of Atg5 display proliferative defects following TCR stimulation, with fewer cells undergoing division, and fewer divisions completed in response to anti-CD3 compared with wildtype T cells. The proliferative defect is not rescued by anti-CD28 or IL-2 and the cells display normal levels of TCR and upregulate T cell activation markers CD69 and CD25. A similar phenotype is observed in Atg7fl/flLck-Cre mice where the specific deletion of Atg7 in the T cell lineage results in mostly normal thymocyte development, however CD4+ and CD8+ T cell numbers are greatly reduced in the peripheral lymphoid compartment. Both subsets of peripheral T cells display evidence of apoptosis; with a large proportion staining with annexin V and exhibiting caspase 9 activity. Impaired T cell survival in the absence of autophagy is likely to arise from its pivotal role in maintaining intracellular organelle integrity. Atg7-deficient T cells possess increased numbers of mitochondria and increased ROS levels (Pua et al., 2009). For the B cell compartment, the absence of Atg5 impairs B cell development in the bone marrow, with reduced survival of pre-B cells and consequently reduced numbers of mature B-1a cells in the periphery (Miller et al., 2008). In some settings autophagy facilitates lymphocyte death, rather than survival. Examples include autophagy-mediated T cell death following growth factor withdrawal (Li et al., 2006) and following binding of the HIV-1 envelope protein to C-X-C chemokine receptor 4 (Espert et al., 2006b). Therefore, autophagy serves as a critical pathway in lymphocytes that facilitates the intricate balance between survival and death.
The critical impact of autophagy on effector functions of innate and adaptive immunity outlined herein, is highlighted by the association of autophagy gene mutations with increased incidence of inflammatory disease, susceptibility to microbial infection and autoimmune disease. A prominent example is Atg16L1, a susceptibility gene for Crohn’s disease (Massey and Parkes, 2007), where the small intestine exhibits chronic inflammation triggered by a breakdown in the clearance of commensal bacteria. Atg16L1-deficient Paneth cells possess elevated pro-inflammatory cytokine transcripts, while Atg16L1-deficient mice develop exacerbated intestinal inflammation following environmental triggers, similar to that observed in human patients (Cadwell et al., 2008). Other examples include cystic fibrosis, a chronic inflammatory disease of the pulmonary airways, where autophagy-mediated maintenance of lung epithelial cell homeostasis is important to limit lung inflammation and disease (Luciani et al., 2010). Autophagy is also postulated to contribute to inflammation-associated responses underlying metabolic diseases, such as obesity (Yang et al., 2010). In infectious disease settings, mouse models illustrate a contribution of autophagy to a growing number of microbial pathogens. IRGM, a human immunity-related GTPase that plays a critical role in regulating autophagy-mediated clearance of mycobacteria, is a genetic risk factor for tuberculosis (Intemann et al., 2009). Finally, a link between single nucleotide polymorphisms in the Atg5 gene and susceptibility to systemic lupus erythematosus, a debilitating autoimmune disease has been reported (Gateva et al., 2009). The emergence of often surprising roles for autophagy in diseases of the immune system means that a wide range of immune-related diseases may now be amenable to targeting by autophagy modulating drugs. As such studies of autophagy in innate and adaptive pathways provide excellent insights into this complex pathway, uncovering new roles for autophagy in disease.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Bernales, S., McDonald, K. L., and Walter, P. (2006). Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4, e423. doi:10.1371/journal.pbio.0040423
Birmingham, C. L., Smith, A. C., Bakowski, M. A., Yoshimori, T., and Brumell, J. H. (2006). Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281, 11374–11383.
Blanchet, F. P., Moris, A., Nikolic, D. S., Lehmann, M., Cardinaud, S., Stalder, R., Garcia, E., Dinkins, C., Leuba, F., Wu, L., Schwartz, O., Deretic, V., and Piguet, V. (2010). Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 32, 654–669.
Brazil, M. I., Weiss, S., and Stockinger, B. (1997). Excessive degradation of intracellular protein in macrophages prevents presentation in the context of major histocompatibility complex class II molecules. Eur. J. Immunol. 27, 1506–1514.
Cadwell, K., Liu, J. Y., Brown, S. L., Miyoshi, H., Loh, J., Lennerz, J. K., Kishi, C., Kc, W., Carrero, J. A., Hunt, S., Stone, C. D., Brunt, E. M., Xavier, R. J., Sleckman, B. P., Li, E., Mizushima, N., Stappenbeck, T. S., and Virgin, H. W. IV. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263.
Chaumorcel, M., Lussignol, M., Mouna, L., Cavignac, Y., Fahie, K., Cotte-Laffitte, J., Geballe, A., Brune, W., Beau, I., Codogno, P., and Esclatine, A. (2012). The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with beclin 1. J. Virol. 86, 2571–2584.
Chicz, R. M., Urban, R. G., Gorga, J. C., Vignali, D. A., Lane, W. S., and Strominger, J. L. (1993). Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J. Exp. Med. 178, 27–47.
Comber, J. D., Robinson, T. M., Siciliano, N. A., Snook, A. E., and Eisenlohr, L. C. (2011). Functional macroautophagy induction by influenza A virus without a contribution to major histocompatibility complex class II-restricted presentation. J. Virol. 85, 6453–6463.
Cooney, R., Baker, J., Brain, O., Danis, B., Pichulik, T., Allan, P., Ferguson, D. J., Campbell, B. J., Jewell, D., and Simmons, A. (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97.
Crisan, T. O., Plantinga, T. S., van de Veerdonk, F. L., Farcas, M. F., Stoffels, M., Kullberg, B. J., van der Meer, J. W., Joosten, L. A., and Netea, M. G. (2011). Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS ONE 6, e18666. doi:10.1371/journal.pone.0018666
Delgado, M. A., Elmaoued, R. A., Davis, A. S., Kyei, G., and Deretic, V. (2008). Toll-like receptors control autophagy. EMBO J. 27, 1110–1121.
Dengjel, J., Schoor, O., Fischer, R., Reich, M., Kraus, M., Muller, M., Kreymborg, K., Altenberend, F., Brandenburg, J., Kalbacher, H., Brock, R., Driessen, C., Rammensee, H. G., and Stevanovic, S. (2005). Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 7922–7927.
Dongre, A. R., Kovats, S., deRoos, P., McCormack, A. L., Nakagawa, T., Paharkova-Vatchkova, V., Eng, J., Caldwell, H., Yates, J. R. III, and Rudensky, A. Y. (2001). In vivo MHC class II presentation of cytosolic proteins revealed by rapid automated tandem mass spectrometry and functional analyses. Eur. J. Immunol. 31, 1485–1494.
Dorfel, D., Appel, S., Grunebach, F., Weck, M. M., Muller, M. R., Heine, A., and Brossart, P. (2005). Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro-transcribed MUC1 RNA. Blood 105, 3199–3205.
Dunn, W. A. Jr., Cregg, J. M., Kiel, J. A., van der Klei, I. J., Oku, M., Sakai, Y., Sibirny, A. A., Stasyk, O. V., and Veenhuis, M. (2005). Pexophagy: the selective autophagy of peroxisomes. Autophagy 1, 75–83.
English, L., Chemali, M., Duron, J., Rondeau, C., Laplante, A., Gingras, D., Alexander, D., Leib, D., Norbury, C., Lippe, R., and Desjardins, M. (2009). Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487.
Espert, L., Denizot, M., Grimaldi, M., Robert-Hebmann, V., Gay, B., Varbanov, M., Codogno, P., and Biard-Piechaczyk, M. (2006a). Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 116, 2161–2172.
Espert, L., Denizot, M., Grimaldi, M., Robert-Hebmann, V., Gay, B., Varbanov, M., Codogno, P., and Biard-Piechaczyk, M. (2006b). Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 116, 2161–2172.
Florey, O., Kim, S. E., Sandoval, C. P., Haynes, C. M., and Overholtzer, M. (2011). Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 13, 1335–1343.
Fujita, K., Maeda, D., Xiao, Q., and Srinivasula, S. M. (2011). Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc. Natl. Acad. Sci. U.S.A. 108, 1427–1432.
Gannage, M., Dormann, D., Albrecht, R., Dengjel, J., Torossi, T., Ramer, P. C., Lee, M., Strowig, T., Arrey, F., Conenello, G., Pypaert, M., Andersen, J., García-Sastre, A., and Münz, C. (2009). Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380.
Gateva, V., Sandling, J. K., Hom, G., Taylor, K. E., Chung, S. A., Sun, X., Ortmann, W., Kosoy, R., Ferreira, R. C., Nordmark, G., Gunnarsson, I., Svenungsson, E., Padyukov, L., Sturfelt, G., Jönsen, A., Bengtsson, A. A., Rantapää-Dahlqvist, S., Baechler, E. C., Brown, E. E., Alarcón, G. S., Edberg, J. C., Ramsey-Goldman, R., McGwin, G. Jr., Reveille, J. D., Vilá, L. M., Kimberly, R. P., Manzi, S., Petri, M. A., Lee, A., Gregersen, P. K., Seldin, M. F., Rönnblom, L., Criswell, L. A., Syvänen, A. C., Behrens, T. W., and Graham, R. R. (2009). A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41, 1228–1233.
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766.
Harris, J., Hartman, M., Roche, C., Zeng, S. G., O’Shea, A., Sharp, F. A., Lambe, E. M., Creagh, E. M., Golenbock, D. T., Tschopp, J., Kornfeld, H., Fitzgerald, K. A., and Lavelle, E. C. (2011). Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 286, 9587–9597.
Henell, F., Berkenstam, A., Ahlberg, J., and Glaumann, H. (1987). Degradation of short- and long-lived proteins in perfused liver and in isolated autophagic vacuoles – lysosomes. Exp. Mol. Pathol. 46, 1–14.
Intemann, C. D., Thye, T., Niemann, S., Browne, E. N., Amanua Chinbuah, M., Enimil, A., Gyapong, J., Osei, I., Owusu-Dabo, E., Helm, S., Rüsch-Gerdes, S., Horstmann, R. D., and Meyer, C. G. (2009). Autophagy gene variant IRGM-261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog. 5, e1000577. doi:10.1371/journal.ppat.1000577
Jagannath, C., Lindsey, D. R., Dhandayuthapani, S., Xu, Y., Hunter, R. L. Jr., and Eissa, N. T. (2009). Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat. Med. 15, 267–276.
Jounai, N., Takeshita, F., Kobiyama, K., Sawano, A., Miyawaki, A., Xin, K. Q., Ishii, K. J., Kawai, T., Akira, S., Suzuki, K., and Okuda, K. (2007). The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A. 104, 14050–14055.
Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., and Kim, D. H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295.
Kageyama, S., Omori, H., Saitoh, T., Sone, T., Guan, J. L., Akira, S., Imamoto, F., Noda, T., and Yoshimori, T. (2011). The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol. Biol. Cell 22, 2290–2300.
Ke, P. Y., and Chen, S. S. (2011). Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 121, 37–56.
Kirkin, V., Lamark, T., Sou, Y. S., Bjorkoy, G., Nunn, J. L., Bruun, J. A., Shvets, E., McEwan, D. G., Clausen, T. H., Wild, P., Bilusic, I., Theurillat, J. P., Øvervatn, A., Ishii, T., Elazar, Z., Komatsu, M., Dikic, I., and Johansen, T. (2009). A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 33, 505–516.
Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., Mizushima, N., Iwata, J., Ezaki, J., Murata, S., Hamazaki, J., Nishito, Y., Iemura, S., Natsume, T., Yanagawa, T., Uwayama, J., Warabi, E., Yoshida, H., Ishii, T., Kobayashi, A., Yamamoto, M., Yue, Z., Uchiyama, Y., Kominami, E., and Tanaka, K. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163.
Kraft, C., Deplazes, A., Sohrmann, M., and Peter, M. (2008). Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 10, 602–610.
Ku, B., Woo, J. S., Liang, C., Lee, K. H., Hong, H. S., E, X., Kim, K. S., Jung, J. U., and Oh, B. H. (2008). Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog. 4, e25. doi:10.1371/journal.ppat.0040025
Kundu, M., Lindsten, T., Yang, C. Y., Wu, J., Zhao, F., Zhang, J., Selak, M. A., Ney, P. A., and Thompson, C. B. (2008). Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112, 1493–1502.
Kyei, G. B., Dinkins, C., Davis, A. S., Roberts, E., Singh, S. B., Dong, C., Wu, L., Kominami, E., Ueno, T., Yamamoto, A., Federico, M., Panganiban, A., Vergne, I., and Deretic, V. (2009). Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186, 255–268.
Lee, H. K., Lund, J. M., Ramanathan, B., Mizushima, N., and Iwasaki, A. (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401.
Lee, H. K., Mattei, L. M., Steinberg, B. E., Alberts, P., Lee, Y. H., Chervonsky, A., Mizushima, N., Grinstein, S., and Iwasaki, A. (2010). In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32, 227–239.
Lee, J. S., Li, Q., Lee, J. Y., Lee, S. H., Jeong, J. H., Lee, H. R., Chang, H., Zhou, F. C., Gao, S. J., Liang, C., and Jung, J. U. (2009). FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11, 1355–1362.
Lelouard, H., Gatti, E., Cappello, F., Gresser, O., Camosseto, V., and Pierre, P. (2002). Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 417, 177–182.
Levine, B. (2005). Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120, 159–162.
Li, B., Lei, Z., Lichty, B. D., Li, D., Zhang, G. M., Feng, Z. H., Wan, Y., and Huang, B. (2010). Autophagy facilitates major histocompatibility complex class I expression induced by IFN-gamma in B16 melanoma cells. Cancer Immunol. Immunother. 59, 313–321.
Li, C., Capan, E., Zhao, Y., Zhao, J., Stolz, D., Watkins, S. C., Jin, S., and Lu, B. (2006). Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 177, 5163–5168.
Li, H., Li, Y., Jiao, J., and Hu, H. M. (2011a). Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat. Nanotechnol. 6, 645–650.
Li, Y. Y., Ishihara, S., Aziz, M. M., Oka, A., Kusunoki, R., Tada, Y., Yuki, T., Amano, Y., Ansary, M. U., and Kinoshita, Y. (2011b). Autophagy is required for toll-like receptor-mediated interleukin-8 production in intestinal epithelial cells. Int. J. Mol. Med. 27, 337–344.
Li, Y., Wang, L. X., Yang, G., Hao, F., Urba, W. J., and Hu, H. M. (2008). Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 68, 6889–6895.
Liang, C., Lee, J. S., Inn, K. S., Gack, M. U., Li, Q., Roberts, E. A., Vergne, I., Deretic, V., Feng, P., Akazawa, C., and Jung, J. U. (2008). Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 10, 776–787.
Luciani, A., Villella, V. R., Esposito, S., Brunetti-Pierri, N., Medina, D., Settembre, C., Gavina, M., Pulze, L., Giardino, I., Pettoello-Mantovani, M., D’Apolito, M., Guido, S., Masliah, E., Spencer, B., Quaratino, S., Raia, V., Ballabio, A., and Maiuri, L. (2010). Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 12, 863–875.
Martinez, J., Almendinger, J., Oberst, A., Ness, R., Dillon, C. P., Fitzgerald, P., Hengartner, M. O., and Green, D. R. (2011). Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. U.S.A. 108, 17396–17401.
Massey, D. C., and Parkes, M. (2007). Genome-wide association scanning highlights two autophagy genes, ATG16L1 and IRGM, as being significantly associated with Crohn’s disease. Autophagy 3, 649–651.
Matsunaga, K., Saitoh, T., Tabata, K., Omori, H., Satoh, T., Kurotori, N., Maejima, I., Shirahama-Noda, K., Ichimura, T., Isobe, T., Akira, S., Noda, T., and Yoshimori, T. (2009). Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396.
Miller, B. C., Zhao, Z., Stephenson, L. M., Cadwell, K., Pua, H. H., Lee, H. K., Mizushima, N. N., Iwasaki, A., He, Y. W., Swat, W., and Virgin, H. W. IV. (2008). The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 4, 309–314.
Mizushima, N., and Klionsky, D. J. (2007). Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19–40.
Nakagawa, I., Amano, A., Mizushima, N., Yamamoto, A., Yamaguchi, H., Kamimoto, T., Nara, A., Funao, J., Nakata, M., Tsuda, K., Hamada, S., and Yoshimori, T. (2004). Autophagy defends cells against invading group A Streptococcus. Science 306, 1037–1040.
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., Englert, J. A., Rabinovitch, M., Cernadas, M., Kim, H. P., Fitzgerald, K. A., Ryter, S. W., and Choi, A. M. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230.
Nakatogawa, H., Ichimura, Y., and Ohsumi, Y. (2007). Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178.
Nedjic, J., Aichinger, M., Emmerich, J., Mizushima, N., and Klein, L. (2008). Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455, 396–400.
Nimmerjahn, F., Milosevic, S., Behrends, U., Jaffee, E. M., Pardoll, D. M., Bornkamm, G. W., and Mautner, J. (2003). Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 33, 1250–1259.
Noda, N. N., Kumeta, H., Nakatogawa, H., Satoo, K., Adachi, W., Ishii, J., Fujioka, Y., Ohsumi, Y., and Inagaki, F. (2008). Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 13, 1211–1218.
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–731.
Orvedahl, A., Alexander, D., Talloczy, Z., Sun, Q., Wei, Y., Zhang, W., Burns, D., Leib, D. A., and Levine, B. (2007). HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1, 23–35.
Orvedahl, A., MacPherson, S., Sumpter, R. Jr., Talloczy, Z., Zou, Z., and Levine, B. (2010). Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127.
Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T., and Munz, C. (2005). Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596.
Pua, H. H., Dzhagalov, I., Chuck, M., Mizushima, N., and He, Y. W. (2007). A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 204, 25–31.
Pua, H. H., Guo, J., Komatsu, M., and He, Y. W. (2009). Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 182, 4046–4055.
Qu, X., Zou, Z., Sun, Q., Luby-Phelps, K., Cheng, P., Hogan, R. N., Gilpin, C., and Levine, B. (2007). Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128, 931–946.
Saitoh, T., Fujita, N., Hayashi, T., Takahara, K., Satoh, T., Lee, H., Matsunaga, K., Kageyama, S., Omori, H., Noda, T., Yamamoto, N., Kawai, T., Ishii, K., Takeuchi, O., Yoshimori, T., and Akira, S. (2009). Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. U.S.A. 106, 20842–20846.
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., Omori, H., Noda, T., Yamamoto, N., Komatsu, M., Tanaka, K., Kawai, T., Tsujimura, T., Takeuchi, O., Yoshimori, T., and Akira, S. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268.
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., and Wang, J. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235.
Sanjuan, M. A., Dillon, C. P., Tait, S. W., Moshiach, S., Dorsey, F., Connell, S., Komatsu, M., Tanaka, K., Cleveland, J. L., Withoff, S., and Green, D. R. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257.
Sanjuan, M. A., and Green, D. R. (2008). Eating for good health: linking autophagy and phagocytosis in host defense. Autophagy 4, 607–611.
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195.
Schmid, D., Pypaert, M., and Munz, C. (2007). Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26, 79–92.
Shi, C. S., Shenderov, K., Huang, N. N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., Sher, A., and Kehrl, J. H. (2012). Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263.
Singh, S. B., Davis, A. S., Taylor, G. A., and Deretic, V. (2006). Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313, 1438–1441.
Tanida, I., Ueno, T., and Kominami, E. (2004). LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518.
Tang, D., Kang, R., Livesey, K. M., Cheh, C. W., Farkas, A., Loughran, P., Hoppe, G., Bianchi, M. E., Tracey, K. J., Zeh, H. J. III, and Lotze, M. T. (2010). Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190, 881–892.
Thorburn, J., Horita, H., Redzic, J., Hansen, K., Frankel, A. E., and Thorburn, A. (2009). Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 16, 175–183.
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221.
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A., and Randow, F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418.
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhaes, J. G., Yuan, L., Soares, F., Chea, E., Le Bourhis, L., Boneca, I. G., Allaoui, A., Jones, N. L., Nuñez, G., Girardin, S. E., and Philpott, D. J. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62.
Uhl, M., Kepp, O., Jusforgues-Saklani, H., Vicencio, J. M., Kroemer, G., and Albert, M. L. (2009). Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 16, 991–1005.
Weidberg, H., Shvets, E., and Elazar, Z. (2011). Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 80, 125–156.
Wild, P., Farhan, H., McEwan, D. G., Wagner, S., Rogov, V. V., Brady, N. R., Richter, B., Korac, J., Waidmann, O., Choudhary, C., Dötsch, V., Bumann, D., and Dikic, I. (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233.
Xu, Y., Jagannath, C., Liu, X. D., Sharafkhaneh, A., Kolodziejska, K. E., and Eissa, N. T. (2007). Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27, 135–144.
Yang, L., Li, P., Fu, S., Calay, E. S., and Hotamisligil, G. S. (2010). Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478.
Yano, T., Mita, S., Ohmori, H., Oshima, Y., Fujimoto, Y., Ueda, R., Takada, H., Goldman, W. E., Fukase, K., Silverman, N., Yoshimori, T., and Kurata, S. (2008). Autophagic control of listeria through intracellular innate immune recognition in Drosophila. Nat. Immunol. 9, 908–916.
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., Mimuro, H., Nakagawa, I., Yanagawa, T., Ishii, T., Kakizuka, A., Sztul, E., Chakraborty, T., and Sasakawa, C. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240.
Zhong, Y., Wang, Q. J., Li, X., Yan, Y., Backer, J. M., Chait, B. T., Heintz, N., and Yue, Z. (2009). Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11, 468–476.
Keywords: autophagy, innate immunity, adaptive immunity, antigen presentation, T lymphocytes
Citation: Mintern JD and Villadangos JA (2012) Autophagy and mechanisms of effective immunity. Front. Immun. 3:60. doi: 10.3389/fimmu.2012.00060
Received: 23 January 2012; Accepted: 08 March 2012;
Published online: 04 April 2012.
Edited by:
Irina Caminschi, Burnet Institute, AustraliaCopyright: © 2012 Mintern and Villadangos. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Justine D. Mintern, Department of Biochemistry and Molecular Biology, The University of Melbourne, Parkville, VIC 3010, Australia. e-mail:am1pbnRlcm5AdW5pbWVsYi5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.