 Loredana Saveanu1,2*
Loredana Saveanu1,2* Peter van Endert1,2*
Peter van Endert1,2*- 1 Institut National de la Santé et de le Recherche Médicale, Paris, France
- 2 Faculté de Médecine, Université Paris Descartes, Paris, France
Production of MHC-I ligands from antigenic proteins generally requires multiple proteolytic events. While the proteolytic steps required for antigen processing in the endogenous pathway are clearly established, persisting gaps of knowledge regarding putative cross-presentation compartments have made it difficult to map the precise proteolytic events required for generation of cross-presented antigens. It is only in the past decade that the importance of aminoterminal trimming as the final step in the endogenous presentation pathway has been recognized and that the corresponding enzymes have been described. This review focuses on the aminoterminal trimming of exogenous cross-presented peptides, with particular emphasis on the identification of insulin responsive aminopeptidase (IRAP) as the principal trimming aminopeptidase in endosomes and phagosomes.
History
Insulin responsive aminopeptidase (IRAP) was initially identified due to its abundance in adipocytes, in specialized endosomes called Glut4 storage vesicles (GSV). The discovery of the glucose transporter Glut4 in 1989 (Birnbaum, 1989; James et al., 1989) was followed by sustained efforts to identify the biochemical composition of GSV, which revealed an abundant protein with a MW of around 160–165 kDa that was constantly associated with isolated GSV and called vp 165. Finally, Keller et al. (1995) identified several tryptic peptides from vp 165, designed oligonucleotide probes based on these peptide sequences and succeeded in cloning full-length IRAP cDNA from an adipose tissue cDNA library. Thus, intracellular localization of IRAP in GSV was the basis for its initial identification. Analysis of IRAP in the context of the regulation of glucose homeostasis has greatly advanced our knowledge about the cellular biology of IRAP endosomes, which are recognized as ubiquitous storage vesicles whose dynamics is regulated by cell-specific stimuli. However, why an aminopeptidase activity is present in GSV remains yet a mystery.
The situation was completely different for the second identification of the aminopeptidase, this time in the context of antigen presentation. In the early 2000s, evidence about the requirement of aminoterminal trimming of MHC class I ligand precursors accumulated (Lauvau et al., 1999; Fruci et al., 2001; Serwold et al., 2001) and prompted several laboratories, including ours, to search for the enzymes performing this final antigen-processing step. Searching for aminopeptidase activities in fractionated protein extracts of crude human B cell microsomes, we identified IRAP (Saveanu et al., 2009) as well as ERAP1–ERAP2 complexes (Saveanu et al., 2005b). The similarity between IRAP and the two ERAP proteins and the co-purification of MHC class I molecules with IRAP suggested that the enzyme was involved in antigen presentation. While the involvement of IRAP in endogenous presentation can not be entirely ruled out, the experimental data available today strongly support the conclusion that IRAP is required for trimming of epitope precursors exclusively in MHC class I antigen cross-presentation.
IRAP as a Member of the M1 Metallopeptidase Family
The genomic structure of the human IRAP gene (synonyms LNPEP, oxytocinase, P-LAP) is very similar to the ERAP1 and ERAP2 genes (Hattori et al., 2000; Rasmussen et al., 2000; Tanioka et al., 2005). These three genes are located contiguously on the human chromosome 5q15 suggesting the possibility of their divergence from a common ancestral gene. Interestingly, rodents have only two of these enzymes: IRAP (located on murine chromosome 17) and ERAP1 (located on the murine chromosome 13).
While both ER aminopeptidases are strongly induced by cell exposure to interferon gamma (IFN-γ), IRAP protein levels do not change upon IFN-γ stimulation. However, while the ERAP genes have the features of house keeping genes without any TATA or CAAT boxes, the IRAP gene is probably regulated during cell differentiation. Studies performed on the 5′ untranslated region of the IRAP gene in BeWo cells indicated that the transcription factors AP2 and Ikaros cooperatively up-regulate IRAP transcription during differentiation into trophoblastic cells and directly bind to the gene promoter (Iwanaga et al., 2003).
The human IRAP gene codes for a type II transmembrane protein with three domains: a cytoplasmic N-terminal domain of 109-amino-acid, a transmembrane domain of 23-amino-acid, and an intraluminal (or extracellular) domain of 893 amino acids, which include 16 potential N-glycosylation sites (Keller et al., 1995). The long C-terminal, intra-endosomal domain contains a Zn-binding motif HEXXH(X)18E and the exopeptidase motif GAMEN, which are encoded by exons 6 and 7. These two motifs are also found in ERAP1 and ERAP2 and are shared by all members of the M1 family of aminopeptidases (Tsujimoto and Hattori, 2005).
The high percentage of protein sequence identity between IRAP and ERAP1 or ERAP2 (43 or 49% identity, respectively), in correlation with the phylogenetic analysis (Saveanu et al., 2005a) indicates that these enzymes can be classified in a sub-family of M1 aminopeptidases referred to, using an alternative designation of IRAP, “oxytocinase family” (Tsujimoto and Hattori, 2005). The principal difference between the protein sequences of IRAP and the other members of M1 metallopeptidase family, including ERAP1 and 2, is the N-terminal cytoplasmic IRAP domain, which is required for the enzyme localization and its complex intracellular trafficking (see below).
Enzymatic Activity and Natural Peptide Substrates
Synthetic Analogs of Peptide Substrates
Several studies analyzed the substrate specificity of IRAP using fluorogenic analogs of peptide substrates (e.g., aminoacyl 7-amino-4-methylcoumarin; Saveanu et al., 2009; Georgiadou et al., 2010; Zervoudi et al., 2011) and demonstrated that the enzyme removes efficiently the following N-terminal aminoacids: Cys, Arg, Lys, Leu, Met, Tyr, Phe, Ala, and Gln. Although IRAP, ERAP1, and ERAP2 display substantial sequence identity and similarity, their substrate specificity as measured using fluorogenic substrates is quite different. Interestingly, the substrate specificity of IRAP is very similar to the specificity of the ERAP1–ERAP2 heterodimer with the difference that IRAP is active in a broader pH range than ERAPs. While 80% of maximal activity of IRAP is conserved at pH 5 or 8, ERAP1 preserved only 30% activity at these pH values (Georgiadou et al., 2010). The functional role of the differences in substrate specificity between IRAP and ERAPs will be discussed later.
Hormones and Vasoactive Peptides are Natural IRAP Substrates
Prior to its characterization as an epitope trimming aminopeptidase, IRAP has been described to inactivate several hormones, vasoactive peptides, and neuropeptides such as oxytocin, somatostatin, cholecystokinin, angiotensin III, Lys-bradikinin, vasopressin, Met-and Leu-enkephalin, neurokinin A, and dynorphin A (Tsujimoto et al., 1992; Herbst et al., 1997; Matsumoto et al., 2001).
A soluble form of human IRAP (starting at residue Ala155) can be detected in human serum during pregnancy. This form is generated by shedding of IRAP from the cell membrane by members of the ADAM (a disintegrin and metalloproteinase) family of endopeptidases (Ito et al., 2004). Increased levels of soluble IRAP in human serum during pregnancy and the ability of IRAP to inactivate the hormone oxytocin by cleavage between the N-terminal cysteine and the adjacent tyrosine residue initially designated IRAP as the enzyme regulating the oxytocin levels to prevent premature delivery. However, it is likely that other serum enzymes can also inactivate oxytocin since murine IRAP does not have the Phe154-Ala155 sequence where cleavage occurs to produce the soluble IRAP enzyme, and since IRAP deficient mice display normal reproductive and maternal behavior (Pham et al., 2009). In support of this conclusion, Keller’s group evaluated the oxytocin clearance from the circulation of wt and IRAP deficient mice and showed that oxytocin is inactivated in vivo in the absence of IRAP (Wallis et al., 2007). The same authors demonstrated that vasopressin is cleaved in vivo exclusively by IRAP and found that IRAP deficient mice have a decreased vasopressin synthesis, probably due to a negative feedback effect. In conclusion, until now, there is only one peptide hormone identified as a specific substrate for IRAP in vivo, vasopressin. A genetic study on patients undergoing septic shock further suggested that inactivation of vasopressin by IRAP is physiologically relevant (Nakada et al., 2011). In this study, an increased 28-day mortality in sepsis, which was accompanied by an increased vasopressin clearance, was associated with a SNP (single nucleotide polymorphism: rs4869317) located in a regulatory region of the IRAP gene that may alter the transcription of the gene.
IRAP Action on Neuropeptides Involved in Learning and Memory
Next to hormones and vasoactive peptides, neuropeptides involved in learning and memory represent an important group of IRAP substrates. Given that memory can be improved by angiotensin IV (AT4) administration (Braszko et al., 1988), the surprising identification of IRAP as a high affinity AT4 receptor in the brain (Albiston et al., 2001) prompted many efforts aiming at characterizing the role of IRAP role in memory and learning. The group of Albiston and Chai showed first that AT4 is a high-affinity inhibitor of IRAP by binding to the active site of the enzyme (Lew et al., 2003) and developed later a new class of IRAP inhibitors which are cognitive enhancers (Albiston et al., 2008). Several hypothetical mechanisms were initially considered for memory enhancement induced by AT4 (Stragier et al., 2008). However, it remains difficult today to draw a conclusion concerning the role of IRAP as an AT4 receptor and in memory mechanisms. Even the analysis of IRAP deficient mice did not shed much light on this issue. A clear result obtained using these mice is that in the absence of IRAP the high affinity-binding site for AT4 is lost in the brain and other tissues, providing strong evidence that IRAP is the AT4 receptor (Albiston et al., 2010). This result is important, considering that the AT4 receptor identity was controversial until recently (De Bundel et al., 2008; Stragier et al., 2008; Wright et al., 2008). Nevertheless, the analysis of the memory phenotype of IRAP deficient mice displayed unexpected results. Despite the fact that acute administration of IRAP inhibitors increases memory, constitutive IRAP deletion in mice does not improve memory (Albiston et al., 2010).
Tissue Distribution and Intracellular Localization
Even though IRAP was first identified and thus extensively studied in adipocytes, the enzyme is expressed in a very large array of tissues. Initially, IRAP was detected by immunoblot at similarly high levels in heart, spleen, brain, lung, and adipose tissues and at low level in kidney and muscles. The only tissue in which IRAP amounts were below the limit of immunoblot detection was the liver (Keller et al., 1995). Subsequently a more detailed analysis was performed on brain, placenta, and spleen cells, showing that the enzyme has a cell-specific expression pattern. In the brain its expression is restricted to hippocampus, neocortex, and motor neurons (Fernando et al., 2005). In the placenta, IRAP is expressed mainly in differentiated cells in the trophoblast and the syncytiotrophoblast (Nomura et al., 2005). In the spleen the enzyme is detected in dendritic cells (DCs), B cells, T cells (with highest level of expression observed in conventional CD11chigh DCs), and it is absent from granulocytes (Saveanu et al., 2009).
In all the studied cells the enzyme resides in endosomal vesicles. IRAP vesicles recycle slowly in the basal state and can be rapidly and massively translocated to the plasma membrane upon cell-specific stimulation. These vesicles are often called GSV or insulin responsive compartment (IRC) in adipocytes and muscle cells. Since there is no obvious difference between IRAP-containing endosomes in different cell types, we prefer to name the IRAP vesicles “cell-specific storage endosomes.” It is now well established that the entire information required for the endosomal localization of IRAP, for its slow recycling and its sensitivity to cell-specific regulation is encoded by the N-terminal cytosolic tail of the protein. For example, a chimera between the N-terminal IRAP domain and the transferrin receptor (TfR) displayed the same intracellular localization and trafficking as the full-length IRAP (Subtil et al., 2000; Hou et al., 2006). Fusion proteins composed by the IRAP cytosolic tail and TfR or EGFP were extensively used as tools to analyze the consequences of different mutations in the targeting motifs that are present in the cytosolic tail of IRAP. The most studied targeting sequences are the two dileucine motifs: LL53,54 and LL76,77. The LL53,54 signal seems to be a form of the (D/E)XXXL(L/I) type signal in which the acidic D or E residue is substituted with a basic R residue (RXXXLL; Bonifacino and Traub, 2003). Interestingly, GLUT4 also contains a RXXXLL sequence, and in this context the RXXXLL signal seems to be important for routing of endocytosed GLUT4 from the plasma membrane to the insulin responsive storage vesicles (Sandoval et al., 2000). However, in the case of IRAP, the substitution of LL53,54 by AA53,54 did not affect the enzyme trafficking in differentiated 3T3-L1 adipocytes (Hou et al., 2006). Unlike this mutation, the substitution of LL76,77 by AA76,77 had a strong impact on IRAP localization and trafficking and resulted in rapid transport of newly synthesized IRAP to the plasma membrane, in a manner indistinguishable from other proteins constitutively directed to the plasma membrane such as vesicular stomatitis virus G coat protein (VSV-G), GLUT1 or syntaxin3 (Watson et al., 2008).
Transient expression of IRAP–GFP demonstrated that newly synthetized IRAP accumulates in storage endosomes as early as 3 h after transfection (Watson et al., 2004). The acquisition of insulin sensitivity starts at 6 h post transfection and is fully accomplished after 9 h. During the first 3 h after enzyme synthesis, IRAP trafficking is brefeldin A sensitive, but once it enters the storage endosomes its translocation to the plasma membrane becomes insensitive to brefeldin A treatment (Watson et al., 2008). In adipocytes, the sorting of newly synthesized IRAP from the trans-Golgi network (TGN) to the storage endosomes requires the clathrin adaptor GGA1, a member of the γ-ear-containing, ADP-ribosylation factor (Arf)-binding (GGA) family of clathrin adaptors (Figure 1; Liu et al., 2005; Hou et al., 2006). A chimera between the cytosolic IRAP tail bearing the AA76,77 mutation and the TfR bypasses the GGA1 dependent sorting step in the TGN, but once it arrives at the plasma membrane it can be endocytosed normally, reaches the storage endosomes and is even able to translocate back to the cell surface upon insulin treatment (Watson et al., 2008).
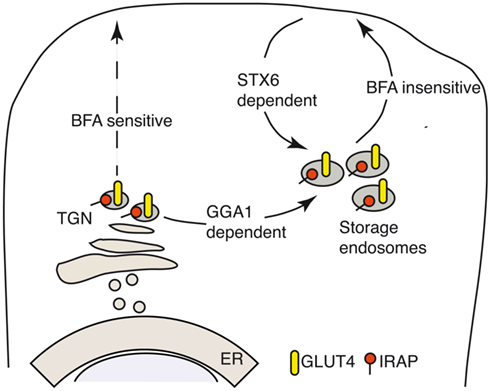
Figure 1. IRAP storage vesicles are slow recycling endosomes. Newly synthetized IRAP accumulates in storage endosomes as early as 3 h after transfection (Watson et al., 2004) and acquires insulin responsiveness after 6–9 h. During the first 3 h after enzyme synthesis, IRAP sorting from the ER is brefeldin A sensitive. Once the enzyme reaches the storage endosomes its translocation to the plasma membrane becomes insensitive to brefeldin A treatment (Watson et al., 2008). Sorting of newly synthetized IRAP from the TGN to the storage endosomes in adipocytes requires the GGA1 clathrin adaptor (Liu et al., 2005; Hou et al., 2006) and the LL76,77 sequence in the cytosolic domain of IRAP (Watson et al., 2008).
Regulation of IRAP Trafficking
Signaling Pathways Involved in IRAP Trafficking Regulation
The regulation of IRAP trafficking is understood by far in most detail in insulin responsive tissues, mainly in adipocytes, where it has been studied for nearly two decades. As already mentioned before, in these cells, IRAP and GLUT4 are co-localizing in storage endosomes called GSV. Insulin stimulates glucose uptake into adipocytes by stimulating translocation of GLUT4 from the storage endosomes to the plasma membrane (Antonescu et al., 2009). In the absence of insulin, GLUT4 is stored by active mechanisms and sequestered away from the common recycling endosomes (Martin et al., 2006). GLUT4 exocytosis to the plasma membrane from the storage endosomes is very slow compared with general exocytosis from recycling endosomes (Zeigerer et al., 2004). Insulin accelerates GLUT4 and IRAP transport to the cell surface and simultaneously reduces GLUT4 internalization having as a global effect a rapid increase of GLUT4 in the plasma membrane of insulin-stimulated adipocytes. The insulin receptor is a member of the tyrosine kinase receptor family. Binding of insulin to its receptor induces receptor autophosphorylation on several cytoplasmic tyrosines and the rapid recruitment and phosphorylation of the effector proteins insulin receptor substrate 1 and 2 (IRS1, IRS2), and Shc (Goalstone and Draznin, 1997). Tyrosine phosphorylation of IRSs induces the binding of SH2 domains of the regulatory subunits of class I-A PI3Ks, which initiate the subsequent intracellular signaling. Briefly, the PI3K activity produces the lipid PtdIns(3,4,5)P3, which will recruit to membranes the phosphoinositide-dependent kinase-1 (PDK1) and PDK1 substrates, including the kinases of Akt/PKB family (Siddle, 2011). There are three Akt proteins: Akt1, Akt2, and Akt3, which are encoded by separate genes and which have different functional specificities.
Production of different Akt deficient mice demonstrated that Akt1 is important for cell survival, while Akt2 deficient mice have impaired glucose homeostasis and develop type II diabetes and Akt3 deficiency induces abnormal brain development (Gonzalez and McGraw, 2009a). Even though both Akt1 and Akt2 can be recruited to the plasma membrane after PI3K activation, it has been shown by TIRF microscopy that it is mainly Akt2 that is recruited to the plasma membrane upon insulin stimulation. Moreover, siRNA knockdown of Akt2 demonstrated that the protein is required for GLUT4 translocation in response to insulin stimulation (Gonzalez and McGraw, 2009b). These in vitro data correlated with the phenotype of Akt2 deficient mice and suggest that Akt2 is the essential Akt isoform controlling trafficking of IRAP–GLUT4 storage endosomes in adipocytes. Nevertheless, a functional overlap among the three Akt isoforms exists. This is suggested by the fact that mice with a single Akt isoform deletion are viable, while mice with double knockouts for Akt are not. The simultaneous deletion of Akt1 and Akt2 leads to death immediately after birth (Peng et al., 2003) and Akt1 and Akt3 double knockout mice are embryonic lethal (Yang et al., 2005). The overlapping roles of Akt1 and Akt2 could also be involved in particular situations in IRAP–GLUT4 trafficking in adipocytes. For example, forced localization of Akt1 to the plasma membrane [via E17K mutation of Akt1 (Gonzalez and McGraw, 2009b) or by addition of a myristoyl group to Akt1 (Kohn et al., 1996)] was demonstrated to induce an Akt2-like signaling and translocation of GLUT4 vesicles (and probably IRAP) to the plasma membrane.
Protein Interactions of the Cytosolic Domain of IRAP
In adipocytes and muscles PI3K-PDK1-Akt are the protein kinases that are activated in a cascade after insulin receptor stimulation and Rab8, Rab10, and Rab14 the small GTPases that drive GSV translocation to the cell surface. Much of the work leading to the description of this signaling pathway (Figure 2) was carried out using GLUT4 or IRAP as markers of GSV, without discriminating between the function of these proteins as simple cargo or active players in GSV trafficking. It is important to note that several findings suggest that both proteins, and especially IRAP, might have more complex roles than simple cargos in vesicular trafficking. First, it was observed that deletion of either of these proteins affects the stability of the other. Mice deficient for IRAP (Keller et al., 2002) displayed a reduced level of GLUT4 protein (50–80% reduction) and mice deficient for GLUT4 had a redistribution of IRAP to the plasma membrane (Jiang et al., 2001). These in vivo data, which reflect the situation in primary adipocytes isolated from mice, were only partially confirmed by in vitro experiments using siRNA knockdown of GLUT4 and IRAP in 3T3-L1 adipocytes (Jordens et al., 2010). In 3T3-L1 adipocytes, GLUT4 knockdown does not change the distribution of IRAP between the plasma membrane and intracellular vesicles, indicating that IRAP traffic is independent of GLUT4. However, the IRAP knockdown in 3T3-L1 differentiated adipocytes affected GLUT4 trafficking, increasing three times its level at the plasma membrane. At the same time, intracellular GLUT4 was partially rerouted to constitutive endosomes that contain the TfR. When insulin was added to IRAP knockdown adipocytes, the level of GLUT4 at the plasma membrane increased further to a similar extent as in wt adipocytes.
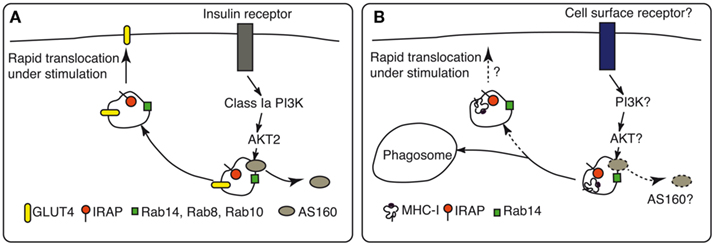
Figure 2. Cell-specific regulation of IRAP storage vesicles. (A) In insulin-responsive tissues, such as adipocytes and muscles, IRAP trafficking is regulated by insulin. Upon insulin binding to its receptor, PI3K-PDK1-Akt protein kinases are activated in a cascade. The most important effector in these cells seems to be the RabGAP AS160, which is phosphorylated by Akt2. Phosphorylation of AS160 leads to its dissociation from the storage endosomes and activation of Rab8, Rab10, and Rab14 that drive the endosome translocation to the cell surface. The biological effect is the increase at cell surface of the principal vesicle cargo, the glucose transporter GLUT4. (B) In other cells, the stimuli that regulate IRAP endosome trafficking and the signaling pathways involved in this process are poorly characterized. In DCs, IRAP endosomes are recruited rapidly to the phagosomal membrane. The phagocytic receptors and the signaling molecules responsible for this phenomenon are not yet identified.
Thus, IRAP is required for intracellular retention of GLUT4 but not for sensitivity of its localization to insulin stimulation. The expression of the cytosolic tail of IRAP was sufficient to recover the normal intracellular distribution of GLUT4 in IRAP knockdown adipocytes. The concomitant analysis of the TfR in IRAP knockdown adipocytes showed that its trafficking was not affected by the absence of IRAP, indicating that the role of IRAP is specific to the trafficking of storage endosomes and does not regulate constitutive TfR+ recycling endosomes (Jordens et al., 2010). Thus recruitment of the sorting machinery for GLUT4 targeting to storage endosomes is at least partially accomplished by the cytosolic domain of IRAP. The cytosolic domain of IRAP might recruit sorting machinery components since it interacts with several proteins implicated in protein sorting, vesicle formation, and coupling of the signaling pathways with cytoskeleton remodeling.
Early after its identification, IRAP was found to interact with tankyrase-1 and 2 (Chi and Lodish, 2000), two modular proteins with both poly(ADP)-ribosylation enzymatic activity and scaffolding activities. Tankyrases interact via their ankyrin-repeat domain with diverse partners having multiple biological roles. Among the complex functions attributed to tankyrases, the implication in regulation of Golgi vesicle trafficking and the regulation of protein targeting in response to growth factor signaling are especially relevant in the context of their interaction with IRAP. The identification of the “RXXPDG” sequence as the tankyrase-binding motif of IRAP (Sbodio and Chi, 2002) might allow a better comprehension of the IRAP-tankyrase interaction via mutagenesis experiments.
Another protein involved in the transport between Golgi stacks that was found to interact directly with the cytosolic tail of IRAP is p115 (Hosaka et al., 2005), which assists vesicle fusion by assembling SNARE pin complexes. In addition to these Golgi proteins interacting with IRAP, co-immunoprecipitation experiments and yeast two-hybrid analysis revealed the interaction of the cytosolic domain of IRAP with two cytoskeleton-linked factors: vimentin (Hirata et al., 2011) and FHOS (formin homolog overexpressed in the spleen; Tojo et al., 2003). Vimentin is a part of intermediate filaments in the cytoskeleton, which support and anchor cellular organelles. Formins are multidomain proteins essential for actin polymerization that are involved in the transport of vesicles on actin cables (Goode and Eck, 2007). Since the cytosolic part of IRAP interacts with a protein important in organelle anchoring to the cytoskeleton and another protein involved in guided endosome motility along the cytoskeleton, it is tempting to speculate that the IRAP cytosolic tail regulates GSV trafficking and possibly specialized sorting in several other cell types. It will be of interest to confirm these protein interactions of IRAP in different cell types and assess their function in the regulation of storage endosome trafficking.
Additional indications about a regulatory role of IRAP in GSV trafficking came from two recent studies that reported an interaction between the cytoplasmic domain of IRAP and a well-characterized Akt substrate, the RabGAP AS160, an interaction detected by reciprocal co-immunoprecipitation experiments (Larance et al., 2005; Peck et al., 2006). The IRAP sequence required for the interaction with AS160 was mapped to the region between amino acids 27 and 58 of the IRAP cytosolic domain (Larance et al., 2005). Because the presence of AS160 on GSV is a key player in intracellular retention of GSV in the basal state, the interaction of AS160 with IRAP supports the hypothesis that IRAP is a critical component of GSV retention machinery. However, another recent study concluded that IRAP knockdown in 3T3-L1 adipocytes affect neither AS160 association with GSV nor its regulation by insulin via PI3K-Akt-AS160 (Jordens et al., 2010). It seems therefore that IRAP is not essential for the targeting of AS160 to GSV and even if IRAP binds AS160, additional sites of interaction for AS160 with GSV membranes must exist.
IRAP Trafficking in Immune Cells
Among the cell types of the immune system, IRAP was only studied in mast cells (Liao et al., 2006) and DCs (Saveanu et al., 2009; Segura et al., 2009). In bone marrow-derived mast cells and in rat peritoneal mast cells, IRAP is highly expressed and localizes to intracellular vesicles. IRAP vesicles in mastocytes contain VAMP3 and VAMP2 and are different from the secretory granules since there is no overlap between IRAP and histamine or CD63, which are markers of secretory granules. By cell-surface biotinylation experiments, Liao et al. demonstrated that IRAP translocates rapidly to the plasma membrane upon stimulation of mast cells by antigen/immunoglobulin E (IgE) complexes. While exocytosis of mast cell secretory granules following stimulation by antigen-IgE complexes requires PI3K and PKC activities, export of IRAP endosomes to the cell surface is independent of PKC and PI3K activities but depends on intracellular calcium. Thus, interestingly, in mast cells the same extracellular signal, antigen-IgE complexes, induce exocytosis of two intracellular vesicle pools, secretory granules and IRAP vesicles, although the signaling pathways regulating these transport events are different. Contrary to what would be expected by analogy with adipocytes, the activation of PI3K does not induce IRAP translocation to the plasma membrane. These results highlight the cell-specific behavior of IRAP endosomes and show that results obtained in a given cell type cannot readily be extended to others.
We analyzed the intracellular distribution of IRAP in several types of DCs: human monocytes-derived DC, murine bone marrow-derived DC (BMDCs; Saveanu et al., 2009), splenic DC subsets (conventional CD8+ and CD8− DCs and plasmacytoid DCs, pDCs; Weimershaus et al., 2012). In all DC subsets analyzed, the enzyme colocalized at high levels (more than 50% of the total IRAP signal) with Rab14, syntaxin 6 (STX6), and the type1-mannose receptor (MR, alternatively named CD206) and to lesser extent (20 to 30% of total IRAP signal) with EEA1 and MHC class I. The extracellular stimuli and the signaling pathways that regulate IRAP endosomes trafficking in DCs are yet unknown. By several experimental approaches (immunofluorescence microscopy, cell fractionation, flow cytometry analysis of isolated phagosomes), we have demonstrated that IRAP endosomes are recruited to early phagosomes in DCs (Saveanu et al., 2009; Weimershaus et al., 2012), but the phagocytic receptors that induce IRAP vesicle fusion with the phagosome are still unknown.
Immunological Function of the Enzyme
Our initial observation that connected IRAP with MHC class I antigen presentation was the co-purification of these two proteins during the screening of crude human microsome lysates for enzymes involved in aminoterminal trimming of MHC class I ligands. IRAP and MHC class I were identified from an IFN-γ-induced peak of aminopeptidase activity isolated by anion exchange chromatography. Surprisingly, subsequent analyses showed that while MHC class I protein synthesis was strongly increased, IRAP mRNA was not up-regulated upon IFN-γ treatment of HeLa cells. This suggested that either IRAP changed its intracellular localization upon IFN-γ treatment or that increased recovery of IRAP activity was the consequence of its association with IFN-γ-induced MHC class I. While the former hypothesis remains to be studied in detail, association of IRAP with MHC class I molecules was confirmed by reciprocal co-immunoprecipitation experiments in murine BMDCs (Saveanu et al., 2009), as well as in human lymphoblastoid cell lines (our unpublished data).
Trimming of Antigenic Peptides by IRAP
The ability of IRAP to digest peptide precursors of MHC class I ligands was demonstrated by several teams. Initially we demonstrated that IRAP purified from human microsomes, as well as the recombinant enzyme, efficiently processed two MHC class I ligand precursors: the HIV gag derived peptide, R-SLYNTVATL and the HIV gp160 derived peptide, KIRIQR-GPGRAFVTI (final epitope underlined; Saveanu et al., 2009). Equivalent amounts of isolated ERAP1 or ERAP2 were relatively inefficient in the production of the final epitope from the HIV gp160 precursor, which needs the combined action of ERAP1–ERAP2 (Saveanu et al., 2005b). Notably, IRAP was as efficient as the ERAP1–ERAP2 mixture in the generation of HIV gp160 final epitope, suggesting that the specificity of IRAP resembles that of ERAP1 and ERAP2 combined. The group of Stratikos confirmed this finding in two subsequent reports that analyzed the substrate specificity of ERAP1, ERAP2, and IRAP (Georgiadou et al., 2010; Zervoudi et al., 2011). Using fluorogenic analogs of peptide substrates, they demonstrated that IRAP has broader substrate specificity than isolated ERAP1 or ERAP2 and a broader pH range for optimal activity. However, as it has been demonstrated for ERAP1 (Hearn et al., 2009), the aminopeptidase activity against fluorogenic substrate does not always match the specificity of trimming natural peptides. Therefore, testing IRAP specificity against peptide substrates with systematic variation of the N-terminal residues needs further experimental work. The relative pH independence of IRAP makes the enzyme more appropriate for antigen trimming in endosomes, known for their dynamic pH changes. Taking advantage of the availability of the crystal structure of ERAP1 (Kochan et al., 2011; Nguyen et al., 2011) and the high similarity between these trimming aminopeptidases, the structure of ERAP1 was used to make homology models of the other two aminopeptidases, which allowed the analysis of S1 pocket of all enzymes (Zervoudi et al., 2011). The S1 pocket is defined by 12 amino acids. Six of them are identical for all three enzyme and the other six confer the substrate specificity. In direct correlation with the enzyme ability to cleave basic amino acids, the S1 pocket has different numbers of acidic amino acids: three for ERAP2 (E177, D198, and D888), two for IRAP (E426 and E541), and only one for ERAP1 (E865). Mutagenesis of E541 to R in the S1 pocket of IRAP resulted in a selectivity profile similar to that of ERAP1. The authors concluded that residue E541 is largely responsible for the ability of IRAP to process peptides that are substrates for ERAP2 in the ER.
Among trimming aminopeptidases, only human ERAP1 has been studied extensively with respect to substrate specificity (Chang et al., 2005; Hearn et al., 2009). Using peptide libraries Chang et al. (2005) have demonstrated that human ERAP1 digests optimally peptide with a length of 9–16 amino acids. The transporter associated with antigen presentation (TAP) has an identical length preference for the substrate (van Endert et al., 1994). Given the concordance between ERAP1 and TAP substrate length specificity, Chang et al. proposed the attractive model of “molecular ruler” action for ERAP1. According to this model, ERAP1 is exceptionally adapted to produce MHC class I peptides that have typically 8–10 amino acids length. There are arguments for, and against, the molecular ruler model of ERAP1 mode of action. Several studies in vitro showed that ERAP1, as well as its murine homolog ERAAP, could destroy several epitopes, findings in potential conflict with the molecular ruler model (Serwold et al., 2002; York et al., 2002; Georgiadou et al., 2010). On the other hand, the structural analysis of ERAP1 offered a mechanism for the molecular ruler model. According to Nguyen et al. (2011), the enzyme has a regulatory site close to the catalytic site and only long peptides are capable to bind to the regulatory site, inducing the conformational change required for catalytic activity. Next to the structural analysis, one of the strongest arguments for the molecular ruler mode of action of ERAP1 is perhaps the fact that not only the amino terminal residues, but also the carboxy-terminal residues of the peptide substrate control the trimming efficiency, indicating that the enzyme interacts with both peptide ends at the same time (Chang et al., 2005).
The lack of both a systematic analysis of its trimming activity and of a crystal structure does not allow for predicting if IRAP has substrate length or sequence preferences. Georgiadou et al. (2010) performed the so far most detailed analysis of antigenic peptide trimming activity of IRAP. They evaluated the trimming of 14 peptide precursors by IRAP and ERAP1. ERAP1 produced the final epitopes in 13 out of 14 and IRAP in 10 out of 14 cases. The authors concluded that, like ERAP1, IRAP can trim long antigenic peptides efficiently and, in the majority of cases, it accumulates considerable amounts of final antigenic epitope. It is important to mention that both peptidases destroyed some of the final epitopes analyzed. ERAP1 over-digested 6 and IRAP 9 of the 14 precursors. The number of peptides processed in this study is too small to evaluate the substrate length specificity of IRAP. However, a look at the analyzed peptide sequences suggests that IRAP has preferences unrelated to the N-terminal residue, since the same residues are removed with different efficiency depending on the studied peptide. In conclusion, the enzymatic activity data available today indicate that IRAP is capable to trim MHC class I peptide precursors.
The Role of IRAP in MHC Class I Antigen Processing
Analysis of BMDCs from IRAP deficient mice allowed us to demonstrate that IRAP trimming activity is not required for direct presentation (or endogenous presentation) by MHC class I of two model epitopes: the SIINFEKL peptide, an H-2Kb restricted epitope derived from ovalbumin and the KCSRNRQYL peptide, a Db restricted epitope derived from the SMCY male antigen (Saveanu et al., 2009). The production of these two epitopes in the endogenous processing pathway requires ERAP and proteasome activity, but not IRAP. These results strongly suggested that IRAP is not involved in the endogenous MHC class I processing pathway and are in concordance with the endosomal localization of IRAP. In steady-state conditions, in human monocyte-derived DCs (moDCs) and murine BMDCs, we could not detect colocalization of IRAP with ER-resident proteins such as ERAP1, TAP, calnexin or the KDEL receptor. Using ER-targeted variants of IRAP, we verified that the absence of IRAP colocalization with ER markers was not due to the inability of antibodies to recognize ER-resident IRAP. When the KDEL sequence was added to the C-terminal end of the enzyme, the IRAP-specific antibodies detected a strong colocalization of IRAP with two ER markers: TAP and the KDEL receptor. We concluded that the endogenous newly synthesized IRAP molecules exit rapidly from the ER and travel to endosomes containing Rab14, STX6, and MR.
The endosomes containing IRAP are massively recruited to early phagosomes (Saveanu et al., 2009). This was demonstrated by fluorescence microscopy experiments and immunoblot analysis of isolated phagosomes containing latex beads. In the same experimental settings we could not detect the murine ER peptidase (ERAP1/ERAAP) in the phagosomes. These results were in agreement with the proteomic analysis of latex beads phagosomes carried out by Rogers and Foster (2007) who detected IRAP, but not ERAP, in isolated phagosomes. Thus, in our hands, IRAP was the sole member of the oxytocinase family of potential trimming aminopeptidases present in DC phagosomes. The absence of ERAP in phagosomes is of particular interest considering the previous reports suggesting a fusion between ER membranes and newly formed phagosomes (Ackerman et al., 2003; Guermonprez et al., 2003; Houde et al., 2003) and the later controversy concerning the existence and relevance of this phenomenon (Touret et al., 2005). Our results suggest that the ER-phagosome fusion is a selective process, which can provide integral membrane proteins as TAP to the phagosome, but not soluble proteins such as ERAP.
The endosomal and phagosomal localization of IRAP suggested that IRAP might be involved in MHC class I cross-presentation of exogenous antigens. Indeed, IRAP deficiency partially compromised cross-presentation of ovalbumin antigen internalized through phagocytosis both in vitro (cross-presentation of ovalbumin-coated latex beads and necrotic cells expressing ovalbumin by BMDCs) and in vivo (mice immunized with ovalbumin-polyI:C loaded apoptotic cells). Moreover, IRAP was required for efficient in vivo cross-presentation of ovalbumin internalized by receptor-mediated endocytosis (Saveanu et al., 2009; ovalbumin fusion proteins targeted to TLR2 and MR; Kratzer et al., 2010). Importantly, IRAP was involved in a cross-presentation pathway that also requires proteasome activity. While IRAP deficiency decreased the ability of DCs to cross-present ovalbumin by about half, proteasome inhibition (using conditions strictly controlled to avoid toxic side effects) almost abolished cross-presentation completely. These experiments suggested that the initial steps in processing of cross-presented epitopes are almost exclusively performed by the proteasome, while more than one enzyme can perform the last step of antigen processing, the aminoterminal trimming. Analysis of ERAP-deficient mice and BMDCs had previously identified ERAP as another enzyme that can perform the aminoterminal trimming of cross-presented peptides (Yan et al., 2006; Firat et al., 2007). Consistent with this, we found that the absence of either IRAP or ERAP alone reduced cross-presentation of ovalbumin by about half, while the simultaneous deletion of ERAP and IRAP had an additive effect (Saveanu et al., 2009). This functional redundancy, correlated with the absence of ERAP–IRAP colocalization, led us to the hypothesis that the two enzymes act in two independent pathways of proteasome-dependent cross-presentation.
An Updated View of Cross-Presentation Pathways
Classically, cross-presentation is divided in two pathways: one that is TAP and proteasome-dependent and one that is TAP and proteasome independent. In the proteasome independent pathway, commonly referred to as vacuolar cross-presentation, internalized exogenous antigens remain in endocytic vesicles where they are processed by acidic lysosomal proteases, with a prominent role for the cathepsin S (Shen et al., 2004) and where loading of the MHC class I molecules occurs. In the proteasome-dependent pathway, which is considered more efficient than the vacuolar pathway (Sigal and Rock, 2000), the antigens are shuttled into the cytosol. Several groups demonstrated that intact, functional proteins such as the toxin gelonin, the enzyme horseradish peroxidase (HRP), or ovalbumin internalized by macrophages and DCs are transferred to the cytosol where they are digested by the proteasome (Kovacsovics-Bankowski and Rock, 1995; Norbury et al., 1995; Rodriguez et al., 1999). One possibility is that the resulting peptides are transported by TAP into the perinuclear ER where they bind to newly synthesized MHC class I molecules (Gromme and Neefjes, 2002; Rock and Shen, 2005). In the context of the latter model, ERAP involvement in proteasome-dependent cross-presentation is an expected finding. However, both experimental observations suggesting fusion between the ER and phagosomes and our discovery that endosomal IRAP plays a role in cross-presentation suggest strongly that MHC class I loading with cross-presented peptides is not limited to the perinuclear ER and can occur in endosomal or phagosomal compartments.
Independent of our work, several recent reports have put forward endosomal subpopulations as active players in cross-presentation (Burgdorf et al., 2007, 2008; Kutomi et al., 2009). It has been demonstrated that ovalbumin endocytosed via the MR reaches an early endosomal compartment (EEA1+, Rab5+) distinct from lysosomes, which enables cross-presentation, while ovalbumin ingested via pinocytosis reaches lysosomes and results in antigen presentation via MHC class II (Burgdorf et al., 2007). Burgdorf proposed that MR+ endosomes are likely to be identical or largely overlap with the early static endosomes described earlier (Lakadamyali et al., 2006). By tracking fluorescent Rabs in live cells, these authors demonstrated that early endosomes consist of two distinct populations: one highly mobile on microtubules and maturing rapidly toward lysosomes and a second “static” maturing much more slowly. Since it is now clearly established that the efficiency of antigen cross-presentation is enhanced by limiting proteolysis and maintaining a close to neutral pH in endolysosomes (Savina et al., 2006; Jancic et al., 2007), it would be interesting to test the overlap of mannose receptor with “static” slowly maturating early endosomes. The substantial colocalization of IRAP with the MR (50–75% depending on the DC type analyzed) and the colocalization of the enzyme with EEA1 and endocytosed soluble ovalbumin (Saveanu et al., 2009; Weimershaus et al., 2012) designate IRAP as an optimal candidate for antigen trimming in these endosomes.
We have found that IRAP was strongly recruited to early phagosomes, where it colocalized with internalized MHC class I and phagocytized antigen. At later time points, when the phagosome was converted to a phagolysosome, the enzyme and MHC class I molecules were detected only in isolated endosomes, often adjacent to the phagosome, but not within. Therefore, IRAP storage endosomes seem to accumulate internalized MHC class I molecules. However, we have not explored whether IRAP vesicles can pinch out internalized antigen and MHC class I during the membrane fusion and fission events occurring during phagosomal maturation.
Our data suggest strongly that MHC class I loading with cross-presented peptides takes place in endocytic vesicles, but it is impossible to ascertain whether these vesicles are endosomes or phagosomes. One argument for loading events in the phagosome derives from the phenomenon of ER-phagosome membrane fusion (Ackerman et al., 2003; Guermonprez et al., 2003; Houde et al., 2003). Several groups demonstrated originally that ER-resident proteins, including key factors of the antigen-presenting machinery (TAP, Tapasin, Calnexin) and Sec61 are associated with the phagosome. Based on these results, it was proposed that the phagosome becomes an autonomous cross-presentation compartment according to the following scenario: phagocytized antigen is transported to cytosol via Sec61, processed by the proteasome and the peptides generated in the cytosol are re-imported by phagosomal TAP complexes and loaded on class I molecules within the phagosome. However, this model was challenged later, when Touret et al. (2005) did not detect any significant contribution of the ER to forming or maturing phagosomes in macrophages or DCs. Although the reasons for this discrepancy remain unclear, different experimental conditions may be the explanation: use of opsonized vs non-opsonized antigen, or possible interference from contaminating TLR ligands that could change phagosomal maturation and membrane fusion events. Molecular characterization of the fusion mechanism and of the proteins controlling the fusion event likely will eventually help to settle this controversy and evaluate the relative contribution of ER-phagosome fusion to overall cross-presentation.
In the light of the experimental data available today, we propose that proteasome-dependent cross-presentation can use at least three intracellular compartments for MHC class I loading with cross-presented peptides (Figure 3): the perinuclear ER, phagosomes, and specialized endosomes. MHC class I loading in compartments distinct from the ER in proteasome-dependent cross-presentation is supported also by our recent analysis of TAP deficient BMDCs (Merzougui et al., 2011). We have found that restoration of cell-surface MHC class I molecules on TAP deficient BMDCs by low temperature (26°C) pre-incubation normalizes cross-presentation of phagocytized ovalbumin, but not cross-presentation of receptor targeted soluble ovalbumin. Surprisingly, restored cross-presentation by TAP deficient cells requires antigen degradation by the proteasome. These findings suggest that the principal role of TAP in proteasome-dependent cross-presentation may be to ensure to provide sufficient levels of cell-surface class I molecules that can be loaded during recycling through phagosomal compartments. Our results also indicate that a TAP-independent mechanism for import of antigenic peptides from the cytosol into phagosomes might exist.
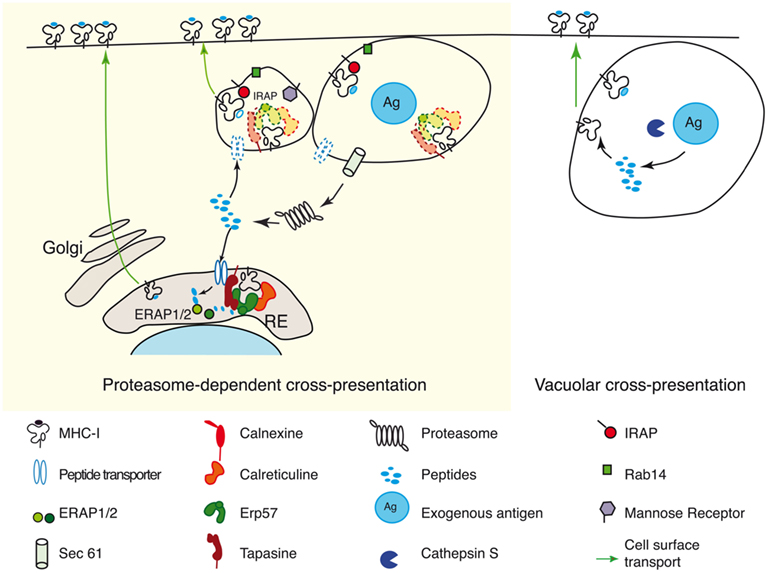
Figure 3. Cross-presentation pathways. According to the involvement of the proteasome in antigen processing cross-presentation is divided in two main pathways: proteasome-dependent cross-presentation (left panel) and vacuolar cross-presentation (right panel). The vacuolar cross-presentation does not require proteasome activity; the entire antigen processing and MHC class I loading with cross-presented peptides occur inside the vacuole. The proteasome-dependent cross-presentation involves the transport of the exogenous antigens (possibly by Sec61) into the DC cytosol of the DC and generation of N-terminal extended precursors of MHC-I ligands in the cytosol. The aminoterminal trimming of these peptide precursors can occur in two different compartments: (i) in a cytosol to ER pathway, epitope precursors will join the endogenous processing pathway after their transport by TAP into the ER (Kovacsovics-Bankowski and Rock, 1995). In this case peptide trimming is carried out by ERAPs; (ii) in a cytosol to endosome pathway, the precursors of class I ligands are retro-transported into specialized endosomes (Burgdorf et al., 2008; Saveanu et al., 2009) or into phagosomes (Guermonprez et al., 2003) and final trimming is performed by IRAP (Saveanu et al., 2009).
Cross-Presentation Pathways and DC Subsets
According to our experimental findings, several proteasome-dependent cross-presentation pathways identified by the nature of the trimming peptidase involved can operate simultaneously in DCs (Figure 3): (i) a cytosol to ER pathway where the trimming aminopeptidase is ERAP and (ii) a cytosol to endosome pathway dependent on IRAP. For antigens that are shuttled in the cytosol the possibility of cytosolic trimming of peptide precursors also exists. Cytosolic trimming, considered to have a minor impact on overall MHC-I presentation (van Endert, 2011), could be essential for specific epitopes, as for example an epitope derived from the EBNA3C protein of Ebstein Barr virus, which requires nardilysin for amino terminal trimming (Kessler et al., 2011). The relative importance of these pathways in antigen processing may depend on a variety of factors including the nature of the antigen but also the DC subset involved. DCs are a heterogeneous cell population (Shortman and Naik, 2007). Leaving aside tissue resident DCs and migratory DCs, the spleen and the lymph nodes contain at least three main DC subsets in steady-state conditions: pDCs, CD8+, and CD8− conventional DCs (cDCs). Equivalents of these splenic DC subsets can be obtained in vitro by FMS-like tyrosine kinase 3 ligand (Flt3L) differentiated cultures of murine BM precursors (Naik et al., 2005). In inflammatory conditions, a new population of DCs, called moDCs arise from blood monocytes. The equivalents of moDCs are obtained in vitro by culturing murine BM precursors or human blood monocytes in the presence of granulocyte macrophage colony stimulating factor (GM-CSF) and Interleukin 4 (IL-4; Inaba et al., 1992). pDCs are best known for their ability to secrete high amounts of type I interferons (Reizis et al., 2011), CD8+ DCs for their competence in antigen cross-presentation (Hildner et al., 2008; Lin et al., 2008) and CD8− DCs for their ability to present antigens via MHC class II (Dudziak et al., 2007). moDCs, which proliferate strongly upon inflammatory stimuli, are responsible in vivo for highly efficient cross-presentation of gram-negative bacteria and soluble antigens in the presence of TLR ligands (Cheong et al., 2010).
A given DC subset could use preferentially the “cytosol to the ER” or the “cytosol to endosome” pathway of proteasome-dependent cross-presentation. An initial report by Segura et al. (2009) suggested that the cross-presentation pathway involving IRAP and MR is functional only in moDCs. The same authors found that all steady-state DCs have similar amounts of IRAP but did not observe IRAP colocalization with internalized ovalbumin in cell fractionation experiments. We have recently analyzed pDCs and CD8+ and CD8− cDCs (ex vivo sorted from the spleens as well as their equivalents obtained from Flt3L cultures) with respect to expression and intracellular localization of IRAP and to the ability to cross-present soluble ovalbumin and yeast cells displaying ovalbumin at cell surface.
Our recent results (Weimershaus et al., 2012) lead to quite different conclusions and indicate that cDCs employ IRAP in cross-presentation. Like Segura and colleagues, we found identical IRAP protein levels in all DC subsets analyzed. Additionally, we looked at the intracellular distribution of IRAP in DC subsets. In steady-state conditions, the enzyme colocalized with previously known markers of storage endosomes: Rab14 and STX6. However, there was a slight but significant increase in Rab14/IRAP colocalization in CD8+ DCs in comparison with CD8− DCs in steady-state. Considering that Rab14 is one of the small GTPases that drives IRAP translocation upon cell activation, the increased IRAP–Rab14 colocalization in CD8+ DCs may be the reason for higher and prolonged recruitment of IRAP in the phagosomal membrane of these cells. It is conceivable that fusion between Rab14–IRAP endosomes and phagosome contributes to a delay in phagosomal maturation since Rab14 recruitment to the phagosomes prevents phagosome fusion with lysosomes (Kyei et al., 2006; Kuijl et al., 2007). Different from the data obtained by Segura et al. (2009) we also readily detected colocalization of internalized ovalbumin with IRAP using cell fractionation and fluorescence microscopy. Moreover, in vitro cross-presentation of soluble ovalbumin and yeasts displaying ovalbumin on the surface was impaired in both CD8+ and CD8− cDCs in the absence of IRAP (Weimershaus et al., 2012).
At first glance, our results on IRAP role in DC subsets are contradictory with those reported by Segura et al. (2009) Examination of the experimental settings, such as for example, the timing of antigen processing, the antigen dose, and the method of DC preparation, can explain some of the observed differences. Multiple cross-presentation pathways can produce the SIINFEKL peptide from internalized ovalbumin. At least four proteolytic enzymes can produce the SIINFEKL epitope from its precursors: cathepsin S in endosomes (Shen et al., 2004), the trimming peptidases ERAP, and IRAP (Serwold et al., 2002; Saveanu et al., 2009) and even cytosolic proteasome complexes (Cascio et al., 2001). It is not surprising in this situation that extended antigen-processing times or high antigen doses overcome the cross-presentation defects observed in the absence of IRAP, ERAP, or even TAP (Weimershaus et al., 2012).
There are several lines of evidence that the cell biology of CD8+ DCs is optimized for cross-presentation. For example, CD8+ DCs have the most efficient transport of internalized antigens to the cytosol (Lin et al., 2008), the closest to neutral phagosomal pH (Savina et al., 2009) and an up-regulated MHC class I loading machinery in comparison with the CD8− cDCs (Dudziak et al., 2007). The maturation state of CD8+ DCs allowing these cells to excel in cross-presentation assays was carefully examined very recently. Using Flt3L BMDCs, Sathe et al. (2011) demonstrated that newly formed CD8+ DC acquire their cross-presentation capacity in a maturation step (accompanied by CD103 cell-surface expression) triggered by inflammatory cytokines such as GM-CSF, IL3, or TLR ligands (CpG). Interestingly, CD8+ DCs from Flt3L cultures treated with TGF-β up-regulated cell-surface CD103, but did not cross-present. This indicated that CD103 is a marker of the maturation stage but does not correlate with cross-presentation capacity. The work of Sathe et al. highlighted that antigen cross-presentation is not an innate feature of the CD8+ DC lineage, but acquired after cell stimulation by cytokines and TLR ligands. Dresch et al. confirmed the fact that splenic CD8+ DCs require stimulation by GM-CSF or CD40 ligand for acquisition of antigen cross-presentation ability. In contrast, cross-presentation of soluble and cell-associated ovalbumin by thymic CD8+ DCs is very efficient in the absence of licensing factors, such as GM-CSF or CD40 ligand (Dresch et al., 2011).
Thus, peripheral CD8+ DCs, but not their thymic equivalents, need maturation by CD40 ligand, GM-CSF or TLR stimulation to activate the cross-presentation machinery. This recent conclusion perfectly agrees with the longstanding concept that immature DCs are poor antigen-presenting cells and that their activation is crucial for the initiation of immunity (Banchereau and Steinman, 1998). It remains to be seen whether the numerous factors that induce DC activation have an influence on the balance between the cross-presentation pathways depicted in the Figure 3.
Concluding Remarks and Future Directions
Current data indicate that the aminopeptidase activity of IRAP is responsible for endosomal aminoterminal trimming of cross-presented peptides. In addition to the peptide trimming activity, the most important IRAP features are its interaction with MHC class I molecules and its presence and role in cell-specific regulation of IRAP endosomes. The substrate specificity of IRAP, as well as the nature of the associated MHC class I molecules will require further investigation. Specifically the IRAP preferences for the length and the internal sequence of peptide substrates are entirely unknown. Another issue of importance is the extent to which the different cross-presentation pathways are used by different DC subsets under various physiologic conditions. Little is known about how the physiological context; e.g., inflammatory cytokines, pathogen-associated danger signals, simultaneous antibody responses, and CD4+ T cell help modulate intracellular cross-presentation pathways.
Another issue that should be addressed in the future is a potential IRAP role in antigen storage by the DCs. van Montfoort et al. (2009) have shown recently that soluble immune complexes can be stored by DCs for several days in lysosome-like organelles thus increasing the potency of cytotoxic T cell priming. Although preliminary results do not suggest a direct presence of IRAP in such compartments, it cannot be ruled out that IRAP plays a role in routing immune complexes and/or phagocytosed antigens to such compartments.
As demonstrated by the study of IRAP trafficking in insulin responsive cells, there are strong indications that the enzyme is not only a cargo of cell-specific storage endosomes but also an active player in the regulation of their trafficking. Our ongoing studies suggest that IRAP is required for normal maturation of phagosomes in BMDCs. We believe that it will be of great interest to characterize the extracellular stimuli and intracellular signaling pathways that regulate the trafficking of IRAP endosomes in DCs. Investigation of the cross-presentation pathways used by thymic DCs may also be of interest. As mentioned above, thymic CD8+ DCs seem to be the only steady-state DC subpopulation with a constitutively activated cross-presentation capacity, a feature that could be important for T cell selection in the thymus and maintenance of immune tolerance in the periphery.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work on IRAP-related projects by the authors was supported by grant IRAP-DC (NT09_522096) from the Agence Nationale de Recherche.
References
Ackerman, A. L., Kyritsis, C., Tampe, R., and Cresswell, P. (2003). Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc. Natl. Acad. Sci. U.S.A. 100, 12889–12894.
Albiston, A. L., Fernando, R. N., Yeatman, H. R., Burns, P., Ng, L., Daswani, D., Diwakarla, S., Pham, V., and Chai, S. Y. (2010). Gene knockout of insulin-regulated aminopeptidase: loss of the specific binding site for angiotensin IV and age-related deficit in spatial memory. Neurobiol. Learn. Mem. 93, 19–30.
Albiston, A. L., Mcdowall, S. G., Matsacos, D., Sim, P., Clune, E., Mustafa, T., Lee, J., Mendelsohn, F. A., Simpson, R. J., Connolly, L. M., and Chai, S. Y. (2001). Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin-regulated aminopeptidase. J. Biol. Chem. 276, 48623–48626.
Albiston, A. L., Morton, C. J., Ng, H. L., Pham, V., Yeatman, H. R., Ye, S., Fernando, R. N., De Bundel, D., Ascher, D. B., Mendelsohn, F. A., Parker, M. W., and Chai, S. Y. (2008). Identification and characterization of a new cognitive enhancer based on inhibition of insulin-regulated aminopeptidase. FASEB J. 22, 4209–4217.
Antonescu, C. N., Foti, M., Sauvonnet, N., and Klip, A. (2009). Ready, set, internalize: mechanisms and regulation of GLUT4 endocytosis. Biosci. Rep. 29, 1–11.
Banchereau, J., and Steinman, R. M. (1998). Dendritic cells and the control of immunity. Nature 392, 245–252.
Birnbaum, M. J. (1989). Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell 57, 305–315.
Bonifacino, J. S., and Traub, L. M. (2003). Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447.
Braszko, J. J., Kupryszewski, G., Witczuk, B., and Wisniewski, K. (1988). Angiotensin II-(3–8)-hexapeptide affects motor activity, performance of passive avoidance and a conditioned avoidance response in rats. Neuroscience 27, 777–783.
Burgdorf, S., Kautz, A., Bohnert, V., Knolle, P. A., and Kurts, C. (2007). Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 316, 612–616.
Burgdorf, S., Scholz, C., Kautz, A., Tampe, R., and Kurts, C. (2008). Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat. Immunol. 9, 558–566.
Cascio, P., Hilton, C., Kisselev, A. F., Rock, K. L., and Goldberg, A. L. (2001). 26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. EMBO J. 20, 2357–2366.
Chang, S. C., Momburg, F., Bhutani, N., and Goldberg, A. L. (2005). The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. Proc. Natl. Acad. Sci. U.S.A. 102, 17107–17112.
Cheong, C., Matos, I., Choi, J. H., Dandamudi, D. B., Shrestha, E., Longhi, M. P., Jeffrey, K. L., Anthony, R. M., Kluger, C., Nchinda, G., Koh, H., Rodriguez, A., Idoyaga, J., Pack, M., Velinzon, K., Park, C. G., and Steinman, R. M. (2010). Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell 143, 416–429.
Chi, N. W., and Lodish, H. F. (2000). Tankyrase is a golgi-associated mitogen-activated protein kinase substrate that interacts with IRAP in GLUT4 vesicles. J. Biol. Chem. 275, 38437–38444.
De Bundel, D., Smolders, I., Vanderheyden, P., and Michotte, Y. (2008). Ang II and Ang IV: unraveling the mechanism of action on synaptic plasticity, memory, and epilepsy. CNS Neurosci. Ther. 14, 315–339.
Dresch, C., Ackermann, M., Vogt, B., De Andrade Pereira, B., Shortman, K., and Fraefel, C. (2011). Thymic but not splenic CD8 DCs can efficiently cross-prime T cells in the absence of licensing factors. Eur. J. Immunol. 41, 2544–2555.
Dudziak, D., Kamphorst, A. O., Heidkamp, G. F., Buchholz, V. R., Trumpfheller, C., Yamazaki, S., Cheong, C., Liu, K., Lee, H. W., Park, C. G., Steinman, R. M., and Nussenzweig, M. C. (2007). Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111.
Fernando, R. N., Larm, J., Albiston, A. L., and Chai, S. Y. (2005). Distribution and cellular localization of insulin-regulated aminopeptidase in the rat central nervous system. J. Comp. Neurol. 487, 372–390.
Firat, E., Saveanu, L., Aichele, P., Staeheli, P., Huai, J., Gaedicke, S., Nil, A., Besin, G., Kanzler, B., Van Endert, P., and Niedermann, G. (2007). The role of endoplasmic reticulum-associated aminopeptidase 1 in immunity to infection and in cross-presentation. J. Immunol. 178, 2241–2248.
Fruci, D., Niedermann, G., Butler, R. H., and Van Endert, P. M. (2001). Efficient MHC class I-independent amino-terminal trimming of epitope precursor peptides in the endoplasmic reticulum. Immunity 15, 467–476.
Georgiadou, D., Hearn, A., Evnouchidou, I., Chroni, A., Leondiadis, L., York, I. A., Rock, K. L., and Stratikos, E. (2010). Placental leucine aminopeptidase efficiently generates mature antigenic peptides in vitro but in patterns distinct from endoplasmic reticulum aminopeptidase 1. J. Immunol. 185, 1584–1592.
Gonzalez, E., and McGraw, T. E. (2009a). The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 8, 2502–2508.
Gonzalez, E., and McGraw, T. E. (2009b). Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 7004–7009.
Goode, B. L., and Eck, M. J. (2007). Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 76, 593–627.
Gromme, M., and Neefjes, J. (2002). Antigen degradation or presentation by MHC class I molecules via classical and non-classical pathways. Mol. Immunol. 39, 181–202.
Guermonprez, P., Saveanu, L., Kleijmeer, M., Davoust, J., Van Endert, P., and Amigorena, S. (2003). ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425, 397–402.
Hattori, A., Kitatani, K., Matsumoto, H., Miyazawa, S., Rogi, T., Tsuruoka, N., Mizutani, S., Natori, Y., and Tsujimoto, M. (2000). Characterization of recombinant human adipocyte-derived leucine aminopeptidase expressed in Chinese hamster ovary cells. J. Biochem. 128, 755–762.
Hearn, A., York, I. A., and Rock, K. L. (2009). The specificity of trimming of MHC class I-presented peptides in the endoplasmic reticulum. J. Immunol. 183, 5526–5536.
Herbst, J. J., Ross, S. A., Scott, H. M., Bobin, S. A., Morris, N. J., Lienhard, G. E., and Keller, S. R. (1997). Insulin stimulates cell surface aminopeptidase activity toward vasopressin in adipocytes. Am. J. Physiol. 272, E600–E606.
Hildner, K., Edelson, B. T., Purtha, W. E., Diamond, M., Matsushita, H., Kohyama, M., Calderon, B., Schraml, B. U., Unanue, E. R., Diamond, M. S., Schreiber, R. D., Murphy, T. L., and Murphy, K. M. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100.
Hirata, Y., Hosaka, T., Iwata, T., Le, C. T., Jambaldorj, B., Teshigawara, K., Harada, N., Sakaue, H., Sakai, T., Yoshimoto, K., and Nakaya, Y. (2011). Vimentin binds IRAP and is involved in GLUT4 vesicle trafficking. Biochem. Biophys. Res. Commun. 405, 96–101.
Hosaka, T., Brooks, C. C., Presman, E., Kim, S. K., Zhang, Z., Breen, M., Gross, D. N., Sztul, E., and Pilch, P. F. (2005). p115 Interacts with the GLUT4 vesicle protein, IRAP, and plays a critical role in insulin-stimulated GLUT4 translocation. Mol. Biol. Cell 16, 2882–2890.
Hou, J. C., Suzuki, N., Pessin, J. E., and Watson, R. T. (2006). A specific dileucine motif is required for the GGA-dependent entry of newly synthesized insulin-responsive aminopeptidase into the insulin-responsive compartment. J. Biol. Chem. 281, 33457–33466.
Houde, M., Bertholet, S., Gagnon, E., Brunet, S., Goyette, G., Laplante, A., Princiotta, M. F., Thibault, P., Sacks, D., and Desjardins, M. (2003). Phagosomes are competent organelles for antigen cross-presentation. Nature 425, 402–406.
Inaba, K., Inaba, M., Romani, N., Aya, H., Deguchi, M., Ikehara, S., Muramatsu, S., and Steinman, R. M. (1992). Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176, 1693–1702.
Ito, N., Nomura, S., Iwase, A., Ito, T., Kikkawa, F., Tsujimoto, M., Ishiura, S., and Mizutani, S. (2004). ADAMs, a disintegrin and metalloproteinases, mediate shedding of oxytocinase. Biochem. Biophys. Res. Commun. 314, 1008–1013.
Iwanaga, K., Nomura, S., Ito, T., Ikoma, Y., Yamamoto, E., Okada, M., Itakura, A., Kikkawa, F., Tsujimoto, M., and Mizutani, S. (2003). Placental leucine aminopeptidase/oxytocinase gene regulation by activator protein-2 in BeWo cell model of human trophoblast differentiation. FEBS Lett. 552, 120–124.
James, D. E., Strube, M., and Mueckler, M. (1989). Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature 338, 83–87.
Jancic, C., Savina, A., Wasmeier, C., Tolmachova, T., El-Benna, J., Dang, P. M., Pascolo, S., Gougerot-Pocidalo, M. A., Raposo, G., Seabra, M. C., and Amigorena, S. (2007). Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat. Cell Biol. 9, 367–378.
Jiang, H., Li, J., Katz, E. B., and Charron, M. J. (2001). GLUT4 ablation in mice results in redistribution of IRAP to the plasma membrane. Biochem. Biophys. Res. Commun. 284, 519–525.
Jordens, I., Molle, D., Xiong, W., Keller, S. R., and Mcgraw, T. E. (2010). Insulin-regulated aminopeptidase is a key regulator of GLUT4 trafficking by controlling the sorting of GLUT4 from endosomes to specialized insulin-regulated vesicles. Mol. Biol. Cell 21, 2034–2044.
Keller, S. R., Davis, A. C., and Clairmont, K. B. (2002). Mice deficient in the insulin-regulated membrane aminopeptidase show substantial decreases in glucose transporter GLUT4 levels but maintain normal glucose homeostasis. J. Biol. Chem. 277, 17677–17686.
Keller, S. R., Scott, H. M., Mastick, C. C., Aebersold, R., and Lienhard, G. E. (1995). Cloning and characterization of a novel insulin-regulated membrane aminopeptidase from Glut4 vesicles. J. Biol. Chem. 270, 23612–23618.
Kessler, J. H., Khan, S., Seifert, U., Le Gall, S., Chow, K. M., Paschen, A., Bres-Vloemans, S. A., De Ru, A., Van Montfoort, N., Franken, K. L., Benckhuijsen, W. E., Brooks, J. M., Van Hall, T., Ray, K., Mulder, A., Doxiadis, I. I., Van Swieten, P. F., Overkleeft, H. S., Prat, A., Tomkinson, B., Neefjes, J., Kloetzel, P. M., Rodgers, D. W., Hersh, L. B., Drijfhout, J. W., Van Veelen, P. A., Ossendorp, F., and Melief, C. J. (2011). Antigen processing by nardilysin and thimet oligopeptidase generates cytotoxic T cell epitopes. Nat. Immunol. 12, 45–53.
Kochan, G., Krojer, T., Harvey, D., Fischer, R., Chen, L., Vollmar, M., Von Delft, F., Kavanagh, K. L., Brown, M. A., Bowness, P., Wordsworth, P., Kessler, B. M., and Oppermann, U. (2011). Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc. Natl. Acad. Sci. U.S.A. 108, 7745–7750.
Kohn, A. D., Summers, S. A., Birnbaum, M. J., and Roth, R. A. (1996). Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 271, 31372–31378.
Kovacsovics-Bankowski, M., and Rock, K. L. (1995). A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 267, 243–246.
Kratzer, R., Mauvais, F. X., Burgevin, A., Barilleau, E., and Van Endert, P. (2010). Fusion proteins for versatile antigen targeting to cell surface receptors reveal differential capacity to prime immune responses. J. Immunol. 184, 6855–6864.
Kuijl, C., Savage, N. D., Marsman, M., Tuin, A. W., Janssen, L., Egan, D. A., Ketema, M., Van Den Nieuwendijk, R., Van Den Eeden, S. J., Geluk, A., Poot, A., Van Der Marel, G., Beijersbergen, R. L., Overkleeft, H., Ottenhoff, T. H., and Neefjes, J. (2007). Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 450, 725–730.
Kutomi, G., Tamura, Y., Okuya, K., Yamamoto, T., Hirohashi, Y., Kamiguchi, K., Oura, J., Saito, K., Torigoe, T., Ogawa, S., Hirata, K., and Sato, N. (2009). Targeting to static endosome is required for efficient cross-presentation of endoplasmic reticulum-resident oxygen-regulated protein 150-peptide complexes. J. Immunol. 183, 5861–5869.
Kyei, G. B., Vergne, I., Chua, J., Roberts, E., Harris, J., Junutula, J. R., and Deretic, V. (2006). Rab14 is critical for maintenance of Mycobacterium tuberculosis phagosome maturation arrest. EMBO J. 25, 5250–5259.
Lakadamyali, M., Rust, M. J., and Zhuang, X. (2006). Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124, 997–1009.
Larance, M., Ramm, G., Stockli, J., Van Dam, E. M., Winata, S., Wasinger, V., Simpson, F., Graham, M., Junutula, J. R., Guilhaus, M., and James, D. E. (2005). Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J. Biol. Chem. 280, 37803–37813.
Lauvau, G., Kakimi, K., Niedermann, G., Ostankovitch, M., Yotnda, P., Firat, H., Chisari, F. V., and Van Endert, P. M. (1999). Human transporters associated with antigen processing (TAPs) select epitope precursor peptides for processing in the endoplasmic reticulum and presentation to T cells. J. Exp. Med. 190, 1227–1240.
Lew, R. A., Mustafa, T., Ye, S., Mcdowall, S. G., Chai, S. Y., and Albiston, A. L. (2003). Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP). J. Neurochem. 86, 344–350.
Liao, H., Keller, S. R., and Castle, J. D. (2006). Insulin-regulated aminopeptidase marks an antigen-stimulated recycling compartment in mast cells. Traffic 7, 155–167.
Lin, M. L., Zhan, Y., Proietto, A. I., Prato, S., Wu, L., Heath, W. R., Villadangos, J. A., and Lew, A. M. (2008). Selective suicide of cross-presenting CD8+ dendritic cells by cytochrome c injection shows functional heterogeneity within this subset. Proc. Natl. Acad. Sci. U.S.A. 105, 3029–3034.
Liu, G., Hou, J. C., Watson, R. T., and Pessin, J. E. (2005). Initial entry of IRAP into the insulin-responsive storage compartment occurs prior to basal or insulin-stimulated plasma membrane recycling. Am. J. Physiol. Endocrinol. Metab. 289, E746–E752.
Martin, O. J., Lee, A., and Mcgraw, T. E. (2006). GLUT4 distribution between the plasma membrane and the intracellular compartments is maintained by an insulin-modulated bipartite dynamic mechanism. J. Biol. Chem. 281, 484–490.
Matsumoto, H., Nagasaka, T., Hattori, A., Rogi, T., Tsuruoka, N., Mizutani, S., and Tsujimoto, M. (2001). Expression of placental leucine aminopeptidase/oxytocinase in neuronal cells and its action on neuronal peptides. Eur. J. Biochem. 268, 3259–3266.
Merzougui, N., Kratzer, R., Saveanu, L., and Van Endert, P. (2011). A proteasome-dependent, TAP-independent pathway for cross-presentation of phagocytosed antigen. EMBO Rep.
Naik, S. H., Proietto, A. I., Wilson, N. S., Dakic, A., Schnorrer, P., Fuchsberger, M., Lahoud, M. H., O’Keeffe, M., Shao, Q. X., Chen, W. F., Villadangos, J. A., Shortman, K., and Wu, L. (2005). Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 174, 6592–6597.
Nakada, T. A., Russell, J. A., Wellman, H., Boyd, J. H., Nakada, E., Thain, K. R., Thair, S. A., Hirasawa, H., Oda, S., and Walley, K. R. (2011). Leucyl/cystinyl aminopeptidase gene variants in septic shock. Chest 139, 1042–1049.
Nguyen, T. T., Chang, S. C., Evnouchidou, I., York, I. A., Zikos, C., Rock, K. L., Goldberg, A. L., Stratikos, E., and Stern, L. J. (2011). Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat. Struct. Mol. Biol. 18, 604–613.
Nomura, S., Ito, T., Yamamoto, E., Sumigama, S., Iwase, A., Okada, M., Shibata, K., Ando, H., Ino, K., Kikkawa, F., and Mizutani, S. (2005). Gene regulation and physiological function of placental leucine aminopeptidase/oxytocinase during pregnancy. Biochim. Biophys. Acta 1751, 19–25.
Norbury, C. C., Hewlett, L. J., Prescott, A. R., Shastri, N., and Watts, C. (1995). Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity 3, 783–791.
Peck, G. R., Ye, S., Pham, V., Fernando, R. N., Macaulay, S. L., Chai, S. Y., and Albiston, A. L. (2006). Interaction of the Akt substrate, AS160, with the glucose transporter 4 vesicle marker protein, insulin-regulated aminopeptidase. Mol. Endocrinol. 20, 2576–2583.
Peng, X. D., Xu, P. Z., Chen, M. L., Hahn-Windgassen, A., Skeen, J., Jacobs, J., Sundararajan, D., Chen, W. S., Crawford, S. E., Coleman, K. G., and Hay, N. (2003). Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 17, 1352–1365.
Pham, V., Burns, P., Albiston, A. L., Yeatman, H. R., Ng, L., Diwakarla, S., and Chai, S. Y. (2009). Reproduction and maternal behavior in insulin-regulated aminopeptidase (IRAP) knockout mice. Peptides 30, 1861–1865.
Rasmussen, T. E., Pedraza-Diaz, S., Hardre, R., Laustsen, P. G., Carrion, A. G., and Kristensen, T. (2000). Structure of the human oxytocinase/insulin-regulated aminopeptidase gene and localization to chromosome 5q21. Eur. J. Biochem. 267, 2297–2306.
Reizis, B., Colonna, M., Trinchieri, G., Barrat, F., and Gilliet, M. (2011). Plasmacytoid dendritic cells: one-trick ponies or workhorses of the immune system? Nat. Rev. Immunol. 11, 558–565.
Rock, K. L., and Shen, L. (2005). Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol. Rev. 207, 166–183.
Rodriguez, A., Regnault, A., Kleijmeer, M., Ricciardi-Castagnoli, P., and Amigorena, S. (1999). Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1, 362–368.
Rogers, L. D., and Foster, L. J. (2007). The dynamic phagosomal proteome and the contribution of the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 104, 18520–18525.
Sandoval, I. V., Martinez-Arca, S., Valdueza, J., Palacios, S., and Holman, G. D. (2000). Distinct reading of different structural determinants modulates the dileucine-mediated transport steps of the lysosomal membrane protein LIMPII and the insulin-sensitive glucose transporter GLUT4. J. Biol. Chem. 275, 39874–39885.
Sathe, P., Pooley, J., Vremec, D., Mintern, J., Jin, J. O., Wu, L., Kwak, J. Y., Villadangos, J. A., and Shortman, K. (2011). The acquisition of antigen cross-presentation function by newly formed dendritic cells. J. Immunol. 186, 5184–5192.
Saveanu, L., Carroll, O., Hassainya, Y., and Van Endert, P. (2005a). Complexity, contradictions, and conundrums: studying post-proteasomal proteolysis in HLA class I antigen presentation. Immunol. Rev. 207, 42–59.
Saveanu, L., Carroll, O., Lindo, V., Del Val, M., Lopez, D., Lepelletier, Y., Greer, F., Schomburg, L., Fruci, D., Niedermann, G., and Van Endert, P. M. (2005b). Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol. 6, 689–697.
Saveanu, L., Carroll, O., Weimershaus, M., Guermonprez, P., Firat, E., Lindo, V., Greer, F., Davoust, J., Kratzer, R., Keller, S. R., Niedermann, G., and Van Endert, P. (2009). IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science 325, 213–217.
Savina, A., Jancic, C., Hugues, S., Guermonprez, P., Vargas, P., Moura, I. C., Lennon-Dumenil, A. M., Seabra, M. C., Raposo, G., and Amigorena, S. (2006). NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126, 205–218.
Savina, A., Peres, A., Cebrian, I., Carmo, N., Moita, C., Hacohen, N., Moita, L. F., and Amigorena, S. (2009). The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity 30, 544–555.
Sbodio, J. I., and Chi, N. W. (2002). Identification of a tankyrase-binding motif shared by IRAP, TAB182, and human TRF1 but not mouse TRF1. NuMA contains this RXXPDG motif and is a novel tankyrase partner. J. Biol. Chem. 277, 31887–31892.
Segura, E., Albiston, A. L., Wicks, I. P., Chai, S. Y., and Villadangos, J. A. (2009). Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20377–20381.
Serwold, T., Gaw, S., and Shastri, N. (2001). ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat. Immunol. 2, 644–651.
Serwold, T., Gonzalez, F., Kim, J., Jacob, R., and Shastri, N. (2002). ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature 419, 480–483.
Shen, L., Sigal, L. J., Boes, M., and Rock, K. L. (2004). Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 21, 155–165.
Shortman, K., and Naik, S. H. (2007). Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7, 19–30.
Siddle, K. (2011). Signalling by insulin and IGF receptors: supporting acts and new players. J. Mol. Endocrinol. 47, R1–R10.
Sigal, L. J., and Rock, K. L. (2000). Bone marrow-derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and -independent pathways of antigen presentation. J. Exp. Med. 192, 1143–1150.
Stragier, B., De Bundel, D., Sarre, S., Smolders, I., Vauquelin, G., Dupont, A., Michotte, Y., and Vanderheyden, P. (2008). Involvement of insulin-regulated aminopeptidase in the effects of the renin-angiotensin fragment angiotensin IV: a review. Heart Fail. Rev. 13, 321–337.
Subtil, A., Lampson, M. A., Keller, S. R., and Mcgraw, T. E. (2000). Characterization of the insulin-regulated endocytic recycling mechanism in 3T3-L1 adipocytes using a novel reporter molecule. J. Biol. Chem. 275, 4787–4795.
Tanioka, T., Hattori, A., Mizutani, S., and Tsujimoto, M. (2005). Regulation of the human leukocyte-derived arginine aminopeptidase/endoplasmic reticulum-aminopeptidase 2 gene by interferon-gamma. FEBS J. 272, 916–928.
Tojo, H., Kaieda, I., Hattori, H., Katayama, N., Yoshimura, K., Kakimoto, S., Fujisawa, Y., Presman, E., Brooks, C. C., and Pilch, P. F. (2003). The Formin family protein, formin homolog overexpressed in spleen, interacts with the insulin-responsive aminopeptidase and profilin IIa. Mol. Endocrinol. 17, 1216–1229.
Touret, N., Paroutis, P., Terebiznik, M., Harrison, R. E., Trombetta, S., Pypaert, M., Chow, A., Jiang, A., Shaw, J., Yip, C., Moore, H. P., Van Der Wel, N., Houben, D., Peters, P. J., De Chastellier, C., Mellman, I., and Grinstein, S. (2005). Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell 123, 157–170.
Tsujimoto, M., and Hattori, A. (2005). The oxytocinase subfamily of M1 aminopeptidases. Biochim. Biophys. Acta 1751, 9–18.
Tsujimoto, M., Mizutani, S., Adachi, H., Kimura, M., Nakazato, H., and Tomoda, Y. (1992). Identification of human placental leucine aminopeptidase as oxytocinase. Arch. Biochem. Biophys. 292, 388–392.
van Endert, P. (2011). Post-proteasomal and proteasome-independent generation of MHC class I ligands. Cell. Mol. Life Sci. 68, 1553–1567.
van Endert, P. M., Tampe, R., Meyer, T. H., Tisch, R., Bach, J. F., and Mcdevitt, H. O. (1994). A sequential model for peptide binding and transport by the transporters associated with antigen processing. Immunity 1, 491–500.
van Montfoort, N., Camps, M. G., Khan, S., Filippov, D. V., Weterings, J. J., Griffith, J. M., Geuze, H. J., Van Hall, T., Verbeek, J. S., Melief, C. J., and Ossendorp, F. (2009). Antigen storage compartments in mature dendritic cells facilitate prolonged cytotoxic T lymphocyte cross-priming capacity. Proc. Natl. Acad. Sci. U.S.A. 106, 6730–6735.
Wallis, M. G., Lankford, M. F., and Keller, S. R. (2007). Vasopressin is a physiological substrate for the insulin-regulated aminopeptidase IRAP. Am. J. Physiol. Endocrinol. Metab. 293, E1092–E1102.
Watson, R. T., Hou, J. C., and Pessin, J. E. (2008). Recycling of IRAP from the plasma membrane back to the insulin-responsive compartment requires the Q-SNARE syntaxin 6 but not the GGA clathrin adaptors. J. Cell. Sci. 121, 1243–1251.
Watson, R. T., Khan, A. H., Furukawa, M., Hou, J. C., Li, L., Kanzaki, M., Okada, S., Kandror, K. V., and Pessin, J. E. (2004). Entry of newly synthesized GLUT4 into the insulin-responsive storage compartment is GGA dependent. EMBO J. 23, 2059–2070.
Weimershaus, M., Maschalidi, S., Sepulveda, F., Manoury, B., Van Endert, P., and Saveanu, L. (2012). Conventional dendritic cells require IRAP-Rab14 endosomes for efficient cross-presentation. J. Immunol. 188, 1840–1846.
Wright, J. W., Yamamoto, B. J., and Harding, J. W. (2008). Angiotensin receptor subtype mediated physiologies and behaviors: new discoveries and clinical targets. Prog. Neurobiol. 84, 157–181.
Yan, J., Parekh, V. V., Mendez-Fernandez, Y., Olivares-Villagomez, D., Dragovic, S., Hill, T., Roopenian, D. C., Joyce, S., and Van Kaer, L. (2006). In vivo role of ER-associated peptidase activity in tailoring peptides for presentation by MHC class Ia and class Ib molecules. J. Exp. Med. 203, 647–659.
Yang, Z. Z., Tschopp, O., Di-Poi, N., Bruder, E., Baudry, A., Dummler, B., Wahli, W., and Hemmings, B. A. (2005). Dosage-dependent effects of Akt1/protein kinase Balpha (PKBalpha) and Akt3/PKBgamma on thymus, skin, and cardiovascular and nervous system development in mice. Mol. Cell. Biol. 25, 10407–10418.
York, I. A., Chang, S. C., Saric, T., Keys, J. A., Favreau, J. M., Goldberg, A. L., and Rock, K. L. (2002). The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8-9 residues. Nat. Immunol. 3, 1177–1184.
Zeigerer, A., Mcbrayer, M. K., and Mcgraw, T. E. (2004). Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol. Biol. Cell 15, 4406–4415.
Keywords: cross-presentation, aminopeptidase, IRAP, ERAP, dendritic cells, MHC class I
Citation: Saveanu L and van Endert P (2012) The role of insulin-regulated aminopeptidase in MHC class I antigen presentation. Front. Immun. 3:57. doi: 10.3389/fimmu.2012.00057
Received: 05 December 2011; Accepted: 03 March 2012;
Published online: 21 March 2012.
Edited by:
Christian Kurts, Friedrich Wilhelms-Universität Bonn, GermanyReviewed by:
Hansjörg Schild, University Medical Center of the Johannes Gutenberg-University, GermanySven Burgdorf, Rheinische Friedrich Wilhelms-Universität Bonn, Germany
Copyright: © 2012 Saveanu and van Endert. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Loredana Saveanu, Faculté de Médecine, Université Paris Descartes, Sorbonne Paris Cité, 75015 Paris, France. e-mail:bG9yZWRhbmEuc2F2ZWFudUBpbnNlcm0uZnI=; Peter van Endert, Institut National de la Santé et de le Recherche Médicale, U1013, Paris 75015, France. e-mail:cGV0ZXIudmFuLWVuZGVydEBpbnNlcm0uZnI=