 Ming-Xiao He
Ming-Xiao He
- Department of Immunology, Duke University, Durham, NC, USA
Macroautophagy (referred to as autophagy) is a fundamental intracellular process characterized by the sequestration of cytoplasmic compartments through double-membrane vesicles, termed autophagosomes. Recent studies have established important roles of autophagy in regulating T lymphocyte development and function. Resting T lymphocytes have basal levels of autophagy that is upregulated by T cell receptor stimulation. Several specific knockout or transgenic models have been developed during the past few years, and it has been revealed that autophagy plays an essential role in regulating thymocyte selection, peripheral T cell survival, and proliferation. The regulation of T cell development and function by autophagy is mediated through its role in regulating self-antigen presentation, intracellular organelle homeostasis, and energy production. Here we will review the current findings concerning how autophagy regulates T cell function, as well as compare different models in studying autophagy in T lymphocytes.
Introduction
Macroautophagy is a cellular process characterized by the sequestration of cytoplasmic compartments through double-membrane vesicles, termed autophagosomes. Macroautophagy (therefore referred as autophagy) starts with the generation of double-membrane bound structures referred as isolation membranes or phagophores. The phagophore then retains intracellular components to form autophagosomes. Consequentially, autophagosomes fuse with lysosomes, in which the inside content is digested (Klionsky and Emr, 2000). Autophagosome biogenesis requires a group of evolutionarily conserved genes, referred to as autophagy-related genes (atgs; Longatti and Tooze, 2009). The initiation of the phagophore requires the class III phosphatidylinositol 3-kinase (PtdINs3K) complex, which further recruits two ubiquitin-like conjugation systems: The Atg12–Atg5 conjugation and LC3 processing (Ohsumi and Mizushima, 2004; He and Klionsky, 2009; Figure 1). Autophagy has been shown to be important for providing nutrients during starvation, clearing long-lived proteins as well as unwanted organelles, and fighting intracellular infections in multiple types of mammalian cells (Mortimore and Poso, 1987; Levine et al., 2011). The primary role of autophagy in certain cells depends on the cell type and environment. To study autophagy in primary T lymphocytes, several specific knockout or transgenic models have been developed during the past few years, and it has been revealed that autophagy plays an essential role in T cell homeostasis and function. The phenotypes of different atg-deficient T cells showed a great deal of similarity, with certain differences, which can be explained as a function of deletion timing or efficiency, accumulating effects, or the molecular functions outside of autophagy. Here we will review the current findings concerning how autophagy regulates T cell function, as well as compare different models in studying autophagy in T lymphocytes.
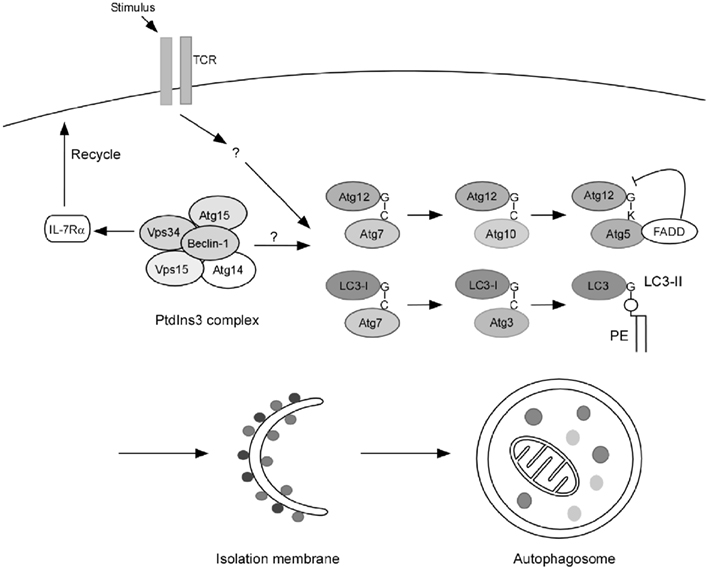
Figure 1. Autophagy in T lymphocytes. Published results indicate that TCR engagement is a strong inducer of autophagy in primary T cells. Although autophagy is induced by the PtdIns3k complex in many types of cells, the role of this complex in autophagy induction in T lymphocytes may not be the same as in other cells. Vps34 is dispensable for autophagy induction in T cells while it promotes T cell survival by regulating IL-7Rα surface expression. The role of Beclin-1 in T cell autophagy induction remains to be further defined. However, the two autophagosome processing pathways: Atg5–Atg12 conjugation and LC3 processing are essential for autophagy induction as deleting Atg3, Atg5, and Atg7 all results in impaired autophagy in T cells. Furthermore, FADD, the death adaptor protein in the extrinsic death pathway, may play a key role in mediating a crosstalk between the apoptosis and autophagy pathways.
Autophagy Induction in T Lymphocytes
Although relatively low, autophagy can be detected in all subsets of thymocytes and freshly isolated naïve T lymphocytes (Li et al., 2006; Pua et al., 2007; Stephenson et al., 2009). These results were fairly surprising, since T cells contain limited cytoplasm. The existence of autophagy in T lymphocytes suggests a regulatory role of intracellular program. Aging T lymphocytes, on the other hand, showed an accumulation of autophagic vacuoles. In a study of long-term in vitro human lymphocyte culture, the percentage of cells with autophagosomes increased during culture, which was associated with the increase in lysosomal mass and accumulation of lipofusion events (Gerland et al., 2004).
The T cell receptor (TCR) mediates activation signal upon interaction with the antigenic peptide presented by the major histocompatibility complex (MHC) on antigen presenting cells. TCR activation is a strong trigger for autophagy in T lymphocytes (Pua et al., 2007, 2009; Hubbard et al., 2010; Jia and He, 2011; Jia et al., 2011; Kovacs et al., 2012). CD4+ T cells upregulate Beclin-1 (Atg6) and LC3 (Atg8) upon TCR stimulation (Arsov et al., 2008). Ultrastructural studies revealed an increase in the number and a decrease in the size of autophagosomes after TCR stimulation. Interestingly, while mitochondria are frequently contained in the autophagosomes of resting T lymphocytes, the autophagic cargo switches to almost exclusively cytosolic material in activated T lymphocytes (Hubbard et al., 2010). Mitochondria undergo morphological changes during autophagy induction to escape autophagic degradation and maintain energy production in mouse embryonic fibroblasts (MEFs; Gomes et al., 2011). It is important to determine whether T lymphocytes apply similar strategies or utilize unique pathways to regulate the mitochondrial content.
T cell receptor-induced autophagy requires the key autophagy machinery, as deleting Atg5, Atg7, and Atg3 can abolish autophagosomal induction (Pua et al., 2007, 2009; Hubbard et al., 2010; Jia and He, 2011). However, the class III PI3K, Vps34, seems to be dispensable for autophagy induction in mature T lymphocytes (McLeod et al., 2011). JNK1/JNK2 is required for TCR-induced autophagy in CD4+ T cells (Li et al., 2006). Beclin-1, a component of PtdINs3K complex, was shown to be crucial for autophagy initiation (Yue et al., 2003). The level of autophagy in Beclin-1 deficient primary T lymphocytes remains to be measured (Kovacs et al., 2012). Nevertheless, overexpression of Beclin-1 in T lymphocytes by a BAC transgene did not change the basal level of autophagy in multiple organs including thymus and spleen (Arsov et al., 2008), suggesting that additional autophagy initiators may be required in T lymphocytes for autophagy induction.
T cell receptor-induced autophagy is compromised in aged CD4+ T lymphocytes (Mattoo et al., 2009). The mechanism by which autophagy induction is defective during aging is unclear. Some evidence suggested that the Rel family member, p65, might be involved, as the nuclear translocation of p65 upon TCR signaling is impaired in aged CD4+ T cells (Mattoo et al., 2009), and p65 has been shown to be essential for autophagy induction by upregulating the transcription of Beclin-1 in multiple cell lines (Copetti et al., 2009). It is also unknown whether defective autophagy induction leads to other defects in aged T lymphocytes, such as increased mitochondrial damage, reduction in glycolysis, or enhanced apoptosis upon primary TCR stimulation. It would be appealing to investigate whether modulating autophagic levels can restore the function of aged T lymphocytes.
Autophagy in T Lymphocyte Homeostasis: Different Genetic Models, Similar Defects
Several tissue-specific knockout models have been developed during the past few years to study the role of autophagy in T lymphocytes. The deficiency in autophagy-related genes leads to a blockage in autophagic flux, as well as impaired T cell homeostasis. In Atg5−/− fetal liver chimeric mice, the thymocytes undergo full maturation, but the thymic cellularity is reduced by half. The peripheral T lymphocyte number is dramatically decreased, which may be the result of both the loss of thymocytes and increased cell death rate (Pua et al., 2007). The Atg5f/fLck-Cre+ mice generated later showed an almost identical phenotype to the Atg5−/− chimera (Stephenson et al., 2009). The question was whether the loss of autophagy was the real reason for the enhanced cell death, since Atg5 interacts with Fas-associated death domain (FADD) protein and the anti-apoptotic protein Bcl-xL (Pyo et al., 2005; Yousefi et al., 2006). Atg5 may regulate apoptosis through the extrinsic or intrinsic cell death pathways (Zhang et al., 2005).
To address whether Atg5 regulates T cell homeostasis through autophagy, several other tissue-specific knockout models have been analyzed since. Autophagosome formation requires two interrelated protein conjugation systems: LC3 processing and Atg5–Atg12 conjugation. During LC3 processing, Atg3, along with Atg7, catalyzes the cleavage and conjugation of LC3-I to generate the membrane bound LC3-phosphatidylethanolamine (LC3-II/LC3-PE; Levine et al., 2011). Tissue-specific knockout of Atg3 using an Lck-Cre system [deletion occurs during double negative (DN) stage in thymus] revealed an almost identical phenotype as the Atg5−/− chimera, characterized by normal frequencies of DN, double positive (DP), and single positive (SP) thymocytes in the thymus, but a reduced cellularity in both the thymus and peripheral T cell pools (Jia and He, 2011). On the other hand, the thymocyte cellularity in Atg7f/fLck-Cre+ mice was barely decreased (Stephenson et al., 2009), with only a minor reduction in SP cells. Despite the difference inside the thymus, T lymphocytes in all these models (Atg5−/− chimera, Atg5f/fLck-Cre+, Atg7f/fLck-Cre+, Atg3f/fLck-Cre+, and BECNf/fCD4-Cre+) showed decreased T cell numbers in the periphery and an enhanced apoptosis (Pua et al., 2007, 2009; Stephenson et al., 2009; Jia and He, 2011; Kovacs et al., 2012).
Ortiz’s group utilized a Rag-1−/− (recombination activating gene-1 null) blastocyte complementation to study the role of Beclin-1 in hematopoietic cells. Beclin-1-deficient (BECN−/−) blastocytes were transferred to a Rag-1−/− host. The data showed that Beclin-1 is required for the maintenance of early thymocyte progenitors. Controversially, survival and TCR-induced proliferation remained normal in BECN−/− T lymphocytes developed in this system. Moreover, autophagy can be detected in those cells, though decreased compared to controls (Arsov et al., 2011). These results indicate that a Beclin-1 independent autophagy-inducing pathway may occur in T lymphocytes. The mechanism of autophagy induction in the absence of Beclin-1 needs to be determined.
Vps34, a key component in PtdINs3K complex, regulates the assembly of phagophore. Surprisingly, Vps34 deficient T cells showed no change in LC3 puncta formation after TCR stimulation or starvation. Moreover, autophagosomes could be detected in naïve Vps34−/− T cells by transmission electronic microscopy (McLeod et al., 2011). These results suggest that Vps34 is not essential for autophagy induction in T lymphocytes. The differences in findings between Vps34 and Beclin-1 knockout T cells can be explained by the accessory molecules associated with the complex. Beclin-1 also has affinity for Bcl-2, and is an interaction partner with Bcl-2 during conditions of stress (Liang et al., 1998), inhibiting the ability of Bcl-2 to repress pro-apoptotic Bax/Bak (Wei et al., 2008). Additionally, other functions of Beclin-1 include recruitment of proteins that bend and flex membranes, including UVRAG and Bif1, to provide the physical machinery for autophagosomal elongation (Takahashi et al., 2007, 2009). Finally, Vps34 can function as a scaffold for the autophagic process, but the lipid kinase function of this molecule can potentially be compensated for by phosphatidylinositol metabolism. This has been recently confirmed by data in MEFs showing that the class Ia PI3K, p110β, is a positive regulator of autophagy (Dou et al., 2010).
Autophagy in Thymocyte Selection
Autophagy plays an important role in the generation and selection of thymocytes which have little autoreactivity. Interestingly, this autophagy occurs in thymic epithelial cells (TECs), which although are not hematopoietic in nature, are the major selecting cells in thymic education and tolerance induction. As thymocytes mature, they undergo a functional rearrangement of VDJ gene segments in various TCR loci (either αβ or γδ TCR subunits) to express a surface TCR receptor (Schatz and Ji, 2011). These maturing thymocytes depend on receiving TCR-mediated survival cues. During this process, DP thymocytes of the αβ lineage are selected by cortical thymic epithelial cells (cTECs) by a process termed “positive selection” (Takahama et al., 2010) and migrate inward to be selected by medullary TECs by “negative selection.” Through this process, TCR clones are generated that have low affinities for self-peptides presented in the context of a MHC. One of the mechanisms that TECs use for this process is the expression of autoimmune regulator (AIRE), which regulates the expression of a host of tissue-specific proteins against which developing thymocytes can be selected (Heino et al., 1999). Another possibility is that thymocytes develop in the presence of antigens derived from peripheral cells that are transported back to the thymus by dendritic cells. However, from studies performed by Nedjic et al. (2008a), it is clear that autophagy is a major mechanism by which self-antigens are processed and presented for selection of developing thymocytes.
Thymic epithelial cells have extraordinarily high levels of constitutive autophagy (Nedjic et al., 2008a). Using Atg5-deficient TECs engrafted into nude mice, Nedjic observed normal thymocyte numbers. However, these cells had undergone a disrupted process of positive and negative selection, despite the normal expression of AIRE, suggesting a role of macroautophagy in the selection. Moreover, when T cells selected by Atg5-deficient TECs were transferred, they recapitulated a wasting disease with colitis and lung infiltration highly reminiscent of typical autoimmune disorders (Nedjic et al., 2008a). These results are consistent with that autophagy is essential for the expression of self peptide–MHC complexes on TECs to foster normal T cell development. When the abnormally low phagocytic activity of TECs is accounted for, the prominence of autophagy in generating peptides for MHC display is enhanced even further (Nedjic et al., 2008b). It would be of great interest to investigate the opposite effect, namely would increasing autophagic activity in TECs promote T cell production?
Conversely, a study showed that autophagic activity in thymocytes (including TECs) was greatly reduced in aged mice, correlating with a reduced pool of mature T cell production in these mice (Uddin et al., 2012). It would be of great interest to determine whether increased levels of autophagy could be stimulated in TECs of aged mice to increase the pool of mature T lymphocytes and maintain clonal diversity into old age.
Autophagy in T Cell Function
Autophagy and Intracellular Organelle Homeostasis
Autophagy selectively degrading mitochondria, termed as mitophagy (Kraft et al., 2009), plays a role in controlling mitochondria content during T cell development (Pua et al., 2009; Stephenson et al., 2009). Transcriptional profiling of Atg5-deficient T lymphocytes revealed a remarkable enrichment in mitochondrion-associated genes (Stephenson et al., 2009). The mitochondrial contents in T lymphocytes are gradually decreased during the transition from thymocytes to peripheral T lymphocytes. The level of mitochondria was shown to be the highest in DP thymocytes, followed by SP thymocytes, and mature T lymphocytes contained the lowest volume (Pua et al., 2009). This contraction of mitochondria requires autophagy, as least partially, as mitochondrial content was higher in Atg5−/−, Atg7−/−, and Atg3−/− than in wild-type T lymphocytes (Pua et al., 2009; Stephenson et al., 2009; Jia et al., 2011). On the other hand, it was reported that cells from BECNf/f CD4-Cre+ mice showed no change in mitochondrial content (Kovacs et al., 2012). One possible explanation is that genomic deletion only starts in DP thymocytes in this model, and persistent level of Beclin-1 protein in cells is enough to manage the autophagic degradation of mitochondria. Still, one cannot rule out the possibility that mitochondrial content regulation and even autophagy are achieved independent of Beclin-1 in T lymphocytes. Current data indicates that autophagy protects T lymphocytes migrating to the periphery by eliminating excessive mitochondria. Mitochondria are assumed to be the major reservoir of toxic reactive oxygen species (ROS; Hildeman et al., 2003). The oxidative tension is much higher in the blood than the thymus (Braun et al., 2001; Sitkovsky and Lukashev, 2005). Therefore, maintaining the same level of mitochondria in peripheral T lymphocytes as in thymocytes may lead to intolerable levels of ROS for T lymphocytes.
Endoplasmic reticulum (ER) levels are as well regulated by autophagy. Selective autophagy toward ER is termed as reticulophagy (or ER-phagy; Kraft et al., 2009). Different from mitochondria, ER volume is downregulated in the transition of DN to DP thymocytes, and maintained to a similar level afterward. An increase in ER volume was observed in autophagy-deficient T lymphocytes (Jia et al., 2011). The accumulation of ER, as well as mitochondria, requires a long period of time to be established (up to 18 days in vitro) once autophagy is abolished by induced-deletion in mature T lymphocytes. Interestingly, lymphocytes did not show elevated cell death until 24 days after inducible deletion of Atg3. These data suggest that the elevated cell death in autophagy-deficient T lymphocytes is possibly the outcome of cellular organelles’ abnormality.
Autophagy in T Cell Activation
Different groups have identified that autophagy-related genes are required for T cell proliferation upon TCR stimulation. T lymphocytes lacking Atg5, Atg7, Atg3, or Beclin-1 all showed impaired proliferation (Pua et al., 2007, 2009; Stephenson et al., 2009; Hubbard et al., 2010; Jia et al., 2011; Kovacs et al., 2012). TCR stimulation induced cell death was also observed in Beclin-1-deficient lymphocytes (Kovacs et al., 2012). The survival defect may contribute to impaired proliferation in Beclin-1-deficient T lymphocytes. Atg7−/− T cells displayed impaired calcium influx upon TCR stimulation, as well as increased calcium efflux from ER to cytosol. The excessive ER in Atg7-deficient T cells may lead to abnormal redistribution of calcium into the ER upon TCR stimulation (Jia et al., 2011). Interestingly, other TCR signaling components such as PLCγ-1(the upstream signal of calcium efflux), p38, ERK, and NF-κB activation all remain unchanged in Atg7-deficient T lymphocytes.
Autophagy is involved in the metabolism of activated T lymphocytes. Upon receipt of a TCR signal, T lymphocytes undergo a metabolic switch and produce more ATP to ensure sufficient energy for protein synthesis, cytokine secretion, and cellular division (Fox et al., 2005). Without autophagy, the ATP production in activated T lymphocytes is reduced to resting levels. The lack of ATP, at least partially, contributes to impaired transcription and production of IFN-γ and IL-2 in those cells, as methyl pyruvate, a cell-permeable intermediate of glucose, incompletely rescues both ATP generation and cytokine production (Hubbard et al., 2010). However, another study showed increased levels of IL-2 secretion in autophagy-deficient T lymphocytes after TCR activation (Jia et al., 2011). This controversy may be the consequence of the autocrine nature of IL-2: IL-2 is consistently produced and utilized by activated T lymphocytes (Cantrell et al., 1988). Therefore, the IL-2 detected in culture medium at a single time point may or may not reflect the capacity of IL-2 production. To settle the debate, careful study in the kinetics of the transcription and secretion of cytokines is needed.
Autophagy is differentially regulated in each T helper subset. T lymphocytes cultured in Th2 polarizing condition in vitro contain more autophagosomes than T lymphocytes induced by Th1 polarizing conditions (Li et al., 2006). Th17 cells are relatively more (Wu et al., 2011) resistant to cell death without Beclin-1, compared to Th0, Th1, and Th2 (Kovacs et al., 2012). Th17 cells, along with Th1 cells, mediate the pathogenesis of experimental autoimmune encephalomyelitis (EAE) (El-behi et al., 2010). Though Th17 cells manage to survive when autophagy is blocked, mice with Beclin-1-deficient T lymphocytes still showed resistance to EAE development. The number of MOG specific Th1 and Th17 cells were reduced in the periphery and were undetectable in the central nerve system (CNS) in Beclin-1 knockout mice (Kovacs et al., 2012). The protective role of Beclin-1-deficient T lymphocytes in EAE induction may be the result of defective antigen-induced proliferation.
Autophagy in HIV Infection
Autophagy was reported to be involved in the progressive decline in the number of CD4+ T lymphocytes during HIV infection. HIV infection induces cell death in both infected and uninfected “bystander” CD4+ T cells (Laurent-Crawford et al., 1993). The repertoire of HIV-1 envelope glycoproteins (Env) is composed of gp41 and gp120 and expressed on infected human lymphocytes. Via CXCR4 and CCR5, Env triggers autophagy in uninfected “bystander” CD4+ T cells, which eventually leads to apoptosis (Espert et al., 2006, 2009). The gp41 fusion to target membranes is required for Env-mediated autophagy (Denizot et al., 2008). Interestingly, autophagy is repressed in HIV-infected CD4+ T cells (Zhou and Spector, 2008). The effect of killing “bystander” T cells through surface protein Env seems to be T cell specific, as Env expressed on macrophages with CXCR4 and CCR5 fails to induce autophagy in uninfected CD4+ T cells in a coculture experiment (Espert et al., 2009). From the perspective of virus, down-regulation of autophagy in host cells may be beneficial as autophagy attacks virus through xenophagic degradation and facilitates antigen presentation (Kim et al., 2010). At the same time, provoking excessive autophagy in uninfected surrounding lymphocytes mediates cell death, further weakening the host defense by reducing potential adaptive immunity against the virus.
Autophagy in T Cell Death
As mentioned above, the loss of autophagy-related genes generally leads to increased cell death (Pua et al., 2007, 2009; Stephenson et al., 2009; Jia et al., 2011). The crosstalk between autophagy and apoptosis has been observed in various eukaryotic cells (Thorburn, 2008), but how autophagy regulates apoptosis in T lymphocytes remains unclear. Atg7−/− T lymphocytes upregulate the expression of Bcl-2 (Pua et al., 2009). Beclin-1-deficient T lymphocytes demonstrate highly increased protein levels, but not mRNA levels of pro-apoptotic pro-caspase-8, pro-caspase-3, and Bim as well as a moderate increase in Bcl-2. Interestingly, Caspase-8 is detected in p62/ubiquitin-containing aggregates in Beclin-1−/− T cells (Kovacs et al., 2012). Although p62 may target caspase-8 for autophagic degradation, pro-caspase-8 level is comparable between wild-type and p62-deficient T cells. Further studies are required to identify whether Beclin-1 controls pro-apoptotic protein levels through Bcl-2 binding, autophagy induction, or other pathways.
Although it is clearly established that autophagy is required for T cell survival, excessive autophagy seems to be destructive for T lymphocytes. Autophagy promotes, rather than protects, growth factor withdrawal-induced cell death in a Th2 cell line (Li et al., 2006). In long-term human CD8+ T cell cultures, cells with higher numbers of autophagosomes died out first. These results might imply that autophagy renders lymphocytes more vulnerable to apoptosis under certain conditions (Gerland et al., 2004). However, it is still unclear whether the high number of autophagosomes reflects an increase in flux or a blockage of autophagosome–lysosome fusion. Whether those cells undergo enhanced autophagy remains to be verified. Other data supporting the pro-death role of autophagy involves Irgm 1 (Interferon-γ inducible, immunity-related GTPase−/−). When exposed to IFN-γ, Irgm 1−/− T cells showed greatly enhanced death in an autophagy-dependent manner (Feng et al., 2008). Consistent with this, Ginsenoside Re suppresses autophagy by inhibiting Irgm 1 and IFN-γ production in human T lymphocytes, which leads to enhanced proliferation and reduced cell death (Son et al., 2010).
Blocking caspase-8 activity was shown to induce autophagy and non-apoptotic death (Yu et al., 2004; Li et al., 2006; Wu et al., 2011). In T lymphocytes, loss of capase-8 activity may lead to necroptosis upon TCR stimulation. Necroptosis is a programmed necrotic cell death, regulated by Rip-1/Rip-3 kinases and independent of the apoptotic pathway (Galluzzi and Kroemer, 2008). It is still controversial whether autophagy is involved in TCR-induced necroptosis in T lymphocytes. Caspase-8 activation requires the adaptor protein FADD (Zhang et al., 1998). FADDdd (dominant negative) T lymphocytes showed hyperactive autophagy and RIP-1-dependent cell death (Figure 1). Inhibiting autophagy by either 3-MA administration or silencing of Atg7, rescued those cells from cell death (Bell et al., 2008). However, studies in FADD−/− and caspase-8−/− T cells suggested that enhanced cell death cannot be rescued by 3-MA treatment or deletion of Atg7 (Osborn et al., 2010; Ch’en et al., 2011). It is imprudent to draw any conclusions from the current results, as thoroughly abolishing autophagy may create additional stress, thus killing instead of protecting T lymphocytes. Careful analyses of cell death pathways in caspase-8-deficient T cells, especially with modest inhibition of autophagy, will provide us valuable information to understand the relationship between autophagy, apoptosis, and necroptosis.
Conclusion
Autophagy, a fundamental cellular process, is required for the homeostasis and function of T lymphocytes. Deletion of different autophagy-related genes results in similar phenotypes in T lymphocytes, indicating that the defects are caused by a loss of autophagy instead of other functions of those proteins. T lymphocytes utilize autophagy to maintain intracellular organelle homeostasis, intact TCR signaling, and metabolic switch upon TCR activation. It is worthy to notice that autophagy plays a dual role in T lymphocytes: autophagy induction is critical for cell survival and T cell response, while too many autophagic vesicles can create intolerable stress. Future studies can focus on how to modulate the levels of autophagy to promote/inhibit T cell responses in various disease models.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in the authors’ laboratory is supported by NIH grants AI074944, AI074754, and AI073947.
References
Arsov, I., Adebayo, A., Kucerova-Levisohn, M., Haye, J., MacNeil, M., Papavasiliou, F. N., Yue, Z., and Ortiz, B. D. (2011). A role for autophagic protein beclin 1 early in lymphocyte development. J. Immunol. 186, 2201–2209.
Arsov, I., Li, X., Matthews, G., Coradin, J., Hartmann, B., Simon, A. K., Sealfon, S. C., and Yue, Z. (2008). BAC-mediated transgenic expression of fluorescent autophagic protein beclin 1 reveals a role for beclin 1 in lymphocyte development. Cell Death Differ. 15, 1385–1395.
Bell, B. D., Leverrier, S., Weist, B. M., Newton, R. H., Arechiga, A. F., Luhrs, K. A., Morrissette, N. S., and Walsh, C. M. (2008). FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc. Natl. Acad. Sci. U.S.A. 105, 16677–16682.
Braun, R. D., Lanzen, J. L., Snyder, S. A., and Dewhirst, M. W. (2001). Comparison of tumor and normal tissue oxygen tension measurements using oxylite or microelectrodes in rodents. Am. J. Physiol. Heart Circ. Physiol. 280, H2533–H2544.
Cantrell, D. A., Collins, M. K., and Crumpton, M. J. (1988). Autocrine regulation of T-lymphocyte proliferation: differential induction of IL-2 and IL-2 receptor. Immunology 65, 343–349.
Ch’en, I. L., Tsau, J. S., Molkentin, J. D., Komatsu, M., and Hedrick, S. M. (2011). Mechanisms of necroptosis in T cells. J. Exp. Med. 208, 633–641.
Copetti, T., Bertoli, C., Dalla, E., Demarchi, F., and Schneider, C. (2009). p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell. Biol. 29, 2594–2608.
Denizot, M., Varbanov, M., Espert, L., Robert-Hebmann, V., Sagnier, S., Garcia, E., Curriu, M., Mamoun, R., Blanco, J., and Biard-Piechaczyk, M. (2008). HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 4, 998–1008.
Dou, Z., Chattopadhyay, M., Pan, J. A., Guerriero, J. L., Jiang, Y. P., Ballou, L. M., Yue, Z., Lin, R. Z., and Zong, W. X. (2010). The class IA phosphatidylinositol 3-kinase p110-beta subunit is a positive regulator of autophagy. J. Cell Biol. 191, 827–843.
El-behi, M., Rostami, A., and Ciric, B. (2010). Current views on the roles of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 5, 189–197.
Espert, L., Denizot, M., Grimaldi, M., Robert-Hebmann, V., Gay, B., Varbanov, M., Codogno, P., and Biard-Piechaczyk, M. (2006). Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 116, 2161–2172.
Espert, L., Varbanov, M., Robert-Hebmann, V., Sagnier, S., Robbins, I., Sanchez, F., Lafont, V., and Biard-Piechaczyk, M. (2009). Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 4, e5787. doi:10.1371/journal.pone.0005787
Feng, C. G., Zheng, L., Jankovic, D., Bafica, A., Cannons, J. L., Watford, W. T., Chaussabel, D., Hieny, S., Caspar, P., Schwartzberg, P. L., Lenardo, M. J., and Sher, A. (2008). The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-gamma-induced cell death. Nat. Immunol. 9, 1279–1287.
Fox, C. J., Hammerman, P. S., and Thompson, C. B. (2005). Fuel feeds function: energy metabolism and the T-cell response. Nat. Rev. Immunol. 5, 844–852.
Galluzzi, L., and Kroemer, G. (2008). Necroptosis: a specialized pathway of programmed necrosis. Cell 135, 1161–1163.
Gerland, L. M., Genestier, L., Peyrol, S., Michallet, M. C., Hayette, S., Urbanowicz, I., Ffrench, P., Magaud, J. P., and Ffrench, M. (2004). Autolysosomes accumulate during in vitro CD8+ T-lymphocyte aging and may participate in induced death sensitization of senescent cells. Exp. Gerontol. 39, 789–800.
Gomes, L. C., Di Benedetto, G., and Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598.
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93.
Heino, M., Peterson, P., Kudoh, J., Nagamine, K., Lagerstedt, A., Ovod, V., Ranki, A., Rantala, I., Nieminen, M., Tuukkanen, J., Scott, H. S., Antonarakis, S. E., Shimizu, N., and Krohn, K. (1999). Autoimmune regulator is expressed in the cells regulating immune tolerance in thymus medulla. Biochem. Biophys. Res. Commun. 257, 821–825.
Hildeman, D. A., Mitchell, T., Kappler, J., and Marrack, P. (2003). T cell apoptosis and reactive oxygen species. J. Clin. Invest. 111, 575–581.
Hubbard, V. M., Valdor, R., Patel, B., Singh, R., Cuervo, A. M., and Macian, F. (2010). Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunol. 185, 7349–7357.
Jia, W., and He, Y. W. (2011). Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J. Immunol. 186, 5313–5322.
Jia, W., Pua, H. H., Li, Q. J., and He, Y. W. (2011). Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J. Immunol. 186, 1564–1574.
Kim, H. J., Lee, S., and Jung, J. U. (2010). When autophagy meets viruses: a double-edged sword with functions in defense and offense. Semin. Immunopathol. 32, 323–341.
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721.
Kovacs, J. R., Li, C., Yang, Q., Li, G., Garcia, I. G., Ju, S., Roodman, D. G., Windle, J. J., Zhang, X., and Lu, B. (2012). Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 19, 144–152.
Kraft, C., Reggiori, F., and Peter, M. (2009). Selective types of autophagy in yeast. Biochim. Biophys. Acta 1793, 1404–1412.
Laurent-Crawford, A. G., Krust, B., Riviere, Y., Desgranges, C., Muller, S., Kieny, M. P., Dauguet, C., and Hovanessian, A. G. (1993). Membrane expression of HIV envelope glycoproteins triggers apoptosis in CD4 cells. AIDS Res. Hum. Retroviruses 9, 761–773.
Levine, B., Mizushima, N., and Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335.
Li, C., Capan, E., Zhao, Y., Zhao, J., Stolz, D., Watkins, S. C., Jin, S., and Lu, B. (2006). Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 177, 5163–5168.
Liang, X. H., Kleeman, L. K., Jiang, H. H., Gordon, G., Goldman, J. E., Berry, G., Herman, B., and Levine, B. (1998). Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72, 8586–8596.
Longatti, A., and Tooze, S. A. (2009). Vesicular trafficking and autophagosome formation. Cell Death Differ. 16, 956–965.
Mattoo, H., Faulkner, M., Kandpal, U., Das, R., Lewis, V., George, A., Rath, S., Durdik, J. M., and Bal, V. (2009). Naive CD4 T cells from aged mice show enhanced death upon primary activation. Int. Immunol. 21, 1277–1289.
McLeod, I. X., Zhou, X., Li, Q. J., Wang, F., and He, Y. W. (2011). The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J. Immunol. 187, 5051–5061.
Mortimore, G. E., and Poso, A. R. (1987). Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu. Rev. Nutr. 7, 539–564.
Nedjic, J., Aichinger, M., Emmerich, J., Mizushima, N., and Klein, L. (2008a). Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455, 396–400.
Nedjic, J., Aichinger, M., and Klein, L. (2008b). Autophagy and T cell education in the thymus: eat yourself to know yourself. Cell Cycle 7, 3625–3628.
Ohsumi, Y., and Mizushima, N. (2004). Two ubiquitin-like conjugation systems essential for autophagy. Semin. Cell Dev. Biol. 15, 231–236.
Osborn, S. L., Diehl, G., Han, S. J., Xue, L., Kurd, N., Hsieh, K., Cado, D., Robey, E. A., and Winoto, A. (2010). Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc. Natl. Acad. Sci. U.S.A. 107, 13034–13039.
Pua, H. H., Dzhagalov, I., Chuck, M., Mizushima, N., and He, Y. W. (2007). A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 204, 25–31.
Pua, H. H., Guo, J., Komatsu, M., and He, Y. W. (2009). Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 182, 4046–4055.
Pyo, J. O., Jang, M. H., Kwon, Y. K., Lee, H. J., Jun, J. I., Woo, H. N., Cho, D. H., Choi, B., Lee, H., Kim, J. H., Mizushima, N., Oshumi, Y., and Jung, Y. K. (2005). Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 280, 20722–20729.
Schatz, D. G., and Ji, Y. (2011). Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 11, 251–263.
Sitkovsky, M., and Lukashev, D. (2005). Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 5, 712–721.
Son, Y. M., Kwak, C. W., Lee, Y. J., Yang, D. C., Park, B. C., Lee, W. K., Han, S. H., and Yun, C. H. (2010). Ginsenoside Re enhances survival of human CD4+ T cells through regulation of autophagy. Int. Immunopharmacol. 10, 626–631.
Stephenson, L. M., Miller, B. C., Ng, A., Eisenberg, J., Zhao, Z., Cadwell, K., Graham, D. B., Mizushima, N. N., Xavier, R., Virgin, H. W., and Swat, W. (2009). Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 5, 625–635.
Takahama, Y., Nitta, T., Mat Ripen, A., Nitta, S., Murata, S., and Tanaka, K. (2010). Role of thymic cortex-specific self-peptides in positive selection of T cells. Semin. Immunol. 22, 287–293.
Takahashi, Y., Coppola, D., Matsushita, N., Cualing, H. D., Sun, M., Sato, Y., Liang, C., Jung, J. U., Cheng, J. Q., Mule, J. J., Pledger, W. J., and Wang, H. G. (2007). Bif-1 interacts with beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9, 1142–1151.
Takahashi, Y., Meyerkord, C. L., and Wang, H. G. (2009). Bif-1/endophilin B1: a candidate for crescent driving force in autophagy. Cell Death Differ. 16, 947–955.
Thorburn, A. (2008). Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis 13, 1–9.
Uddin, M. N., Nishio, N., Ito, S., Suzuki, H., and Isobe, K. (2012). Autophagic activity in thymus and liver during aging. Age (Dordr) 34, 75–85.
Wei, Y., Sinha, S., and Levine, B. (2008). Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951.
Wu, Y. T., Tan, H. L., Huang, Q., Sun, X. J., Zhu, X., and Shen, H. M. (2011). zVAD-induced necroptosis in L929 cells depends on autocrine production of TNFalpha mediated by the PKC-MAPKs-AP-1 pathway. Cell Death Differ. 18, 26–37.
Yousefi, S., Perozzo, R., Schmid, I., Ziemiecki, A., Schaffner, T., Scapozza, L., Brunner, T., and Simon, H. U. (2006). Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 8, 1124–1132.
Yu, L., Alva, A., Su, H., Dutt, P., Freundt, E., Welsh, S., Baehrecke, E. H., and Lenardo, M. J. (2004). Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502.
Yue, Z., Jin, S., Yang, C., Levine, A. J., and Heintz, N. (2003). Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. U.S.A. 100, 15077–15082.
Zhang, J., Cado, D., Chen, A., Kabra, N. H., and Winoto, A. (1998). Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392, 296–300.
Zhang, N., Hartig, H., Dzhagalov, I., Draper, D., and He, Y. W. (2005). The role of apoptosis in the development and function of T lymphocytes. Cell Res. 15, 749–769.
Keywords: autophagy, apoptosis, T lymphocyte, thymocyte selection, organelle homeostasis
Citation: He M-X, McLeod IX, Jia W and He Y-W (2012) Macroautophagy in T lymphocyte development and function. Front. Immun. 3:22. doi: 10.3389/fimmu.2012.00022
Received: 01 December 2011;
Paper pending published: 15 December 2011;
Accepted: 07 February 2012;
Published online: 21 February 2012.
Edited by:
Christian Munz, University of Zurich, SwitzerlandReviewed by:
Joern Dengjel, Albert-Ludwigs-Universität Freiburg, GermanyBinfeng Lu, University of Pittsburgh, USA
Copyright: © 2012 He, McLeod, Jia and He. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: You-Wen He, Department of Immunology, Duke University, Durham, NC 27710, USA. e-mail:he000004@mc.duke.ed