

- 1 Department of Microbiology and Immunology, Keio University School of Medicine, Shinanomachi, Tokyo, Japan
- 2 Division of Cardiovascular Medicine, Kurume University School of Medicine, Kurume, Japan
Cytokines play essential roles in innate and adaptive immunity. However, excess cytokines or dysregulation of cytokine signaling will cause a variety of diseases, including allergies, autoimmune diseases, inflammation, and cancer. Most cytokines utilize the so-called Janus kinase–signal transducers and activators of transcription pathway. This pathway is negatively regulated by various mechanisms including suppressors of cytokine signaling (SOCS) proteins. SOCS proteins bind to JAK or cytokine receptors, thereby suppressing further signaling events. Especially, suppressor of cytokine signaling-1 (SOCS1) and SOCS3 are strong inhibitors of JAKs, because these two contain kinase inhibitory region at the N-terminus. Studies using conditional knockout mice have shown that SOCS proteins are key physiological as well as pathological regulators of immune homeostasis. Recent studies have also demonstrated that SOCS1 and SOCS3 are important regulators of helper T cell differentiation and functions. This review focuses on the roles of SOCS1 and SOCS3 in T cell mediated inflammatory diseases.
The CIS/SOCS Family
Cytokines play essential roles in the development, differentiation, and function of myeloid and lymphoid cells. Some of them, including interleukins, interferons (IFNs), and hematopoietic growth factors, activate the Janus kinase (JAK)–signal transducers and activators of the transcription (STAT) pathway (O’Shea et al., 2002; Adamson et al., 2009). In this pathway, cytokine binding results in receptor oligomerization, which initiates the activation of JAK kinases (JAK1, JAK2, JAK3, and Tyk2). The activated JAKs phosphorylate the receptor cytoplasmic domains, which creates docking sites for SH2-containing signaling proteins, including STATs. It is now known that a large number of cytokines, growth factors, and hormonal factors activate JAK and/or STAT proteins.
The CIS/suppressors of cytokine signaling (SOCS) family of proteins is one of the major mechanisms for regulations of cytokine signaling (Endo et al., 1997; Kubo et al., 2003; Yoshimura et al., 2007). The first member of the family discovered is CIS, cytokine-inducible SH2 protein (Yoshimura et al., 1995). This molecule was identified by subtraction as an immediate-early gene induced by erythropoietin (EPO). CIS is found to be a negative-feedback regulator of the STAT5 pathway; binding to the phosphorylated tyrosine residues of cytokine receptors through the SH2 domain, thereby masking STAT5 docking sites. CIS is a very specific negative regulator of STAT5, and was confirmed in vivo by generating CIS transgenic mice (Matsumoto et al., 1999). The second member, suppressor of cytokine signaling-1/JAK-binding protein (SOCS1/JAB) was identified by three groups by different methods (Endo et al., 1997; Naka et al., 1997; Starr et al., 1997). We have isolated SOCS1/JAB as a JAK-binding protein, and subsequently, we showed that SOCS1/JAB strongly inhibited JAK tyrosine kinase activity.
At the time of their discovery, the SOCS proteins were recognized as an important mechanism in the negative regulation of the cytokine-JAK–STAT pathway, but recent studies using gene-disrupted mice have revealed that they play additional unexpected and important roles in many immunological processes (Chinen et al., 2011; Hiwatashi et al., 2011; Takahashi et al., 2011; Tamiya et al., 2011), atherosclerosis (Taleb et al., 2009), metabolism (Mori et al., 2004; Torisu et al., 2007), and cancer (Yoshida et al., 2004; Ogata et al., 2006a,b; Hiwatashi et al., 2011). In this review, we will focus on the recent progress of SOCS studies on inflammation and helper T cell differentiation.
The CIS/SOCS Family
Overview
The SOCS proteins and CIS (also known as CISH) protein comprise a family of intracellular proteins (Yasukawa et al., 2003; Yoshimura et al., 2007; Tamiya et al., 2011). There are eight CIS/SOCS family proteins: CIS, SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, and SOCS7, each of which has a central SH2 domain, an amino-terminal domain of variable length and sequence, and a carboxy-terminal 40-amino-acid module known as the SOCS box (Figure 1 left; Masuhara et al., 1997).
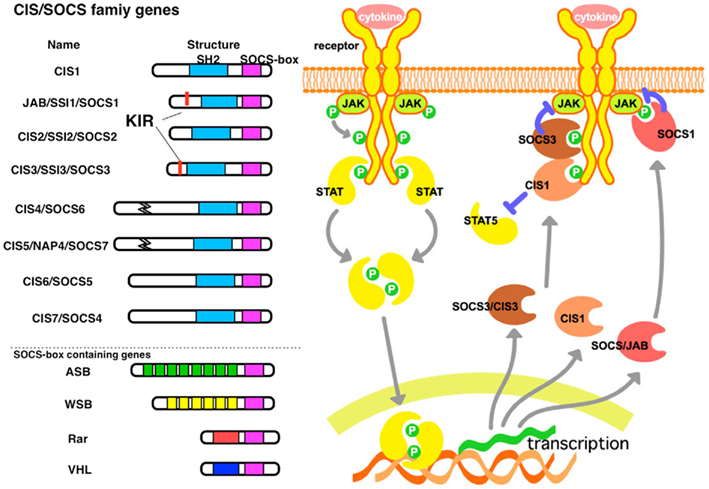
Figure 1. The structure and function of SOCS proteins. (left) Schematic structure of the CIS/SOCS family proteins. The SOCS box is conserved in all CIS/SOCS family proteins. SOCS1 and SOCS3 contain a unique kinase inhibitory region (KIR) immediately upstream of the central SH2 domain, which is proposed to function as a pseudosubstrate. Other SOCS box-containing proteins are illustrated. (right) Mechanism of suppression by CIS, SOCS1, and SOCS3. All of these are induced by cytokine stimulation. CIS binds to the STAT5 activating receptors, thereby suppressing further activation of STAT5 and inducing degradation of the receptor. SOCS1 binds to JAKs, and SOCS3 binds to the receptor through the SH2 domain, but both inhibit JAK activity through KIR. These complexes may be degraded by ubiquitination and proteasomal degradation recruited through the SOCS box.
In addition, both SOCS1 and SOCS3 can inhibit JAK tyrosine kinase activity directly through their kinase inhibitory region (KIR). KIR has been proposed to function as a pseudosubstrate that is essential for the suppression of cytokine signals (Yasukawa et al., 1999). The SH2 domain of SOCS3 does not have a high affinity to the activation loop of JAKs yet the KIR of SOCS3 has a higher affinity to the kinase domain of JAK2 than that of SOCS1 (Sasaki et al., 1999). Because the receptors to which SOCS3 binds mostly activate STAT3, SOCS3 is an inhibitor that is relatively specific to STAT3. SOCS3 also inhibits STAT4, which is activated by IL-12 (Yamamoto et al., 2003). However, because SOCS3 does not bind to the IL-10 receptor, SOCS3 cannot inhibit IL-10 signaling. Therefore, IL-10 induces a robust and prolonged STAT3 activation, whereas IL-6-mediated STAT3 activation is transient in macrophages. This is an important mechanism to distinguish the anti-inflammatory activity of IL-10 and inflammatory activity of IL-6 (Yasukawa et al., 2003).
SOCS1 and SOCS3 inhibit not only STATs but also other signaling pathways such as Ras/ERK and PI3K, which affect cell-proliferation, survival, and differentiation (Lu et al., 2006; Madonna et al., 2008). Interestingly, SOCS3 is tyrosine phosphorylated upon cytokine or growth factor stimulation, and phosphorylated Y221 of SOCS3 interacts with p120-RasGAP, resulting in a sustained activation of ERK. Although SOCS proteins inhibit growth factor responses, tyrosine phosphorylation of SOCS3 can ensure cell survival and proliferation through the Ras pathway (Cacalano et al., 2001).
The SOCS Box and Ubiqutination
The SOCS box is also found in other miscellaneous proteins (Figure 1, left). The SOCS box interacts with elongin B and elongin C, Cullins, and the RING-finger-domain-only protein RBX2 (which recruits E2 ubiquitin-transferase; Kamizono et al., 2001; Kamura et al., 2004). VHL (von Hippel–Lindau) gene product, whose gene product is the principal negative regulator of hypoxia-inducible factor has been shown to bind to SOCS1 and induces the degradation of Jak2. Chuvash polycythemia-associated VHL mutants have altered affinity for SOCS1 and do not engage with and degrade phosphorylated JAK2 (Russell et al., 2011). These results indicate that CIS/SOCS family proteins, as well as other SOCS box-containing molecules, function as E3 ubiquitin ligases and mediate the degradation of proteins that are associated with these family members through their N-terminal regions (Figure 1, left).
The central SH2 domain determines the target of each SOCS and CIS protein. The SH2 domain of SOCS1 directly binds to the activation loop of JAKs (Yasukawa et al., 1999). The SH2 domains of CIS, SOCS2, and SOCS3 bind to phosphorylated tyrosine residues on activated cytokine receptors (Kubo et al., 2003). SOCS3 binds to gp130-related cytokine receptors, including the phosphorylated tyrosine 757 (Tyr757) residue of gp130, the Tyr800 residue of IL-12 receptor β2, and Tyr985 of the leptin receptor (Bjorbak et al., 2000; Sasaki et al., 2000; Schmitz et al., 2000; Lehmann et al., 2003; Yamamoto et al., 2003). Thus, SOCS3 in the brain has been implicated in leptin-resistance (Mori et al., 2004; Zhang et al., 2008). SOCS molecules bind to several tyrosine phosphorylated proteins, including Mal (TLR signaling; Kobayashi et al., 2006) and IRS1/2 (insulin signaling; Torisu et al., 2007). Thus, SOCS proteins generally induce the degradation of the target molecules by binding through the SH2 domain and ubiquitination through the SOCS box (Figure 1, right).
SOCS and Inflammation
SOCS1 and Tissue Inflammation
Although SOCS1 knockout (KO) mice are normal at birth, they exhibit stunted growth and die within 3 weeks of birth, with activation of peripheral T cells, necrosis of the liver, and macrophage infiltration of major organs (Marine et al., 1999). The neonatal defects exhibited by SOCS1−/− mice appear to occur primarily as a result of unbridled IFNγ signaling, since SOCS1−/− mice that also lack the IFNγ gene or the IFNγ receptor gene do not die neonatal (Alexander et al., 1999). Since SOCS1/Rag2-double knockout (DKO) mice survived much longer, SOCS1 has been thought to be an important negative regulator of T cells. This is confirmed by analyzing T cell-specific SOCS1-conditional KO (cKO) mice (Tanaka et al., 2008). T cell specific SOCS1 cKO mice developed several inflammatory diseases with high levels of IFNγ.
In addition, SOCS1 has been demonstrated to be involved in the suppression of inflammation by regulating innate immune cells and non-immune cells. Using liver-specific SOCS1 cKO mice, we demonstrated that SOCS1 deletion in hepatocytes enhanced concanavalin A (ConA)-induced hepatitis due to enhanced pro-apoptotic signals, including STAT1 and JNK, in the SOCS1-deficient liver (Torisu et al., 2008). SOCS1 deletion in NKT cells also enhanced sensitivity to ConA-induced hepatitis. However, the number of iNKT cells was drastically decreased but that of type II NKT cells was increased by SOCS1 deficiency (Hashimoto et al., 2011). The mechanism of imbalance between type I and type II NKT cells by SOCS1 deficiency remains to be clarified. Deficiency of SOCS1 in macrophages resulted in hyper-responses to lipopolysaccharide (LPS; Kinjyo et al., 2002; Hashimoto et al., 2009; Sachithanandan et al., 2011) and SOCS1-deficient dendritic cells (DCs) promoted hyperactivation of Th1, lupus-like autoimmune diseases, and anti-tumor immunity (Hanada et al., 2003, 2005).
We have demonstrated that SOCS1 plays an essential role in intestinal immune homeostasis by regulating prostaglandin E2 (PGE2)-mediated DC and macrophage suppression (Chinen et al., 2011). Although SOCS1/Rag2 DKO mice did not die neonatally, these mice developed severe colitis at 2–6 months of age, mostly due to impairment of the PGE2-mediated anti-inflammatory mechanism. PGE2 has been shown to inhibit TLR signaling by suppressing NF-kB activity through c-Fos (Koga et al., 2009). This suppression system is shown to be impaired in SOCS1-deficeint DCs, due to hyperactivation of STAT1 (Figure 2; Chinen et al., 2011). SOCS1 has been implicated in the mechanism of glucocorticoid-mediated STAT1 suppression (Haffner et al., 2008; Bhattacharyya et al., 2011).
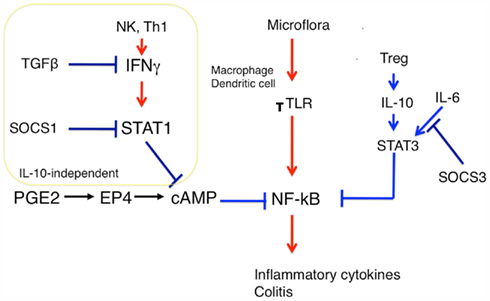
Figure 2. Role of SOCS1 and PGE2 in macrophage/dendritic cell activation. Treg/IL-10/STAT3 and PGE2/cAMP are two independent anti-inflammatory mechanisms, although precise molecular mechanisms have not been clarified yet. SOCS1 is involved in the cAMP suppression system but not in the IL-10 system. SOCS1 prevents the breakdown of cAMP-mediated TLR signaling by suppressing STAT1. On the other hand, SOCS3 may keep proper TLR signaling by inhibiting IL-6-mediated STAT3 activation. SOCS3 has no effect on IL-10 mediated anti-inflammatory effect.
SOCS1 and Infection
SOCS1 is also highly upregulated by M. tuberculosis infection and reduced responses to IL-12, resulting in an impaired IFNγ secretion by macrophages that in turn accounts for deteriorated intracellular mycobacterial control. Thus, SOCS1 expression by macrophages hampered M. tuberculosis clearance early after infection in vivo in an IFNγ-dependent manner. On the other hand, at later time points, SOCS1 expression by non-macrophage cells protected the host from infection-induced detrimental inflammation (Carow et al., 2011). Similarly, SOCS1 is highly induced by Toxoplasma gondii infection, which is a mechanism to escape from IFNγ action (Stutz et al., 2012). Hepatitis C virus (HCV) core protein has been shown to impair IL-12 expression in monocytes/macrophages through interaction with a complement receptor gC1qR, which triggers the expression of SOCS1 (Zhang et al., 2011). SOCS1 is also induced by Ebola virus infection in macrophages (Okumura et al., 2010). These reports suggest that SOCS1 is induced in macrophages by various type of infection and inhibits TLR signaling, IL-12 production and IFNγ responses, which is an important mechanism for microbes to escape from host immunity.
SOCS3 Regulates IL-6-Medaited Pro- and Anti-Inflammatory Reaction
In contrast to SOCS1, the role of SOCS3 in innate inflammation is complex. SOCS3 deficiency in macrophages protects mice from endotoxemia, because of the reduced production of inflammatory cytokines, which is due to the enhanced anti-inflammatory effect of STAT3 (Yasukawa et al., 2003). Furthermore, macrophage-specific SOCS3 cKO mice have reduced IL-12 responses and succumb to toxoplasmosis (Whitmarsh et al., 2011). In the absence of SOCS3, macrophages are hypersensitive to the anti-inflammatory properties of IL-6. Thus, SOCS3 plays a critical role in suppressing IL-6 signals and promoting immune responses to control T. gondii infection (Whitmarsh et al., 2011). On the contrary, mice with a conditional deletion of SOCS3 in hematopoietic cells have been shown to develop lethal inflammatory disease during adult life and develop gross histopathological changes during experimental arthritis, typified by elevated IL-6 levels (Wong et al., 2006). Croker et al. reported that acute responses to IL-1β were lethal to SOCS3 cKO mice but not SOCS3/IL-6-double KO mice, indicating that loss of SOCS3 is pro-inflammatory when IL-6 is required for inflammation. Furthermore, they showed that infection of SOCS3 cKO mice with LCMV induced a lethal inflammatory response that was dependent on IL-6 (Croker et al., 2012). Therefore, SOCS3 is probably both pro- and anti-inflammatory depending on the pro- and anti-inflammatory action of IL-6.
SOCS3 and Macrophage Polarization
SOCS3 in macrophages may regulate macrophage polarization. At least two distinct subpopulations with different functions, the classically (M1) and the alternatively (M2) activated macrophages, have been found (Murray and Wynn, 2011). Macrophages in which SOCS3 was knocked down by short interfering RNA prevented M1 activation, suggesting that SOCS3 is necessary for M1 (Liu et al., 2008). Wang et al. (2010) reported that forced activation of Notch signaling in macrophages enhanced M1 polarization and their anti-tumor capacity through SOCS3 induction. Macrophage-specific SOCS3 cKO mice exhibited resistance to the tumor transplantation model because of reduced tumor-promoting cytokines such as TNFα and IL-6 and enhanced production of anti-tumorigenic chemokine MCP2/CCL8 (Hiwatashi et al., 2011). Thus, SOCS3 is an important modulator of macrophage phase and functions.
SOCS3 and DCs
SOCS3−/− DCs exhibited constitutive activation of STAT3 and expressed low levels of MHC class II molecules, co-stimulatory molecules, and IL-12 (Matsumura et al., 2007). Adoptive transfer of SOCS3−/− DCs suppressed experimental autoimmune encephalomyelitis (EAE). SOCS3−/− DCs produced a higher amount of TGF-β than WT DCs, resulting in a selective expansion of forkhead box P3 (FoxP3)-positive regulatory T cells (Tregs). Thus, in the absence of SOCS3, DCs tends to become tolerogenic DCs. However, SOCS3-transduced DCs also expressed low levels of MHC class II and CD86 molecules and produced high levels of IL-10 but low levels of IL-12, IFNγ, and IL-23 p19 (Li et al., 2006). STAT3 activation was suppressed by SOCS3 overexpression. Although the mechanism has not yet been clarified, SOCS3-transduced DCs efficiently induced Th2-cell differentiation and suppressed Th17 in vitro and in vivo and the adoptive transfer of SOCS3-overexpressing DCs suppressed EAE, just like SOCS3−/− DCs (Li et al., 2006). These results suggest that the status of STAT3 activation levels may determine the balance between Th2 and Tregs induced by DCs.
In addition, SOCS3 is an important negative regulator of granulopoiesis because SOCS3 negatively regulates the G-CSF receptor signaling (Croker et al., 2004; Kimura et al., 2004). Mice in which the SOCS3 gene was deleted in all hematopoietic cells developed a spectrum of inflammatory pathologies with hyper-neutrophilia. SOCS3-deficient mice developed inflammatory neutrophil infiltration into multiple tissues and consequent hind-leg paresis (Wong et al., 2006). SOCS3 has also been shown to inhibit NKT cell activation (Nakaya et al., 2009).
Function of SOCS3 in Non-Immune Cells for Inflammation
In non-immune cells, SOCS3 suppresses inflammatory reactions by inhibiting STAT3. STAT3 activation is found in epithelial and lamina propria cells in the colon of mice with intestinal bowel disease (IBD), as well as in human ulcerative colitis and Crohn’s disease patients (Suzuki et al., 2001) and in synovial fibroblasts of RA patients (Shouda et al., 2001). Forced expression of either SOCS3 or a dominant negative form of STAT3 in mouse arthritis models suppressed the induction/development of the disease, indicating that SOCS3 in non-immune cells is probably anti-inflammatory (Shouda et al., 2001). These findings are consistent with the idea that the IL-6- and IL-6-related cytokines-STAT3 pathway promotes chronic disease progression and SOCS3 is part of this negative-feedback loop (Sawa et al., 2006 #412). This idea is supported by a recent finding that the JAK inhibitor CP-690550 is a potent therapeutic agent for the autoimmune arthritis model by suppressing the IL-6/STAT3 amplification (Mori et al., 2011). However, when STAT3 plays a protective role for tissue injury, such as in ConA-induced hepatitis, deletion of SOCS3 is anti-inflammatory (Ogata et al., 2006b).
SOCS and Helper T Cells
SOCS1 and Effector Helper T Cells
We have recently demonstrated that SOCS1 is an essential regulator for helper T cell differentiation. Most SOCS1−/−CD4 naïve T cells differentiated into Th1, even under Th2 or Th17 skewing conditions, whereas Th17 differentiation was strongly suppressed (Tanaka et al., 2008). This was also dependent on IFNγ, because Th17 was normally developed in SOCS1−/− IFNγ−/− T cells. As a result, T cell-specific SOCS1-deficient mice developed autoimmune inflammatory diseases with age (Takahashi et al., 2011) and were very sensitive to dextran sulfate sodium (DSS)-induced colitis (Horino et al., 2008) and ConA-induced hepatitis (Th1 type disease; Hashimoto et al., 2011), but were resistant to EAE, a typical Th17 type disease (Tanaka et al., 2008).
Th17 suppression by SOCS1 deficiency is probably due to the hyperproduction and signal transduction of IFNγ. Indeed, STAT1 activation in SOCS1−/− T cells was upregulated and strong Th1 skewing was corrected under STAT1+/− conditions (Tanaka et al., 2008). Interestingly, STAT3 activation was reduced in SOCS1-deficient T cells, mostly due to the upregulation of SOCS3 gene expression, which can account for reduced IL-6 responses and Th17 differentiation. Indeed, SOCS3-tg mice were resistant to EAE, and Th17 differentiation of SOCS3-tg T cells was suppressed. The reciprocal regulation of Th1 and Th17 by SOCS1 and SOCS3 is illustrated in Figure 3. In addition, SOCS1−/− T cells were less responsive to TGF-β, although the mechanism has not yet been clarified. Reduced STAT3 activation and TGF-β signaling may explain the suppression of Th17 differentiation in SOCS1-deficient T cells. Our microarray analysis revealed that T-bet, Eomesodermin, and Gfi-1 were upregulated in SOCS1-deficeint T cells under Th17 skewing conditions, all of which have been reported to suppress Th17 differentiation (Ichiyama et al., 2009, 2011). Role of SOCS1 and SOCS3 in Th differentiation is summarized in Figures 3 and 4A.
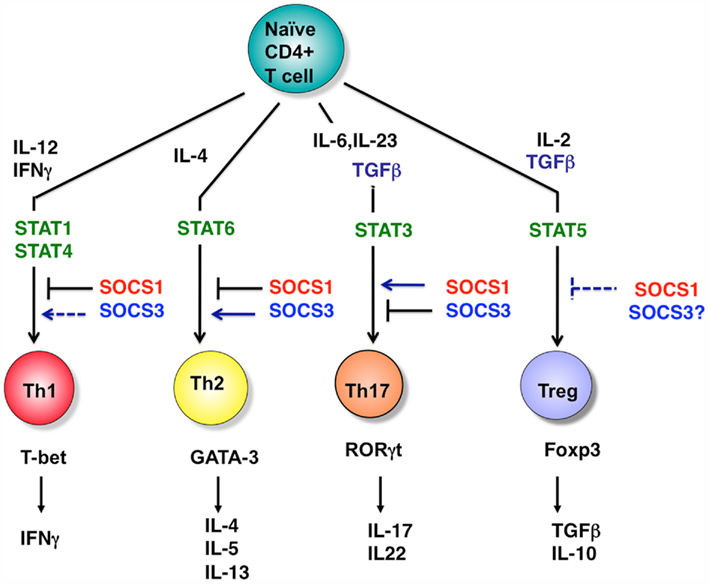
Figure 3. Role of STATs and SOCS proteins in helper T cell differentiation. T cell differentiation from naïve cells into the various functional subtypes (e.g., Th1, Th2, Th17, and Treg cells) primarily depends on the action of cytokines as indicated. SOCS1 and SOCS3 regulate the cytokine pathways indicated, and thereby dictate CD4+ T cell differentiation. Note that SOCS3 inhibits Th1 through overexpression by suppressing IL-12 mediated STAT4 activation. However, SOCS3 deletion also inhibits Th1 by promoting the production of IL-10 and TGF-β. In Tregs, SOCS1 deletion promotes Treg expansion; however, these are functionally impaired. Thus, SOCS1 negatively regulates the Treg number, but is necessary for the function of Tregs. The role of SOCS3 in Tregs has not yet been clarified. For Th17, RORγt induction by TGF-β has been demonstrated to be independent of Smad2/3/4.
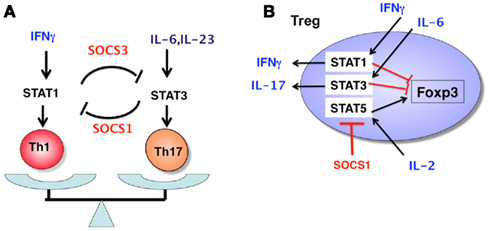
Figure 4. Role of SOCS1 and SOCS3 in Th1/Th17 balance (A) and in nTreg integrity (B). In Th1 conditions, activated STAT1 induces SOCS3 expression, which in turn inhibits STAT3 activity, thereby suppressing Th17 development. In Th17 conditions, SOCS1 is highly expressed and inhibits IFNγ-mediated Th1 development. In nTregs, the SOCS1 level is constantly high, and suppresses STAT1, STAT3, and STAT5 in inflammatory conditions (high levels of cytokines such as IL-2, IL-6, and IFNγ). The higher activation of STAT5 results in an increase in the number of Tregs, because STAT5 directly upregulates Foxp3 expression. The higher activation of STAT1 and STAT3 induces loss of Foxp3 expression by unknown mechanisms and promotes IFNγ and IL-17 production, respectively. Thus, SOCS1 is a “guardian” of Tregs.
SOCS1 and Tregs
Suppressor of cytokine signaling-1 also plays an important role in the regulation of regulatory T cells. Higher numbers of Tregs are observed in the thymus and spleen of T cell-specific SOCS1-deficient mice (Lu et al., 2009). This is probably due to higher IL-2 responses, because IL-2 enhances the proliferation of Tregs. Importantly, SOCS1 has been shown to be a target of miRNA-155 in Tregs (Lu et al., 2009). During thymic differentiation, the upregulation of Foxp3 drives the high expression of miR155, which in turn promotes the expansion of Treg cells by targeting SOCS1. However, SOCS1 has recently been found to play more important functional roles in Tregs. Various studies have suggested that Tregs may become harmful effector T cells in inflammatory conditions. Lu et al. (2010) observed that SOCS1 deletion specifically in Tregs induced the development of spontaneous dermatitis, splenomegaly, and lymphadenopathy, suggesting a defective Treg function in these mice. The defective suppression activity of SOCS1-deficient Tregs was confirmed through the failure to suppress colitis in Rag2−/− mice by the co-transfer of naïve T cells and Tregs (Takahashi et al., 2011). In the absence of SOCS1, Tregs easily lost Foxp3 expression, and became pathogenic T cells that induced severe colitis (Lu et al., 2010). In addition, SOCS1 plays an important role in preventing inflammatory cytokine production from Tregs. Normally, Tregs do not secrete inflammatory cytokines even in inflammatory conditions. In the absence of SOCS1, Tregs secrete IFNγ and IL-17 by hyperactivation of STAT1 and STAT3, respectively (Takahashi et al., 2011). Thus, SOCS1 is a “guardian” of Tregs, since SOCS1 inhibits loss of Foxp3 and conversion of Tregs to Th1- or Th17-like cells (Figure 4B).
Role of SOCS3 in Helper T Cells
The degree to which SOCS3 expression in T cells is increased is correlated to the severity of human allergic diseases such as asthma and atopic dermatitis (Seki et al., 2003). The enhanced action of SOCS3 may promote allergic responses, since transgenic SOCS3 expression in T cells inhibits Th1 development and promotes Th2 development (Seki et al., 2003). Enhanced Th2 development may be due to the suppression of Th1 because IL-12 mediated Th1 differentiation by SOCS3 overexpression. Therefore, SOCS3-tg mice were sensitive to L. Major infection, where Th1 is necessary for eradication of this microbe (Nakaya et al., 2011). As described before, SOCS3-expressing T cells differentiated into Th17 cells less efficiently than WT T cells (Tanaka et al., 2008). In contrast, mice lacking SOCS3 in T cells result in reduced allergen-induced eosinophilia in the airways (Kinjyo et al., 2006; Figure 3). SOCS3 silencing with small interfering RNA (siRNA) in primary CD4+ T cells attenuated the Th2 response in vitro and in vivo (Moriwaki et al., 2011). SOCS3 deficiency promoted Th17 differentiation in T cells (Chen et al., 2006). Using VavCre-SOCS3 cKO mice, Wong et al. (2006) reported that the IL-1-induced inflammatory joint disease model was severely deteriorated in the absence of SOCS3 accompanying the enhanced IL-17 production from CD4+ T cells. SOCS3 deficiency in T cells reduced atherosclerotic lesion development and vascular inflammation, which was dependent on IL-17, whereas the overexpression of SOCS3 in T cells reduced IL-17 and accelerated atherosclerosis (Taleb et al., 2009). The absence of SOCS3 in helper T cells therefore generally inhibits Th1 and Th2 by producing IL-10 and TGF-β, but had dramatic pro-inflammatory effects under Th17 conditions (Tamiya et al., 2011). Recently, leukemia inhibitory factor (LIF) has been shown to inhibit Th17 differentiation by inducing SOCS3 (Cao et al., 2011). The paradoxical effect of SOCS3 on T cell regulation is mostly due to the dual function of STAT3; it promotes the production of both inflammatory IL-17 and anti-inflammatory IL-10 and TGF-β.
In the LCMC clone 13 infection model, SOCS3 is highly induced in T cells, and T cell-specific SOCS3-deficient mice exhibit a profound augmentation of immunity and are protected from severe organ pathology, with an increase in the number of virus-specific CD8 + T cells and an increase in the ability of CD4 + T cells to secrete TNF-α and IL-17. This T cell-intrinsic SOCS3 induction has been implicated as a major factor contributing to immunological failure in the setting of chronic active infection (Pellegrini et al., 2011).
SOCS and Human Inflammatory Diseases
It has been estimated that more than 20% of all malignancies are initiated or exacerbated by inflammation; for example, most human hepatocellular carcinomas (HCCs) are a consequence of HCV infection (Yoshimura, 2006). The expression of SOCS1 is often silenced in these tumors by hypermethylation of CpG islands including HCCs. We found that silencing of SOCS1 was frequently observed even in pre-malignant HCV-infected patients (Yoshida et al., 2004). Liver injury is associated with hyperactivation of STAT1 and reduced activation of STAT3. Therefore, the reduced expression of SOCS1 may enhance tissue injury and inflammation through the hyperactivation of STAT1, promoting the turnover of epithelial cells and enhancing their susceptibility to oncogenesis. Therefore, SOCS1 is a unique anti-oncogene that prevents carcinogenesis by suppressing chronic inflammation (Hanada et al., 2006; Torisu et al., 2008).
SOCS3 may also be involved in the development and progression of malignancies. SOCS3 expression levels were reduced in tumor areas of patients infected with HCV compared with non-tumor regions (Ogata et al., 2006b). Hyperactivation of STAT3 by SOCS3 repression may contribute to tumorigenesis by inducing multiple tumor-promoting genes (Ogata et al., 2006a).
As mentioned before, levels of SOCS3 in T cells are correlated to allergic diseases (Seki et al., 2003). Several genomic SNPs in the human SOCS1 gene were found to be associated with serum IgE levels (Mostecki et al., 2011), asthma (Harada et al., 2007), and leukemia (Guillem et al., 2012). SOCS1 mutations were found in human lymphomas (Mottok et al., 2009).
Concluding Remarks
Over the past decade, following the discovery of the SOCS protein families, we have extended our understanding of the structure and function of these proteins. SOCS proteins act as simple negative-feedback regulators, and they also play a part in the fine tuning of the immune response and inflammation. Therapeutic trials using SOCS anti-sense oligonucleotides, shRNA, and peptide mimetics are currently underway in animal models. SOCS1 and SOCS3 are ideal therapeutic targets for autoimmune diseases and inflammatory diseases, including cancer.
Funding
This work was supported by special Grants-in-Aid from the Ministry of Education, Science, Technology, Sports and Culture of Japan, the Program for the Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO), and the Uehara Memorial Science Foundation, the SENSHIN Foundation, the Mochida Memorial Foundation, and the Takeda Science Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adamson, A. S., Collins, K., Laurence, A., and O’Shea, J. J. (2009). The Current STATus of lymphocyte signaling: new roles for old players. Curr. Opin. Immunol. 21, 161–166.
Alexander, W. S., Starr, R., Fenner, J. E., Scott, C. L., Handman, E., Sprigg, N. S., Corbin, J. E., Cornish, A. L., Darwiche, R., Owczarek, C. M., Kay, T. W., Nicola, N. A., Hertzog, P. J., Metcalf, D., and Hilton, D. J. (1999). SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 98, 597–608.
Bhattacharyya, S., Zhao, Y., Kay, T. W., and Muglia, L. J. (2011). Glucocorticoids target suppressor of cytokine signaling 1 (SOCS1) and type 1 interferons to regulate Toll-like receptor-induced STAT1 activation. Proc. Natl. Acad. Sci. U.S.A. 108, 9554–9559.
Bjorbak, C., Lavery, H. J., Bates, S. H., Olson, R. K., Davis, S. M., Flier, J. S., and Myers, M. G. Jr. (2000). SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J. Biol. Chem. 275, 40649–40657.
Cacalano, N. A., Sanden, D., and Johnston, J. A. (2001). Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat. Cell Biol. 3, 460–465.
Cao, W., Yang, Y., Wang, Z., Liu, A., Fang, L., Wu, F., Hong, J., Shi, Y., Leung, S., Dong, C., and Zhang, J. Z. (2011). Leukemia inhibitory factor inhibits T helper 17 cell differentiation and confers treatment effects of neural progenitor cell therapy in autoimmune disease. Immunity 35, 273–284.
Carow, B., Ye, X., Gavier-Widen, D., Bhuju, S., Oehlmann, W., Singh, M., Skold, M., Ignatowicz, L., Yoshimura, A., Wigzell, H., and Rottenberg, M. E. (2011). Silencing suppressor of cytokine signaling-1 (SOCS1) in macrophages improves Mycobacterium tuberculosis control in an interferon-gamma (IFN-gamma)-dependent manner. J. Biol. Chem. 286, 26873–26887.
Chen, Z., Laurence, A., Kanno, Y., Pacher-Zavisin, M., Zhu, B. M., Tato, C., Yoshimura, A., Hennighausen, L., and O’Shea, J. J. (2006). Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. U.S.A. 103, 8137–8142.
Chinen, T., Komai, K., Muto, G., Morita, R., Inoue, N., Yoshida, H., Sekiya, T., Yoshida, R., Nakamura, K., Takayanagi, R., and Yoshimura, A. (2011). Prostaglandin E2 and SOCS1 have a role in intestinal immune tolerance. Nat. Commun. 2, 190.
Croker, B. A., Kiu, H., Pellegrini, M., Toe, J., Preston, S., Metcalf, D., O’Donnell, J. A., Cengia, L. H., McArthur, K., Nicola, N. A., Alexander, W. S., and Roberts, A. W. (2012). IL-6 promotes acute and chronic inflammatory disease in the absence of SOCS3. Immunol. Cell Biol. 90, 124–129.
Croker, B. A., Metcalf, D., Robb, L., Wei, W., Mifsud, S., DiRago, L., Cluse, L. A., Sutherland, K. D., Hartley, L., Williams, E., Zhang, J. G., Hilton, D. J., Nicola, N. A., Alexander, W. S., and Roberts, A. W. (2004). SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity 20, 153–165.
Endo, T. A., Masuhara, M., Yokouchi, M., Suzuki, R., Sakamoto, H., Mitsui, K., Matsumoto, A., Tanimura, S., Ohtsubo, M., Misawa, H., Miyazaki, T., Leonor, N., Taniguchi, T., Fujita, T., Kanakura, Y., Komiya, S., and Yoshimura, A. (1997). A new protein containing an SH2 domain that inhibits JAK kinases. Nature 387, 921–924.
Guillem, V., Amat, P., Cervantes, F., Alvarez-Larran, A., Cervera, J., Maffioli, M., Bellosillo, B., Collado, M., Marugan, I., Martinez-Ruiz, F., and Hernández-Boluda, J. C. (2012). Functional polymorphisms in SOCS1 and PTPN22 genes correlate with the response to imatinib treatment in newly diagnosed chronic-phase chronic myeloid leukemia. Leuk. Res. 36, 174–181.
Haffner, M. C., Jurgeit, A., Berlato, C., Geley, S., Parajuli, N., Yoshimura, A., and Doppler, W. (2008). Interaction and functional interference of glucocorticoid receptor and SOCS1. J. Biol. Chem. 283, 22089–22096.
Hanada, T., Kobayashi, T., Chinen, T., Saeki, K., Takaki, H., Koga, K., Minoda, Y., Sanada, T., Yoshioka, T., Mimata, H., Kato, S., and Yoshimura, A. (2006). IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J. Exp. Med. 203, 1391–1397.
Hanada, T., Tanaka, K., Matsumura, Y., Yamauchi, M., Nishinakamura, H., Aburatani, H., Mashima, R., Kubo, M., Kobayashi, T., and Yoshimura, A. (2005). Induction of hyper Th1 cell-type immune responses by dendritic cells lacking the suppressor of cytokine signaling-1 gene. J. Immunol. 174, 4325–4332.
Hanada, T., Yoshida, H., Kato, S., Tanaka, K., Masutani, K., Tsukada, J., Nomura, Y., Mimata, H., Kubo, M., and Yoshimura, A. (2003). Suppressor of cytokine signaling-1 is essential for suppressing dendritic cell activation and systemic autoimmunity. Immunity 19, 437–450.
Harada, M., Nakashima, K., Hirota, T., Shimizu, M., Doi, S., Fujita, K., Shirakawa, T., Enomoto, T., Yoshikawa, M., Moriyama, H., Matsumoto, K., Saito, H., Suzuki, Y., Nakamura, Y., and Tamari, M. (2007). Functional polymorphism in the suppressor of cytokine signaling 1 gene associated with adult asthma. Am. J. Respir. Cell Mol. Biol. 36, 491–496.
Hashimoto, M., Ayada, T., Kinjyo, I., Hiwatashi, K., Yoshida, H., Okada, Y., Kobayashi, T., and Yoshimura, A. (2009). Silencing of SOCS1 in macrophages suppresses tumor development by enhancing antitumor inflammation. Cancer Sci. 100, 730–736.
Hashimoto, M., Hiwatashi, K., Ichiyama, K., Morita, R., Sekiya, T., Kimura, A., Sugiyama, Y., Sibata, T., Kuroda, K., Takahashi, R., and Yoshimura, A. (2011). SOCS1 regulates type I/type II NKT cell balance by regulating IFNgamma signaling. Int. Immunol. 23, 165–176.
Hiwatashi, K., Tamiya, T., Hasegawa, E., Fukaya, T., Hashimoto, M., Kakoi, K., Kashiwagi, I., Kimura, A., Inoue, N., Morita, R., Yasukawa, H., and Yoshimura, A. (2011). Suppression of SOCS3 in macrophages prevents cancer metastasis by modifying macrophage phase and MCP2/CCL8 induction. Cancer Lett. 308, 172–180.
Horino, J., Fujimoto, M., Terabe, F., Serada, S., Takahashi, T., Soma, Y., Tanaka, K., Chinen, T., Yoshimura, A., Nomura, S., Kawase, I., Hayashi, N., Kishimoto, T., and Naka, T. (2008). Suppressor of cytokine signaling-1 ameliorates dextran sulfate sodium-induced colitis in mice. Int. Immunol. 20, 753–762.
Ichiyama, K., Hashimoto, M., Sekiya, T., Nakagawa, R., Wakabayashi, Y., Sugiyama, Y., Komai, K., Saba, I., Moroy, T., and Yoshimura, A. (2009). Gfi1 negatively regulates T(h)17 differentiation by inhibiting RORgammat activity. Int. Immunol. 21, 881–889.
Ichiyama, K., Sekiya, T., Inoue, N., Tamiya, T., Kashiwagi, I., Kimura, A., Morita, R., Muto, G., Shichita, T., Takahashi, R., and Yoshimura, A. (2011). Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-beta is mediated by suppression of eomesodermin. Immunity 34, 741–754.
Kamizono, S., Hanada, T., Yasukawa, H., Minoguchi, S., Kato, R., Minoguchi, M., Hattori, K., Hatakeyama, S., Yada, M., Morita, S., Kitamura, T., Kato, H., Nakayama, K. I., and Yoshimura, A. (2001). The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J. Biol. Chem. 276, 12530–12538.
Kamura, T., Maenaka, K., Kotoshiba, S., Matsumoto, M., Kohda, D., Conaway, R. C., Conaway, J. W., and Nakayama, K. I. (2004). VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 18, 3055–3065.
Kimura, A., Kinjyo, I., Matsumura, Y., Mori, H., Mashima, R., Harada, M., Chien, K. R., Yasukawa, H., and Yoshimura, A. (2004). SOCS3 is a physiological negative regulator for granulopoiesis and granulocyte colony-stimulating factor receptor signaling. J. Biol. Chem. 279, 6905–6910.
Kinjyo, I., Hanada, T., Inagaki-Ohara, K., Mori, H., Aki, D., Ohishi, M., Yoshida, H., Kubo, M., and Yoshimura, A. (2002). SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity 17, 583–591.
Kinjyo, I., Inoue, H., Hamano, S., Fukuyama, S., Yoshimura, T., Koga, K., Takaki, H., Himeno, K., Takaesu, G., Kobayashi, T., and Yoshimura, A. (2006). Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J. Exp. Med. 203, 1021–1031.
Kobayashi, T., Takaesu, G., and Yoshimura, A. (2006). Mal-function of TLRs by SOCS. Nat. Immunol. 7, 123–124.
Koga, K., Takaesu, G., Yoshida, R., Nakaya, M., Kobayashi, T., Kinjyo, I., and Yoshimura, A. (2009). Cyclic adenosine monophosphate suppresses the transcription of proinflammatory cytokines via the phosphorylated c-Fos protein. Immunity 30, 372–383.
Kubo, M., Hanada, T., and Yoshimura, A. (2003). Suppressors of cytokine signaling and immunity. Nat. Immunol. 4, 1169–1176.
Lehmann, U., Schmitz, J., Weissenbach, M., Sobota, R. M., Hortner, M., Friederichs, K., Behrmann, I., Tsiaris, W., Sasaki, A., Schneider-Mergener, J., Yoshimura, A., Neel, B. G., Heinrich, P. C., and Schaper, F. (2003). SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J. Biol. Chem. 278, 661–671.
Li, Y., Chu, N., Rostami, A., and Zhang, G. X. (2006). Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J. Immunol. 177, 1679–1688.
Liu, Y., Stewart, K. N., Bishop, E., Marek, C. J., Kluth, D. C., Rees, A. J., and Wilson, H. M. (2008). Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J. Immunol. 180, 6270–6278.
Lu, L. F., Boldin, M. P., Chaudhry, A., Lin, L. L., Taganov, K. D., Hanada, T., Yoshimura, A., Baltimore, D., and Rudensky, A. Y. (2010). Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 142, 914–929.
Lu, Y., Fukuyama, S., Yoshida, R., Kobayashi, T., Saeki, K., Shiraishi, H., Yoshimura, A., and Takaesu, G. (2006). Loss of SOCS3 gene expression converts STAT3 function from anti-apoptotic to pro-apoptotic. J. Biol. Chem. 281, 36683–36690.
Lu, L. F., Thai, T. H., Calado, D. P., Chaudhry, A., Kubo, M., Tanaka, K., Loeb, G. B., Lee, H., Yoshimura, A., Rajewsky, K., and Rudensky, A. Y. (2009). Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity 30, 80–91.
Madonna, S., Scarponi, C., De Pita, O., and Albanesi, C. (2008). Suppressor of cytokine signaling 1 inhibits IFN-gamma inflammatory signaling in human keratinocytes by sustaining ERK1/2 activation. FASEB J. 22, 3287–3297.
Marine, J. C., Topham, D. J., McKay, C., Wang, D., Parganas, E., Stravopodis, D., Yoshimura, A., and Ihle, J. N. (1999). SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell 98, 609–616.
Masuhara, M., Sakamoto, H., Matsumoto, A., Suzuki, R., Yasukawa, H., Mitsui, K., Wakioka, T., Tanimura, S., Sasaki, A., Misawa, H., Yokouchi, M., Ohtsubo, M., and Yoshimura, A. (1997). Cloning and characterization of novel CIS family genes. Biochem. Biophys. Res. Commun. 239, 439–446.
Matsumoto, A., Seki, Y., Kubo, M., Ohtsuka, S., Suzuki, A., Hayashi, I., Tsuji, K., Nakahata, T., Okabe, M., Yamada, S., and Yoshimura, A. (1999). Suppression of STAT5 functions in liver, mammary glands, and T cells in cytokine-inducible SH2-containing protein 1 transgenic mice. Mol. Cell. Biol. 19, 6396–6407.
Matsumura, Y., Kobayashi, T., Ichiyama, K., Yoshida, R., Hashimoto, M., Takimoto, T., Tanaka, K., Chinen, T., Shichita, T., Wyss-Coray, T., Sato, K., and Yoshimura, A. (2007). Selective expansion of foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J. Immunol. 179, 2170–2179.
Mori, H., Hanada, R., Hanada, T., Aki, D., Mashima, R., Nishinakamura, H., Torisu, T., Chien, K. R., Yasukawa, H., and Yoshimura, A. (2004). Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 10, 739–743.
Mori, T., Miyamoto, T., Yoshida, H., Asakawa, M., Kawasumi, M., Kobayashi, T., Morioka, H., Chiba, K., Toyama, Y., and Yoshimura, A. (2011). IL-1{beta} and TNF{alpha}-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int. Immunol. 23, 701–712.
Moriwaki, A., Inoue, H., Nakano, T., Matsunaga, Y., Matsuno, Y., Matsumoto, T., Fukuyama, S., Kan, O. K., Matsumoto, K., Tsuda-Eguchi, M., Nagakubo, D., Yoshie, O., Yoshimura, A., Kubo, M., and Nakanishi, Y. (2011). T cell treatment with small interfering RNA for suppressor of cytokine signaling 3 modulates allergic airway responses in a murine model of asthma. Am. J. Respir. Cell Mol. Biol. 44, 448–455.
Mostecki, J., Cassel, S. L., Klimecki, W. T., Stern, D. A., Knisz, J., Iwashita, S., Graves, P., Miller, R. L., van Peer, M., Halonen, M., Martinez, F. D., Vercelli, D., and Rothman, P. B. (2011). A SOCS-1 promoter variant is associated with total serum IgE levels. J. Immunol. 187, 2794–2802.
Mottok, A., Renne, C., Seifert, M., Oppermann, E., Bechstein, W., Hansmann, M. L., Kuppers, R., and Brauninger, A. (2009). Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B-cell lymphoma entities. Blood 114, 4503–4506.
Murray, P. J., and Wynn, T. A. (2011). Obstacles and opportunities for understanding macrophage polarization. J. Leukoc. Biol. 89, 557–563.
Naka, T., Narazaki, M., Hirata, M., Matsumoto, T., Minamoto, S., Aono, A., Nishimoto, N., Kajita, T., Taga, T., Yoshizaki, K., Akira, S., and Kishimoto, T. (1997). Structure and function of a new STAT-induced STAT inhibitor. Nature 387, 924–929.
Nakaya, M., Hamano, S., Kawasumi, M., Yoshida, H., Yoshimura, A., and Kobayashi, T. (2011). Aberrant IL-4 production by SOCS3-over-expressing T cells during infection with Leishmania major exacerbates disease manifestations. Int. Immunol. 23, 195–202.
Nakaya, M., Hashimoto, M., Nakagawa, R., Wakabayashi, Y., Ishizaki, T., Takada, I., Komai, K., Yoshida, H., and Yoshimura, A. (2009). SOCS3 in T and NKT cells negatively regulates cytokine production and ameliorates ConA-induced hepatitis. J. Immunol. 183, 7047–7053.
Ogata, H., Chinen, T., Yoshida, T., Kinjyo, I., Takaesu, G., Shiraishi, H., Iida, M., Kobayashi, T., and Yoshimura, A. (2006a). Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene 25, 2520–2530.
Ogata, H., Kobayashi, T., Chinen, T., Takaki, H., Sanada, T., Minoda, Y., Koga, K., Takaesu, G., Maehara, Y., Iida, M., and Yoshimura, A. (2006b). Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology 131, 179–193.
Okumura, A., Pitha, P. M., Yoshimura, A., and Harty, R. N. (2010). Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J. Virol. 84, 27–33.
O’Shea, J. J., Gadina, M., and Schreiber, R. D. (2002). Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 109(Suppl.), S121–S131.
Pellegrini, M., Calzascia, T., Toe, J. G., Preston, S. P., Lin, A. E., Elford, A. R., Shahinian, A., Lang, P. A., Lang, K. S., Morre, M., Assouline, B., Lahl, K., Sparwasser, T., Tedder, T. F., Paik, J. H., DePinho, R. A., Basta, S., Ohashi, P. S., and Mak, T. W. (2011). IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell 144, 601–613.
Russell, R. C., Sufan, R. I., Zhou, B., Heir, P., Bunda, S., Sybingco, S. S., Greer, S. N., Roche, O., Heathcote, S. A., and Chow, V. W. (2011). Loss of JAK2 regulation via a heterodimeric VHL-SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat. Med. 17, 845–853.
Sachithanandan, N., Graham, K. L., Galic, S., Honeyman, J. E., Fynch, S. L., Hewitt, K. A., Steinberg, G. R., and Kay, T. W. (2011). Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes 60, 2023–2031.
Sasaki, A., Yasukawa, H., Shouda, T., Kitamura, T., Dikic, I., and Yoshimura, A. (2000). CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J. Biol. Chem. 275, 29338–29347.
Sasaki, A., Yasukawa, H., Suzuki, A., Kamizono, S., Syoda, T., Kinjyo, I., Sasaki, M., Johnston, J. A., and Yoshimura, A. (1999). Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 4, 339–351.
Sawa, S., Kamimura, D., Jin, G. H., Morikawa, H., Kamon, H., Nishihara, M., Ishihara, K., Murakami, M., and Hirano, T. (2006) Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J. Exp. Med. 203, 1459–1470.
Schmitz, J., Weissenbach, M., Haan, S., Heinrich, P. C., and Schaper, F. (2000). SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 275, 12848–12856.
Seki, Y., Inoue, H., Nagata, N., Hayashi, K., Fukuyama, S., Matsumoto, K., Komine, O., Hamano, S., Himeno, K., Inagaki-Ohara, K., Cacalano, N., O’Garra, A., Oshida, T., Saito, H., Johnston, J. A., Yoshimura, A., and Kubo, M. (2003). SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat. Med. 9, 1047–1054.
Shouda, T., Yoshida, T., Hanada, T., Wakioka, T., Oishi, M., Miyoshi, K., Komiya, S., Kosai, K., Hanakawa, Y., Hashimoto, K., Nagata, K., and Yoshimura, A. (2001). Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J. Clin. Invest. 108, 1781–1788.
Starr, R., Willson, T. A., Viney, E. M., Murray, L. J., Rayner, J. R., Jenkins, B. J., Gonda, T. J., Alexander, W. S., Metcalf, D., Nicola, N. A., and Hilton, D. J. (1997). A family of cytokine-inducible inhibitors of signalling. Nature 387, 917–921.
Stutz, A., Kessler, H., Kaschel, M. E., Meissner, M., and Dalpke, A. H. (2012). Cell invasion and strain dependent induction of suppressor of cytokine signaling-1 by Toxoplasma gondii. Immunobiology 217, 28–36.
Suzuki, A., Hanada, T., Mitsuyama, K., Yoshida, T., Kamizono, S., Hoshino, T., Kubo, M., Yamashita, A., Okabe, M., Takeda, K., Akira, S., Matsumoto, S., Toyonaga, A., Sata, M., and Yoshimura, A. (2001). CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 193, 471–481.
Takahashi, R., Nishimoto, S., Muto, G., Sekiya, T., Tamiya, T., Kimura, A., Morita, R., Asakawa, M., Chinen, T., and Yoshimura, A. (2011). SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-{gamma} and IL-17A production. J. Exp. Med. 208, 2055–2067.
Taleb, S., Romain, M., Ramkhelawon, B., Uyttenhove, C., Pasterkamp, G., Herbin, O., Esposito, B., Perez, N., Yasukawa, H., Van Snick, J., Yoshimura, A., Tedgui, A., and Mallat, Z. (2009). Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J. Exp. Med. 206, 2067–2077.
Tamiya, T., Kashiwagi, I., Takahashi, R., Yasukawa, H., and Yoshimura, A. (2011). Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler. Thromb. Vasc. Biol. 31, 980–985.
Tanaka, K., Ichiyama, K., Hashimoto, M., Yoshida, H., Takimoto, T., Takaesu, G., Torisu, T., Hanada, T., Yasukawa, H., Fukuyama, S., Inoue, H., Nakanishi, Y., Kobayashi, T., and Yoshimura, A. (2008). Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J. Immunol. 180, 3746–3756.
Torisu, T., Nakaya, M., Watanabe, S., Hashimoto, M., Yoshida, H., Chinen, T., Yoshida, R., Okamoto, F., Hanada, T., Torisu, K., Takaesu, G., Kobayashi, T., Yasukawa, H., and Yoshimura, A. (2008). Suppressor of cytokine signaling 1 protects mice against concanavalin A-induced hepatitis by inhibiting apoptosis. Hepatology 47, 1644–1654.
Torisu, T., Sato, N., Yoshiga, D., Kobayashi, T., Yoshioka, T., Mori, H., Iida, M., and Yoshimura, A. (2007). The dual function of hepatic SOCS3 in insulin resistance in vivo. Genes Cells 12, 143–154.
Wang, Y. C., He, F., Feng, F., Liu, X. W., Dong, G. Y., Qin, H. Y., Hu, X. B., Zheng, M. H., Liang, L., Feng, L., Liang, Y. M., and Han, H. (2010). Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 70, 4840–4849.
Whitmarsh, R. J., Gray, C. M., Gregg, B., Christian, D. A., May, M. J., Murray, P. J., and Hunter, C. A. (2011). A Critical Role for SOCS3 in Innate Resistance to Toxoplasma gondii. Cell Host Microbe 10, 224–236.
Wong, P. K., Egan, P. J., Croker, B. A., O’Donnell, K., Sims, N. A., Drake, S., Kiu, H., McManus, E. J., Alexander, W. S., Roberts, A. W., and Wicks, I. P. (2006). SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J. Clin. Invest. 116, 1571–1581.
Yamamoto, K., Yamaguchi, M., Miyasaka, N., and Miura, O. (2003). SOCS-3 inhibits IL-12-induced STAT4 activation by binding through its SH2 domain to the STAT4 docking site in the IL-12 receptor beta2 subunit. Biochem. Biophys. Res. Commun. 310, 1188–1193.
Yasukawa, H., Misawa, H., Sakamoto, H., Masuhara, M., Sasaki, A., Wakioka, T., Ohtsuka, S., Imaizumi, T., Matsuda, T., Ihle, J. N., and Yoshimura, A. (1999). The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 18, 1309–1320.
Yasukawa, H., Ohishi, M., Mori, H., Murakami, M., Chinen, T., Aki, D., Hanada, T., Takeda, K., Akira, S., Hoshijima, M., Hirano, T., Chien, K. R., and Yoshimura, A. (2003). IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 4, 551–556.
Yoshida, T., Ogata, H., Kamio, M., Joo, A., Shiraishi, H., Tokunaga, Y., Sata, M., Nagai, H., and Yoshimura, A. (2004). SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J. Exp. Med. 199, 1701–1707.
Yoshimura, A. (2006). Signal transduction of inflammatory cytokines and tumor development. Cancer Sci. 97, 439–447.
Yoshimura, A., Naka, T., and Kubo, M. (2007). SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 7, 454–465.
Yoshimura, A., Ohkubo, T., Kiguchi, T., Jenkins, N. A., Gilbert, D. J., Copeland, N. G., Hara, T., and Miyajima, A. (1995). A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 14, 2816–2826.
Zhang, R., Dhillon, H., Yin, H., Yoshimura, A., Lowell, B. B., Maratos-Flier, E., and Flier, J. S. (2008). Selective inactivation of Socs3 in SF1 neurons improves glucose homeostasis without affecting body weight. Endocrinology 149, 5654–5661.
Zhang, Y., Ma, C. J., Ni, L., Zhang, C. L., Wu, X. Y., Kumaraguru, U., Li, C. F., Moorman, J. P., and Yao, Z. Q. (2011). Cross-talk between programmed death-1 and suppressor of cytokine signaling-1 in inhibition of IL-12 production by monocytes/macrophages in hepatitis C virus infection. J. Immunol. 186, 3093–3103.
Keywords: cytokine, signal transduction, immunity, STAT, helper T cell
Citation: Yoshimura A, Suzuki M, Sakaguchi R, Hanada T and Yasukawa H (2012) SOCS, inflammation, and autoimmunity. Front. Immun. 3:20. doi: 10.3389/fimmu.2012.00020
Received: 31 October 2011; Paper pending published: 16 November 2011;
Accepted: 03 February 2012; Published online: 12 March 2012.
Edited by:
Anna Rubartelli, National Cancer Research Institute, ItalyReviewed by:
Masaaki Murakami, Osaka University, JapanSeth Lucian Masters, Walter and Eliza Hall Institute, Australia
Cristina Albanesi, Istituo Dermopatico dell’Immacolata-IRCCS, Italy
Copyright: © 2012 Yoshimura, Suzuki, Sakaguchi, Hanada and Yasukawa. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Akihiko Yoshimura, Department of Microbiology and Immunology, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo, 160-8582, Japan. e-mail:eW9zaGltdXJhQGE2LmtlaW8uanA=