


- 1 Immunology Division, The Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia
- 2 Department of Microbiology and Immunology, The University of Melbourne, Melbourne, VIC, Australia
- 3 CSL Limited, Melbourne, VIC, Australia
Monoclonal antibodies that recognize cell surface molecules have been used deliver antigenic cargo to dendritic cells (DC) for induction of immune responses. The encouraging anti-tumor immunity elicited using this immunization strategy suggests its suitability for clinical trials. This review discusses the complex network of DC, the functional specialization of DC subsets, the immunological outcomes of targeting different DC subsets and their cell surface receptors, and the requirements for the induction of effective anti-tumor CD4 and CD8 T cell responses that can recognize tumor-specific antigens. Finally, we review preclinical experiments and the progress toward targeting human DC in vivo.
Introduction
Dendritic cells (DC) are superior antigen presenting cells (APC) that are critical for the initiation of immune responses. They are uniquely equipped to take-up, process, and present antigens (Ag) for the activation and expansion of naïve and memory T cells. The idea of harnessing the immunogenicity of DC to induce immune responses, coupled with the capacity to generate large numbers of DC ex vivo, gave rise to the exploration of DC-based vaccines. Since the initial clinical trials in the 1990s, there have been a plethora of DC-based vaccine trials employing diverse methodologies for the propagation, antigenic loading, and administration of these DC (Tacken et al., 2007). Though DC-based vaccines can induce T cell responses, objective clinical responses are anecdotal and DC-based vaccines have largely failed to live up to their expectation as an effective means of treating cancer (Lesterhuis et al., 2008; Eubel and Enk, 2009).
To enhance the efficacy of DC-based vaccines, there is a need to identify the parameters that potentially thwart positive clinical outcomes (Lesterhuis et al., 2008; Robson et al., 2010). Whilst the ability of Ag-loaded DC to raise measurable anti-tumor responses has been demonstrated, the question of whether optimal priming has been achieved remains unanswered. Certainly the observation that only a small proportion of the injected Ag-loaded DC ever reach the lymph node (Morse et al., 1999; De Vries et al., 2003) where naïve T cells require priming, suggests that only sub-optimal immune activation is ever achieved (Lesterhuis et al., 2008). From an immunological perspective, it is not clear if the ex vivo Ag-loaded DC directly prime immunity or whether they simply shuttle their Ag to the draining lymph node where it is acquired by resident DC that subsequently prime recipient T cells. Even if the ex vivo Ag-loaded DC directly activate naïve T cells, it remains to be determined if they do so as efficiently as DC that naturally acquire Ag in vivo.
An alternative strategy to vaccinating with ex vivo Ag-loaded DC, is to directly deliver the Ag to DC in vivo. Since DC express a unique pattern of cell surface receptors, monoclonal antibodies (mAb) against these receptors can be used as vehicles to deliver Ag to the DC (Figure 1). Numerous studies in animal models have exploited this strategy to elicit potent immune responses, and in some instances, effective anti-tumor responses. A multitude of factors govern the efficiency and type of immune response evoked by targeting DC in vivo. On the one hand, one needs to consider the biological function of the receptor, its expression pattern, and the nature of the interaction between the targeting mAb and the receptor (agonist/antagonist). On the other hand, the DC subset or subsets expressing this receptor and their maturation state, may be critical to the type of immune response evoked.
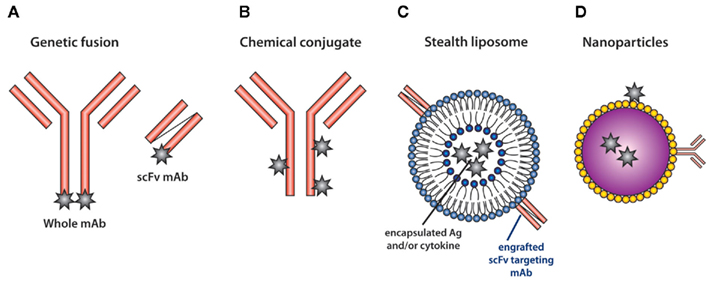
Figure 1. Methods for targeting antigen to DC in vivo. (A) The heavy and light chain of the whole mAb or the single-chain fragments of the variable regions (scFv) are sequenced and genetically engineered to carry model Ag (star). Alternatively, (B) Ag can be chemically conjugated to whole mAb. (C) The scFv of the mAb recognizing DC-specific molecules can also be engrafted into stealth liposomes. The liposomes can carry both antigen and cytokines (van Broekhoven et al., 2004). (D) Nanoparticles carrying Ag are modified to permit the attachment of DC-specific mAb and have been used to target Ag (Cruz et al., 2010).
This review discusses the network of DC, the outcomes of delivering Ag in vivo to DC subsets in experimental models, and the implications that this may have for DC-based vaccines in the clinical setting.
DC Diversity
Dendritic cells are not a homogenous population of cells, but represent a complex network of subsets that differ in ontology and specialized functions (Figure 2). A major division, seen both in mouse and man, occurs between plasmacytoid DC (pDC) and myeloid DC, the latter of which are commonly referred to as conventional DC (cDC; Shortman and Liu, 2002). The pDC are the most effective producers of type I IFN (Asselin-Paturel et al., 2001; Hochrein et al., 2001) and provide an innate defense against viral infections, but their role in Ag presentation and priming of naïve T cells remains unclear (Liu, 2005). By contrast, cDC are potent APC that specialize in activating adaptive immune responses and consequently, are the focus of this review.
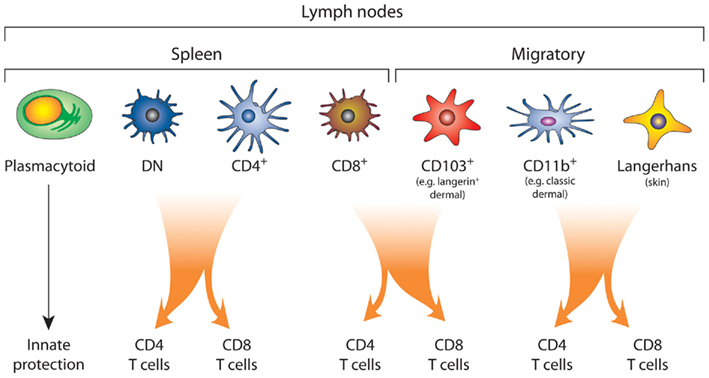
Figure 2. The complex network of DC subsets. Plasmacytoid DC provide an innate barrier against pathogens by the efficient production of type I interferon. Conventional DC, which include both the lymphoid tissue-resident DC and migratory DC, drive the adaptive immune response. In the mouse spleen, the lymphoid tissue-resident DC are divided into those that express of CD8α (CD8+), CD4 (CD4+), or those that express neither CD4 or CD8α, the double negative (DN) DC subset. The lymph nodes also contain migratory DC, which can be further segregated into at least three subsets: the CD103+ DC, CD11b+ (dermal) DC, and Langerhans’ cells. There is functional specialization between the DC subsets, where the CD103+ DC and CD8+ DC are most proficient at cross-presentation and activation of CD8+ T cells. By contrast, splenic DN and CD4+ DC and lymphoid CD11b+ DC and Langerhans’ cells are more efficient at driving CD4+ T cell responses. Although, under certain conditions both CD4+ DC, DN DC, and Langerhans’ cells have been shown to cross present antigen (Pooley et al., 2001; Flacher et al., 2010).
In the mouse, blood-borne precursors seed the spleen and develop into immature cDC (Naik et al., 2003, 2006; Wilson et al., 2003; Liu et al., 2007, 2009) that sample the blood for pathogens. These lymphoid tissue-resident cDC are usually divided into subsets based on their expression of CD8α and CD4. The CD8+ DC subset expresses CD8α but lacks CD4, the CD4+ DC expresses CD4 but lacks CD8α, and the double negative (DN) DC expresses neither CD4 nor CD8 (Vremec et al., 2000; Figure 2). The CD4+ DC and DN DC are often collectively referred to the CD8− DC. Precursor–product studies have shown that CD8+ DC and CD8− DC are not directly related, supporting the view that they represent different sublineages (Kamath et al., 2000, 2002; Naik et al., 2003, 2006).
Blood-borne DC precursors also seed the lymph nodes giving rise to the immature lymphoid tissue-resident CD8+ DC and CD8− DC subsets in these secondary lymphoid organs (Liu et al., 2007, 2009). In addition to these resident DC, however, the lymph nodes also contain migratory subsets (Figure 2). These migratory DC, unlike the resident DC, do not develop from precursors within the lymph nodes, but arrive via the afferent lymphatics in a mature state (Henri et al., 2001, 2010; Turnbull and MacPherson, 2001). In the steady state, and at an increased rate upon activation, migratory DC travel from the peripheral tissues that they survey, to the draining lymph nodes (Wilson et al., 2008), where they share Ag with the lymph node-resident cDC (Allan et al., 2006) or present their Ag directly to T cells (Bedoui et al., 2009). There are several subsets of migratory DC and their presence varies depending on the peripheral tissues they monitor.
In the lung (Sung et al., 2006; Bursch et al., 2007; Desch et al., 2011) and the mediastinal LN draining the lungs (Belz et al., 2004b; Sung et al., 2006; GeurtsvanKessel et al., 2008), at least two migratory DC subsets have been characterized; a CD11b+ DC and a CD103+ DC. The CD103+ DC in the lung express langerin (CD207) and only low levels of CD11b, consequently considered CD11b− (Sung et al., 2006). Such CD11b− DC are also found in the liver (Bursch et al., 2007), muscular layer of the small intestine (Flores-Langarica et al., 2005), and the LN draining the liver (Belz et al., 2004b), kidney (Belz et al., 2004b), gut (mesenteric), and payer patches (Iwasaki and Kelsall, 2001). In fact, the CD103+ DC and CD11b+ DC are found in many tissues, suggesting that most organs probably contain at least one CD11b−(CD103+) and one CD11b+ DC population (Ginhoux et al., 2009). The profile in the skin and its draining lymph nodes is more complex. In the last few years, three DC subsets were identified: the Langerhans’ cells (CD207+CD11b+CD103−), the classical dermal DC (CD207−CD11b+CD103−), and the CD103+ dermal DC (CD207+CD11b−CD103+; Heath and Carbone, 2009). It has been suggested that there are at least two other DC subsets in the skin and its associated lymph nodes, but whether these demonstrate functional specializations remains to be elucidated (Henri et al., 2010).
DC Subset Functional Specialization
All cDC subsets share the capacity to take-up, process, and present Ag, and activate naïve T cells. However, the biological relevance of the numerous DC subsets resides in their functional specialization (i.e., performing specific functions). The CD8+ DC are the most potent producers of IL-12p70 (Hochrein et al., 2001), and perhaps because of this they can direct T helper 1 (Th1) responses (Maldonado-Lopez et al., 1999; Pulendran et al., 1999). The defining characteristic of CD8+ DC is their strong capacity to constitutively present exogenous Ag in the MHC class I pathway, a process known as “cross-presentation” (den Haan et al., 2000; Pooley et al., 2001; Belz et al., 2002). That is, CD8+ DC can take-up exogenous Ag and present it on MHC class I for efficient stimulation of CD8+ T cells (Figure 2). This critical function is probably the reasons that CD8+ DC are the major subset involved in presentation of viral Ag (Allan et al., 2003; Smith et al., 2003; Belz et al., 2004a). Importantly for tumor immunity, where Ag may be acquired from necrotic tumor mass, the CD8+ DC are also superior at taking up dead and dying cells (Iyoda et al., 2002). This coupled with their ability to cross-present, results in CD8+ DC being superior at cross-presenting dead-cell-derived Ag (Shortman and Heath, 2010). More recently, the migratory CD103+ DC have also been shown to efficiently cross-present apoptotic cell-associated Ag, soluble Ag, self-Ag, and viral Ag (del Rio et al., 2007; Bedoui et al., 2009; Kim and Braciale, 2009; Desch et al., 2011). Indeed the migratory CD103+ DC resemble lymphoid tissue-resident CD8+ DC not only in their ability to cross-present, but also in expressing langerin, XCR1 (Crozat et al., 2011), their lack of CD11b expression, and their developmental requirement for fms-like tyrosine kinase-3 (Flt3) ligand (Ginhoux et al., 2009) and the transcription factors BatF3 (Hildner et al., 2008), Irf8 (Edelson et al., 2010), and Id2 (Ginhoux et al., 2009). The similar gene expression profile of these two subsets also supports the notion of a developmental relationship (Ginhoux et al., 2009; Edelson et al., 2010; Crozat et al., 2011). The role of CD103+ DC in tumor immunity remains to be investigated.
The ability of CD8− DC to cross-present Ag is somewhat more controversial and there is evidence to support (Schulz and Reis e Sousa, 2002; Kamphorst et al., 2010) and dispute (den Haan et al., 2000; Belz et al., 2005; Schnorrer et al., 2006; Hildner et al., 2008; Lin et al., 2008; Farrand et al., 2009; Segura et al., 2009) their cross-presentation capacity. Despite the controversy, there appear to be three scenarios that promote cross-presentation in CD8− DC. Firstly, “activated” CD8− DC have an enhanced ability to cross-present soluble Ag (Pooley et al., 2001; Kamphorst et al., 2010). Secondly, receptor mediated-endocytosis via DEC-205 promotes cross-presentation. Thus, Ag delivered via DEC-205 to CD8− DC and even B cells, is cross-presented (Kamphorst et al., 2010). Thirdly, Ag taken-up by CD8− DC via the FcγR is cross-presented (den Haan and Bevan, 2002). Indeed enforced expression of FcRγIIA endowed non-professional APC with the capacity to cross-present (Giodini et al., 2009). More recently it was shown that in CD8− DC, the neonatal FcR, a non-classical FcR collaborates with FcγR to facilitate cross-presentation of immuno-complexed Ag by directing intracellular sorting (Baker et al., 2011). The ability of the FcγR to promote cross-presentation was exploited to induce anti-tumor responses, though even in a prophylactic immunization setting only mild anti-tumor immunity was evoked whether this reached statistical significance was not clear (Akiyama et al., 2003).
The role of other cell types in cross-presentation and activation of naïve CD8 T cells is yet to be established. Macrophages (CD169+CD11c+) were shown to effectively cross-present apoptotic material, though interestingly, a large proportion of these macrophages expressed high levels of CD8α. The authors suggest that these CD169+CD11c+ cells are macrophages not DC, because in CD169-diphtheria toxin (DT) receptor (DTR) mice treatment with DT depletes macrophages but not CD8+DC. However, upon close inspection of their data two things become apparent: (1) There is indeed a small drop in the proportion of CD8+ DC after DT treatment and (2) CD169+CD11c+ cells only constitute a minute proportion of the total DC, therefore depletion of CD8+CD169+CD11c+ would barely be detectable. Thus, since this study did not rule out that DT treatment in CD169-DTR mice depletes a proportion of CD8+DC (i.e., the CD8+CD169+CD11c+ DC) it is not clear whether depletion of CD169+ cells prevents cross-presentation by removing macrophages or a subpopulation of CD8+ DC (Asano et al., 2011). However, macrophages have been shown to be involved in cross-presentation – albeit in an indirect manner. Marginal metallophilic macrophages transfer captured Ag to CD8+ DC, which then cross present the Ag (Backer et al., 2010). Thus targeting Ag to marginal metallophilic macrophages can result in cross-presentation, albeit in an indirect manner that requires Ag transfer to CD8+ DC.
Highly inflammatory conditions such as those generated by immunization with completes Freund’s adjuvant induce monocytes that are extremely effective at cross-presentation (Kamphorst et al., 2010). Therefore in an immunization setting where inflammation may be caused, targeting Ag to monocytes may be a way to further boost cross-presentation.
Ultimately that capacity of a particular cell type to cross-present does not equate to this cell playing a central role in cross-priming. For example, though CD8− DC can cross-present via FcγR, in vivo they are redundant for the activation of CD8 T cell responses (den Haan and Bevan, 2002). The advent of the BatF3 deficient mice, which lack CD8+ DC has also corroborated the critical role CD8+ DC play in cross-presentation; CD8+ DC deficient mice are inferior at cross-presenting viral and tumor antigen (Hildner et al., 2008). The BatF3 deficient mice are not completely devoid of CD8+ DC, indeed enough Clec9A+ DC are present to allow MHC class II presentation. However, cross-presentation upon targeting Clec9A was significantly inhibited in BatF3−/− re-iterating the important role of CD8+ DC in activating CD8 T cells in vivo (Caminschi et al., 2011).
Less controversial is the generalization that the CD8− DC are more efficient at the presentation of Ag via MHC class II pathway (Pooley et al., 2001; den Haan and Bevan, 2002; Carter et al., 2006; Dudziak et al., 2007; Kamphorst et al., 2010). CD8− DC have also have been shown to induce of Th2 type responses (Pulendran et al., 1999). Similar to the CD8− DC, the migratory CD11b+ DC appear proficient at MHC class II restricted presentation (Figure 2). In a disease model of herpes simplex, it is the migratory dermal DC (CD11b+CD103−) in the draining LN that preferentially present Ag in the MHC class II pathway (Zhao et al., 2003; Bedoui et al., 2009). In this viral diseases model, Langerhans’ cells only play a minor or negligible role in MHC class II presentation. However, in a skin infection model of Candida albicans, Langerhans’ cells are essential for the generation of Th17 cells, but unable to induce cytotoxic T cells, illustrating their importance in the presentation of MHC class II Ag but limited role in cross-presentation (Igyarto et al., 2011). There is evidence to suggest that the function of migratory DC may also involve “ferrying” Ag from the site of infection to the draining LN where it is handed it over to CD8+ DC for cross-presentation (Allan et al., 2006).
In conclusion, with some exceptions (Kim and Braciale, 2009; Lukens et al., 2009), most experimental models support the view that the lymphoid tissue-resident CD8+ DC and migratory CD103+ DC dominate MHC class I-restricted cross-presentation, whilst the migratory CD11b+ DC and Langerhans’ cells are more efficient at MHC class II presentation (Figure 2). The most effective DC subset capable of priming anti-tumor T cell immunity remains to be elucidated.
Though DC subsets have functional specializations, and theoretically these specializations can be harnessed to manipulate the immune responses generated, in reality the technology has not reached the stage of being able to deliver Ag to just one DC subset. Most mAb that have been used to target Ag to DC, deliver the Ag to multiple subsets and even other cell types. The interplay between DC subsets, receptors, adjuvants, and immunological outcomes of such targeting strategies is discussed below.
Targeting DC in vivo
DC-Specific Receptors
When attempting to deliver Ag to DC in vivo via the specific surface receptors that they express, an immediate concern is the consequence of simultaneously targeting other cell types expressing the same receptor and therefore also acquiring the Ag. Particularly when Ag presentation by certain cells, i.e., B cells, has been shown to induce tolerance (Bennett et al., 1998). In this regard, very few cell surface molecules are exclusively expressed by DC. Even CD11c, the classical DC-marker in the mouse, is expressed on activated CD8 T cells (Huleatt and Lefrancois, 1995), NK cells (Laouar et al., 2005), and macrophages (Vallon-Eberhard et al., 2006). Interestingly, however, there appears to be no obvious detrimental effect on the immune response generated, when cell types in addition to DC receive the Ag. Indeed, CD11c (Wang et al., 2000; Castro et al., 2008; Wei et al., 2009), DEC-205 (Hawiger et al., 2001; Bonifaz et al., 2004), CD36 (Tagliani et al., 2008), LOX1 (Delneste et al., 2002), mannose receptor (MR; He et al., 2007), Clec12A (Lahoud et al., 2009) have all been successfully used to elicit immune responses, despite their expression by non-DC (Witmer-Pack et al., 1993; Inaba et al., 1995). Furthermore, whilst promiscuous receptor expression does not prevent the induction of immunity, it appears that it is the DC, and not the other APC, that are directly responsible for the priming of T cells (He et al., 2007; Nchinda et al., 2008; Lahoud et al., 2009). Thus, Ag presentation by DC appears to dominate the immunological outcome.
The Requirements for Inducing Immunity
The current paradigm of DC biology is that immature DC efficiently take-up Ag, but are poor at interacting with T cells. DC require some level of maturation to tolerize, or to effectively stimulate and expand Ag-specific T cells (Albert et al., 2001). Upon maturation/activation, DC shutdown Ag acquisition but progressively increase Ag presentation and expression of appropriate co-stimulatory molecules required for efficiently priming T cells (Villadangos and Schnorrer, 2007). Importantly, mature DC retain the ability to capture and present Ag via endocytic receptors (Platt et al., 2010). This is particularly relevant to the delivery Ag to DC-receptors via mAb because it takes advantage of the retained capacity of mature DC to endocytose and present Ag taken-up via endocytic receptors. In the vaccine setting, it is considered critical to mature (or activate) DC, since Ag presentation by immature DC is likely to induce tolerance rather than immunity. DC can undergo maturation in response to pathogens and their by-products and experimentally this is mimicked by the administration of adjuvants that mature DC (i.e., anti-CD40 mAb, poly I:C, CpG). Whilst the requirement for adjuvant for the induction of humoral responses is variable (Caminschi et al., 2009; Shortman et al., 2009), the induction of cellular immunity and particularly the generation cytotoxic T lymphocyte (CTL) responses clearly requires adjuvants. The most comprehensively studied molecule utilized for the delivery of Ag to DC is DEC-205 (Hawiger et al., 2001; Bonifaz et al., 2002, 2004). DEC-205 is a multi-lectin endocytic receptor (Mahnke et al., 2000) and though its primary function remains unclear, it has been reported to bind dead cells (Shrimpton et al., 2009) and bacterial components (Zhang et al., 2008). DEC-205 is expressed highly on CD8+ DC in the spleen, but is also found on Langerhans’ cells, dermal DC, and interstitial DC in lymph nodes. When anti-DEC-205 mAb is conjugated to model Ag and co-administered with DC-maturation agents such as anti-CD40 mAb and poly I:C, vigorous cellular and humoral immune responses are generated (Bonifaz et al., 2004; van Broekhoven et al., 2004; Mahnke et al., 2005; Boscardin et al., 2006; Charalambous et al., 2006; Trumpfheller et al., 2006, 2008; Soares et al., 2007; Do et al., 2008; Johnson et al., 2008). But in the absence of these maturation signals, targeting Ag to DEC-205 can lead to T cell tolerance, by activation induced cell death of responding T cells and by regulatory T cell (Treg) generation (Hawiger et al., 2001; Bonifaz et al., 2002, 2004; Mahnke et al., 2003; Bruder et al., 2005; Kretschmer et al., 2005; Mukhopadhaya et al., 2008; Yamazaki et al., 2008), emphasizing the importance of creating the ideal priming environment. Similarly, targeting Ag to DC via DCIR2 (Dudziak et al., 2007; Soares et al., 2007), Dectin-1 (Carter et al., 2006), MR (He et al., 2007), CD11c (Castro et al., 2008; Wei et al., 2009), and MHC class II (Dickgreber et al., 2009) also requires co-administration of activation stimuli to induce robust cellular immunity. This suggests that targeting Ag to DC-receptors in vivo will require the co-administration of activating agents (or adjuvants) for optimal induction of anti-tumor CTL and T helper cells.
Biasing the Type of Immune Response Generated
A fundamental difference between the multiple DC subsets appears to be their ability to cross-present Ag. In several experimental settings, the lymphoid tissue-resident CD8+ DC and the migratory CD103+ DC appear most efficient at cross-presentation, whereas CD8− DC and other CD11b+ DC such as dermal DC are more efficient at MHC class II presentation (den Haan et al., 2000; Pooley et al., 2001; Zhao et al., 2003; del Rio et al., 2007; Bedoui et al., 2009; Kim and Braciale, 2009). Indeed, when the model Ag ovalbumin (OVA) is delivered to CD8+ DC or CD8− DC using mAb, in the presence of maturation signals, a similar Ag presentation hierarchy is observed: CD8+ DC targeted using anti-DEC-205-OVA mAb are more efficient at inducing OVA-specific CD8 T cell responses (Dudziak et al., 2007), whilst CD8− DC targeted using anti-DCIR-2-OVA mAb or anti-Dectin-1-OVA mAb are more efficient at inducing OVA-specific CD4+ T cell responses (Carter et al., 2006; Dudziak et al., 2007). This evidence supports the view that CD8− DC are inherently better at MHC class II presentation while CD8+ DC favor cross-presentation.
The nature of the DC-surface molecule targeted can also influence the immune outcome and override DC subset specific Ag presentation biases, a case-in-point demonstrated by targeting antigen to Clec9A. This molecule is selectively expressed by CD8+ DC and to a lesser extent pDC but is not expressed by any other cell types (Caminschi et al., 2008; Huysamen et al., 2008; Sancho et al., 2008). Clec9A has an extracellular C-type lectin-like domain and an intracellular hemITAM motif involved in signaling (Huysamen et al., 2008; Sancho et al., 2009) and facilitates the cross-presentation of dead-cell-associated Ag (Sancho et al., 2009). When Ag is delivered to CD8+ DC using an anti-Clec9A mAb, even in the absence of adjuvant, strong CD4+ T cell responses are induced, superior to those obtained by targeting DEC-205 (Lahoud et al., 2011); clearly CD8+ DC can be targeted to induce potent CD4+ T cell responses. Interestingly though, targeting Ag to Clec9A induces T follicular helpers (TFH) and whilst they are critical in supporting B cell responses (Caminschi et al., 2008), their role in supporting CTL responses is unclear. Indeed, targeting Ag to Clec9A in the absence of DC-activating factors induced potent B cell responses, presumably aided by the strong TFH response, but failed to induce CTL (Lahoud et al., 2011). Either, the TFH provided inadequate help to support CTL induction, or alternatively, effective CTL priming requires an additional DC-signal that is not provided when Ag is delivered to Clec9A in the absence of DC-activating factors. From a tumor-vaccine perspective, strong CTL responses are desirable, therefore optimal T helper responses are sought. One way to ensure strong T cells responses is to administer targeted Ag in the presence of adjuvant. To this end, when mice are immunized multiple times with targeting mAb carrying HIV gag p24 in the presence of potent adjuvants (anti-CD40 and/or poly IC or ICLC), targeting either DEC-205, Clec9A, or Langerin facilitate strong endogenous anti-HIV gag p24 CD4+ and CD8+ T cell responses (Idoyaga et al., 2011).
It is important to emphasize that CD8+ DC are clearly capable of inducing effective CD4+ T cell responses. When CD8+ DC and CD8− DC are respectively targeted with anti-DEC-205 or anti-DCIR2 mAb engineered to carry the LACK protein of Leishmania major, both DC subsets induce transgenic LACK-specific CD4+ T cells to proliferate, albeit CD8− DC are more efficient (Soares et al., 2007). Interestingly, however, CD8+ DC induce the CD4+ T cells to produce exclusively IFN-γ, whereas the CD8− DC induce both IFN-γ and IL-4. Furthermore, while both DC subsets induce CD4+ T cell production of IFN-γ, the CD8+ DC required expression of CD70 but not IL-12p40, whereas the CD8− DC relied on IL-12p40 but not on CD70 (Soares et al., 2007).
An alternative method of skewing the type of immune response is through the type of adjuvant used. In a prime-boost model where HIV gag p24 was targeted to the DEC-205 receptor, potent Th1 type responses were induced when poly I:C was used as an adjuvant (Trumpfheller et al., 2006; Longhi et al., 2009). The HIV gag-specific CD4+ T cells produced IFN-γ, IL-2, and TNF-α (Trumpfheller et al., 2006; Longhi et al., 2009). Compared with other toll-like receptors (TLR) [i.e., MALP-2 (TLR/2TLR6), Pam3Cys (TLR1/TLR2), lipopolysaccharide (TLR4), R848 (TLR7/TLR8), and CpG (TLR9)], poly I:C was the most efficient inducer of Th1 CD4+ T cells (Longhi et al., 2009). Poly I:C also induced a Th1 response when anti-Clec9A mAb was used to deliver OVA peptide to CD8+ DC (Joffre et al., 2010). Interestingly, when curdlan (a β-glucan) was used to activate DC, immunization with anti-Clec9A mAb resulted in the induction of Th17 response (Joffre et al., 2010). In the case of targeting Ag to Clec9A it appears that the targeting mAb themselves may also affect the immunological outcome (Caminschi et al., 2008; Joffre et al., 2010): as previously mentioned, in the absence of adjuvant one anti-Clec9A mAb induces TFH (Lahoud et al., 2011), whereas another induces Tregs (Joffre et al., 2010). Importantly, both anti-Clec9A mAb induced robust CTL when co-administered with adjuvant (Sancho et al., 2008; Lahoud et al., 2011).
Although both Th1 and Th2 helper cells have been associated with effective anti-tumor responses, the type of T helper cell response induced may become an important consideration when choosing which DC subset to target in vivo. The interplay between the targeted DC subsets and the receptor used to deliver the Ag has been investigated in several tumor models and is detailed below.
Inducing Anti-Tumor Immunity by Targeting DC in vivo
A variety of molecules expressed on DC have been used to deliver Ag and elicit anti-tumor responses, including DEC-205 (Bonifaz et al., 2004; Mahnke et al., 2005; Johnson et al., 2008), CD11c (van Broekhoven et al., 2004; Wei et al., 2009), Clec9A (Sancho et al., 2008), MHC class II (Dickgreber et al., 2009), LOX1 (Delneste et al., 2002), MR (He et al., 2007), CD36 (Tagliani et al., 2008), and even Bst2 (CD317), a molecules expressed on pDC (Loschko et al., 2011). In the majority of studies OVA is employed as a model tumor Ag, and mice are inoculated with OVA transfectants of the melanoma cell line B16 (Bonifaz et al., 2004; van Broekhoven et al., 2004; He et al., 2007; Sancho et al., 2008; Dickgreber et al., 2009; Loschko et al., 2011), or EL4 (Delneste et al., 2002; Tagliani et al., 2008). However, bona fide tumor Ag such as the tyrosine related protein (TRP)-2 (Mahnke et al., 2005) and gp100 (Johnson et al., 2008) have been also successfully delivered to DC using the anti-DEC-205 mAb. With the exception of CD36 (Tagliani et al., 2008) and LOX1 (Delneste et al., 2002), delivery of Ag to all other DC molecules, required the provision of activation signals (e.g., CpG, poly I:C, anti-CD40 mAb, or a combination of these) to mature DC (Bonifaz et al., 2004; van Broekhoven et al., 2004; Mahnke et al., 2005; He et al., 2007; Johnson et al., 2008; Sancho et al., 2008; Dickgreber et al., 2009; Wei et al., 2009). In the absence of these activation signals, the targeted Ag failed to elicit effective anti-tumor immunity (van Broekhoven et al., 2004; Mahnke et al., 2005; Dickgreber et al., 2009; Wei et al., 2009).
Prophylactic immunization with anti-CD36 (Tagliani et al., 2008), anti-MR (He et al., 2007), anti-DEC-205 (Bonifaz et al., 2004; Mahnke et al., 2005), anti-Clec9A (Sancho et al., 2008), anti-CD11c (Wei et al., 2009), and anti-Bst2 mAb delivering tumor Ag, could prevent or inhibit tumor growth. More importantly, in therapeutic settings, established tumors have been shown to be rejected, or to display delayed growth when mice were immunized with Ag targeted using anti-DEC-205 (Bonifaz et al., 2004; Mahnke et al., 2005; Johnson et al., 2008), Clec9A (Sancho et al., 2008), or anti-CD11c (Wei et al., 2009) mAb.
Protective tumor immunity correlated with the induction of strong CD4+ and CD8+ T cell responses (Mahnke et al., 2005; He et al., 2007; Johnson et al., 2008). The CD4+ T cells displayed a Th1 phenotype producing IFN-γ (Bonifaz et al., 2004) and TNF-α (Wei et al., 2009), whilst the CD8+ T cells produced IFN-γ (Wei et al., 2009) and possessed cytolytic capacity (Bonifaz et al., 2004; Sancho et al., 2008; Wei et al., 2009). T cell subset depletion studies demonstrated a strong dependence of anti-tumor activity on CD8+ T cells (Bonifaz et al., 2004; Mahnke et al., 2005), whilst the role of CD4+ T cells was more variable (Bonifaz et al., 2004; Mahnke et al., 2005). In the B16 tumor model (Mahnke et al., 2005), mice immunized with anti-DEC-205 mAb carrying the tumor Ag TRP-2, were critically dependent on CD4+ T cells to prevent tumor growth. By contrast, mice immunized with anti-DEC-205 mAb carrying OVA and challenged with B16-OVA were less reliant upon CD4+ T cells to reject tumors (Bonifaz et al., 2004). The basis for differences in CD4+ T cell dependence for these tumor models is unclear, but may relate to the nature of the antigen; with OVA providing foreign antigenic determinants dominated by a single epitope versus TRP-2 representing an over-expressed self antigen containing multiple weakly immunogenic CD4 and CD8 epitopes.
Tumor Ag are often self-Ag over-expressed in malignancies, and raising immunity against a self-Ag may face the additional complication of pre-existing tolerance. In a study by Wei et al. (2009), single-chain-fragment variable region (scFv) mAb specific for CD11c were used to deliver the HER-2 protein and induce protection against HER-2 expressing tumors. In an interesting twist, these authors attempted to mimic the human scenario of pre-existing tolerance to the tumor self-Ag. Transgenic BALB/c mice expressing the rat HER-2/neu oncogenes (Balb–neuT) are tolerant to the rat HER-2/neu protein and develop spontaneous mammary carcinoma. When these mice are vaccinated in the presence of CpG with scFv-anti-CD11c carrying the rat-neu protein, they are protected from subsequent challenge with neu-expressing TUBO tumor cells. Furthermore, this vaccination strategy also delayed the onset of the spontaneously arising mammary tumors (Wei et al., 2009). Though the mechanisms of tumor protection were not investigated in detail, CD8+ T cells are unlikely to have been directly involved in tumor cell lysis, as only low anti-tumor cytotoxicity was detected. However, a distinct anti-neu antibody response was induced and this may have provided anti-tumor protection. Certainly this was the case in an alternative vaccination strategy of the Balb–neuT mice, where the induced antibody response was both necessary and sufficient to protect against tumors (Park et al., 2005).
So far only one group appears to have compared in vivo DC-targeting to priming with ex vivo Ag-loaded DC (Bonifaz et al., 2004). Mice were inoculated with B16 expressing OVA, then 7 days later, vaccinated with anti-DEC-205-OVA in the presence of anti-CD40 mAb, or with ex vivo OVA-loaded and matured DC. In this experimental setting, vaccinating with the ex vivo OVA-loaded DC did not inhibit tumor growth. By contrast, vaccinating with anti-DEC-205-OVA effectively prevented tumor growth (Bonifaz et al., 2004). If this single comparative study is predictive of the clinical experience, then in vivo targeting of DC may indeed prove a more effective way of eliciting anti-tumor responses.
Translating Mouse DC Biology to Clinical Application
Although vaccinating patients with ex vivo Ag-loaded DCs has yet to emerge as an effective means of treating cancer patients, it has shown promise in some cancer settings. Hormone refractory prostate cancer appears particularly amenable to vaccine-based immunotherapy. Dendreon’s Sipuleucel-T therapy (Provenge), which utilizes blood DCs pulsed with full length, prostatic acid phosphatase–GM-CSF fusion protein, has demonstrated improved overall survival in cancer patients in two independent Phase III clinical trials. Provenge has recently been approved by the FDA for the treatment of asymptomatic or minimally symptomatic metastatic, castration-resistant prostate cancer making it the first in its class therapeutic cancer vaccine approved (Higano et al., 2009). Interestingly, the complex process involved in Sipuleucel-T therapy may reflect what is required to generate DC-mediated immunity in cancer patients.
Vaccinating with ex vivo Ag-loaded DC remains a cumbersome, labor intensive, and expensive procedure that depends on the establishment and utilization of vaccine manufacturing facilities that operate under clinical Good Manufacturing Practice guidelines. Targeting DC in vivo using engineered Ag–mAb constructs should substantially simplify the therapeutic procedures that cancer patients would be subjected to, eliminating the need for leukophoresis and prolonged hospital stays whilst awaiting the manufacture and quality control release of vaccine batches. The challenge is to translate the positive experimental findings from animal models into clinical outcomes. The first obstacle will be to ensure that the candidate molecules chosen for targeting of the vaccine Ag based on preclinical studies in mice retain the same immunogenic properties when tested in cancer patients. Ideally, the expression pattern and function of the DC-specific molecules should be conserved between species. Targeting to DEC-205 has been extensively utilized in the mouse to promote cross-presentation and immunity, and encouragingly, the ability of DEC-205 to facilitate cross-presentation was mirrored by human monocyte-derived DC (Bozzacco et al., 2007) leading to its clinical evaluation. Celldex Therapeutics are currently evaluating DC-targeting vaccines in Phase I/II clinical trials for the treatment of several types of solid cancers. CDX 1401 is a fully humanized anti-DEC-205 mAb conjugated to the NY-ESO-1 tumor Ag. Vaccination is combined with topical administration of R848 gel (resiquimod™ TLR7/8 agonist) at the injection site to activate local DCs. Another therapeutic, CDX-1307, is a fusion protein composed of a fully human MR-specific mAb fused to human chorionic gonadotropin (hCG-β) protein. Vaccination again is combined with various immunostimulators such as topical resiquimod™ gel, Poly IC:LC (Hiltonol™, TLR 3 agonist), or GM-CSF to activate DC (Ramakrishna et al., 2007). The potential drawback of targeting Ag to DEC-205 (or macrophage MR or CD11c for that matter), is that the expression pattern of these molecules is not restricted to DC in humans, rather these molecules are also expressed on monocytes and several types of lymphocytes. Thus, although Ag acquisition by DC appears to control immunogenicity, ubiquitous expression of a receptor could potentially result in the targeting mAb being mopped-up by other cells thereby reducing its targeting efficiency.
Targeting molecules that are restrictively expressed on human DC would therefore seem advantageous. Such a candidate could be Clec9A, which is exclusively expressed on DC: it is expressed at high levels on CD8α+ DC and at lesser levels on pDC (Caminschi et al., 2008; Huysamen et al., 2008; Sancho et al., 2008). Strikingly, Clec9A also appears to be exclusively expressed on human DC, where only the CD141+ DC (Caminschi et al., 2008; Huysamen et al., 2008; Sancho et al., 2008), considered to be the equivalent of the mouse CD8+ DC (Bachem et al., 2010; Crozat et al., 2010; Jongbloed et al., 2010), express this molecule. Similarly, the function of Clec9A is conserved between species: both mouse and human Clec9A binds necrotic material (Sancho et al., 2009). Furthermore, targeting Ag in vivo to Clec9A via anti-Clec9A-mAb-Ag conjugates induces anti-tumor responses in mice (Sancho et al., 2008). The observation that Clec9A function and expression pattern appear to be conserved between mouse and human and the demonstration of proof-of-concept in animal models of tumor immunity, make Clec9A an ideal candidate for clinical evaluation.
Other candidate molecules considered for the delivery of Ag to human DC in vivo include DC-SIGN (a C-type lectin expressed on human DC), DCIR (a C-type lectin expressed on various APC), and the MR (He et al., 2004; Ramakrishna et al., 2004; Tacken et al., 2005; Keler et al., 2007; Pereira et al., 2007; Gurer et al., 2008; Meyer-Wentrup et al., 2008; Klechevsky et al., 2010; Flynn et al., 2011). There are three types of preclinical tests designed to assess the suitability of using receptors to deliver Ag to DC: primate studies, humanized mice, and in vitro targeting assays.
Primate studies demonstrated that administration of anti-DC-SIGN mAb targets APC (Pereira et al., 2007). In a more detailed study in Rhesus macaques, the impact of targeting HIV gag p24 to DEC-205 was assessed. Targeting the viral protein to DEC-205, in the presence adjuvant (poly ICLC), resulted in better cross-priming of CD8+ T cells (Flynn et al., 2011). However, the CD4+ T cell responses of primates immunized with anti-DEC-205 mAb or the non-targeted HIV gag p24 protein were comparable (Flynn et al., 2011), as were the anti-Gag Ab responses. Surprisingly, the avidity of the anti-Gag Ab was approximately 10-fold higher in primates immunized with the non-targeted Gag p24. The authors suggest that the aberrant expression of DEC-205 on all germinal center B cells, may have resulted in the processing and presentation of Gag protein by Gag-non-specific B cells thereby diverting the T helper cells required for high-affinity Ab generation (Flynn et al., 2011). Clearly these findings may have ramification for future clinical trials and assessing the suitability of utilizing DEC-205 in Ag delivery: DEC-205 strongly promotes cross-presentation and the activation of CD8+ T cells, but may not promote the strongest CD4+ T cell and B cell responses.
An alternative model of in vivo targeting utilizes “humanized mice,” that is mice reconstituted with human immune cells. Humanized mice have been used to show that delivering Ag via anti-DC-SIGN or anti-human DEC-205 mAb in vivo enhanced T cell responses (Kretz-Rommel et al., 2007; Gurer et al., 2008).
In lieu of delivering Ag to DC in vivo, many preclinical tests assess the outcomes of delivering Ag to human DC via receptor-specific mAb in vitro. Targeting Ag to human DC using mAb against DC-SIGN, DEC-205, DCIR, or the MR have been used to induce Ag-specific T cell responses (He et al., 2004; Ramakrishna et al., 2004; Tacken et al., 2005; Bozzacco et al., 2007; Birkholz et al., 2010; Klechevsky et al., 2010; Tel et al., 2011). So far only one study has directly compared the efficiency of targeting Ag to these molecules (Bozzacco et al., 2007). When anti-DEC-205, anti-DC-SIGN, and the anti-MR mAb, were compared in their ability to target HIV p24 gag protein to human DC, it was found all mAb bound their target molecules on monocyte-derived DC and were internalized. However, anti-DEC-205 induced more IFN-γ producing memory CD8+ T cells (Bozzacco et al., 2007). It is unclear to what extent these in vitro findings reflect the potential efficacy of targeting these distinct molecules in vivo.
Second-Generation Vaccination Approaches and Combination Therapies
One of the potential complications of targeting Ag to DC in vivo using mAb, is that the mAb have to be engineered to carry the Ag. So far researchers have either chemically conjugated Ag to the mAb, or genetically fused the DNA sequence of the Ag to the sequence encoding the mAb (Figure 1). A more versatile approach involved creating a bifunctional vector containing the single-chain antibody recognizing DEC-205 fused to a core-streptavidin domain (Wang et al., 2009). The streptavidin domain was used to attach a diverse group of biotinylated Ag, and this bifunctional vector injected in conjunction with a DC-maturation agent (anti-CD40) successfully induced cellular and humoral responses (Wang et al., 2009). In an alternative strategy, a fusion protein was designed to contain an Ag attached to tandem Ig binding domains of protein G. These Ig binding domains bound a wide range of mAb and provided a generic approach to attach any desired Ab to the fusion protein. This strategy allowed the comparison of different receptors in their ability to promote cellular and humoral immunity. Targeting Ag to APC was vastly more efficient at inducing T and B cell responses than free Ag, and certain receptors were inherently more efficient at promoting cross-presentation (Kratzer et al., 2010).
New approaches have emerged that build on the idea of targeting more complex Ag to DC using mAb that recognize DC-specific molecules (Figure 1; Reddy et al., 2006). Stealth liposomes or plasma membrane vesicles from tumor cells have been engineered to encapsulate Ag as well as cytokines or TLR ligands (i.e., lipopolysaccharide) whilst also being engrafted with single-chain fragments of mAb that recognize DC-specific molecules. Liposomes and plasma membrane vesicles carrying Ag and IFN-γ have successfully been targeted to the DC using either anti-CD11c or anti-DEC-205 mAb and induced potent anti-tumor responses (van Broekhoven et al., 2004). In an alternative approach, poly DL-lactic acid biodegradable nanoparticles were concomitantly conjugated to anti-tumor (rat-neu) mAb and anti-CD40 mAb (Dominguez and Lustgarten, 2010). Mice injected with nanoparticles bearing both anti-tumor and anti-CD40 mAb were protected against rat-neu tumor, whilst mice injected with nanoparticles bearing either mAb alone were not protected. The authors propose that the formation of conjugates between DC and tumor cells facilitated by the mAb-coated nanoparticles, promotes the anti-tumor response (Dominguez and Lustgarten, 2010). The notion of targeting nanoparticles to DC using DC-specific mAb in a quest to enhance vaccine efficacy is actively being investigated (Kwon et al., 2005; Cruz et al., 2010, 2011a,b; Joshi et al., 2011).
Ultimately Ag can be targeted to DC exploiting other proteins that can also bind cell surface receptors. The Shiga toxin is composed of an A-subunit and B-subunit. The B-subunit is non-toxic and binds glycolipid Gb3 found on APC such as DC and B cells. Coupling antigen such as OVA to B-subunit toxin results in DC-dependent priming of CTL, which inturn inhibits tumor growth (Vingert et al., 2006). Similarly, the phenol-soluble modulins released by Staphylococcus epidermidis, which bind TLR2 have been exploited to deliver Ag and elicit anti-tumor responses (Durantez et al., 2010). This type of targeting approach has reached the stage of clinical trials: adenylate cyclase of Bordetella pertussis binds αMβ2 integrin (CD11b/CD18) and can deliver Ag to CD11b+ DC (Guermonprez et al., 2002). Adenylate cyclase has been utilized to deliver Ag and induces strong CTL responses that inhibited tumor growth in animal models (Fayolle et al., 1999) and is now being trialed in a phase I/II study in melanoma patients for its ability to induce immunity against an A2 epitope of the melanoma-Ag tyrosinase (http://www.clinicaltrials.gov).
Priming an effective anti-tumor response utilizing DC-based vaccines is the first step in tumor eradication. Optimal priming can minimize the development of Treg that can undermine anti-tumor responses, and maximize the quantity and quality of effector T cells. However, other obstacles such as the immunosuppressive microenvironment of the tumor may also need to be overcome to make it more permissive to recruitment and functional activation of the vaccine-induced CTL. Consequently combining DC-based vaccines with other therapies that aim to antagonize tumor-induced suppressive factors (i.e., IL-10, IL-13, TGF-β, IDO, PGE2, VEGF), or dysregulate the emergence of T regulatory cell networks (e.g., immunogenic chemotherapy such as doxorubicin, cyclophosphamide or IL-2-DT ONTAK, or anti-CTLA-4 mAb), or enhance the ongoing immune response (e.g., TLR ligands) should improve vaccine efficacy and clinical outcomes (Palucka et al., 2008; Robson et al., 2010).
Conclusion/Future
Delivering Ag to DC in vivo using DC-targeting mAb has emerged as an exiting new approach to DC-based vaccines. In mouse models, priming immunity by targeting Ag to DC in vivo has resulted in effective anti-tumor immunity and regression of established cancer. The challenge will be to translate this promising experimental data into positive clinical outcomes for patients. Fundamental immunological differences between mouse and man, the genetic diversity of humans and the impact of environmental factors, add to the inherent complication of translational research. One step toward aligning experimental and clinical outcomes may be to identify candidate molecules that are conserved between species and have retained their functional properties when evaluated in humans. Ultimately, empirical data from clinical trials will assess this promising new approach to immune modulation and vaccination.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Prof. Shortman and Dr. Lahoud for advice and critical review of this manuscript, and Peter Maltezos for the production of the depicted graphics. Supported by the Australian Research Council (William Ross Heath), the Howard Hughes Medical Institute (William Ross Heath), Bill and Belinda and Gates Foundation (Irina Caminschi), and the National Health and Medical Research Council of Australia (William Ross Heath and Irina Caminschi). Eugene Maraskovsky is an employe of CSL Limited.
Abbreviations
Ag, antigen; APC, antigen presenting cells; cDC, conventional DC; DC, dendritic cells; mAb, monoclonal antibody; OVA, ovalbumin; pDC, plasmacytoid DC; TLR, toll-like receptor; Treg, T regulatory cells.
References
Akiyama, K., Ebihara, S., Yada, A., Matsumura, K., Aiba, S., Nukiwa, T., and Takai, T. (2003). Targeting apoptotic tumor cells to Fc gamma R provides efficient and versatile vaccination against tumors by dendritic cells. J. Immunol. 170, 1641–1648.
Albert, M., Jegathesan, M., and Darnell, R. (2001). Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat. Immunol. 2, 1010–1017.
Allan, R. S., Smith, C. M., Belz, G. T., van Lint, A. L., Wakim, L. M., Heath, W. R., and Carbone, F. R. (2003). Epidermal viral immunity induced by CD8alpha+ dendritic cells but not by Langerhans cells. Science 301, 1925–1928.
Allan, R. S., Waithman, J., Bedoui, S., Jones, C. M., Villadangos, J. A., Zhan, Y., Lew, A. M., Shortman, K., Heath, W. R., and Carbone, F. R. (2006). Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity 25, 153–162.
Asano, K., Nabeyama, A., Miyake, Y., Qiu, C. H., Kurita, A., Tomura, M., Kanagawa, O., Fujii, S., and Tanaka, M. (2011). CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity 34, 85–95.
Asselin-Paturel, C., Boonstra, A., Dalod, M., Durand, I., Yessaad, N., Dezutter-Dambuyant, C., Vicari, A., O’Garra, A., Biron, C., Briere, F., and Trinchieri, G. (2001). Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2, 1144–1150.
Bachem, A., Guttler, S., Hartung, E., Ebstein, F., Schaefer, M., Tannert, A., Salama, A., Movassaghi, K., Opitz, C., Mages, H. W., Henn, V., Kloetzel, P. M., Gurka, S., and Kroczek, R. A. (2010). Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 207, 1273–1281.
Backer, R., Schwandt, T., Greuter, M., Oosting, M., Jungerkes, F., Tuting, T., Boon, L., O’Toole, T., Kraal, G., Limmer, A., and den Haan, J. M. (2010). Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc. Natl. Acad. Sci. U.S.A. 107, 216–221.
Baker, K., Qiao, S. W., Kuo, T. T., Aveson, V. G., Platzer, B., Andersen, J. T., Sandlie, I., Chen, Z., de Haar, C., Lencer, W. I., Fiebiger, E., and Blumberg, R. S. (2011). Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8-CD11b+ dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 108, 9927–9932.
Bedoui, S., Whitney, P. G., Waithman, J., Eidsmo, L., Wakim, L., Caminschi, I., Allan, R. S., Wojtasiak, M., Shortman, K., Carbone, F. R., Brooks, A. G., and Heath, W. R. (2009). Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat. Immunol. 10, 488–495.
Belz, G. T., Behrens, G. M., Smith, C. M., Miller, J. F., Jones, C., Lejon, K., Fathman, C. G., Mueller, S. N., Shortman, K., Carbone, F. R., and Heath, W. R. (2002). The CD8alpha(+) dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J. Exp. Med. 196, 1099–1104.
Belz, G. T., Shortman, K., Bevan, M. J., and Heath, W. R. (2005). CD8alpha+ dendritic cells selectively present MHC class I-restricted noncytolytic viral and intracellular bacterial antigens in vivo. J. Immunol. 175, 196–200.
Belz, G. T., Smith, C. M., Eichner, D., Shortman, K., Karupiah, G., Carbone, F. R., and Heath, W. R. (2004a). Cutting edge: conventional CD8alpha(+) dendritic cells are generally involved in priming CTL immunity to viruses. J. Immunol. 172, 1996–2000.
Belz, G. T., Smith, C. M., Kleinert, L., Reading, P., Brooks, A., Shortman, K., Carbone, F. R., and Heath, W. R. (2004b). Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc. Natl. Acad. Sci. U.S.A. 101, 8670–8675.
Bennett, S. R., Carbone, F. R., Toy, T., Miller, J. F., and Heath, W. R. (1998). B cells directly tolerize CD8(+) T cells. J. Exp. Med. 188, 1977–1983.
Birkholz, K., Schwenkert, M., Kellner, C., Gross, S., Fey, G., Schuler-Thurner, B., Schuler, G., Schaft, N., and Dorrie, J. (2010). Targeting of DEC-205 on human dendritic cells results in efficient MHC class II-restricted antigen presentation. Blood 116, 2277–2285.
Bonifaz, L., Bonnyay, D., Mahnke, K., Rivera, M., Nussenzweig, M. C., and Steinman, R. M. (2002). Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196, 1627–1638.
Bonifaz, L. C., Bonnyay, D. P., Charalambous, A., Darguste, D. I., Fujii, S., Soares, H., Brimnes, M. K., Moltedo, B., Moran, T. M., and Steinman, R. M. (2004). In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 199, 815–824.
Boscardin, S. B., Hafalla, J. C., Masilamani, R. F., Kamphorst, A. O., Zebroski, H. A., Rai, U., Morrot, A., Zavala, F., Steinman, R. M., Nussenzweig, R. S., and Nussenzweig, M. C. (2006). Antigen targeting to dendritic cells elicits long-lived T cell help for antibody responses. J. Exp. Med. 203, 599–606.
Bozzacco, L., Trumpfheller, C., Siegal, F. P., Mehandru, S., Markowitz, M., Carrington, M., Nussenzweig, M. C., Piperno, A. G., and Steinman, R. M. (2007). DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc. Natl. Acad. Sci. U.S.A. 104, 1289–1294.
Bruder, D., Westendorf, A. M., Hansen, W., Prettin, S., Gruber, A. D., Qian, Y., von Boehmer, H., Mahnke, K., and Buer, J. (2005). On the edge of autoimmunity: T-cell stimulation by steady-state dendritic cells prevents autoimmune diabetes. Diabetes 54, 3395–3401.
Bursch, L. S., Wang, L., Igyarto, B., Kissenpfennig, A., Malissen, B., Kaplan, D. H., and Hogquist, K. A. (2007). Identification of a novel population of Langerin+ dendritic cells. J. Exp. Med. 204, 3147–3156.
Caminschi, I., Lahoud, M. H., and Shortman, K. (2009). Enhancing immune responses by targeting antigen to DC. Eur. J. Immunol. 39, 931–938.
Caminschi, I., Proietto, A. I., Ahmet, F., Kitsoulis, S., Shin Teh, J., Lo, J. C., Rizzitelli, A., Wu, L., Vremec, D., van Dommelen, S. L., Campbell, I. K., Maraskovsky, E., Braley, H., Davey, G. M., Mottram, P., van de Velde, N., Jensen, K., Lew, A. M., Wright, M. D., Heath, W. R., Shortman, K., and Lahoud, M. H. (2008). The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 112, 3264–3273.
Caminschi, I., Vremec, D., Ahmet, F., Lahoud, M. H., Villadangos, J. A., Murphy, K. M., Heath, W. R., and Shortman, K. (2011). Antibody responses initiated by Clec9A-bearing dendritic cells in normal and Batf3(-/-) mice. Mol. Immunol. doi: org/10.1016/j.molimm.2011.11.008. [Epub ahead of print].
Carter, R. W., Thompson, C., Reid, D. M., Wong, S. Y., and Tough, D. F. (2006). Preferential induction of CD4+ T cell responses through in vivo targeting of antigen to dendritic cell-associated C-type lectin-1. J. Immunol. 177, 2276–2284.
Castro, F. V., Tutt, A. L., White, A. L., Teeling, J. L., James, S., French, R. R., and Glennie, M. J. (2008). CD11c provides an effective immunotarget for the generation of both CD4 and CD8 T cell responses. Eur. J. Immunol. 38, 2263–2273.
Charalambous, A., Oks, M., Nchinda, G., Yamazaki, S., and Steinman, R. M. (2006). Dendritic cell targeting of survivin protein in a xenogeneic form elicits strong CD4+ T cell immunity to mouse survivin. J. Immunol. 177, 8410–8421.
Crozat, K., Guiton, R., Contreras, V., Feuillet, V., Dutertre, C. A., Ventre, E., Vu Manh, T. P., Baranek, T., Storset, A. K., Marvel, J., Boudinot, P., Hosmalin, A., Schwartz-Cornil, I., and Dalod, M. (2010). The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J. Exp. Med. 207, 1283–1292.
Crozat, K., Tamoutounour, S., Vu Manh, T. P., Fossum, E., Luche, H., Ardouin, L., Guilliams, M., Azukizawa, H., Bogen, B., Malissen, B., Henri, S., and Dalod, M. (2011). Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8α+ type. J. Immunol. 187, 4411–4415.
Cruz, L. J., Tacken, P. J., Bonetto, F., Buschow, S. I., Croes, H. J., Wijers, M., de Vries, I. J., and Figdor, C. G. (2011a). Multimodal imaging of nanovaccine carriers targeted to human dendritic cells. Mol. Pharm. 8, 520–531.
Cruz, L. J., Tacken, P. J., Fokkink, R., and Figdor, C. G. (2011b). The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials 32, 6791–6803.
Cruz, L. J., Tacken, P. J., Fokkink, R., Joosten, B., Stuart, M. C., Albericio, F., Torensma, R., and Figdor, C. G. (2010). Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J. Control Release 144, 118–126.
De Vries, I. J., Krooshoop, D. J., Scharenborg, N. M., Lesterhuis, W. J., Diepstra, J. H., Van Muijen, G. N., Strijk, S. P., Ruers, T. J., Boerman, O. C., Oyen, W. J., Adema, G. J., Punt, C. J., and Figdor, C. G. (2003). Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 63, 12–17.
del Rio, M. L., Rodriguez-Barbosa, J. I., Kremmer, E., and Forster, R. (2007). CD103- and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J. Immunol. 178, 6861–6866.
Delneste, Y., Magistrelli, G., Gauchat, J., Haeuw, J., Aubry, J., Nakamura, K., Kawakami-Honda, N., Goetsch, L., Sawamura, T., Bonnefoy, J., and Jeannin, P. (2002). Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity 17, 353–362.
den Haan, J. M., and Bevan, M. J. (2002). Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(-) dendritic cells in vivo. J. Exp. Med. 196, 817–827.
den Haan, J. M., Lehar, S. M., and Bevan, M. J. (2000). CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192, 1685–1696.
Desch, A. N., Randolph, G. J., Murphy, K., Gautier, E. L., Kedl, R. M., Lahoud, M. H., Caminschi, I., Shortman, K., Henson, P. M., and Jakubzick, C. V. (2011). CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J. Exp. Med. 208, 1789–1797.
Dickgreber, N., Stoitzner, P., Bai, Y., Price, K. M., Farrand, K. J., Manning, K., Angel, C. E., Dunbar, P. R., Ronchese, F., Fraser, J. D., Bäckström, B. T., and Hermans, I. F. (2009). Targeting antigen to MHC class II molecules promotes efficient cross-presentation and enhances immunotherapy. J. Immunol. 182, 1260–1269.
Do, Y., Park, C. G., Kang, Y. S., Park, S. H., Lynch, R. M., Lee, H., Powell, B. S., and Steinman, R. M. (2008). Broad T cell immunity to the LcrV virulence protein is induced by targeted delivery to DEC-205/CD205-positive mouse dendritic cells. Eur. J. Immunol. 38, 20–29.
Dominguez, A. L., and Lustgarten, J. (2010). Targeting the tumor microenvironment with anti-neu/anti-CD40 conjugated nanoparticles for the induction of antitumor immune responses. Vaccine 28, 1383–1390.
Dudziak, D., Kamphorst, A. O., Heidkamp, G. F., Buchholz, V. R., Trumpfheller, C., Yamazaki, S., Cheong, C., Liu, K., Lee, H. W., Park, C. G., Steinman, R. M., and Nussenzweig, M. C. (2007). Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111.
Durantez, M., Fayolle, C., Casares, N., Belsue, V., Riezu-Boj, J. I., Sarobe, P., Prieto, J., Borras-Cuesta, F., Leclerc, C., and Lasarte, J. J. (2010). Tumor therapy in mice by using a tumor antigen linked to modulin peptides from Staphylococcus epidermidis. Vaccine 28, 7146–7154.
Edelson, B. T., Kc, W., Juang, R., Kohyama, M., Benoit, L. A., Klekotka, P. A., Moon, C., Albring, J. C., Ise, W., Michael, D. G., Bhattacharya, D., Stappenbeck, T. S., Holtzman, M. J., Sung, S. S., Murphy, T. L., Hildner, K., and Murphy, K. M. (2010). Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J. Exp. Med. 207, 823–836.
Eubel, J., and Enk, A. H. (2009). Dendritic cell vaccination as a treatment modality for melanoma. Expert Rev. Anticancer Ther. 9, 1631–1642.
Farrand, K. J., Dickgreber, N., Stoitzner, P., Ronchese, F., Petersen, T. R., and Hermans, I. F. (2009). Langerin+ CD8alpha+ dendritic cells are critical for cross-priming and IL-12 production in response to systemic antigens. J. Immunol. 183, 7732–7742.
Fayolle, C., Ladant, D., Karimova, G., Ullmann, A., and Leclerc, C. (1999). Therapy of murine tumors with recombinant Bordetella pertussis adenylate cyclase carrying a cytotoxic T cell epitope. J. Immunol. 162, 4157–4162.
Flacher, V., Tripp, C. H., Stoitzner, P., Haid, B., Ebner, S., Del Frari, B., Koch, F., Park, C. G., Steinman, R. M., Idoyaga, J., and Romani, N. (2010). Epidermal Langerhans cells rapidly capture and present antigens from C-type lectin-targeting antibodies deposited in the dermis. J. Invest. Dermatol. 130, 755–762.
Flores-Langarica, A., Meza-Perez, S., Calderon-Amador, J., Estrada-Garcia, T., Macpherson, G., Lebecque, S., Saeland, S., Steinman, R. M., and Flores-Romo, L. (2005). Network of dendritic cells within the muscular layer of the mouse intestine. Proc. Natl. Acad. Sci. U.S.A. 102, 19039–19044.
Flynn, B. J., Kastenmuller, K., Wille-Reece, U., Tomaras, G. D., Alam, M., Lindsay, R. W., Salazar, A. M., Perdiguero, B., Gomez, C. E., Wagner, R., Esteban, M., Park, C. G., Trumpfheller, C., Keler, T., Pantaleo, G., Steinman, R. M., and Seder, R. (2011). Immunization with HIV Gag targeted to dendritic cells followed by recombinant New York vaccinia virus induces robust T-cell immunity in nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 108, 7131–7136.
GeurtsvanKessel, C. H., Willart, M. A., van Rijt, L. S., Muskens, F., Kool, M., Baas, C., Thielemans, K., Bennett, C., Clausen, B. E., Hoogsteden, H. C., Osterhaus, A. D., Rimmelzwaan, G. F., and Lambrecht, B. N. (2008). Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J. Exp. Med. 205, 1621–1634.
Ginhoux, F., Liu, K., Helft, J., Bogunovic, M., Greter, M., Hashimoto, D., Price, J., Yin, N., Bromberg, J., Lira, S. A., Stanley, E. R., Nussenzweig, M., and Merad, M. (2009). The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 206, 3115–3130.
Giodini, A., Rahner, C., and Cresswell, P. (2009). Receptor-mediated phagocytosis elicits cross-presentation in nonprofessional antigen-presenting cells. Proc. Natl. Acad. Sci. U.S.A. 106, 3324–3329.
Guermonprez, P., Fayolle, C., Rojas, M. J., Rescigno, M., Ladant, D., and Leclerc, C. (2002). In vivo receptor-mediated delivery of a recombinant invasive bacterial toxoid to CD11c+CD8 alpha -CD11bhigh dendritic cells. Eur. J. Immunol. 32, 3071–3081.
Gurer, C., Strowig, T., Brilot, F., Pack, M., Trumpfheller, C., Arrey, F., Park, C. G., Steinman, R. M., and Munz, C. (2008). Targeting the nuclear antigen 1 of Epstein-Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood 112, 1231–1239.
Hawiger, D., Inaba, K., Dorsett, Y., Guo, M., Mahnke, K., Rivera, M., Ravetch, J. V., Steinman, R. M., and Nussenzweig, M. C. (2001). Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194, 769–779.
He, L. Z., Crocker, A., Lee, J., Mendoza-Ramirez, J., Wang, X. T., Vitale, L. A., O’Neill, T., Petromilli, C., Zhang, H. F., Lopez, J., Rohrer, D., Keler, T., and Clynes, R. (2007). Antigenic targeting of the human mannose receptor induces tumor immunity. J. Immunol. 178, 6259–6267.
He, L. Z., Ramakrishna, V., Connolly, J. E., Wang, X. T., Smith, P. A., Jones, C. L., Valkova-Valchanova, M., Arunakumari, A., Treml, J. F., Goldstein, J., Wallace, P. K., Keler, T., and Endres, M. J. (2004). A novel human cancer vaccine elicits cellular responses to the tumor-associated antigen, human chorionic gonadotropin beta. Clin. Cancer Res. 10, 1920–1927.
Heath, W. R., and Carbone, F. R. (2009). Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 10, 1237–1244.
Henri, S., Poulin, L. F., Tamoutounour, S., Ardouin, L., Guilliams, M., de Bovis, B., Devilard, E., Viret, C., Azukizawa, H., Kissenpfennig, A., and Malissen, B. (2010). CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J. Exp. Med. 207, 189–206.
Henri, S., Vremec, D., Kamath, A., Waithman, J., Williams, S., Benoist, C., Burnham, K., Saeland, S., Handman, E., and Shortman, K. (2001). The dendritic cell populations of mouse lymph nodes. J. Immunol. 167, 741–748.
Higano, C. S., Schellhammer, P. F., Small, E. J., Burch, P. A., Nemunaitis, J., Yuh, L., Provost, N., and Frohlich, M. W. (2009). Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 115, 3670–3679.
Hildner, K., Edelson, B. T., Purtha, W. E., Diamond, M., Matsushita, H., Kohyama, M., Calderon, B., Schraml, B. U., Unanue, E. R., Diamond, M. S., Schreiber, R. D., Murphy, T. L., and Murphy, K. M. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100.
Hochrein, H., Shortman, K., Vremec, D., Scott, B., Hertzog, P., and O’Keeffe, M. (2001). Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J. Immunol. 166, 5448–5455.
Huleatt, J. W., and Lefrancois, L. (1995). Antigen-driven induction of CD11c on intestinal intraepithelial lymphocytes and CD8+ T cells in vivo. J. Immunol. 154, 5684–5693.
Huysamen, C., Willment, J. A., Dennehy, K. M., and Brown, G. D. (2008). CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol. Chem. 283, 16693–16701.
Idoyaga, J., Lubkin, A., Fiorese, C., Lahoud, M. H., Caminschi, I., Huang, Y., Rodriguez, A., Clausen, B. E., Park, C. G., Trumpfheller, C., and Steinman, R. M. (2011). Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc. Natl. Acad. Sci. U.S.A. 108, 2384–2389.
Igyarto, B. Z., Haley, K., Ortner, D., Bobr, A., Gerami-Nejad, M., Edelson, B. T., Zurawski, S. M., Malissen, B., Zurawski, G., Berman, J., and Kaplan, D. H. (2011). Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 35, 260–272.
Inaba, K., Swiggard, W. J., Inaba, M., Meltzer, J., Mirza, A., Sasagawa, T., Nussenzweig, M. C., and Steinman, R. M. (1995). Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. I. Expression on dendritic cells and other subsets of mouse leukocytes. Cell. Immunol. 163, 148–156.
Iwasaki, A., and Kelsall, B. L. (2001). Unique functions of CD11b+, CD8 alpha+, and double-negative Peyer’s patch dendritic cells. J. Immunol. 166, 4884–4890.
Iyoda, T., Shimoyama, S., Liu, K., Omatsu, Y., Akiyama, Y., Maeda, Y., Takahara, K., Steinman, R. M., and Inaba, K. (2002). The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195, 1289–1302.
Joffre, O. P., Sancho, D., Zelenay, S., Keller, A. M., and Reis, E. S. C. (2010). Efficient and versatile manipulation of the peripheral CD4(+) T-cell compartment by antigen targeting to DNGR-1/CLEC9A. Eur. J. Immunol. 40, 1255–1265.
Johnson, T. S., Mahnke, K., Storn, V., Schonfeld, K., Ring, S., Nettelbeck, D. M., Haisma, H. J., Le Gall, F., Kontermann, R. E., and Enk, A. H. (2008). Inhibition of melanoma growth by targeting of antigen to dendritic cells via an anti-DEC-205 single-chain fragment variable molecule. Clin. Cancer Res. 14, 8169–8177.
Jongbloed, S. L., Kassianos, A. J., McDonald, K. J., Clark, G. J., Ju, X., Angel, C. E., Chen, C. J., Dunbar, P. R., Wadley, R. B., Jeet, V., Vulink, A. J., Hart, D. N., and Radford, K. J. (2010). Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 207, 1247–1260.
Joshi, M. D., Unger, W. W., van Beelen, A. J., Bruijns, S. C., Litjens, M., van Bloois, L., Kalay, H., van Kooyk, Y., and Storm, G. (2011). DC-SIGN mediated antigen-targeting using glycan-modified liposomes: formulation considerations. Int. J. Pharm. 416, 426–432.
Kamath, A. T., Henri, S., Battye, F., Tough, D. F., and Shortman, K. (2002). Developmental kinetics and lifespan of dendritic cells in mouse lymphoid organs. Blood 100, 1734–1741.
Kamath, A. T., Pooley, J., O’Keeffe, M. A., Vremec, D., Zhan, Y., Lew, A. M., D’Amico, A., Wu, L., Tough, D. F., and Shortman, K. (2000). The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 165, 6762–6770.
Kamphorst, A. O., Guermonprez, P., Dudziak, D., and Nussenzweig, M. C. (2010). Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J. Immunol. 185, 3426–3435.
Keler, T., He, L., Ramakrishna, V., and Champion, B. (2007). Antibody-targeted vaccines. Oncogene 26, 3758–3767.
Kim, T. S., and Braciale, T. J. (2009). Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS ONE 4, e4204. doi:10.1371/journal.pone.0004204
Klechevsky, E., Flamar, A. L., Cao, Y., Blanck, J. P., Liu, M., O’Bar, A., Agouna-Deciat, O., Klucar, P., Thompson-Snipes, L., Zurawski, S., Reiter, Y., Palucka, A. K., Zurawski, G., and Banchereau, J. (2010). Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood 116, 1685–1697.
Kratzer, R., Mauvais, F. X., Burgevin, A., Barilleau, E., and van Endert, P. (2010). Fusion proteins for versatile antigen targeting to cell surface receptors reveal differential capacity to prime immune responses. J. Immunol. 184, 6855–6864.
Kretschmer, K., Apostolou, I., Hawiger, D., Khazaie, K., Nussenzweig, M. C., and von Boehmer, H. (2005). Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 6, 1219–1227.
Kretz-Rommel, A., Qin, F., Dakappagari, N., Torensma, R., Faas, S., Wu, D., and Bowdish, K. S. (2007). In vivo targeting of antigens to human dendritic cells through DC-SIGN elicits stimulatory immune responses and inhibits tumor growth in grafted mouse models. J. Immunother. 30, 715–726.
Kwon, Y. J., James, E., Shastri, N., and Frechet, J. M. (2005). In vivo targeting of dendritic cells for activation of cellular immunity using vaccine carriers based on pH-responsive microparticles. Proc. Natl. Acad. Sci. U.S.A. 102, 18264–18268.
Lahoud, M. H., Ahmet, F., Kitsoulis, S., Wan, S. S., Vremec, D., Lee, C. N., Phipson, B., Shi, W., Smyth, G. K., Lew, A. M., Kato, Y., Mueller, S. N., Davey, G. M., Heath, W. R., Shortman, K., and Caminschi, I. (2011). Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J. Immunol. 187, 842–850.
Lahoud, M. H., Proietto, A. I., Ahmet, F., Kitsoulis, S., Eidsmo, L., Wu, L., Sathe, P., Pietersz, S., Chang, H. W., Walker, I. D., Maraskovsky, E., Braley, H., Lew, A. M., Wright, M. D., Heath, W. R., Shortman, K., and Caminschi, I. (2009). The C-type lectin Clec12A present on mouse and human dendritic cells can serve as a target for antigen delivery and enhancement of antibody responses. J. Immunol. 182, 7587–7594.
Laouar, Y., Sutterwala, F. S., Gorelik, L., and Flavell, R. A. (2005). Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat. Immunol. 6, 600–607.
Lesterhuis, W. J., Aarntzen, E. H., De Vries, I. J., Schuurhuis, D. H., Figdor, C. G., Adema, G. J., and Punt, C. J. (2008). Dendritic cell vaccines in melanoma: from promise to proof? Crit. Rev. Oncol. Hematol. 66, 118–134.
Lin, M. L., Zhan, Y., Proietto, A. I., Prato, S., Wu, L., Heath, W. R., Villadangos, J. A., and Lew, A. M. (2008). Selective suicide of cross-presenting CD8+ dendritic cells by cytochrome c injection shows functional heterogeneity within this subset. Proc. Natl. Acad. Sci. U.S.A. 105, 3029–3034.
Liu, K., Victora, G. D., Schwickert, T. A., Guermonprez, P., Meredith, M. M., Yao, K., Chu, F. F., Randolph, G. J., Rudensky, A. Y., and Nussenzweig, M. (2009). In vivo analysis of dendritic cell development and homeostasis. Science 324, 392–397.
Liu, K., Waskow, C., Liu, X., Yao, K., Hoh, J., and Nussenzweig, M. (2007). Origin of dendritic cells in peripheral lymphoid organs of mice. Nat. Immunol. 8, 578–583.
Liu, Y. J. (2005). IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 23, 275–306.
Longhi, M. P., Trumpfheller, C., Idoyaga, J., Caskey, M., Matos, I., Kluger, C., Salazar, A. M., Colonna, M., and Steinman, R. M. (2009). Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 206, 1589–1602.
Loschko, J., Schlitzer, A., Dudziak, D., Drexler, I., Sandholzer, N., Bourquin, C., Reindl, W., and Krug, A. B. (2011). Antigen delivery to plasmacytoid dendritic cells via BST2 induces protective T cell-mediated immunity. J. Immunol. 186, 6718–6725.
Lukens, M. V., Kruijsen, D., Coenjaerts, F. E., Kimpen, J. L., and van Bleek, G. M. (2009). Respiratory syncytial virus-induced activation and migration of respiratory dendritic cells and subsequent antigen presentation in the lung-draining lymph node. J. Virol. 83, 7235–7243.
Mahnke, K., Guo, M., Lee, S., Sepulveda, H., Swain, S. L., Nussenzweig, M., and Steinman, R. M. (2000). The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 151, 673–684.
Mahnke, K., Qian, Y., Fondel, S., Brueck, J., Becker, C., and Enk, A. H. (2005). Targeting of antigens to activated dendritic cells in vivo cures metastatic melanoma in mice. Cancer Res. 65, 7007–7012.
Mahnke, K., Qian, Y., Knop, J., and Enk, A. H. (2003). Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood 101, 4862–4869.
Maldonado-Lopez, R., De Smedt, T., Michel, P., Godfroid, J., Pajak, B., Heirman, C., Thielemans, K., Leo, O., Urbain, J., and Moser, M. (1999). CD8alpha+ and CD8alpha- subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J. Exp. Med. 189, 587–592.
Meyer-Wentrup, F., Benitez-Ribas, D., Tacken, P. J., Punt, C. J., Figdor, C. G., de Vries, I. J., and Adema, G. J. (2008). Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-alpha production. Blood 111, 4245–4253.
Morse, M. A., Coleman, R. E., Akabani, G., Niehaus, N., Coleman, D., and Lyerly, H. K. (1999). Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 59, 56–58.
Mukhopadhaya, A., Hanafusa, T., Jarchum, I., Chen, Y. G., Iwai, Y., Serreze, D. V., Steinman, R. M., Tarbell, K. V., and DiLorenzo, T. P. (2008). Selective delivery of {beta} cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in NOD mice. Proc. Natl. Acad. Sci. U.S.A. 105, 6374–6379.
Naik, S., Vremec, D., Wu, L., O’Keeffe, M., and Shortman, K. (2003). CD8alpha+ mouse spleen dendritic cells do not originate from the CD8alpha- dendritic cell subset. Blood 102, 601–604.
Naik, S. H., Metcalf, D., van Nieuwenhuijze, A., Wicks, I., Wu, L., O’Keeffe, M., and Shortman, K. (2006). Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat. Immunol. 7, 663–671.
Nchinda, G., Kuroiwa, J., Oks, M., Trumpfheller, C., Park, C. G., Huang, Y., Hannaman, D., Schlesinger, S. J., Mizenina, O., Nussenzweig, M. C., Uberla, K., and Steinman, R. M. (2008). The efficacy of DNA vaccination is enhanced in mice by targeting the encoded protein to dendritic cells. J. Clin. Invest. 118, 1427–1436.
Palucka, A. K., Ueno, H., Fay, J., and Banchereau, J. (2008). Dendritic cells: a critical player in cancer therapy? J. Immunother. 31, 793–805.
Park, J. M., Terabe, M., Sakai, Y., Munasinghe, J., Forni, G., Morris, J. C., and Berzofsky, J. A. (2005). Early role of CD4+ Th1 cells and antibodies in HER-2 adenovirus vaccine protection against autochthonous mammary carcinomas. J. Immunol. 174, 4228–4236.
Pereira, C. F., Torensma, R., Hebeda, K., Kretz-Rommel, A., Faas, S. J., Figdor, C. G., and Adema, G. J. (2007). In vivo targeting of DC-SIGN-positive antigen-presenting cells in a nonhuman primate model. J. Immunother. 30, 705–714.
Platt, C. D., Ma, J. K., Chalouni, C., Ebersold, M., Bou-Reslan, H., Carano, R. A., Mellman, I., and Delamarre, L. (2010). Mature dendritic cells use endocytic receptors to capture and present antigens. Proc. Natl. Acad. Sci. U.S.A. 107, 4287–4292.
Pooley, J., Heath, W. R., and Shortman, K. (2001). Intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells but cross-presented to CD8+ T cells by CD8+ dendritic cells. J. Immunol. 166, 5327–5330.
Pulendran, B., Smith, J. L., Caspary, G., Brasel, K., Pettit, D., Maraskovsky, E., and Maliszewski, C. R. (1999). Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc. Natl. Acad. Sci. U.S.A. 96, 1036–1041.
Ramakrishna, V., Treml, J. F., Vitale, L., Connolly, J. E., O’Neill, T., Smith, P. A., Jones, C. L., He, L. Z., Goldstein, J., Wallace, P. K., Keler, T., and Endres, M. J. (2004). Mannose receptor targeting of tumor antigen pmel17 to human dendritic cells directs anti-melanoma T cell responses via multiple HLA molecules. J. Immunol. 172, 2845–2852.
Ramakrishna, V., Vasilakos, J. P., Tario, J. D. Jr., Berger, M. A., Wallace, P. K., and Keler, T. (2007). Toll-like receptor activation enhances cell-mediated immunity induced by an antibody vaccine targeting human dendritic cells. J. Transl. Med. 5, 5.
Reddy, S. T., Swartz, M. A., and Hubbell, J. A. (2006). Targeting dendritic cells with biomaterials: developing the next generation of vaccines. Trends Immunol. 27, 573–579.
Robson, N. C., Hoves, S., Maraskovsky, E., and Schnurr, M. (2010). Presentation of tumour antigens by dendritic cells and challenges faced. Curr. Opin. Immunol. 22, 137–144.
Sancho, D., Joffre, O. P., Keller, A. M., Rogers, N. C., Martinez, D., Hernanz-Falcon, P., Rosewell, I., and Reis e Sousa, C. (2009). Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458, 899–903.
Sancho, D., Mourao-Sa, D., Joffre, O. P., Schulz, O., Rogers, N. C., Pennington, D. J., Carlyle, J. R., and Reis e Sousa, C. (2008). Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Invest. 118, 2098–2110.
Schnorrer, P., Behrens, G. M., Wilson, N. S., Pooley, J. L., Smith, C. M., El-Sukkari, D., Davey, G., Kupresanin, F., Li, M., Maraskovsky, E., Belz, G. T., Carbone, F. R., Shortman, K., Heath, W. R., and Villadangos, J. A. (2006). The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc. Natl. Acad. Sci. U.S.A. 103, 10729–10734.
Schulz, O., and Reis e Sousa, C. (2002). Cross-presentation of cell-associated antigens by CD8alpha+ dendritic cells is attributable to their ability to internalize dead cells. Immunology 107, 183–189.
Segura, E., Albiston, A. L., Wicks, I. P., Chai, S. Y., and Villadangos, J. A. (2009). Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20377–20381.
Shortman, K., Lahoud, M. H., and Caminschi, I. (2009). Improving vaccines by targeting antigens to dendritic cells. Exp. Mol. Med. 41, 61–66.
Shortman, K., and Liu, Y. (2002). Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2, 151–161.
Shrimpton, R. E., Butler, M., Morel, A. S., Eren, E., Hue, S. S., and Ritter, M. A. (2009). CD205 (DEC-205): a recognition receptor for apoptotic and necrotic self. Mol. Immunol. 46, 1229–1239.
Smith, C. M., Belz, G. T., Wilson, N. S., Villadangos, J. A., Shortman, K., Carbone, F. R., and Heath, W. R. (2003). Cutting edge: conventional CD8alpha(+) dendritic cells are preferentially involved in CTL priming after footpad infection with herpes simplex virus-1. J. Immunol. 170, 4437–4440.
Soares, H., Waechter, H., Glaichenhaus, N., Mougneau, E., Yagita, H., Mizenina, O., Dudziak, D., Nussenzweig, M. C., and Steinman, R. M. (2007). A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J. Exp. Med. 204, 1095–1106.
Sung, S. S., Fu, S. M., Rose, C. E. Jr., Gaskin, F., Ju, S. T., and Beaty, S. R. (2006). A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 176, 2161–2172.
Tacken, P. J., de Vries, I. J., Gijzen, K., Joosten, B., Wu, D., Rother, R. P., Faas, S. J., Punt, C. J., Torensma, R., Adema, G. J., and Figdor, C. G. (2005). Effective induction of naive and recall T-cell responses by targeting antigen to human dendritic cells via a humanized anti-DC-SIGN antibody. Blood 106, 1278–1285.
Tacken, P. J., de Vries, I. J., Torensma, R., and Figdor, C. G. (2007). Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 7, 790–802.
Tagliani, E., Guermonprez, P., Sepulveda, J., Lopez-Bravo, M., Ardavin, C., Amigorena, S., Benvenuti, F., and Burrone, O. R. (2008). Selection of an antibody library identifies a pathway to induce immunity by targeting CD36 on steady-state CD8alpha+ dendritic cells. J. Immunol. 180, 3201–3209.
Tel, J., Benitez-Ribas, D., Hoosemans, S., Cambi, A., Adema, G. J., Figdor, C. G., Tacken, P. J., and de Vries, I. J. (2011). DEC-205 mediates antigen uptake and presentation by both resting and activated human plasmacytoid dendritic cells. Eur. J. Immunol. 41, 1014–1023.
Trumpfheller, C., Caskey, M., Nchinda, G., Longhi, M. P., Mizenina, O., Huang, Y., Schlesinger, S. J., Colonna, M., and Steinman, R. M. (2008). The microbial mimic poly IC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. Proc. Natl. Acad. Sci. U.S.A. 105, 2574–2579.
Trumpfheller, C., Finke, J. S., Lopez, C. B., Moran, T. M., Moltedo, B., Soares, H., Huang, Y., Schlesinger, S. J., Park, C. G., Nussenzweig, M. C., Granelli-Piperno, A., and Steinman, R. M. (2006). Intensified and protective CD4+ T cell immunity in mice with anti-dendritic cell HIV gag fusion antibody vaccine. J. Exp. Med. 203, 607–617.
Turnbull, E., and MacPherson, G. (2001). Immunobiology of dendritic cells in the rat. Immunol. Rev. 184, 58–68.
Vallon-Eberhard, A., Landsman, L., Yogev, N., Verrier, B., and Jung, S. (2006). Transepithelial pathogen uptake into the small intestinal lamina propria. J. Immunol. 176, 2465–2469.
van Broekhoven, C. L., Parish, C. R., Demangel, C., Britton, W. J., and Altin, J. G. (2004). Targeting dendritic cells with antigen-containing liposomes: a highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 64, 4357–4365.
Villadangos, J. A., and Schnorrer, P. (2007). Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 7, 543–555.
Vingert, B., Adotevi, O., Patin, D., Jung, S., Shrikant, P., Freyburger, L., Eppolito, C., Sapoznikov, A., Amessou, M., Quintin-Colonna, F., Fridman, W. H., Johannes, L., and Tartour, E. (2006). The Shiga toxin B-subunit targets antigen in vivo to dendritic cells and elicits anti-tumor immunity. Eur. J. Immunol. 36, 1124–1135.
Vremec, D., Pooley, J., Hochrein, H., Wu, L., and Shortman, K. (2000). CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164, 2978–2986.
Wang, H., Griffiths, M. N., Burton, D. R., and Ghazal, P. (2000). Rapid antibody responses by low-dose, single-step, dendritic cell-targeted immunization. Proc. Natl. Acad. Sci. U.S.A. 97, 847–852.
Wang, W. W., Das, D., and Suresh, M. R. (2009). A versatile bifunctional dendritic cell targeting vaccine vector. Mol. Pharm. 6, 158–172.
Wei, H., Wang, S., Zhang, D., Hou, S., Qian, W., Li, B., Guo, H., Kou, G., He, J., Wang, H., and Guo, Y. (2009). Targeted delivery of tumor antigens to activated dendritic cells via CD11c molecules induces potent antitumor immunity in mice. Clin. Cancer Res. 15, 4612–4621.
Wilson, N. S., El-Sukkari, D., Belz, G. T., Smith, C. M., Steptoe, R. J., Heath, W. R., Shortman, K., and Villadangos, J. A. (2003). Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood 102, 2187–2194.
Wilson, N. S., Young, L. J., Kupresanin, F., Naik, S. H., Vremec, D., Heath, W. R., Akira, S., Shortman, K., Boyle, J., Maraskovsky, E., Belz, G. T., and Villadangos, J. A. (2008). Normal proportion and expression of maturation markers in migratory dendritic cells in the absence of germs or Toll-like receptor signaling. Immunol. Cell Biol. 86, 200–205.
Witmer-Pack, M. D., Crowley, M. T., Inaba, K., and Steinman, R. M. (1993). Macrophages, but not dendritic cells, accumulate colloidal carbon following administration in situ. J. Cell. Sci. 105, 965–973.
Yamazaki, S., Dudziak, D., Heidkamp, G. F., Fiorese, C., Bonito, A. J., Inaba, K., Nussenzweig, M. C., and Steinman, R. M. (2008). CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J. Immunol. 181, 6923–6933.
Zhang, S. S., Park, C. G., Zhang, P., Bartra, S. S., Plano, G. V., Klena, J. D., Skurnik, M., Hinnebusch, B. J., and Chen, T. (2008). Plasminogen activator Pla of Yersinia pestis utilizes murine DEC-205 (CD205) as a receptor to promote dissemination. J. Biol. Chem. 283, 31511–31521.
Keywords: dendritic cells, vaccine, targeting, dendritic cell subsets, antigen delivery
Citation: Caminschi I, Maraskovsky E and Heath WR (2012) Targeting dendritic cells in vivo for cancer therapy. Front. Immun. 3:13. doi: 10.3389/fimmu.2012.00013
Received: 26 October 2011;
Accepted: 20 January 2012;
Published online: 07 February 2012.
Edited by:
Peter M. Van Endert, Université Paris Descartes/INSERM, FranceReviewed by:
Peter M. Van Endert, Université Paris Descartes/INSERM, FranceEdda Fiebiger, Children’s Hospital Boston and Harvard Medical School, USA
Copyright: © 2012 Caminschi, Maraskovsky and Heath. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Irina Caminschi, Immunology Division, The Walter and Eliza Hall Institute of Medical Research, 1G, Royal Parade, Parkville, VIC 3050, Australia. e-mail:Y2FtaW5zY2hpQGJ1cm5ldC5lZHUuYXU=