
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 08 December 2011
Sec. Antigen Presenting Cell Biology
volume 2 - 2011 | https://doi.org/10.3389/fimmu.2011.00071
 Isabelle Bouvier1,2 Hélène Jusforgues-Saklani1,2 Annick Lim3 Fabrice Lemaître4,5 Brigitte Lemercier3 Charlotte Auriau1,2 Marie-Anne Nicola6 Sandrine Leroy7 Helen K. Law8 Antonio Bandeira9 James J. Moon10† Philippe Bousso4,5
Isabelle Bouvier1,2 Hélène Jusforgues-Saklani1,2 Annick Lim3 Fabrice Lemaître4,5 Brigitte Lemercier3 Charlotte Auriau1,2 Marie-Anne Nicola6 Sandrine Leroy7 Helen K. Law8 Antonio Bandeira9 James J. Moon10† Philippe Bousso4,5 Matthew L. Albert1,2*
Matthew L. Albert1,2*Delivery of cell-associated antigen represents an important strategy for vaccination. While many experimental models have been developed in order to define the critical parameters for efficient cross-priming, few have utilized quantitative methods that permit the study of the endogenous repertoire. Comparing different strategies of immunization, we report that local delivery of cell-associated antigen results in delayed T cell cross-priming due to the increased time required for antigen capture and presentation. In comparison, delivery of disseminated antigen resulted in rapid T cell priming. Surprisingly, local injection of cell-associated antigen, while slower, resulted in the differentiation of a more robust, polyfunctional, effector response. We also evaluated the combination of cell-associated antigen with poly I:C delivery and observed an immunization route-specific effect regarding the optimal timing of innate immune stimulation. These studies highlight the importance of considering the timing and persistence of antigen presentation, and suggest that intradermal injection with delayed adjuvant delivery is the optimal strategy for achieving CD8+ T cell cross-priming.
CD8 T cell responses are key components of the adaptive immune system. These cells are considered particularly important in the host response to microorganisms and cells undergoing malignant transformation (Heemels and Ploegh, 1995). To carry out their effector function, they must first be activated by dendritic cells (DCs) presenting MHC I/peptide complexes (Mellman and Steinman, 2001). In instances of direct infection of DCs, antigen presentation via the endogenous pathway may account for CD8+ T cell priming; however, for many infections and most tumors, an indirect pathway (referred to as cross-priming) is utilized for the loading of antigen onto the MHC I of DCs (Albert et al., 1998; Albert, 2004). The cross-priming pathway has also been targeted for purposes of prophylactic and therapeutic vaccination (Amigorena, 2000; Palucka et al., 2006; Mitchell et al., 2007; Weide et al., 2008). While of potential value in therapeutic strategies, there is a need to optimize strategies for antigen and adjuvant delivery, taking care that conditions mimic those present during treatment of humans (Russo et al., 2007; Fontana et al., 2009). Herein, we investigate the impact of different routes of immunization when employing cell-associated antigen for cross-priming by host DC.
Over the last 10 years, it has been shown that several factors participate in efficient cross-priming: (i) the presence of high affinity CD8+ T cells (Zehn et al., 2009); (ii) CD4+ T cell help, acting to “license” DCs via CD40L/CD40 engagement, along with other activation stimuli (Bennett et al., 1997, 1998; Ridge et al., 1998; Schoenberger et al., 1998; Albert et al., 2001); (iii) DC maturation, often achieved by delivery of adjuvant (Longhi et al., 2009; Tewari et al., 2010; Flynn et al., 2011); (iv) sufficient antigen capture, thus allowing for high occupancy of MHC I (Buckwalter and Srivastava, 2008); and (v) the persistence of cell-associated antigen, which achieves sustained presentation and TCR stimulation (Prlic et al., 2006; Jusforgues-Saklani et al., 2008). While several of these parameters have been well characterized, experimental models typically do not reflect the conditions present during vaccination of humans. In much of the in vivo experimental work, strategies have been taken to increase the probability of initial encounter between antigen-specific T cells and DCs presenting their cognate antigen. For example, adoptive transfer has been used to artificially increase the precursor frequency of monoclonal, antigen responsive T cells (Kearney et al., 1994; Kurts et al., 1996; den Haan et al., 2000). The trend, however, is moving toward physiologic situations with low cell precursor frequency of responding T cells, and recent data has conclusively demonstrated that all phases of T cell activation are influenced by artificially increasing the precursor frequency: they are easier to activate, they expand more rapidly and typically result in greater memory cell differentiation (Marzo et al., 2005; Badovinac et al., 2007; van Heijst et al., 2009). Newly described assays have made it possible to measure low numbers of antigen-specific T cells in naïve mice or during the first days following immunization (Moon et al., 2007; Obar et al., 2008). Nonetheless, consideration has not been given to the artificial dosing of antigen used in these studies (e.g., LPS + peptide), which remain supra-threshold and do not accurately reflect typical vaccination protocols where antigen is limited. Moreover, the question of cross-priming polyfunctional T cells has not been fully evaluated, and again, optimization of vaccine delivery may help enhance therapeutic strategies aimed at the clearance of chronic infection or malignancies.
We report that following injection of cell-associated antigen, targeting of cross-presenting antigen presenting cells (APCs) for the generation of MHC I/peptide complexes is a limiting factor during the priming of the endogenous repertoire. Strikingly, due to the kinetics of antigen capture, local delivery of antigen resulted in a delayed yet ultimately more robust effector T cell activation as compared to systemic delivery of antigen. Our findings also have important implications for the formulation of vaccines combined with adjuvants, thus providing insight into how to best prime an effector CD8+ T cell response.
To determine optimal conditions for achieving cross-priming, we compared the effects of immunizing with a local versus systemic dissemination of cell-associated antigen. C57BL/6 mice were injected intradermally (i.d.) or intravenously (i.v.) with splenocytes from H-2 Kbm1 mice engineered to express a membrane-bound form of chicken ovalbumin in all tissues (referred to as Kbm1mOva). Use of membrane associated Ova (mOva) ensured that our model was not confounded by secreted protein captured by endocytosis (Nierkens et al., 2008); and an altered Kb molecule (known as Kbm1) ensured a role for host APCs in the cross-priming of CD8+ T cells. In order to precisely monitor the priming of the endogenous T cell repertoire, we utilized Kb–SIINFEKL tetramer-based enrichment, thus allowing precise enumeration and phenotypic analysis of Ovalbumin peptide-specific T cells at early time points after immunization (gating strategy shown in Figure 1A). Accumulation of tetramer-positive cells could be observed as early as day 5 for i.v. immunization (Figure 1B), with cells showing downregulation of CD62L and expression of CD25 (data not depicted). In contrast, the kinetics of T cell priming was delayed when cell-associated antigen was delivered via the i.d. route. In the latter condition, accumulation of Ova-specific CD8+ T cells was not observed until day 7 post-immunization. For both routes of immunization, antigen-specific T cells accumulated over time, with day 9–12 being the peak of the response (Figure 1B).
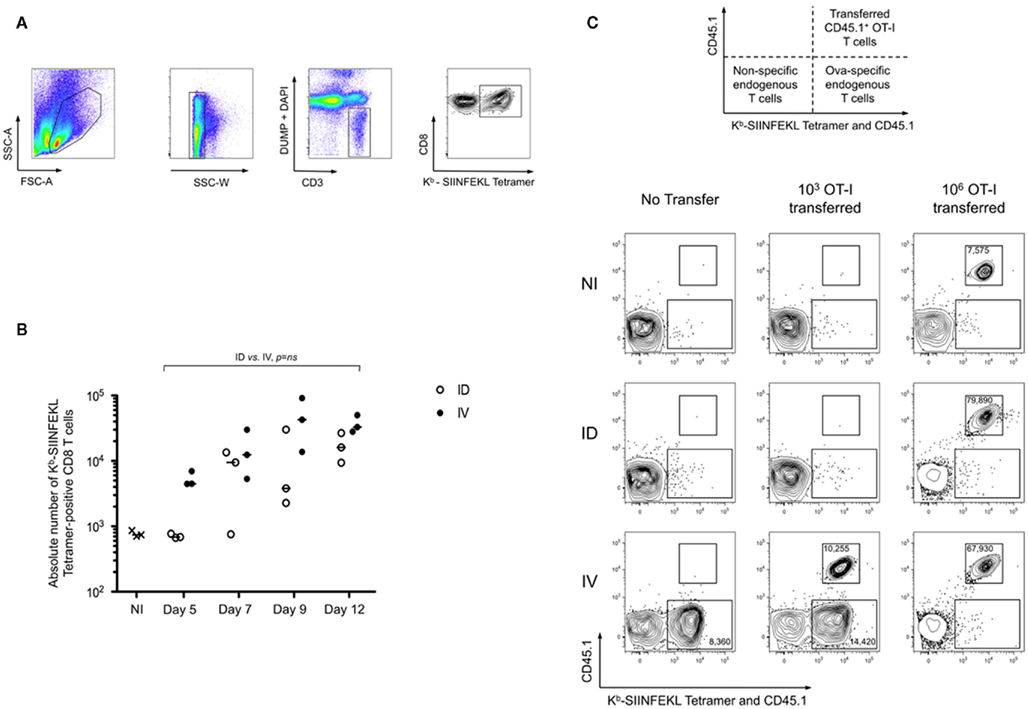
Figure 1. Route of immunization influences the timing of peak T cell cross-priming. (A,B) Mice were immunized intradermally (ID) or intravenously (IV) with 5 × 105 Kbm1mOva splenocytes. On days 5, 7, 9, and 12, 15 macroscopic lymph nodes and the spleen were harvested and a Kb–SIINFEKL tetramer-based enrichment was performed. The gating strategy used for tetramer-based enrichment described in the methods is shown (A). Single cells were selected using SSC-W. Then cells were stained with a mixture of antibodies to lineage markers and DAPI to exclude cells that are not of interest (DUMP gate). CD3+ cells were selected and CD8 and Kb–SIINFEKL tetramer labeling were used to detect antigen-specific CD8+ T cells. (B) Absolute numbers of Ova-specific CD8 T cells at each of the time points were determined. Data points indicate a single mouse. Results are representative of four independent experiments. The distributions according to the two immunization routes were not significantly different over time employing a general linear modeling analysis. (C) To evaluate the skewing of cross-priming responses by adoptive transfer of monoclonal T cells, 103 or 106 CD45.1 OT-I splenocytes were transferred into CD45.2 recipients prior to immunization. Use of congenic markers allowed simultaneous assessment of transferred and endogenous Ova-specific T cells (schematic representation). On day 5 post-immunization enrichment was performed using both Kb–SIINFEKL tetramer and CD45.1 antibody to distinguish endogenous tetramer-positive cells and OT-I cells. Live CD3+ CD8+ DUMP− cells are shown. The upper region highlights the transferred OT-I and the lower region marks the endogenous Ova-reactive CD8+ T cells. Absolute cell numbers are indicated for the respective cell populations. Plots were selected from an experiment with three mice per group; Data are representative of three independent experiments.
While prior studies suggest that the precursor frequency of Ova-specific T cells is similar across individual C57BL/6 mice (Obar et al., 2008), it is true that each mouse possesses distinct T cell repertoires (Bousso et al., 1998). In addition, we wanted to confirm that the delayed priming was not a result of the inability to access high affinity Ova-specific T cells. Thus we employed the strategy of adoptive transfer of low numbers (103) of monoclonal OT-I cells (Badovinac et al., 2007), transferred 1 day prior to immunization. On day 5, tetramer-based enrichment was performed using a combination of anti-CD45.1 and Kb–SIINFEKL tetramer, thus permitting simultaneous assessment of the transferred CD45.1+ OT-I T cells and endogenous Ova-specific T cells. As shown, only the i.v. immunization resulted in the early priming of Ova-specific T cells. Representative plots are shown, indicating that both the OT-I and the endogenous T cells behaved similarly, and that responses were comparable to those observed in animals that had not received OT-I (Figure 1C). Analysis of later time points supported the conclusion that priming is delayed when mice are immunized via the i.d. route (data not shown). Furthermore, we demonstrated that T cell precursor frequency influences the kinetics of priming. Transfer of 106 OT-I prior to immunization, in contrast to low transfer conditions, resulted in the robust and rapid expansion of Ova-specific T cells in both i.v. and i.d. conditions (Figure 1C). Also evident, the transferred cells outcompeted the endogenous repertoire. These data indicate that there exists a qualitative difference between i.v. and i.d. immunization, which is masked when using adoptive transfer of high numbers of monoclonal T cells.
To further define the impact of early dissemination of antigen (i.v. immunization) as compared to the establishment of an antigen depot (i.d. immunization), we monitored T cell effector functions. First, we performed an in vivo cytotoxicity assay to determine if the expanded T cells possessed cytolytic effector function. At different time points following immunization, mice received targets cells pulsed with SIINFEKL peptide and specific killing was determined (Figure 2). We observed a rapid induction of CTL activity after i.v. immunization that began to wane by day 12. Consistent with the delayed expansion after local immunization, we observed a stronger response on Day 12 following i.d. immunization. While both routes of immunization elicit CTL induction, this assay system does not provide per cell information about effector activity. To achieve such an analysis, we combined tetramer-based enrichment with intracellular staining. Using this approach, it was possible to determine the absolute number of tetramer-positive CD8+ T cells (Figure 3A); as well as the percentage of those cells producing IFNγ (Figure 3B). Of note, the absolute number of cells observed in this experiment is lower than those reported in Figure 1B, a consequence of performing intracellular cytokine stain, which requires additional washing and fixation steps. By day 7, the number of Ova-specific T cells was similar for the two routes of immunization, with the contraction phase beginning after day 15.
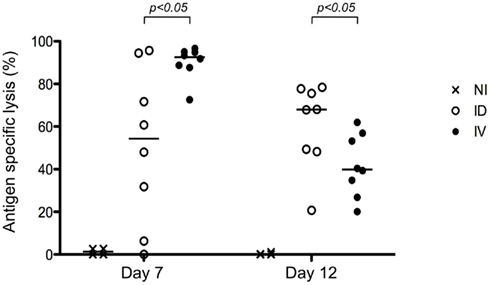
Figure 2. Both i.v. and i.d. immunization result in CTL induction. Mice were immunized i.d. or i.v. with 5 × 105 Kbm1mOva splenocytes. At day 7 or 12 post-immunization, an in vivo cytotoxicity assay was performed. Antigen-specific killing is reported. p-Values were calculated using a Mann–Whitney test (comparing ID versus IV). NI, non-immunized mice, shown here to indicate baseline killing responses.
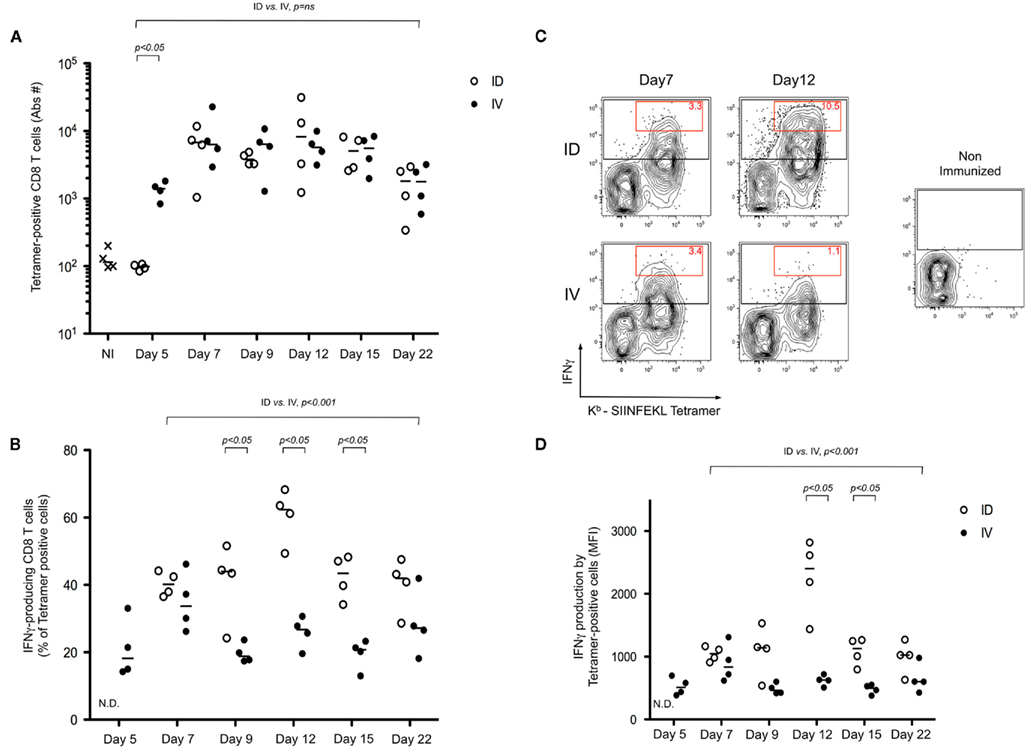
Figure 3. Intradermal immunization results in a more robust differentiation of effector CD8+ T cells. (A–D) Mice were immunized i.d. or i.v. with 5 × 105 Kbm1mOva splenocytes. Three hours prior to the defined time point, mice were re-stimulated in vivo by injecting 5 μg of CpG/DOTAP formulated as a mixture with 1 μg SIINFEKL peptide. Kb–SIINFEKL tetramer-based enrichment combined with an intracellular staining for IFNγ was performed. The absolute number of tetramer-positive cells is reported (A); and the percentage of IFNγ-producing cells among the population of tetramer-positive cells was determined (B). Representative plots of enriched tetramer-positive cells and the respective IFNγ production, per cell, is shown. Data from live, CD3+ CD8+ DUMP− cells are shown. The red gate highlights the tetramer-positive cells with the higher IFNγ staining and the numbers correspond to the percentage of these cells among the tetramer-positive cells population (C). To represent the respective per cell production of IFNγ, tetramer-positive cells were gated and the geometric mean fluorescent intensity (MFI) is shown (D). Data points indicate a single mouse. N.D., not determined, due to low absolute numbers of cells. Results are representative of two independent experiments. Individual pairings of ID versus IV were assessed by Mann–Whitney test and p-values are shown (A,B,D). The global distributions were also evaluated using time as a continuous variable (general linear modeling) (A,B,D).
Consistent with the delayed T cell expansion and cytotoxicity test, IFNγ production following i.v. immunization peaked at day 7, as compared to the i.d. route where the peak response was on day 12. Remarkably, comparing the peak responses indicated that 25–45% of the Ova-specific T cells were producing IFNγ after i.v. injection; whereas 50–70% of the cells were effector CD8+ T cells at the peak of the i.d. response (Figure 3B). Representative FACS plots highlight that not only did we achieve a higher percentage of IFNγ producing cells, but also, on a per cell basis, many of the effector T cells were making 10-fold more cytokine as compared to those isolated after i.v. immunization (Figure 3C, red gate day 12). This was also evident using a population-based analysis – as shown, the geometric mean fluorescent intensity (MFI) of tetramer-positive cells was significantly higher in the i.d. condition on days 9–15 (Figure 3D).
Next, we were interested in characterizing the quality of the T cell response. Prior studies have indicated that cells producing high levels of IFNγ have the unique capacity to secrete multiple cytokines, leading to their being referred to as polyfunctional T cells (Seder et al., 2008). In our model system, we evaluated the simultaneous production of IFNγ, IL-2, and TNFα. Mice were primed using the strategies discussed in Figure 1 and ex vivo restimulation of the tetramer-enriched fraction was performed prior to intracellular staining. As anticipated, the cells producing high levels of IFNγ also expressed TNFα and IL-2 (Figure 4A, IL-2 producing cells are shown in red). The response was evaluated throughout the kinetics of T cell priming (Figure 4A), and for purposes of comparing i.d. versus i.v. immunization, we focused on the peak of the response: Day 7 for i.v. immunization; and Day 12 for i.d. immunization. The percentages of IFNγ+ cells producing the three cytokines – IFNγ, IL-2, and TNFα – was significantly higher after i.d. immunization (Figure 4B). The converse was also true – the percentage of cells producing only IFNγ was higher following i.v. immunization (Figure 4C). Thus, we conclude that cross-priming via the i.d. route establishes a stronger, polyfunctional response.
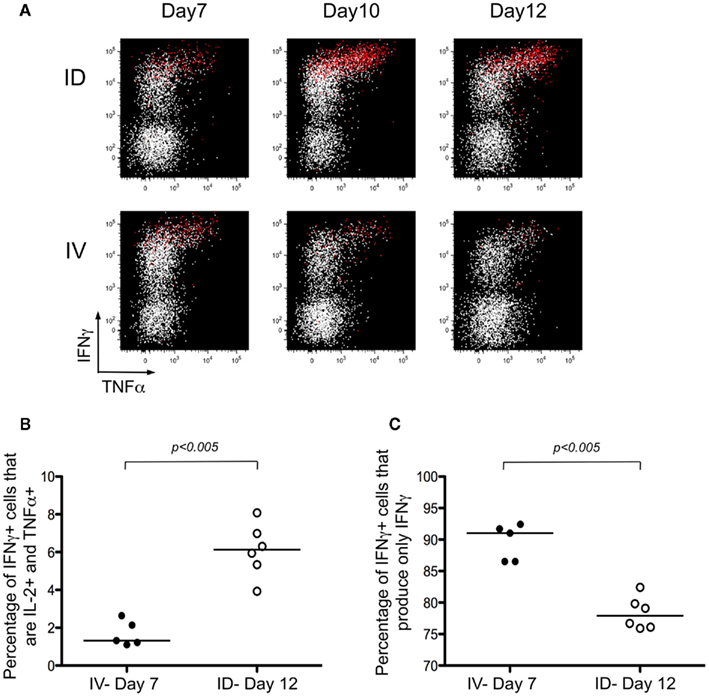
Figure 4. Intradermal immunization induces polyfunctional T cells. (A–C) Mice were immunized i.d. or i.v. with 5 × 105 Kbm1mOva splenocytes. At the different time points, lymph nodes and spleen were harvested and a Kb–SIINFEKL tetramer-based enrichment was performed. The enriched fraction was incubated for 4 h with SIINFEKL-pulsed splenocytes, followed by surface and intracellular staining. (A) Data from live, CD3+, CD8+ T cells are shown. Cells producing IL-2 are highlighted in red. The percentages of IFNγ+ cells that produce either the three cytokines – IFNγ, IL-2, and TNFα (B) or only one cytokine – IFNγ (C) were calculated. p-values were calculated using a Mann–Whitney test.
One potential caveat for the differences observed is that the rate and means of antigen dissemination might influence the diversity of the responding T cell population, with possible consequence on the relative avidity for MHC/peptide complexes (Catron et al., 2006; Zehn et al., 2009). To test this possibility, Ova tetramer-positive CD8+ T cells were FACS sorted, followed by TCR gene amplification and characterization of the distribution of Vβ–Jβ CDR3 length. This method accurately evaluates TCR diversity. 5 × 103 cells per mouse, isolated from five mice per group, were pooled for the analysis. As a control, we purified 25,000 bulk CD3+ CD8+ T cells from a non-immunized animal. Twenty-two Vβ families were detected in both the non-immunized and immunized animals. Data are represented as a profile of the Vβ–Jβ products obtained, plotted in arbitrary intensity units as a function of the size of the DNA fragment (Pannetier et al., 1993). As expected, analysis of the expanded antigen-specific cells in immunized animals showed a non-Gaussian distribution of the peaks as compared to the naïve bulk CD8+ population (Figure 5A). Notably, the Vβ 12.1 and 13.1 families were highly represented in the immunized animals, consistent with prior reports (Dillon et al., 1994). (Please note the change in nomenclature – the populations found here correspond with Vβ 5 and Vβ 8, respectively). To determine the diversity of the T cell responses, the number of distinct peaks detected in all immunoscope profiles were determined (Figure A1 in Appendix). As shown, the number of peaks was significantly reduced in immunized mice with comparable results in the i.v. and i.d. conditions. Given that these results were obtained from pooled mice, there exists the possibility that differences were homogenized and thus not detected; we therefore repeated the experiment using tetramer-positive cells purified from individual animals. Vβ families represented in the primed responses are shown (Figure A2 in Appendix), and the number of peaks per mouse is plotted (Figure 5B).
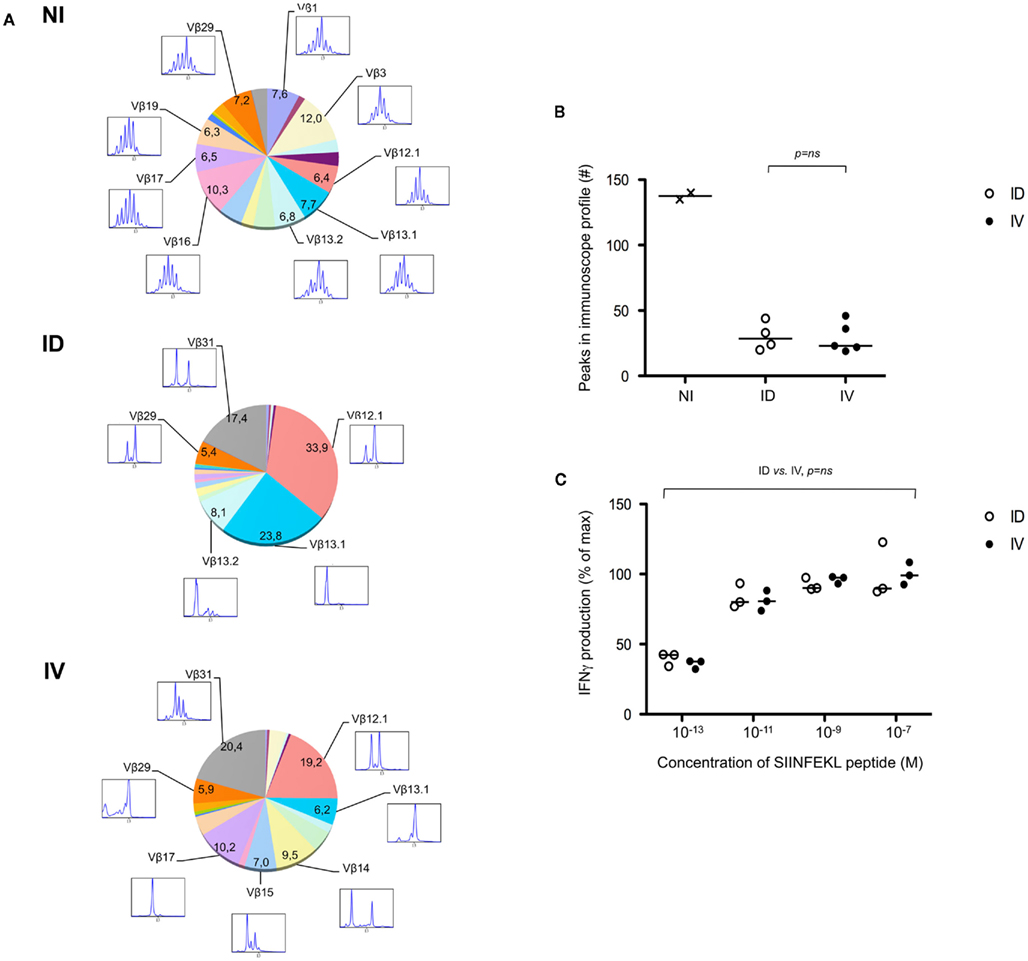
Figure 5. Route of immunization does not influence T cell diversity. (A) Mice were immunized i.d. or i.v. with 5 × 105 Kbm1mOva splenocytes. On day 9, tetramer-based enrichment was performed followed by FACS sorting. For the non-immunized (NI) condition, bulk tetramer negative CD8+ T cells were sorted. For immunized animals, cells were sorted from individual mice and 5,000 cells per animal were pooled to obtain 25,000 cells per condition. Immunoscope analysis was performed to define the length of the CDR3 loop in the 24 Vβ families (IMGT nomenclature). Each color represents a distinct Vβ family. The numbers correspond to the estimated percentage of total population. The immunoscope profile is presented for families that represent more than 5% of the total population. (B) Immunoscope was performed on cells sorted from individual mice (3,000–5,000 cells sorted/mouse). The total number of peaks detected on all the Vβ profiles was enumerated and represented. Data points indicate a single mouse. Vβ family representation for each animal is shown in Figure A2 in Appendix. Statistical analysis comparing ID and IV was assessed by Mann–Whitney U-test and p-values are shown. NI, non-immunized mice, and is shown to indicate baseline diversity of TCR. (C) To assess functional avidity of the responding T cells, the draining lymph node and spleen of five mice were pooled, day 8 post-immunization. CD8+ T cells were purified and analyzed by IFNγ ELISPOT. SIINFEKL peptide pulsed DCs were used to re-stimulate CD8+ T cells. SIINFEKL peptide concentrations are indicated. Results are represented as the percentage of maximal IFNγ production. Data points indicate a replicates for each condition. Results are representative of two independent experiments. Percentage of maximum IFN-γ production was analyzed using peptide concentration as a continuous variable (general linear modeling).
Next, we evaluated the avidity of the responding T cells by determining their ability to produce IFNγ after restimulation with limiting concentrations of SIINFEKL peptide. Responses were in the linear range for peptide concentrations 10−13–10−9, after which maximal IFNγ production was achieved. No differences were observed when comparing T cells isolated from mice that had been primed via the i.d. versus i.v. route (Figure 5C). Based on these data, we concluded that neither the diversity nor the avidity of the Ova-specific CD8+ T cells was influenced by the route of antigen delivery.
To further evaluate the differences observed, we determined the relationship between antigen dissemination and antigen presentation by host accessory cells. First, we assessed the establishment of an antigen depot following i.d. immunization. Luciferase-expressing splenocytes isolated from transgenic animals were injected into wild-type recipients. Due to the strain constraints, FvB male mice were used as a source of donor splenocytes, harboring minor histocompatibility differences with the female recipients. Cells delivered via the i.d. route remained primarily localized within the injection site (Figure A3A in Appendix). Kinetic studies suggested persistence of donor cells for greater than 13 days (Figure A3B in Appendix). Moreover we observed live injected splenocytes in the draining lymph node of i.d. immunized mice and in the spleen of i.v. immunized animals, indicating that there remains intact cell-associated antigen several days after immunization (Figures A3C,D).
Functional studies were used to confirm these findings. As described above, mice were immunized with Kbm1mOva splenocytes and at different time points, CFSE-labeled CD45.1+ OT-I splenocytes were transferred as a means of assessing cross-presentation by host APCs (Figure 6). OT-I transferred prior to immunization and analyzed 3 days later showed significant dilution of CFSE, indicating that cell-associated antigen injected via the i.v. route had already been cross-presented in spleen and lymph nodes (Figure 6, cohort 1). Given that up to seven cell divisions could be observed and that the first cell division is thought to require >24 h post-engagement by host DCs (Celli et al., 2005), we suggest that cross-presentation must have occurred immediately following immunization. Antigen presentation persisted from days 3–6 as the second cohort of OT-I also showed dilution of CFSE (Figure 6, cohort 2). In contrast to the i.v. condition, for i.d. immunization only minimal OT-I divisions were observed for the first cohort of transferred cells. By day 3–6, the response increased and significant OT-I proliferation could be observed in the draining lymph node, with minor responses in the spleen. These data confirm the local versus systemic dissemination of antigen via the two routes, and helps to explain the delayed kinetics of T cell priming after i.d. immunization.
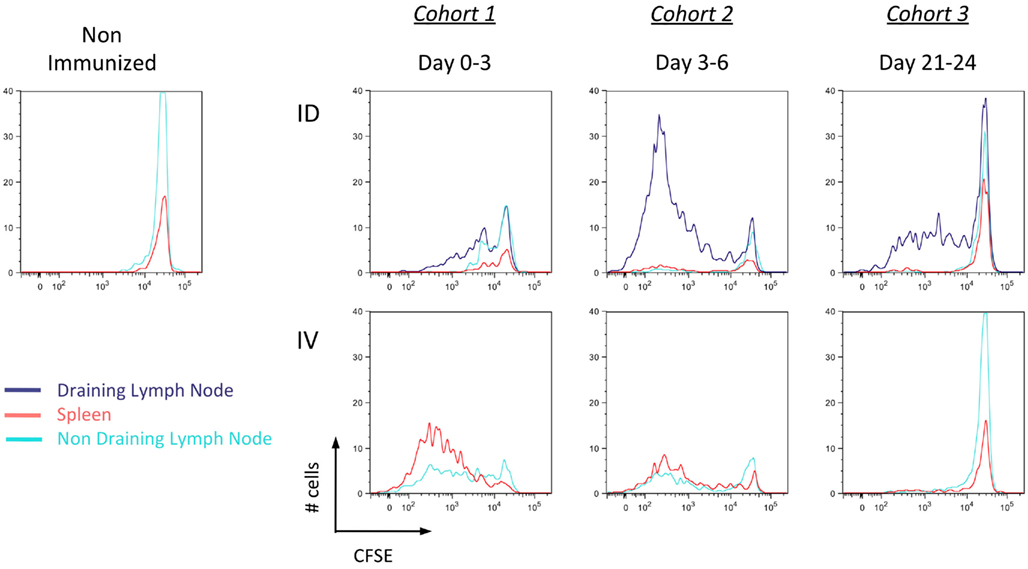
Figure 6. Delayed but persistent antigen presentation in the local draining lymph node after intradermal immunization. Mice were immunized i.d. or i.v. with 5 × 105 Kbm1mOva splenocytes. On days 0, 3, or 21, 5 × 106 CD45.1 CFSE-labeled OT-I splenocytes were adoptively transferred into immunized recipients. Three days later, the spleen, draining lymph node and a non-draining lymph node were harvested and the dilution of CFSE staining of OT-I was determined. Results are representative of three independent experiments.
Unexpectedly, the transfer of a third cohort of OT-I at 21 days post-immunization indicated that when delivered via the i.d. route, antigen was still being presented within the draining lymph node (Figure 6, cohort 3). This was not observed in the i.v. condition, suggesting the absence of APCs presenting Ova-peptide. Based on these findings, we conclude that the localized administration of cell-associated antigen impacts the timing of cross-presentation. While i.d. immunization is slightly slower due to the need for antigen to be captured and cross-presented in local lymphoid organs, the sustained presentation of MHC I/peptide complexes could influence effector and memory response.
In instances where microbial associated molecular patterns are absent (e.g., cell-associated antigen), it is common practice to formulate the vaccine with an adjuvant. Following from the result of delayed cross-presentation after i.d. immunization (Figure 6, cohort 1), we predicted that the optimal timing of adjuvant delivery will depend on the route of immunization. While adjuvants have been shown to be useful for enhancing the response to an antigen, our hypothesis is based on the observation that DC maturation prior to immunization can have the opposite effect – inhibiting T cell priming due to a failure to phagocytose cell-associated antigen (Wilson et al., 2006). To test our prediction, mice were stimulated using poly I:C, injected at different time relative to immunization with antigen. The absolute number of antigen-specific T cells was determined at the respective time of peak response (day 7 for i.v. and day 9 for i.d. immunization). When poly I:C was injected 1 day prior, or the day of i.v. immunization with the Kbm1mOva cells, T cell priming was greatly reduced (Figure 7A). Strikingly, injection of poly I:C 1 day after immunization enhanced T cell priming for the i.v. route. For the i.d. immunization, poly I:C injection 1 day prior to, the day of, or even 1 day after immunization, resulted in inhibited T cell priming (Figure 7B). As shown, it was necessary to wait until day 3 post-immunization to inject poly I:C in order to observe an enhancement of T cell priming (Figure 7B). Following from the results in Figures 6 and 7, we suggest that 1 day of antigen capture is sufficient to permit T cell priming after i.v. but that additional time is required for antigen capture after i.d. immunization.
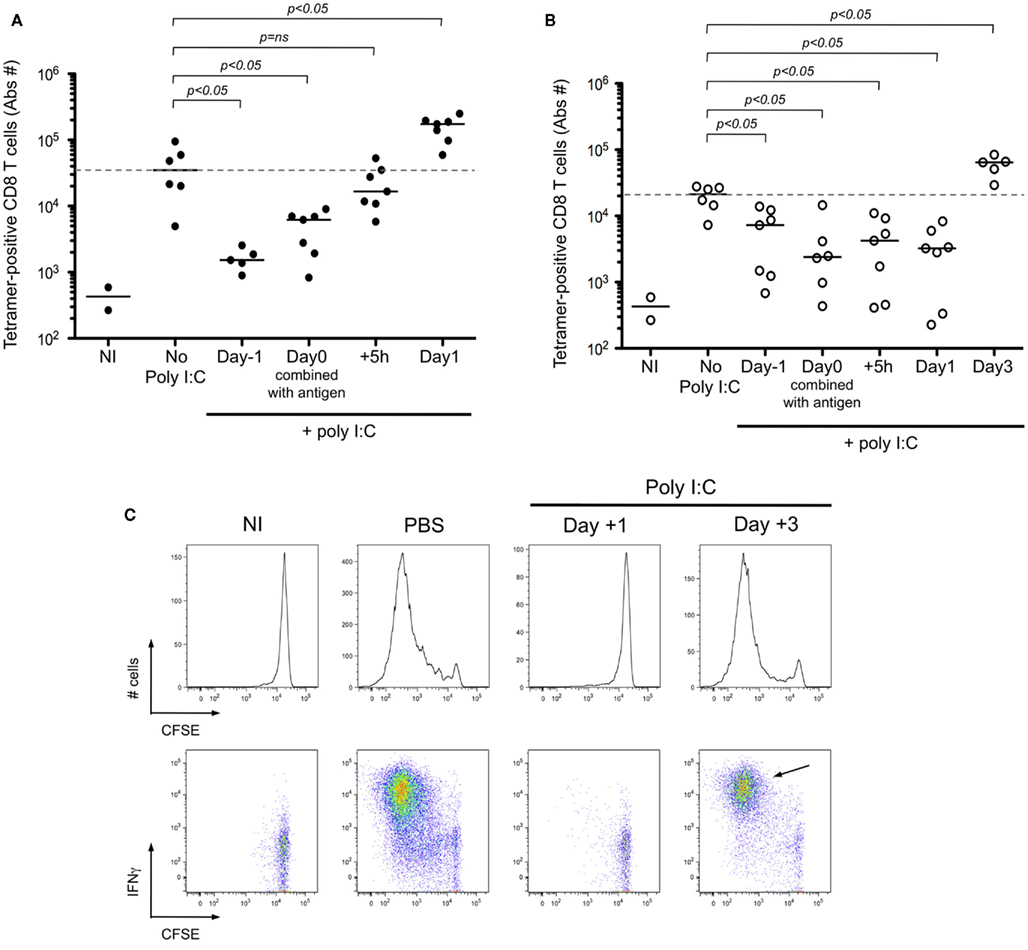
Figure 7. Adjuvant delivery must occur after antigen capture in order to achieve CD8+ T cell priming. (A,B) Mice were immunized i.d. or i.v. with 5 × 105 Kbm1mOva splenocytes and received 100 μg of poly I:C at indicated time points. For mice immunized i.v., they received poly I:C i.v. either: 1 day before immunization; the day of immunization combined with antigen; 5 h or 1 day post-immunization. The spleen and 15 macroscopic lymph nodes were harvested on day 7, which corresponds to the peak of the CD8+ T cell response. Kb–SIINFEKL tetramer-based enrichment was performed and the absolute numbers of tetramer-positive CD8+ T cells is reported (A). For mice immunized i.d., they received poly I:C at the same time points and one additional group was added, 3 days post-immunization. Poly I:C was administrated i.v. except for the mice injected on day 0 with poly I:C formulated with the antigen. Analysis was performed on day 9 post-immunization, again corresponding with peak CD8+ T cell response (B). p-values were calculated using a Mann–Whitney test, comparing in a two-way test, adjuvant condition to no poly I:C treatment. Dotted lines correspond to median number of responding cells in the absence of poly I:C. NI, non-immunized mice are shown to indicate baseline responses. (C) Mice were immunized i.d. with 5 × 105 Kbm1mOva splenocytes. On day 1 or day 3 post-immunization, 50 μg of Poly I:C or PBS was injected i.v. On day 3 post-immunization, 5 × 106 CFSE-labeled CD45.1 OT-I splenocytes were transferred i.v. Three days later the draining lymph node was harvested and the dilution of CFSE staining of OT-I was determined, represented by the histograms. Intracellular staining for IFNγ was performed at the same time, shown in the corresponding FACS plots. CD3+ CD8+ CD45.1+ cells were gated for the analysis shown. Data are representative of three independent experiments. NI, non-immunized mice.
To confirm that early delivery of adjuvant inhibited priming due to a failure to capture and present cell-associated antigen, we again utilized adoptively transferred CFSE-labeled OT-I as a read-out. Administration of poly I:C 1 day after i.d. immunization completely blocked OT-I proliferation and IFNγ production (Figure 7C). If instead we waited until day 3 post-immunization to administer the poly I:C, we no longer observed a blockade and in fact a greater percentage of OT-I showed maximal cell division and effector function (Figure 7C, arrow). To examine precisely the action of poly I:C on host DCs, we performed an in vivo kinetic study, enumerating and phenotyping DC populations in the spleen and lymph nodes. We focused on CD8α+ DCs and CD103+ DCs, as these two subsets are known to express TLR3 and have been shown to be required for antigen cross-presentation (Edelson et al., 2010). Following poly I:C injection, we observed a striking decrease in the total number of splenic CD8α+ DCs (Figure 8A). Analysis of the remaining cells indicated that CD86 and MHC-II molecules are upregulated within 15 h of injection, indicating that maturation is a rapid process (Figure 8B). In contrast to the spleen, DC number in lymph nodes increased after poly I:C injection; and again the cells demonstrated a mature phenotype within 1 day of poly I:C administration (Figures 8A,B).
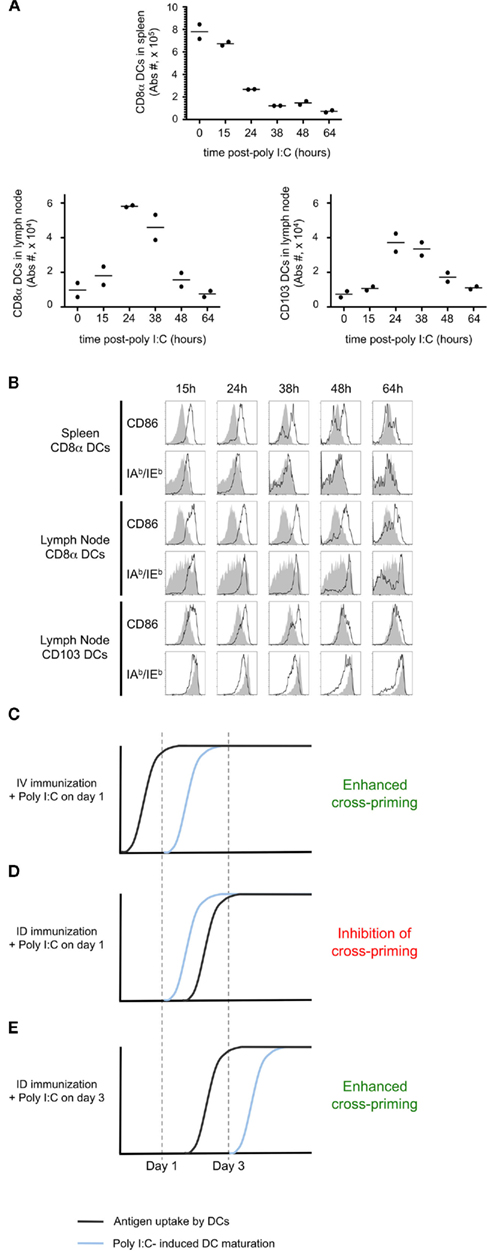
Figure 8. Poly I:C induces rapid DC maturation. Mice were injected i.v. with 100 μg of Poly I:C. At defined time points, the spleen and a lymph node were harvested. The total numbers of CD8α+ DCs and CD103+ DCs per organ (A) and the expression of CD86 and IAb/IEb(B) were determined. In (B) the gray histograms indicate the level of expression in untreated animals and the black line corresponds to poly I:C-injected mice. (C–E) Proposed model to explain the different effects of Poly I:C depending on the timing of delivery. The proposed timing of antigen uptake (black line) and the kinetic of DC maturation upon Poly I:C injection (blue line) are represented for three different conditions.
Comparing the timing of poly I:C induced DC maturation (Figures 8A,B), with the kinetics of antigen cross-presentation (Figure 6), we propose a model to explain the differential impact of adjuvant delivery, with regards to the route of immunization. Systemic dissemination of cell-associated antigen allows for capture and cross-presentation within 1 day. As such, administration of poly I:C on day 1 serves to stimulate cross-presenting DCs and enhance priming (Figure 8C). In contrast, localized delivery of cell-associated antigen requires 3 days for antigen uptake and presentation. Consequently, administration of poly I:C on day 1 results in early maturation of DCs, which are unable to cross-present cell-associated antigen (Figure 8D). If instead, adjuvant administration is performed on day 3 it is possible to achieve the beneficial effects of DC maturation, and enhancement of cross-priming is achieved (Figure 8E).
To test our model, we evaluated the timing of adjuvant delivery using an infectious model. Mice were immunized i.d. with Kbm1mOva splenocytes, and poly I:C was either co-administered on the day of immunization or given 3 days post-immunization. On day 9, mice were challenged with Ova-expressing Listeria and 2 days later, the bacterial load was determined in the spleen (Figure 9A) and in the liver (Figure 9B). We observed that immunization with Kbm1mOva splenocytes alone conferred partial protection to Listeria challenge. If mice received poly I:C on the day of immunization, this basal level of protection was completely abrogated. In contrast, the protection was significantly improved when poly I:C was administered 3 days after immunization. Indeed, the optimization of adjuvant delivery enhanced priming and resulted in a 2–3 log reduction in bacterial load.
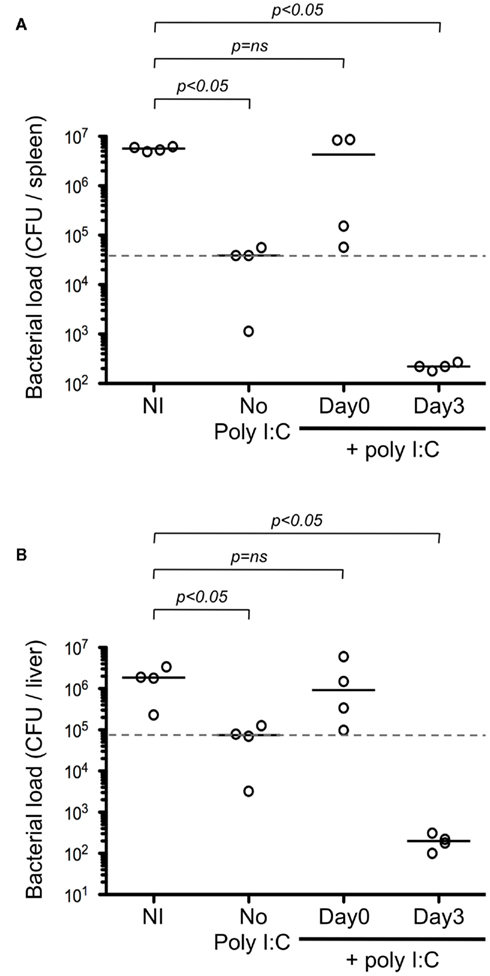
Figure 9. The differential effects of adjuvant impact protection against Listeria. Mice were immunized i.d. with 5 × 105 Kbm1mOva splenocytes. Poly I:C was administered either the day of immunization or 3 days later. On day 9 post-immunization, mice were challenged with 5 × 105 CFU of Ova-expressing Listeria. Two days later, the spleen (A) and the liver (B) were harvested and bacterial load per organ was determined. NI, non-immunized mice. Dotted lines correspond to median CFU in the absence of poly I:C. Mann–Whitney test p-values were calculated, comparing immunization condition to the NI control.
In sum, our study reinforces the need to understand the basis of therapeutic and prophylactic vaccination strategies, taking care to appropriately time the administration of adjuvant in order to effectively coordinate innate and adaptive immune response.
There is considerable interest in the development of vaccine strategies for the priming of CD8+ T cell responses. Stymieing the development of strategies that can be translated to humans is the fact that most experimental models utilize adoptive transfer of T cells and/or delivery of extremely high doses of antigen. Recent advances have solved the problem of detecting rare antigen-specific cells within the endogenous repertoire. Most notably, Moon et al. (2007) combined tetramer labeling and magnetic bead-based enrichment, which permitted the enumeration of T cells with a precursor frequency of 10−7 (equivalent to ∼10 cells per mouse). This approach has now been applied for the study of both CD4+ and CD8+ T cells, however in these studies the priming conditions used trigger maximal activation of the endogenous repertoire. In our study, we have utilized tetramer-based enrichment to evaluate vaccination strategies that more closely reflect what is done for immunotherapy in humans. Specifically, we evaluated the efficiency of CD8+ T cell cross-priming using cell-associated antigen, testing two important parameters that face investigators interested in initiating adaptive immune responses – the route of vaccination and the use of adjuvants. Importantly, there already exist therapeutic vaccines that closely reflect the model system studied herein (Fontana et al., 2009).
We chose to administer donor splenocytes derived from Kbm1mOva mice as the source of antigen: this ensured the need for antigen transfer to host DCs; excluded the possibility that secreted antigen or peptide exchange could account for the generation of MHC I/peptide complexes; and obviated the requirement for a danger signal as live cells expressing a mutated Kb are efficient sources of antigen for cross-priming (Krebs et al., 2009). While we support a role for phagocytosis of donor cells as a means of antigen transfer, an alternative possibility is the spread of antigen via exosomes produced by living cells (Wolfers et al., 2001). In our study, comparison of the intradermal and intravenous routes permitted us to determine the outcome of local versus systemic dissemination of antigen. As expected, systemically disseminated antigen resulted in rapid cross-presentation (Figures 1 and 6, cohort 1), which correlated with early differentiation of effector antigen-specific T cells (Figures 2 and 3). This was in contrast to locally administered antigen, which showed delayed cross-presentation and expansion of responding T cells.
Although delayed, one of the interesting features of locally administered antigen is that it acted as an antigen depot (Figure A3 in Appendix). Our data indicates that persistent antigen cross-presentation by host DCs (Figure 6, cohort 3) correlates and likely is the mechanism for inducing a more robust priming of polyfunctional effector CD8+ T cells (Figures 3 and 4). Interestingly, the magnitude of the T cell response following i.v. immunization was similar to that of the i.d. route. Thus, we conclude that the route of immunization impacted T cell quality but not primary expansion, highlighting the importance of providing in-depth study of vaccine candidates using the endogenous repertoire as a read-out for successful priming. Based on prior patient studies and experimental models of HIV, Leishmania major and Mycobacteria tuberculosis the T cell quality appears important for efficient host response and control of the infectious agent (Almeida et al., 2007; Darrah et al., 2007; Precopio et al., 2007).
Concerning the timing of cross-priming via the i.d. and i.v routes, we do not argue that the observed differences are not simply academic, nor do we consider that achieving T cell cross-priming 2 days earlier is going to improve vaccination strategies. Instead, it is our contention that the timing of antigen capture and T cell engagement has a profound impact on the appropriate timing for adjuvant delivery. Clearly, there is interest to coordinate both innate and adaptive responses, but a careful evaluation of how to optimally administer adjuvant and antigen is required. In our studies we chose to evaluate poly I:C, a synthetic double-stranded RNA (dsRNA) that engages endosomal TLR3 and MDA/5 on stromal cells (Longhi et al., 2009). It can induce IFNα/β and IL-12p70 by DCs and has been reported to be a superior adjuvant for T cell priming (Longhi et al., 2009). In addition, the similar expression pattern of host sensors in mice versus human make poly I:C a more attractive adjuvant for study in experimental models as compared to CpG (Rehli, 2002). Poly I:C has been tested as a direct therapeutic agent in the setting of viral infection and cancer; and has also been used as an ex vivo maturation agent for DC adoptive cell therapy trials. Several formulations of poly I:C are under late stage testing, including Ampligen (HemispheRx) and Hiltonol (Oncovir, Inc.) (Nicodemus and Berek, 2010; Rosenfeld et al., 2010; Flynn et al., 2011; Okada et al., 2011).
While poly I:C is considered a proinflammatory adjuvant, previously studies have also reported that pre-treatment of animals with poly I:C inhibited antigen cross-presentation (Wilson et al., 2006). The contrasting action of poly I:C remains poorly understood and the mechanism of action governing these polar phenomena has not been explored. Herein, we demonstrated that administration of poly I:C 1 day post-intravenous immunization resulted in enhanced cross-priming, however the same timing of administration resulted in a blockade for intradermal injection (Figure 7A). Consistent with the need for DCs to capture antigen prior to adjuvant administration, poly I:C given on day three enhanced the cross-priming of CD8+ T cells following local antigen delivery (Figure 7B). We showed that poly I:C induces DC activation as established by upregulation of CD86 and MHC-II expression 1 day after administration (Figure 8). Together, these data establish that in order to enhance cross-priming poly I:C must be delivered at a time point after the host DCs have captured the injected cell-associated antigen (Figures 8C–E).
Results of recent clinical trials that combine delivery of antigen and adjuvant indicate the importance of defining the optimal time of innate immune stimulation. Using the same NY-ESO-1 protein preparation, delivered locally in the skin, it was observed that co-administration of adjuvant permitted efficient cross-priming, whereas pre-conditioning of the injection site diminished the ability to stimulate antigen-specific T cells (Valmori et al., 2007; Adams et al., 2008). There have also been studies showing that injection of RNA vaccine in combination with innate stimulation is not always the best strategy to achieve efficient priming. First, Carralot and colleagues showed that the delivery of GM–CSF 24 h after RNA injection enhanced T cell priming (Carralot et al., 2004). Importantly, this adjuvant effect was not observed when GM–CSF was delivered in combination with RNA. Moreover, in a follow-up study from Diken et al., it was shown in experiments comparable to ours that subcutaneous delivery of 20 μg poly I:C, 1 day prior to RNA injection intranodally, abrogated the uptake of RNA vaccine (Diken et al., 2011). Taken together with the observations we have made in mice, we suggest that there is a trade-off between stimulating innate receptors in immature DCs for purposes of triggering an inflammatory response and the resulting decrease in antigen capture that is due to the induction of DC maturation. One option might be the use of agonists that bind receptors selectively expressed on mature DCs (e.g., CD40L; Lanzavecchia, 1998).
Our studies highlight the importance of considering the timing and persistence of antigen presentation, and suggest intradermal injection with delayed adjuvant delivery to be the optimal strategy for achieving CD8+ T cell cross-priming. While many studies of CD8+ T cell priming conclude with a remark about how important their findings are for predicting efficient means of vaccinating humans, our efforts have a true possibility to be translated into practice. For example, Russo and Fontana have conducted pre-clinical and clinical studies utilizing peripheral blood lymphocytes genetically modified to express tumor antigens as a strategy for inducing tumor immunity in cancer patients (Russo et al., 2007; Fontana et al., 2009). In their treatment protocols, patients received five bi-weekly, i.v. infusions of escalating numbers of autologous lymphocytes, reaching doses of 5 × 108 total lymphocytes infused (range: 2–7 × 108). Their clinical trial was not designed to assess efficacy; nonetheless, it was possible to observe clinical responses in 3/10 patients, which correlated with priming of Mage-3 specific CD8+ T cells (Fontana et al., 2009). These studies, as well as others, highlight the feasibility of utilizing cell-associated antigen as a means of immunizing patients. It also points to the need for relevant mouse models aimed at optimizing strategies for achieving robust CD8+ T cell cross-priming.
In sum, we demonstrate that, while slower, local injection of cell-associated antigen resulted in the differentiation of a polyfunctional effector cell response during the T cell priming. Our studies also highlight the importance of considering the timing and persistence of antigen presentation, and suggest intradermal injection with delayed adjuvant delivery to be the optimal strategy for achieving CD8+ T cell cross-priming. While we hope this study will impact vaccine design for prophylaxis and therapy, it is clear that in the latter situation additional investigations will be required in order to overcome intrinsic suppressive and/or regulatory mechanisms that limit the success of immunotherapy strategies.
C57BL/6J wild-type mice were obtained from Charles River. PtprcaPepcb/BoyJ (CD45.1) and Tg(TcraTcrb) 1100Mjb (OT-I Rag+/+) mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Mice expressing membrane-bound full-length Ova under an actin promoter were a gift from Dr. M. Jenkins (University of Minnesota, USA) and the cross onto the H-2Kbm1 line was performed by Dr. S. Schoenberger (LIAI, USA). All mice were maintained and bred in a SPF helicobacter-negative facility, and used under approved protocols. In all experiments, 6- to 12-week-old mice were used.
Antibodies for FACS analysis were obtained from BD Biosciences, Biolegend, or eBiosciences (Table A1 in Appendix). Antibodies used in the IFNγ–ELISPOT assays were purchased from Mabtech. The Ovalbumin H-2Kb epitope SIINFEKL peptide was obtained from Polypeptide Group. Monomers were prepared using a modified version of that described (Altman et al., 1996) and tetramerization was performed prior to use, using PE–Streptavidin (Invitrogen), added for 1 h at 25°C. Intracellular cytokine staining was done using the Cytofix/Cytoperm/Brefeldin-A kit (BD Biosciences). Poly I:C and CpG ODN2216 were purchased from Invivogen. DOTAP was obtained from Roche. Labeling with carboxyfluorescein diacetate succinimidyl ester (CFSE) was performed using the Vybrant cell tracer kit from Invitrogen. To label dead cells, DAPI or Aqua-Live/Dead Fixable Dead Cell Stain kits from Invitrogen were used after tetramer-based enrichment.
Splenocytes used for immunization were isolated from Kbm1mOva mice. 5 × 105 cells in a volume of 100 μl were injected intradermally (i.d.) or intravenously (i.v.). The intradermal injection was performed in the right flank with the inguinal lymph node being the draining lymph node. For OT-I transfer, bulk splenocytes were isolated from CD45.1 OT-I mice. 103, 106, or 5 × 106 splenocytes were transferred i.v. depending on the experiment. For injection of Poly I:C, 100 μg of Poly I:C was injected i.v. in a final volume of 100 μl. For in vivo restimulation before intracellular staining, 5 μg of CpG is diluted in PBS and DOTAP; this was then formulated with 1 μg of SIINFEKL peptide and injected i.v. in a volume of 100 μl.
Leukocytes were harvested from 15 lymph nodes and the spleen. Cells were Fc-Blocked with anti-CD16/CD32 antibody and stained with PE-labeled Kb–SIINFEKL tetramers in PBS containing 2% FCS and 0.1% of Sodium Azide for 30 min at 4°C. It was followed by an incubation with anti-PE magnetic microbeads (Miltenyi). Cells were passed over a magnetic LS column to enrich tetramer-positive cells. Bound cells were eluted (“enriched” fraction). Five microliter aliquot was collected for precise counting of the bound fraction. Cells were stained with a mixture of antibodies (CD11c, CD11b, CD4, NK1.1, F4/80, B220, CD3, and CD8) to exclude cells (DUMP gate) and focus on CD8+ T cells (see Figure 1A). Prior to analysis, DAPI was added to mark dead cells. Cells were analyzed using a FACS Canto II (BD Biosciences). Live, non-clumped, CD3+ CD8+ tetramer-positive cells were gated. The percentage of tetramer-positive cells was multiplied by the total number of cells in the enriched fraction to obtain the total number of tetramer-positive CD8+ T cells.
For in vivo restimulation, mice were injected with 5 μg of CpG/DOTAP formulated as a mixture with 1 μg SIINFEKL peptide 3 h prior to leukocyte harvest. Next, the tetramer-based enrichment was performed with the addition of Brefeldin-A during each incubation step. After the elution step, enriched cells were stained with Aqua as a dead cell marker, incubated with surface staining antibodies and fixed. Next, cells were permeabilized and stained using anti-IFNγ as per the manufacturer’s instructions (BD Biosciences). For ex vivo restimulation, the tetramer-based enrichment was performed first and the eluted fraction was incubated 4 h with SIINFEKL-pulsed splenocytes at 37°C. Then cells were stained for IFNγ, IL-2, and TNFα as per the manufacturer’s instructions (BD Biosciences).
CD45.1 OT-I splenocytes were isolated and stained using 5 μM CFSE in PBS. After washing with ice-cold PBS 5 × 106 OT-I splenocytes were injected i.v. into immunized mice. Three days later the draining and non-draining lymph nodes, and the spleen were harvested. Organs were processed independently and cells were labeled with CD8β and CD45.1 antibodies allowing for the identification of the transferred CD8 OT-I T cells and the determination of CFSE intensity.
Spleen and the draining lymph node were harvested and CD8+ T cells were purified using anti-CD8 microbeads and MS columns (Miltenyi). IFNγ ELISPOT assays were performed as previously described (Blachere et al., 2006). The ELISPOT plate evaluation was performed in a blinded fashion by an independent evaluation service (Zellnet Consulting) using an automated ELISPOT reader (Carl Zeiss).
Kb–SIINFEKL tetramer-positive CD8+ T cells were sorted using a FACS Aria-II. Total RNA was prepared from sorted T cells using the Total RNA Miniprep kit (Sigma), and cDNA was synthesized using the SuperScript™ II Reverse Transcriptase (Invitrogen). The different Vβ germline genes can be clustered in 24 families according to their level of homology (IMGT nomenclature). For quantitative repertoire, PCR reactions were carried out by combining a reverse primer and a specific fluorophore-labeled probe for the constant region (MGB–TaqMan probe) with 1 of 24 primers covering the different Vβ chains (Table A2 in Appendix). Real-time PCR reactions were subsequently carried out with a final concentrations of 400 nmol/L of each oligonucleotide primer, 200 nmol/L of the fluorogenic probe, and FastStart master Mix (Roche). Thermal cycling conditions comprised Taq DNA Polymerase activation at 95°C for 10 min, then subjected to 40 cycles of denaturation at 95°C for 15 s, annealing and extension at 60°C for 1 min. For all these different reactions, real-time quantitative PCR was then performed on an ABI-7300 system (Applied Biosystems). The relative usage of each Vβ family was calculated according to the formula:
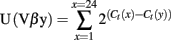
Ct(x) is the fluorescent threshold cycle number measured for the Vβy family. For immunoscope profiles, products were then subjected to run-off reactions with a nested fluorescent primer specific for the constant region (Table A2 in Appendix: Fam-primer) – run for a total of three cycles. The fluorescent products were separated and analyzed using an ABI-PRISM 3730 DNA analyzer. The size and intensity of each band were analyzed with “Immunoscope software” (Pannetier et al., 1993), which has been adapted to the capillary sequencer. Fluorescence intensities were plotted in arbitrary units on the y-axis, and CDR3 lengths (in amino acids) on the x-axis.
At different time points following the immunization, mice were injected i.v. with 5 × 106 CD45.1 splenocytes stained with 0.5 μM CFSE and pulsed with SIINFEKL peptide, and 5 × 106 CD45.1 splenocytes stained with 5 μM CFSE and left unpulsed. Fifteen hours later, spleen was harvested and cells were stained with an anti-CD45.1 antibody. The lysis of injected splenocytes was determined using the CFSE staining and the percentage of specific lysis was calculated.
Spleen and lymph node were digested with Collagenase D (Roche) and Dnase (Invitrogen). Cells were stained for CD11c, CD11b, CD8α, CD103, CD86, IAb/IEb, and analyzed by flow cytometry. An aliquot was used to determine the absolute number of cells per organ.
The Ovalbumin-expressing Listeria is a kind gift from N. Glaichenhaus. Mice were infected i.v. with 5 × 105 colony forming units (CFU). Two days later, the spleen and the liver were harvested and mashed in NP-40 0.2% in water, and serial dilutions were plated to determine the CFU per organ.
Data was plotted with bars representing median value. We used non-parametric (two-tailed) Mann–Whitney test to compare the distributions between two conditions. In some instances, selective comparisons between two groups within a multi-parameter experiment were also performed using non-parametric Mann–Whitney test. Continuous measurements were studied over time or according to the peptide concentration using general linear modeling. Statistical analysis was performed using Stata 11 software (StataCorp, College Station, TX, USA) and Prism 5 (GraphPad Software Inc., La Jolla, CA, USA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Philippe Kourilsky for support of the Immunoscope profiling (College de France). This work was supported by La Ligue contre le cancer and the EURYI scheme (Matthew L. Albert). Isabelle Bouvier is supported by the Association pour la Recherche sur le Cancer (project DOC20110603227). We would also like to thank members of the ICD laboratory for their review of the manuscript and helpful comments throughout the project. The β-actin/luciferase-expressing transgenic FVB/N mice were generous given by Christopher Contag (Stanford University).
Adams, S., O’Neill, D. W., Nonaka, D., Hardin, E., Chiriboga, L., Siu, K., Cruz, C. M., Angiulli, A., Angiulli, F., Ritter, E., Holman, R. M., Shapiro, R. L., Berman, R. S., Berner, N., Shao, Y., Manches, O., Pan, L., Venhaus, R. R., Hoffman, E. W., Jungbluth, A., Gnjatic, S., Old, L., Pavlick, A. C., and Bhardwaj, N. (2008). Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using TLR7 agonist imiquimod as vaccine adjuvant. J. Immunol. 181, 776–784.
Albert, M. L. (2004). Death-defying immunity: do apoptotic cells influence antigen processing and presentation? Nat. Rev. Immunol. 4, 223–231.
Albert, M. L., Jegathesan, M., and Darnell, R. B. (2001). Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat. Immunol. 2, 1010–1017.
Albert, M. L., Sauter, B., and Bhardwaj, N. (1998). Dendritic cells acquire antigen from apoptotic cells and induce class I- restricted CTLs. Nature 392, 86–89.
Almeida, J. R., Price, D. A., Papagno, L., Arkoub, Z. A., Sauce, D., Bornstein, E., Asher, T. E., Samri, A., Schnuriger, A., Theodorou, I., Costagliola, D., Rouzioux, C., Agut, H., Marcelin, A. G., Douek, D., Autran, B., and Appay, V. (2007). Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 204, 2473–2485.
Altman, J. D., Moss, P. A., Goulder, P. J., Barouch, D. H., Mcheyzer-Williams, M. G., Bell, J. I., Mcmichael, A. J., and Davis, M. M. (1996). Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–96.
Amigorena, S. (2000). Cancer immunotherapy using dendritic cell-derived exosomes. Medicina (B Aires) 60(Suppl. 2), 51–54.
Badovinac, V. P., Haring, J. S., and Harty, J. T. (2007). Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity 26, 827–841.
Bennett, S. R., Carbone, F. R., Karamalis, F., Flavell, R. A., Miller, J. F., and Heath, W. R. (1998). Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393, 478–480.
Bennett, S. R., Carbone, F. R., Karamalis, F., Miller, J. F., and Heath, W. R. (1997). Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J. Exp. Med. 186, 65–70.
Blachere, N. E., Morris, H. K., Braun, D., Saklani, H., Di Santo, J. P., Darnell, R. B., and Albert, M. L. (2006). IL-2 is required for the activation of memory CD8+ T cells via antigen cross-presentation. J. Immunol. 176, 7288–7300.
Bousso, P., Casrouge, A., Altman, J. D., Haury, M., Kanellopoulos, J., Abastado, J. P., and Kourilsky, P. (1998). Individual variations in the murine T cell response to a specific peptide reflect variability in naive repertoires. Immunity 9, 169–178.
Buckwalter, M. R., and Srivastava, P. K. (2008). “It is the antigen(s), stupid” and other lessons from over a decade of vaccitherapy of human cancer. Semin. Immunol. 20, 296–300.
Carralot, J. P., Probst, J., Hoerr, I., Scheel, B., Teufel, R., Jung, G., Rammensee, H. G., and Pascolo, S. (2004). Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell. Mol. Life Sci. 61, 2418–2424.
Catron, D. M., Rusch, L. K., Hataye, J., Itano, A. A., and Jenkins, M. K. (2006). CD4+ T cells that enter the draining lymph nodes after antigen injection participate in the primary response and become central-memory cells. J. Exp. Med. 203, 1045–1054.
Celli, S., Garcia, Z., and Bousso, P. (2005). CD4 T cells integrate signals delivered during successive DC encounters in vivo. J. Exp. Med. 202, 1271–1278.
Darrah, P. A., Patel, D. T., De Luca, P. M., Lindsay, R. W., Davey, D. F., Flynn, B. J., Hoff, S. T., Andersen, P., Reed, S. G., Morris, S. L., Roederer, M., and Seder, R. A. (2007). Multifunctional TH1 cells define a correlate of vaccine-mediated protection against leishmania major. Nat. Med. 13, 843–850.
den Haan, J. M., Lehar, S. M., and Bevan, M. J. (2000). CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192, 1685–1696.
Diken, M., Kreiter, S., Selmi, A., Britten, C. M., Huber, C., Tureci, O., and Sahin, U. (2011). Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. 18, 702–708.
Dillon, S. R., Jameson, S. C., and Fink, P. J. (1994). V beta 5+ T cell receptors skew toward OVA+ H-2Kb recognition. J. Immunol. 152, 1790–1801.
Edelson, B. T., Kc, W., Juang, R., Kohyama, M., Benoit, L. A., Klekotka, P. A., Moon, C., Albring, J. C., Ise, W., Michael, D. G., Bhattacharya, D., Stappenbeck, T. S., Holtzman, M. J., Sung, S. S., Murphy, T. L., Hildner, K., and Murphy, K. M. (2010). Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J. Exp. Med. 207, 823–836.
Flynn, B. J., Kastenmuller, K., Wille-Reece, U., Tomaras, G. D., Alam, M., Lindsay, R. W., Salazar, A. M., Perdiguero, B., Gomez, C. E., Wagner, R., Esteban, M., Park, C. G., Trumpfheller, C., Keler, T., Pantaleo, G., Steinman, R. M., and Seder, R. (2011). Immunization with HIV Gag targeted to dendritic cells followed by recombinant New York vaccinia virus induces robust T-cell immunity in nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 108, 7131–7136.
Fontana, R., Bregni, M., Cipponi, A., Raccosta, L., Rainelli, C., Maggioni, D., Lunghi, F., Ciceri, F., Mukenge, S., Doglioni, C., Colau, D., Coulie, P. G., Bordignon, C., Traversari, C., and Russo, V. (2009). Peripheral blood lymphocytes genetically modified to express the self/tumor antigen MAGE-A3 induce antitumor immune responses in cancer patients. Blood 113, 1651–1660.
Heemels, M. T., and Ploegh, H. (1995). Generation, translocation, and presentation of MHC class I-restricted peptides. Annu. Rev. Biochem. 64, 463–491.
Jusforgues-Saklani, H., Uhl, M., Blachere, N., Lemaitre, F., Lantz, O., Bousso, P., Braun, D., Moon, J. J., and Albert, M. L. (2008). Antigen persistence is required for dendritic cell licensing and CD8+ T cell cross-priming. J. Immunol. 181, 3067–3076.
Kearney, E. R., Pape, K. A., Loh, D. Y., and Jenkins, M. K. (1994). Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity 1, 327–339.
Krebs, P., Barnes, M. J., Lampe, K., Whitley, K., Bahjat, K. S., Beutler, B., Janssen, E., and Hoebe, K. (2009). NK-cell-mediated killing of target cells triggers robust antigen-specific T-cell-mediated and humoral responses. Blood 113, 6593–6602.
Kurts, C., Heath, W. R., Carbone, F. R., Allison, J., Miller, J. F., and Kosaka, H. (1996). Constitutive class I-restricted exogenous presentation of self antigens in vivo. J. Exp. Med. 184, 923–930.
Longhi, M. P., Trumpfheller, C., Idoyaga, J., Caskey, M., Matos, I., Kluger, C., Salazar, A. M., Colonna, M., and Steinman, R. M. (2009). Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 206, 1589–1602.
Marzo, A. L., Klonowski, K. D., Le Bon, A., Borrow, P., Tough, D. F., and Lefrancois, L. (2005). Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat. Immunol. 6, 793–799.
Mellman, I., and Steinman, R. M. (2001). Dendritic cells: specialized and regulated antigen processing machines. Cell 106, 255–258.
Mitchell, M. S., Abrams, J., Thompson, J. A., Kashani-Sabet, M., Deconti, R. C., Hwu, W. J., Atkins, M. B., Whitman, E., Ernstoff, M. S., Haluska, F. G., Jakowatz, J. G., DAS Gupta, T. K., Richards, J. M., Samlowski, W. E., Costanzi, J. J., Aronson, F. R., Deisseroth, A. B., Dudek, A. Z., and Jones, V. E. (2007). Randomized trial of an allogeneic melanoma lysate vaccine with low-dose interferon Alfa-2b compared with high-dose interferon Alfa-2b for resected stage III cutaneous melanoma. J. Clin. Oncol. 25, 2078–2085.
Moon, J. J., Chu, H. H., Pepper, M., Mcsorley, S. J., Jameson, S. C., Kedl, R. M., and Jenkins, M. K. (2007). Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27 203–213.
Nicodemus, C. F., and Berek, J. S. (2010). TLR3 agonists as immunotherapeutic agents. Immunotherapy 2, 137–140.
Nierkens, S., Den Brok, M. H., Sutmuller, R. P., Grauer, O. M., Bennink, E., Morgan, M. E., Figdor, C. G., Ruers, T. J., and Adema, G. J. (2008). In vivo colocalization of antigen and CpG (corrected) within dendritic cells is associated with the efficacy of cancer immunotherapy. Cancer Res. 68, 5390–5396.
Obar, J. J., Khanna, K. M., and Lefrancois, L. (2008). Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity 28, 859–869.
Okada, H., Kalinski, P., Ueda, R., Hoji, A., Kohanbash, G., Donegan, T. E., Mintz, A. H., Engh, J. A., Bartlett, D. L., Brown, C. K., Zeh, H., Holtzman, M. P., Reinhart, T. A., Whiteside, T. L., Butterfield, L. H., Hamilton, R. L., Potter, D. M., Pollack, I. F., Salazar, A. M., and Lieberman, F. S. (2011). Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 29, 330–336.
Palucka, A. K., Ueno, H., Connolly, J., Kerneis-Norvell, F., Blanck, J. P., Johnston, D. A., Fay, J., and Banchereau, J. (2006). Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J. Immunother. 29, 545–557.
Pannetier, C., Cochet, M., Darche, S., Casrouge, A., Zoller, M., and Kourilsky, P. (1993). The sizes of the CDR3 hypervariable regions of the murine T-cell receptor beta chains vary as a function of the recombined germ-line segments. Proc. Natl. Acad. Sci. U.S.A. 90, 4319–4323.
Precopio, M. L., Betts, M. R., Parrino, J., Price, D. A., Gostick, E., Ambrozak, D. R., Asher, T. E., Douek, D. C., Harari, A., Pantaleo, G., Bailer, R., Graham, B. S., Roederer, M., and Koup, R. A. (2007). Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J. Exp. Med. 204, 1405–1416.
Prlic, M., Hernandez-Hoyos, G., and Bevan, M. J. (2006). Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J. Exp. Med. 203, 2135–2143.
Rehli, M. (2002). Of mice and men: species variations of Toll-like receptor expression. Trends Immunol. 23, 375–378.
Ridge, J. P., Di Rosa, F., and Matzinger, P. (1998). A conditioned dendritic cell can be a temporal bridge between a CD4+ T- helper and a T-killer cell. Nature 393, 474–478.
Rosenfeld, M. R., Chamberlain, M. C., Grossman, S. A., Peereboom, D. M., Lesser, G. J., Batchelor, T. T., Desideri, S., Salazar, A. M., and Ye, X. (2010). A multi-institution phase II study of poly-ICLC, and radiotherapy with concurrent, and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol. 12, 1071–1077.
Russo, V., Cipponi, A., Raccosta, L., Rainelli, C., Fontana, R., Maggioni, D., Lunghi, F., Mukenge, S., Ciceri, F., Bregni, M., Bordignon, C., and Traversari, C. (2007). Lymphocytes genetically modified to express tumor antigens target DCs in vivo and induce antitumor immunity. J. Clin. Invest. 117, 3087–3096.
Schoenberger, S. P., Toes, R. E., Van Der Voort, E. I., Offringa, R., and Melief, C. J. (1998). T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393, 480–483.
Seder, R. A., Darrah, P. A., and Roederer, M. (2008). T-cell quality in memory and protection: implications for vaccine design. Nat. Rev. Immunol. 8, 247–258.
Tewari, K., Flynn, B. J., Boscardin, S. B., Kastenmueller, K., Salazar, A. M., Anderson, C. A., Soundarapandian, V., Ahumada, A., Keler, T., Hoffman, S. L., Nussenzweig, M. C., Steinman, R. M., and Seder, R. A. (2010). Poly(I:C) is an effective adjuvant for antibody and multi-functional CD4+ T cell responses to Plasmodium falciparum circumsporozoite protein (CSP) and alphaDEC-CSP in non human primates. Vaccine 28, 7256–7266.
Valmori, D., Souleimanian, N. E., Tosello, V., Bhardwaj, N., Adams, S., O’Neill, D., Pavlick, A., Escalon, J. B., Cruz, C. M., Angiulli, A., Angiulli, F., Mears, G., Vogel, S. M., Pan, L., Jungbluth, A. A., Hoffmann, E. W., Venhaus, R., Ritter, G., Old, L. J., and Ayyoub, M. (2007). Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc. Natl. Acad. Sci. U.S.A. 104, 8947–8952.
van Heijst, J. W., Gerlach, C., Swart, E., Sie, D., Nunes-Alves, C., Kerkhoven, R. M., Arens, R., Correia-Neves, M., Schepers, K., and Schumacher, T. N. (2009). Recruitment of antigen-specific CD8+ T cells in response to infection is markedly efficient. Science 325, 1265–1269.
Weide, B., Carralot, J. P., Reese, A., Scheel, B., Eigentler, T. K., Hoerr, I., Rammensee, H. G., Garbe, C., and Pascolo, S. (2008). Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J. Immunother. 31, 180–188.
Wilson, N. S., Behrens, G. M., Lundie, R. J., Smith, C. M., Waithman, J., Young, L., Forehan, S. P., Mount, A., Steptoe, R. J., Shortman, K. D., De Koning-Ward, T. F., Belz, G. T., Carbone, F. R., Crabb, B. S., Heath, W. R., and Villadangos, J. A. (2006). Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol. 7, 165–172.
Wolfers, J., Lozier, A., Raposo, G., Regnault, A., Thery, C., Masurier, C., Flament, C., Pouzieux, S., Faure, F., Tursz, T., Angevin, E., Amigorena, S., and Zitvogel, L. (2001). Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 7, 297–303.
Zehn, D., Lee, S. Y., and Bevan, M. J. (2009). Complete but curtailed T-cell response to very low-affinity antigen. Nature 458, 211–214.
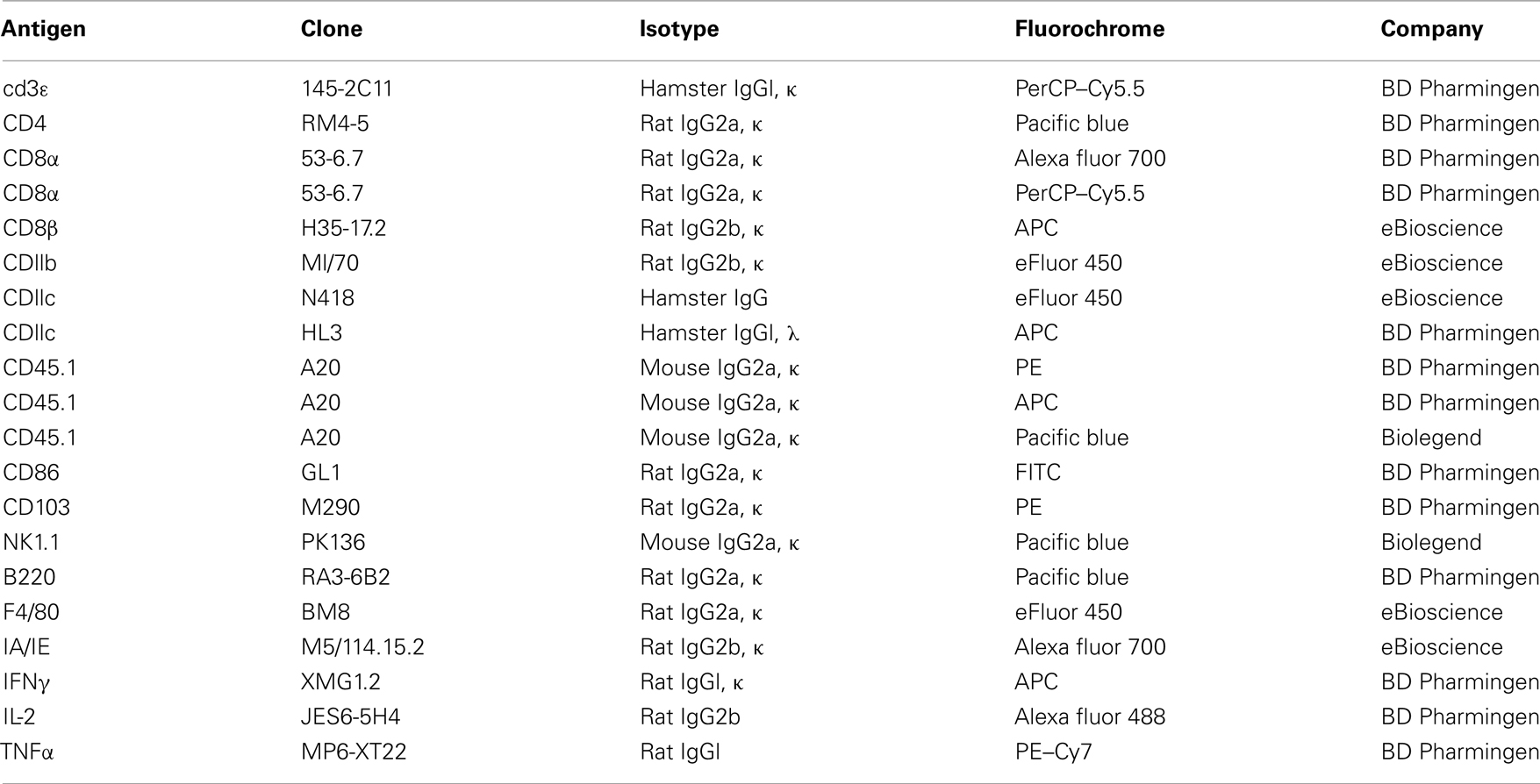
Table A1. Antibodies used for flow cytometry experiments.

Table A2. Sequences of the primers used for the immunoscope analysis.
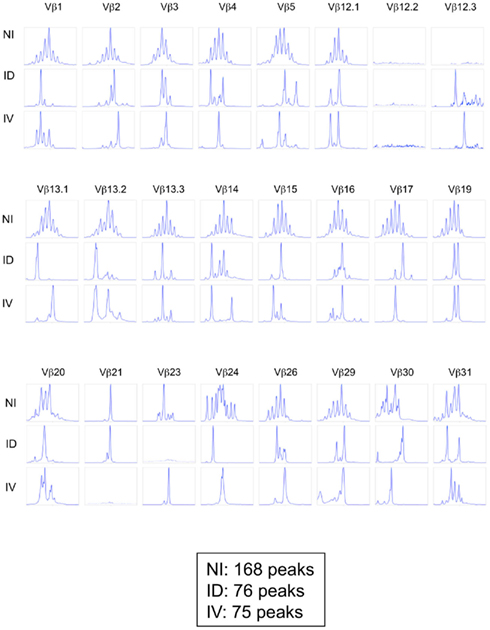
Figure A1. Immunoscope profiles from mice immunized i.d. or i.v. Mice were immunized i.d. or i.v. with 5 × 105 Kbm1mOva splenocytes. On day 9, 15 macroscopic lymph nodes and the spleen were harvested and a Kb–SIINFEKL tetramer-based enrichment was performed for each mouse. CD8 Kb–SIINFEKL tetramer-positive cells were sorted and pooled to obtain 25,000 cells per condition (corresponding to five mice per group). For the non-immunized condition (NI), CD8+ T cells were sorted. An immunoscope was performed to detect the 24 Vβ families (IMGT nomenclature). The immunoscope profile was shown for each Vβ family. The total number of peaks is indicated for each condition. Of note the values indicated in this Figure are higher than those reported in Figure 5B as the former represent pooled mice.
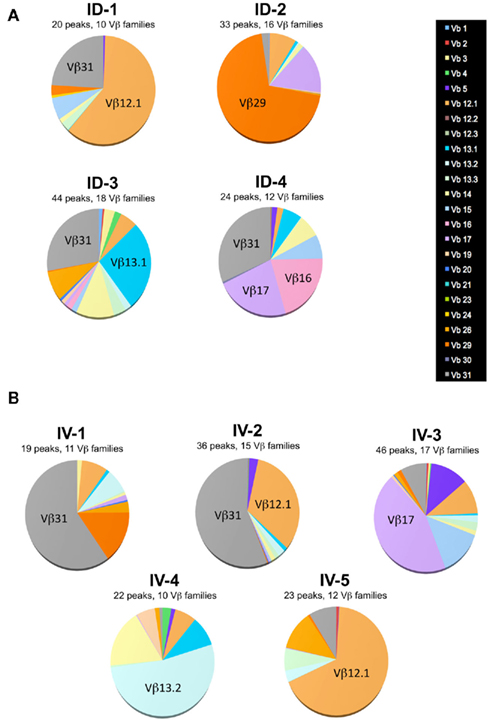
Figure A2. Single mouse analysis confirms that route of immunization does not influence T cell diversity. Mice were immunized i.d. (A) or i.v. (B) with 5 × 105 Kbm1mOva splenocytes. On day 9, 15 macroscopic lymph nodes and the spleen were harvested and a Kb–SIINFEKL tetramer-based enrichment was performed for each mouse. CD8 Kb–SIINFEKL tetramer-positive cells were sorted. Immunoscope was performed on cells sorted from individual mice (3,000–5,000 cells sorted per mouse) to define the length of the CDR3 loop in the 24 Vβ families (IMGT nomenclature). Each color represents a distinct Vβ family.
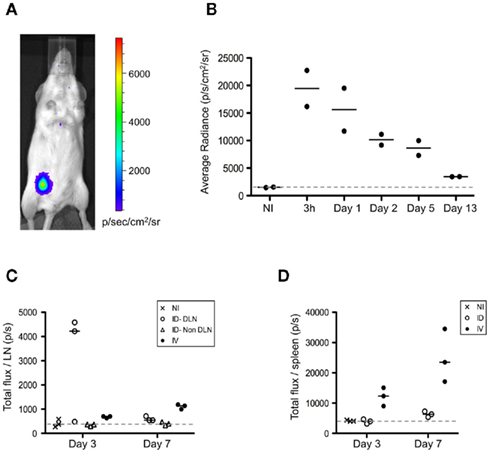
Figure A3. Intradermal injection of splenocytes results in a local depot of donor cells. (A,B) FVB/N female recipients were immunized i.d. or i.v. with 5 × 106 FVB/N-luciferase+ male splenocytes. (A,B) Following immunization, mice injected i.d. were evaluated at 3 h and then on day 1, 2, 5, and 13. Prior to imaging, mice were injected i.p. with 3 mg of D-luciferin (Synchem), followed by isoflurane inhalation to keep animals sedated during analysis. Bioluminescence imaging was performed by using an IVIS Lumina II system (Caliper Life Sciences). Images from mice were acquired over 10 min. Quantification of the light emission was analyzed using Living Image Software version 3.1 (Xenogen Corporation), expressed in photons/s/cm2/steradian. (A) Representative bioluminescence analysis performed at 3 h is shown. (B) Kinetic analysis of the bioluminescent signal is shown. (C,D) On days 3 and 7, the LNs (C) and the spleen (D) from mice immunized i.d. or i.v. were harvested and placed in wells containing PBS and D-luciferin to determine the total bioluminescent signal from each organ. The bioluminescence is expressed as the total flux/organ in photons/s. DLN, draining lymph node.
Keywords: dentritic cells, cross-priming, polyfunctional T cells, adjuvant delivery
Citation: Bouvier I, Jusforgues-Saklani H, Lim A, Lemaître F, Lemercier B, Auriau C, Nicola M-A, Leroy S, Law HK, Bandeira A, Moon JJ, Bousso P and Albert ML (2011) Immunization route dictates cross-priming efficiency and impacts the optimal timing of adjuvant delivery. Front. Immun. 2:71. doi: 10.3389/fimmu.2011.00071
Received: 05 October 2011;
Accepted: 17 November 2011;
Published online: 08 December 2011.
Edited by:
Ken J. Ishii, National Institute of Biomedical Innovation, JapanCopyright: © 2011 Bouvier, Jusforgues-Saklani, Lim, Lemaître, Lemercier, Auriau, Nicola, Leroy, Law, Bandeira, Moon, Bousso and Albert. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Matthew L. Albert, Institut Pasteur, 25, Rue du Dr. Roux, Paris 75724, France. e-mail:YWxiZXJ0bUBwYXN0ZXVyLmZy
†Present address: James J. Moon, Center for Immunology and Inflammatory Diseases, and Pulmonary and Critical Care Unit, Massachusetts General Hospital; and Harvard Medical School, 149 13th Street, Charlestown, MA 02129. USA.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.