 Xiaokuan Hao1†
Xiaokuan Hao1† Ziqi Liu
Ziqi Liu Yanru Wang
Yanru Wang Shihao He
Shihao He Rong Wang
Rong Wang- 1Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
- 2Department of Neurosurgery, Peking University International Hospital, Beijing, China
- 3Department of Neurosurgery, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
- 4China National Clinical Research Center for Neurological Diseases, Beijing, China
Unilateral moyamoya disease (U-MMD) is a chronic vascular disease characterized by progressive stenosis and occlusion of the terminal end of the internal carotid artery and its main branches, resulting in the appearance of moyamoya-like blood vessels at the base of the brain. The etiology of U-MMD is unknown, it accounts for 9.7–17.8% of all moyamoya disease, and the family incidence is 5.5–13.3%. The clinical characteristics are similar to those of typical moyamoya disease, but there are some differences. U-MMD can progress to bilateral moyamoya disease with a median probability of 29.01% (ranging from 6.3 to 58.8%), and there are many risk factors that promote its development. Surgical treatment can effectively reduce the incidence of ischemic stroke and improve prognosis. However, the timing and indications for surgery require further investigation. This article reviews the latest research progress on the etiology, epidemiology, clinical and radiological characteristics, progression, treatment, and prognosis of U-MMD.
Introduction and background
Moyamoya disease (MMD) is a chronic cerebrovascular occlusive disease characterized by the development of stenosis or occlusion at the terminal portion of the internal carotid artery (ICA) and/or initial part of the anterior cerebral artery (ACA) and middle cerebral artery (MCA), accompanied by the formation of abnormally intensive moyamoya-like vessels (Suzuki and Takaku, 1969). In 2012, the Japanese guidelines for the diagnosis and treatment of MMD (On the Pathology, Research Committee, 2012) introduced the concept of unilateral MMD (U-MMD), in which one side exhibits typical MMD-like changes and the opposite side exhibits normal or mild narrowing of the blood vessels (Figure 1). Since 2021, the new guidelines (Kuroda et al., 2022) include U-MMD as a possible manifestation of MMD. U-MMD can progress to typical MMD (Baba et al., 2008), there is a controversy about its development process. Some scholars believe that U-MMD is an independent subtype of MMD (Houkin et al., 1996; Seol et al., 2006), while others believe that U-MMD is an early manifestation of typical MMD (Tian et al., 2022; Kim et al., 2012). This review focuses on recent novel research regarding the etiology, epidemiology, clinical characteristics, radiological characteristics, progression, treatment, and prognosis of U-MMD. The relevant literature over the past 20 years is presented in Table 1.
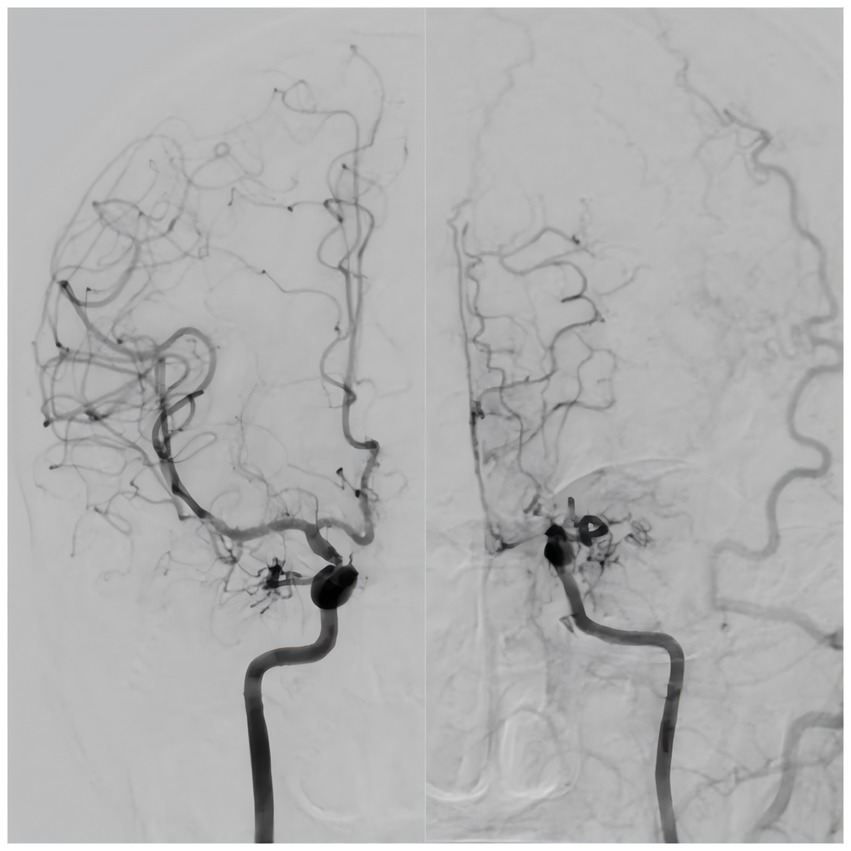
Figure 1. Typical unilateral moyamoya disease.

Table 1. Relevant literature published in the past 20 years.
Epidemiology and clinical characteristics
In recent years, a large number of studies have been conducted on the epidemiology of U-MMD, and the number of cases has increased. However, there is some variation in research data between different countries and ethnic groups.
Kelly et al. (2006) reviewed MMD patients at the Stanford University Medical Center, among whom U-MMD accounted for 17.8%, which is the highest proportion among all studies; however, their study population included almost all races except Black individuals. Yeon et al. (2011) reported that U-MMD accounted for 11.4% of the total number of children and adolescents with MMD in the Korean population. Kim et al. (2012) showed that adults with U-MMD accounted for 14.9% of the total number of Japanese population. Duan et al. (2012) conducted a single-center study with the largest number of cases to date (802), showing that U-MMD accounts for 9.7% of the total number of patients with MMD in the Chinese population. Hayashi et al. (2013) conducted an epidemiological study on MMD across Japan and showed that U-MMD accounted for 10.5% of the total number of patients with MMD in the Japanese population. All of the above studies reported prevalence rates; as can be seen, the proportion of U-MMD in patients with MMD is not high, at approximately 15% on average.
Most studies have reported that the number of female patients with U-MMD is higher than that of male patients, and the female-to-male ratio is approximately 1.5:1 (Ikezaki et al., 1997; Hayashi et al., 2010; Chen et al., 2016), which is consistent with the sex ratio of typical MMD (Scott and Smith, 2009). Some studies have shown no sex differences in U-MMD (Zhang et al., 2016; Smith and Scott, 2008; Wang et al., 2024a; Strunk et al., 2023).
Most studies have shown that the age distribution of patients with U-MMD is bimodal; one group is children around 10 years old, and the other is adults around 40 years old, which is not significantly different from the age distribution of typical MMD (Kelly et al., 2006; Hayashi et al., 2014; Ogata et al., 2008). Some research results show that the other peak of onset is at approximately 50 years of age, which is slightly different (Duan et al., 2012; Wang et al., 2024a). However, a large retrospective study by Zhang et al. (2016) in China showed that age distribution was unimodal, ranging from 30 to 39 years.
Most studies have reported that patients with U-MMD have a family history of the disease. Ikezaki et al. (1997) retrospectively analyzed 180 patients with U-MMD and reported a family incidence rate of 6.7%. Zhang et al. (2016) reported the familial occurrence was 5.5%, whereas in 2024, Wang et al. (2024a) reported it was 13.3%. Most scholars believe that the initial symptoms of U-MMD are similar to those of bilateral MMD, particularly cerebral ischemia (Zhang et al., 2016; Hirotsune et al., 1997; Acker et al., 2015). Transient ischemic attack (39.4%) was the most common symptom at onset (Hayashi et al., 2014). Interestingly, U-MMD demonstrates different characteristics in terms of bleeding patterns compared to bilateral MMD. Yu et al. (2019) reported that subarachnoid hemorrhage was more common in patients with U-MMD whereas intraventricular hemorrhage was more common in patients with bilateral MMD, and the Suzuki staging of acute intracranial hemorrhage was lower in patients with U-MMD than in patients with bilateral MMD. Additionally, the incidence of cerebral hemorrhage was higher in adults with U-MMD (32.8%) than in those with bilateral MMD (27.0%) (Ikezaki et al., 1997).
Etiology
The etiology of U-MMD remains unknown. Previous studies have focused on genetic factors, immune inflammation, and susceptibility genes in U-MMD (Baba et al., 2008; Zhang et al., 2016). With regard to genetics, the question of whether U-MMD is hereditary and whether it represents an early manifestation of MMD or a distinct pathological entity has been controversial. Houkin et al. (1996) reported that U-MMD was not inherited by studying 10 cases of pediatric U-MMD. However, in recent years, an increasing number of studies have shown that it has a certain familial inheritance. Kusaka et al. (2006) reported a case of U-MMD with typical MMD occurring in the same family, suggesting that they are different phenotypes of the same gene. In the same year, Mineharu et al. (2006) conducted chromosomal analyses of 6 patients with a family history of U-MMD and demonstrated that familial U-MMD was inherited in an incomplete autosomal dominant pattern. The above study showed that the heritability of U-MMD was 5.5–10%, which was lower than the 15% heritability of typical bilateral MMD (Scott and Smith, 2009), suggesting that there may be hitherto unknown inheritance patterns. In the area of metabolism and immunology, Houkin et al. (1996) studied basic fibroblast growth factor (bFGF) in cerebrospinal fluid of 10 children with U-MMD, and found that a decrease in bFGF was closely related to U-MMD. However, Jeon et al. (2015) performed a metabolomic analysis of cerebrospinal fluid in adults with U-MMD using a larger sample size; however, the results showed that there was no difference in cerebrospinal fluid metabolite levels between patients with U-MMD and those with bilateral MMD. Hence, future studies on cerebrospinal fluid metabolites of U-MMD are warranted.
Immune disorders may also be a cause of U-MMD. Chen et al. (2015) followed up 25 MMD patients with Graves’ disease and found more frequent progression to U-MMD during the 106.4 ± 48.6 month follow-up period. Soon after, Chen et al. (2016) retrospectively analyzed the clinical characteristics of 316 patients with bilateral MMD and 68 patients with U-MMD and found that the overall prevalence of autoimmune diseases in patients with U-MMD was higher than that in patients with bilateral MMD. Because U-MMD shows a higher association with autoimmune diseases than bilateral MMD, different pathogenic mechanisms may be involved in the formation of diseased blood vessels. To date, no mechanistic studies have examined how immune factors contribute to the pathological angiogenesis that occurs in U-MMD; however, advances in gene sequencing technology has allowed the identification many susceptibility genes, which has helped to advance the study of the disease mechanisms. The RNF213 (ring finger protein 213) gene is currently the most studied susceptibility gene in U-MMD. Ok et al. (2022) performed sequencing analysis on 123 patients with U-MMD and detected an RNF213 p.R4810K gene mutation in 72 patients, all of which were heterozygous. The allele frequency of RNF213 p.R4810K was significantly higher in the U-MMD group than in the control group. Mineharu et al. (2024) analyzed a cohort of 93 patients with U-MMD and found that posterior cerebral artery (PCA) involvement in U-MMD was exclusively ipsilateral and was influenced by the RNF213 mutation gene dosage. In the same year, Suo et al. (2024) identified candidate genes by performing whole-genome sequencing of a family with U-MMD, and their results suggested that the FOXM1 c.1205 C > A mutation potentially contributes to U-MMD pathogenesis. In the most recent study in 2024, Wang et al. (2024a) followed up 83 cases of U-MMD for 7.9 ± 2.0 years and found that heterozygous RNF213 p.R4810K mutations occurred significantly more frequently in U-MMD patients with contralateral progression. In conclusion, these susceptibility genes have an impact both in the pathogenesis and progression of U-MMD, and future studies will focus on the downstream pathways and cellular function of the genes.
Progression
It is important to study the progression of U-MMD by identifying the risk factors for progression so that personalized follow-up and treatment can be provided before a stroke occurs. So far, there have been 18 studies on the development of U-MMD into bilateral MMD [1 from the USA, 1 from Germany, 3 from China, 6 from Japan, and 7 from South Korea (Houkin et al., 1996; Seol et al., 2006; Tian et al., 2022; Kim et al., 2012; Kelly et al., 2006; Yeon et al., 2011; Hayashi et al., 2010; Zhang et al., 2016; Wang et al., 2024a; Strunk et al., 2023; Hirotsune et al., 1997; Ok et al., 2022; Kawano et al., 1994; Kuroda et al., 2005; Joo et al., 2011; Matsushima et al., 1994; Park et al., 2011; Lee et al., 2020)], all of which focused on vascular progression evident on imaging rather than progression in clinical symptoms. Three studies focused only on adult patients, six studies focused solely on pediatric patients, and the rest were not age-specific. However, only 4 studies on U-MMD had a sample size >100. The median rate of progression in each study was 29.01% (range, 6.3–58.8%), and the average progression time varied from 1 to 78 months (Tian et al., 2022). Some scholars (Kim et al., 2012; Duan et al., 2012) believe that among the risk factors for predicting progression, patients with young age, slight stenosis of the contralateral vessels, and familial factors are more likely to have contralateral progression. Smith and Scott (2008) reported that contralateral vascular abnormalities, congenital heart malformations, previous brain irradiation, Asian lineage, and familial MMD syndrome in patients with U-MMD increased the risk of disease progression.
Previous studies have shown that U-MMD in pediatric patients is more likely to progress to the contralateral side than U-MMD in adult patients. Smith and Scott (2008) retrospectively analyzed 33 patients with U-MMD, including 4 adults and 29 children. The results showed that children younger than 7 years were prone to rapid progression, with an average time of 10.8 months, whereas patients older than 7 years showed slow progression, with an average of 37.2 months. Park et al. (2011) studied 34 pediatric patients and concluded that U-MMD progression was related to age. Patients younger than 8 years easily experienced rapid progression, with an average time of 14.18 months. Patients older than 8 years showed slower progression, with an average time of 22.38 months. The rates of progression in the two studies described above were 30 and 58.8% respectively; these ratios seem to be quite high, which may be because the study included children with mild contralateral vascular stenosis, thus creating mixed factors in the target population. Yeon et al. (2011) enrolled 45 patients with typical U-MMD from a cohort of 391 children using strict inclusion criteria and excluded children with mild contralateral vascular stenosis. The results showed that the progression rate was only 17.8% during the 53.4 month follow-up period. They reported that among the 45 children with U-MMD, those younger than 9 years were prone to progression, while those older than 9 years had slower progression. Compared with Smith, the results showed that there was no significant difference in the incidence of contralateral progression between patients younger than 7 years and those younger than 9 years, and contralateral progression is likely to occur within 1.8 to 3.1 years after initial diagnosis, regardless of age.
As the understanding of U-MMD continues to grow, and the sample sizes of studies increase, the evidence becomes more compelling. The first large study was a retrospective review of 109 cases by Zhang et al. (2016), which excluded children with mild contralateral ACA, MCA, or ICA stenoses. The results showed that the incidence of contralateral progression in all cases was only 16.5% over a mean follow-up of 43.8 ± 21.3 months, which was lower than previous studies. To date, the largest sample size was a study of 217 cases of U-MMD performed by Church et al. (2020), with an average follow-up time of 5.8 years (range 1–22 years). The results showed that only 8% of patients experienced contralateral progression. Tian et al. (2022) followed 89 adults with U-MMD for 63 months and found that only 9% experienced contralateral progression. Their study also excluded patients with mild contralateral ACA, MCA, or ICA stenoses. With the improvement of scientific inclusion criteria, it has been proven that the progression of U-MMD in 5 years is not as high as previously reported. It is worth mentioning that in 2024, Wang et al. reported 19/83 (22.9%) of patients with U-MMD in the study experienced contralateral progression during a mean follow-up duration of 7.9 ± 2.0 years (range 2.0–13.9 years). This result is also reasonable because the follow-up period is the longest thus far, and the progression rate is expected to increase over time.
Several studies have reported various risk factors for contralateral progression. Younger age was confirmed as a predictive factor for contralateral progression. From the above discussion, we can see that for pediatric U-MMD patients aged less than 9 years, it is necessary to conduct more frequent clinical follow-up and timely disease management. Contralateral stenosis of the ACA, MCA, or ICA has been identified as a risk factor for contralateral progression (Zhang et al., 2016; Smith and Scott, 2008), whereas Yeon et al. (2011) reported no association between stenosis of the contralateral A1 segment and contralateral progression.
Mineharu et al. (2024) reported that PCA involvement in U-MMD is exclusively ipsilateral and influenced by RNF213 mutation gene dosage. Wang et al. (2024a) reported that RNF213 p.R4810K mutations and PCA involvement were predictors of contralateral progression. Previous studies have reported that patients with the RNF213 p.R4810K mutation (Wang et al., 2020) or PCA involvement (Mugikura et al., 2002; Hishikawa et al., 2013) in classic MMD exhibit more severe disease and more extensive vascular involvement, which can reasonably explain the results of Mineharu and Wang’s study on susceptibility to contralateral progression. Strunk et al. (2023) also reported that PCA involvement and dizziness are significantly more frequent in patients with progressive MMA. In addition, Church et al. (2020) found that hyperlipidemia was a predictor of contralateral progression, and Strunk et al. (2023) and Mineharu et al. (2024) confirmed this phenomenon; they believed that elevated lipids have a deleterious effect on the underlying moyamoya vasculopathy.
Few studies have examined the mechanisms of how U-MMD develops; only Lee et al. (2020) reported a mechanism by which endothelial shear stress causes contralateral progression of U-MMD. Endothelial shear stress parameters were mean and maximum signal intensity gradients at the vessel boundary in time-of-flight sequences from brain magnetic resonance imaging; they believed that the sharp bends and accelerated flow rate at the terminal end of the ICA lead to high spatial variability of endothelial shear stress. Subsequently, this upregulates transcription factors and modulates endothelial gene expression.
Radiologic characteristics
Ogata et al. (2008) studied the pattern of collateral circulation in patients with U-MMD and found that there was no collateral circulation via the ocular artery in patients with U-MMD, but there was increased collateral circulation from the contralateral normal vessels. Other sources of collateral circulation were not significantly different from those in patients with typical MMD. In 2014, a retrospective study of 203 U-MMD cases in Japan showed that U-MMD patients had lower Suzuki stages (Hayashi et al., 2014). In 2016, a study of 109 patients with U-MMD by Acker et al. (2015) showed that 70% of patients with U-MMD had a lower Suzuki stage, which was significantly higher than that of patients with typical MMD (50%). PCA involvement occurs in approximately 24.5–48.6% of patients with typical MMD and is often bilateral (Wang et al., 2020; Miyatake et al., 2012). However, in 2024, a study of 93 cases of U-MMD by Mineharu et al. (2024) reported that only 11.8% of patients with U-MMD manifested PCA involvement, which was significantly lower than that in patients with typical MMD, and that PCA involvement in U-MMD was exclusively ipsilateral.
Although only one hemisphere is involved in U-MMD, there is also a decrease in regional cerebral blood flow (rCBF). Patients with U-MMD have unique perfusion characteristics, as evidenced by reduced local rCBF in the frontotemporal lobe and elevated rCBF in the occipital lobe. The rCBF of U-MMD patients after acetazolamide stimulation was significantly higher than that of typical MMD patients, and their ability to respond to acetazolamide was also higher (Ogawa et al., 1987). A previous study showed that smoky vessels gradually developed with a decrease in rCBF, which also explains the lower Suzuki grade of patients with U-MMD (Ogawa et al., 1990). These results are consistent with those of previous studies (Mugikura et al., 2002), showing that the frequency of PCA involvement is higher in the hemisphere with an advanced Suzuki stage. These observations seem to support a previous hypothesis that MMD belongs to a clinical entity of neurocristopathy, which suggest that neural crest cell migration predominantly occurs ipsilaterally (Tucker and Lumsden, 2004).
Treatment and prognosis
Surgery can improve cerebral perfusion, reduce the incidence of cerebral ischemia, and improve the prognosis of U-MMD (Fung et al., 2005). Zhang et al. (2016) reported that among 109 patients with U-MMD, most had a good functional prognosis after surgery, and approximately 91.7% had a modified Rankin Scale score of 0–2 during follow-up, while the percentage of overall MMD patients who could achieve this score was 84.6%. Zhang et al. (2022) studied the hemodynamic characteristics of 45 patients with U-MMD after bypass grafting, and the results showed that the cerebral blood flow, cerebral blood volume, mean transit time, and time to peak were significantly improved; furthermore, the neurological function of all patients was good. Wang et al. (2024a) reported that of 19/83 (22.9%) patients with U-MMD achieved significant neurological improvements at Matsushima stages A and B after surgery. In conclusion, for U-MMD, surgical plans should be actively evaluated to reduce the risk of stroke. In addition, given its unique PCA involvement pattern, increased attention should be given to follow-up of PCA involvement development (Bao et al., 2023), and ipsilateral posterior circulation revascularization should also be considered if necessary (Wang et al., 2024).
Whether the contralateral normal hemisphere in patients with U-MMD requires surgery remains unclear. An early study by Matsushima et al. (1988) recommended that patients with U-MMD younger than 2 years should undergo bilateral reconstruction because the incidence of normal-side progression is high. The asymptomatic side in patients with U-MMD should be treated surgically, particularly in children. However, some studies (Nagata et al., 2006) have indicated that surgical treatment should be postponed until symptoms appear on the asymptomatic side. As discussed previously, an in-depth study of the natural history of U-MMD has shown that the probability of contralateral progression is low; therefore, an increasing number of scholars believe that prophylactic contralateral revascularization is not warranted in U-MMD cases (Tian et al., 2022; Zhang et al., 2016). Instead, careful and long-term follow-up is more conducive for the early detection of contralateral progression, thereby determining the surgical strategy in a timely manner, especially for patients with risk factors. It is worth mentioning that U-MMD should be strictly distinguished from atherosclerotic disease in future studies (Psychogios et al., 2022), as they differ in clinical features and surgical prognosis (Wang et al., 2024b).
Prospects and challenges
With the increase in U-MMD research and the development of imaging technology, the incidence of U-MMD has increased annually, so it is receiving increasing attention. However, the studies are still not sufficiently thorough, and many problems have not been solved. First, does U-MMD demonstrate a unique pattern in terms of epidemiology, such as clinical symptoms and heritability, compared to typical bilateral MMD? What factors contribute to this development? The reason for the variable results across studies is that there are no prospective cohort studies on the natural course of U-MMD and only a few large retrospective studies. Therefore, the next step is to conduct large multicenter prospective clinical trials. In addition, the high number of risk factors for U-MMD progression to bilateral MMD and the lack of evidence for their use in clinical diagnosis and treatment requires more evidence-based medical studies to support the indications and methods of surgical treatment. Finally, there remains a gap in the study of U-MMD vascular progression at the cellular and organ levels, and further basic proteomic and organ-focused studies are urgently required.
Author contributions
XH: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization. CT: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Investigation, Methodology. ZL: Investigation, Methodology, Writing – review & editing. YT: Investigation, Writing – review & editing, Methodology. YW: Investigation, Writing – review & editing, Methodology. SH: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Investigation, Methodology, Supervision, Validation. RD: Writing – original draft, Writing – review & editing, Investigation, Methodology, Supervision, Validation. RW: Writing – original draft, Writing – review & editing, Investigation, Methodology, Supervision, Validation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the National Natural Science Foundation of China (no. 82171887 to RW).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACA, anterior cerebral artery; bFGF, basic fibroblast growth factor; ICA, internal carotid artery; MCA, middle cerebral artery; MMD, moyamoya disease; PCA, posterior cerebral artery; rCBF, regional cerebral blood flow; RNF213, ring finger protein 213; TIA, transient ischemic attack; U-MMD, unilateral moyamoya disease.
References
Acker, G., Goerdes, S., Schneider, U. C., Schmiedek, P., Czabanka, M., and Vajkoczy, P. (2015). Distinct clinical and radiographic characteristics of moyamoya disease amongst European Caucasians. Eur. J. Neurol. 22, 1012–1017. doi: 10.1111/ene.12702
Baba, T., Houkin, K., and Kuroda, S. (2008). Novel epidemiological features of moyamoya disease. J. Neurol. Neurosurg. Psychiatry 79, 900–904. doi: 10.1136/jnnp.2007.130666
Bao, X. Y., Tong, H. Y., Wang, Q. N., Wang, X. P., Gao, G., Zhang, Q., et al. (2023). A long-term study of posterior circulation changes after revascularization in patients with moyamoya disease. J. Neurosurg. 139, 1281–1286. doi: 10.3171/2023.2.JNS222649
Chen, J. B., Lei, D., He, M., Sun, H., Liu, Y., Zhang, H., et al. (2015). Clinical features and disease progression in moyamoya disease patients with graves disease. J. Neurosurg. 123, 848–855. doi: 10.3171/2014.10.JNS141140
Chen, J. B., Liu, Y., Zhou, L. X., Sun, H., He, M., and You, C. (2016). Increased prevalence of autoimmune disease in patients with unilateral compared with bilateral moyamoya disease. J. Neurosurg. 124, 1215–1220. doi: 10.3171/2015.4.JNS142936
Church, E. W., Bell-Stephens, T. E., Bigder, M. G., Gummidipundi, S., Han, S. S., and Steinberg, G. K. (2020). Clinical course of unilateral Moyamoya disease. Neurosurgery 87, 1262–1268. doi: 10.1093/neuros/nyaa284
Duan, L., Bao, X. Y., Yang, W. Z., Shi, W. C., Li, D. S., Zhang, Z. S., et al. (2012). Moyamoya disease in China: its clinical features and outcomes. Stroke 43, 56–60. doi: 10.1161/STROKEAHA.111.621300
Fung, L. W., Thompson, D., and Ganesan, V. (2005). Revascularisation surgery for paediatric moyamoya: a review of the literature. Childs Nerv. Syst. 21, 358–364. doi: 10.1007/s00381-004-1118-9
Hayashi, K., Horie, N., Izumo, T., and Nagata, I. (2014). A nationwide survey on unilateral moyamoya disease in Japan. Clin. Neurol. Neurosurg. 124, 1–5. doi: 10.1016/j.clineuro.2014.06.010
Hayashi, K., Horie, N., Suyama, K., and Nagata, I. (2013). An epidemiological survey of moyamoya disease, unilateral moyamoya disease and quasi-moyamoya disease in Japan. Clin. Neurol. Neurosurg. 115, 930–933. doi: 10.1016/j.clineuro.2012.09.020
Hayashi, K., Suyama, K., and Nagata, I. (2010). Clinical features of unilateral moyamoya disease. Neurol. Med. Chir. (Tokyo) 50, 378–385. doi: 10.2176/nmc.50.378
Hirotsune, N., Meguro, T., Kawada, S., Nakashima, H., and Ohmoto, T. (1997). Long-term follow-up study of patients with unilateral moyamoya disease. Clin. Neurol. Neurosurg. 99, S178–S181. doi: 10.1016/S0303-8467(97)00043-7
Hishikawa, T., Tokunaga, K., Sugiu, K., and Date, I. (2013). Assessment of the difference in posterior circulation involvement between pediatric and adult patients with moyamoya disease. J. Neurosurg. 119, 961–965. doi: 10.3171/2013.6.JNS122099
Houkin, K., Abe, H., Yoshimoto, T., and Takahashi, A. (1996). Is "unilateral" moyamoya disease different from moyamoya disease? J. Neurosurg. 85, 772–776. doi: 10.3171/jns.1996.85.5.0772
Ikezaki, K., Inamura, T., Kawano, T., and Fukui, M. (1997). Clinical features of probable moyamoya disease in Japan. Clin. Neurol. Neurosurg. 99, S173–S177. doi: 10.1016/S0303-8467(97)00053-X
Jeon, J. P., Yun, T., Jin, X., Cho, W. S., Son, Y. J., Bang, J. S., et al. (2015). 1H-NMR-based metabolomic analysis of cerebrospinal fluid from adult bilateral moyamoya disease: comparison with unilateral moyamoya disease and atherosclerotic stenosis. Medicine (Baltimore) 94:e629. doi: 10.1097/MD.0000000000000629
Joo, S. P., Kim, T. S., Lee, I. K., Kim, J. T., Park, M. S., and Cho, K. H. (2011). A genome-wide study of moyamoya-type cerebrovascular disease in the korean population. J. Korean Neurosurg. Soc. 50, 486–491. doi: 10.3340/jkns.2011.50.6.486
Kawano, T., Fukui, M., Hashimoto, N., and Yonekawa, Y. (1994). Follow-up study of patients with "unilateral" moyamoya disease. Neurol. Med. Chir. (Tokyo) 34, 744–747. doi: 10.2176/nmc.34.744
Kelly, M. E., Bell-Stephens, T. E., Marks, M. P., do, H. M., and Steinberg, G. K. (2006). Progression of unilateral moyamoya disease: a clinical series. Cerebrovasc. Dis. 22, 109–115. doi: 10.1159/000093238
Kim, J., Kim, K., Kim, J., Kang, H. S., Bang, J., Son, Y. J., et al. (2012). Clinical features of adult moyamoya disease with special reference to the diagnosis. Neurol. Med. Chir. (Tokyo) 52, 311–317. doi: 10.2176/nmc.52.311
Kuroda, S., Fujimura, M., Takahashi, J., Kataoka, H., Ogasawara, K., Iwama, T., et al. (2022). Diagnostic criteria for Moyamoya disease—2021 revised version. Neurol. Med. Chir. (Tokyo) 62, 307–312. doi: 10.2176/jns-nmc.2022-0072
Kuroda, S., Ishikawa, T., Houkin, K., Nanba, R., Hokari, M., and Iwasaki, Y. (2005). Incidence and clinical features of disease progression in adult moyamoya disease. Stroke 36, 2148–2153. doi: 10.1161/01.STR.0000182256.32489.99
Kusaka, N., Tamiya, T., Adachi, Y., Katayama, S., Namba, S., Tokunaga, K., et al. (2006). Adult unilateral moyamoya disease with familial occurrence in two definite cases: a case report and review of the literature. Neurosurg. Rev. 29, 82–87. doi: 10.1007/s10143-005-0406-5
Lee, W. J., Jeong, S. K., Han, K. S., Lee, S. H., Ryu, Y. J., Sohn, C. H., et al. (2020). Impact of endothelial shear stress on the bilateral progression of unilateral Moyamoya disease. Stroke 51, 775–783. doi: 10.1161/STROKEAHA.119.028117
Matsushima, T., Inoue, T., Natori, Y., Fujii, K., Fukui, M., Hasuo, K., et al. (1994). Children with unilateral occlusion or stenosis of the ICA associated with surrounding Moyamoya vessels — “unilateral” Moyamoya disease. Acta Neurochir. 131, 196–202. doi: 10.1007/BF01808612
Matsushima, T., Take, S., Fujii, K., Fukui, M., Hasuo, K., Kuwabara, Y., et al. (1988). A case of moyamoya disease with progressive involvement from unilateral to bilateral. Surg. Neurol. 30, 471–475. doi: 10.1016/0090-3019(88)90034-1
Mineharu, Y., Takagi, Y., Koizumi, A., Morimoto, T., Funaki, T., Hishikawa, T., et al. (2024). Posterior cerebral artery involvement in unilateral moyamoya disease is exclusively ipsilateral and influenced by RNF213 mutation gene dose: the SUPRA Japan study: PCA involvement in unilateral moyamoya. J. Stroke Cerebrovasc. Dis. 33:107513. doi: 10.1016/j.jstrokecerebrovasdis.2023.107513
Mineharu, Y., Takenaka, K., Yamakawa, H., Inoue, K., Ikeda, H., Kikuta, K. I., et al. (2006). Inheritance pattern of familial moyamoya disease: autosomal dominant mode and genomic imprinting. J. Neurol. Neurosurg. Psychiatry 77, 1025–1029. doi: 10.1136/jnnp.2006.096040
Miyatake, S., Miyake, N., Touho, H., Nishimura-Tadaki, A., Kondo, Y., Okada, I., et al. (2012). Homozygous c.14576G>a variant of RNF213 predicts early-onset and severe form of moyamoya disease. Neurology 78, 803–810. doi: 10.1212/WNL.0b013e318249f71f
Mugikura, S., Takahashi, S., Higano, S., Shirane, R., Sakurai, Y., and Yamada, S. (2002). Predominant involvement of ipsilateral anterior and posterior circulations in moyamoya disease. Stroke 33, 1497–1500. doi: 10.1161/01.STR.0000016828.62708.21
Nagata, S., Matsushima, T., Morioka, T., Matsukado, K., Mihara, F., Sasaki, T., et al. (2006). Unilaterally symptomatic moyamoya disease in children: long-term follow-up of 20 patients. Neurosurgery 59, 830–837. doi: 10.1227/01.NEU.0000227527.69766.43
Ogata, T., Yasaka, M., Inoue, T., Yasumori, K., Ibayashi, S., Iida, M., et al. (2008). The clinical features of adult unilateral moyamoya disease: does it have the same clinical characteristics as typical moyamoya disease? Cerebrovasc. Dis. 26, 244–249. doi: 10.1159/000147451
Ogawa, A., Nakamura, N., Sakurai, Y., Kayama, T., Wada, T., and Suzuki, J. (1987). Cerebral blood flow in moyamoya disease. No To Shinkei 39, 199–203
Ogawa, A., Yoshimoto, T., Suzuki, J., and Sakurai, Y. (1990). Cerebral blood flow in moyamoya disease. Part 1: correlation with age and regional distribution. Acta Neurochir. 105, 30–34. doi: 10.1007/BF01664854
Ok, T., Jung, Y. H., Kim, J., Park, S. K., Park, G., Lee, S., et al. (2022). RNF213 R4810K variant in suspected unilateral Moyamoya disease predicts contralateral progression. J. Am. Heart Assoc. 11:e025676. doi: 10.1161/JAHA.122.025676
On the Pathology, Research Committee (2012). Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol. Med. Chir. (Tokyo) 52, 245–266. doi: 10.2176/nmc.52.245
Park, E. K., Lee, Y. H., Shim, K. W., Choi, J. U., and Kim, D. S. (2011). Natural history and progression factors of unilateral moyamoya disease in pediatric patients. Childs Nerv. Syst. 27, 1281–1287. doi: 10.1007/s00381-011-1469-y
Psychogios, M., Brehm, A., López-Cancio, E., Marco de Marchis, G., Meseguer, E., Katsanos, A. H., et al. (2022). European stroke organisation guidelines on treatment of patients with intracranial atherosclerotic disease. Eur. Stroke J. 7:XLII. doi: 10.1177/23969873221099715
Scott, R. M., and Smith, E. R. (2009). Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 360, 1226–1237. doi: 10.1056/NEJMra0804622
Seol, H. J., Wang, K. C., Kim, S. K., Lee, C. S., Lee, D. S., Kim, I. O., et al. (2006). Unilateral (probable) moyamoya disease: long-term follow-up of seven cases. Childs Nerv. Syst. 22, 145–150. doi: 10.1007/s00381-005-1234-1
Smith, E. R., and Scott, R. M. (2008). Progression of disease in unilateral moyamoya syndrome. Neurosurg. Focus. 24:E17. doi: 10.3171/FOC/2008/24/2/E17
Strunk, D., Diehl, R. R., Veltkamp, R., Meuth, S. G., and Kraemer, M. (2023). Progression of initially unilateral Moyamoya angiopathy in Caucasian Europeans. J. Neurol. 270, 4415–4422. doi: 10.1007/s00415-023-11793-0
Suo, S., Fang, C., Liu, W., Liu, Q., Zhang, Z., Chang, J., et al. (2024). FOXM1 c.1205 C > a mutation is associated with unilateral Moyamoya disease and inhibits angiogenesis in human brain endothelial cells. Hum. Genet. 143, 939–953. doi: 10.1007/s00439-024-02685-y
Suzuki, J., and Takaku, A. (1969). Cerebrovascular "moyamoya" disease. Disease showing abnormal net-like vessels in base of brain. Arch. Neurol. 20, 288–299. doi: 10.1001/archneur.1969.00480090076012
Tian, X., Hu, M., and Zhang, J. (2022). The contralateral progression in a cohort of Chinese adult patients with unilateral moyamoya disease after revascularization: a single-center long-term retrospective study. Acta Neurochir. 164, 1837–1844. doi: 10.1007/s00701-022-05153-6
Tucker, A. S., and Lumsden, A. (2004). Neural crest cells provide species-specific patterning information in the developing branchial skeleton. Evol. Dev. 6, 32–40. doi: 10.1111/j.1525-142X.2004.04004.x
Wang, H., Hao, F., Feng, J., Zhang, Q., Zhang, Z., Li, B., et al. (2024). Clinical course, therapy, and long-term outcomes of children with Moyamoya disease and posterior cerebral artery involvement. Neurology 103:e209658. doi: 10.1212/WNL.0000000000209658
Wang, X. P., Ren, B., Wang, Q. N., Li, J. J., Liu, J. Q., Yu, D., et al. (2024b). Encephaloduroarteriosynangiosis for symptomatic intracranial atherosclerotic arterial Steno-occlusive disease: clinical and radiological outcomes. J. Am. Heart Assoc. 13:e034707. doi: 10.1161/JAHA.124.034707
Wang, Y., Zhang, Z., Wei, L., Zhang, Q., Zou, Z., Yang, L., et al. (2020). Predictive role of heterozygous p.R4810K of RNF213 in the phenotype of Chinese moyamoya disease. Neurology 94, e678–e686. doi: 10.1212/WNL.0000000000008901
Wang, X. P., Zou, Z. X., Bao, X. Y., Wang, Q. N., Ren, B., Yu, D., et al. (2024a). Clinical and genetic factors associated with contralateral progression in unilateral moyamoya disease: longitudinal and cross-sectional study. Heliyon 10:e26108. doi: 10.1016/j.heliyon.2024.e26108
Yeon, J. Y., Shin, H. J., Kong, D. S., Seol, H. J., Kim, J. S., Hong, S. C., et al. (2011). The prediction of contralateral progression in children and adolescents with unilateral moyamoya disease. Stroke 42, 2973–2976. doi: 10.1161/STROKEAHA.111.622522
Yu, Z., Zheng, J., Guo, R., Li, H., You, C., and Ma, L. (2019). Patterns of acute intracranial hemorrhage in adult patients with bilateral and unilateral Moyamoya disease. Curr. Neurovasc. Res. 16, 202–207. doi: 10.2174/1567202616666190621093652
Zhang, Q., Wang, R., Liu, Y., Zhang, Y., Wang, S., Cao, Y., et al. (2016). Clinical features and long-term outcomes of unilateral Moyamoya disease. World Neurosurg. 96, 474–482. doi: 10.1016/j.wneu.2016.09.018
Keywords: unilateral moyamoya disease, etiology, epidemiology, clinical course, progression, surgery
Citation: Hao X, Tan C, Liu Z, Tie Y, Wang Y, He S, Duan R and Wang R (2025) Research progress in unilateral moyamoya disease. Front. Hum. Neurosci. 19:1503639. doi: 10.3389/fnhum.2025.1503639
Edited by:
Mario Teo, University of Bristol, United KingdomReviewed by:
Jose Eduardo Leon-Rojas, University of the Americas, EcuadorXiao-Peng Wang, Chinese PLA General Hospital, China
Copyright © 2025 Hao, Tan, Liu, Tie, Wang, He, Duan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shihao He, aGVzaGloYW9vQG91dGxvb2suY29t; Ran Duan, ZHVhbnJhbnBrdWloQDEyNi5jb20=; Rong Wang, cm9uZ2VyMDkwNjE0QDEyNi5jb20=
†These authors share first authorship