
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Hum. Neurosci. , 18 March 2024
Sec. Brain Health and Clinical Neuroscience
Volume 18 - 2024 | https://doi.org/10.3389/fnhum.2024.1352118
This article is part of the Research Topic Cognition, Neurodegeneration and Immunity: from Observational Data to Molecular Mechanisms View all 5 articles
 George D. Vavougios1*
George D. Vavougios1* Vasilis-Spyridon Tseriotis2
Vasilis-Spyridon Tseriotis2 Andreas Liampas1
Andreas Liampas1 Theodore Mavridis3
Theodore Mavridis3 Gabriel A. de Erausquin4†
Gabriel A. de Erausquin4† Georgios Hadjigeorgiou1†
Georgios Hadjigeorgiou1†COVID-19’s effects on the human brain reveal a multifactorial impact on cognition and the potential to inflict lasting neuronal damage. Type I interferon signaling, a pathway that represents our defense against pathogens, is primarily affected by COVID-19. Type I interferon signaling, however, is known to mediate cognitive dysfunction upon its dysregulation following synaptopathy, microgliosis and neuronal damage. In previous studies, we proposed a model of outside-in dysregulation of tonic IFN-I signaling in the brain following a COVID-19. This disruption would be mediated by the crosstalk between central and peripheral immunity, and could potentially establish feed-forward IFN-I dysregulation leading to neuroinflammation and potentially, neurodegeneration. We proposed that for the CNS, the second-order mediators would be intrinsic disease-associated molecular patterns (DAMPs) such as proteopathic seeds, without the requirement of neuroinvasion to sustain inflammation. Selective vulnerability of neurogenesis sites to IFN-I dysregulation would then lead to clinical manifestations such as anosmia and cognitive impairment. Since the inception of our model at the beginning of the pandemic, a growing body of studies has provided further evidence for the effects of SARS-CoV-2 infection on the human CNS and cognition. Several preclinical and clinical studies have displayed IFN-I dysregulation and tauopathy in gene expression and neuropathological data in new cases, correspondingly. Furthermore, neurodegeneration identified with a predilection for the extended olfactory network furthermore supports the neuroanatomical concept of our model, and its independence from fulminant neuroinvasion and encephalitis as a cause of CNS damage. In this perspective, we summarize the data on IFN-I as a plausible mechanism of cognitive impairment in this setting, and its potential contribution to Alzheimer’s disease and its interplay with COVID-19.
Our current understanding of COVID-19 is that long term consequences may arise following the acute phase, even in milder cases. Collectively, these syndromes and symptoms exist within what is defined as the long COVID or post-acute COVID-19 condition (PASC) spectrum (Parotto et al., 2023). Of note, neuropsychiatric manifestations and specifically cognitive decline are consistently reported in a subset of long COVID patients (Deer et al., 2021). In 2022, a joint international task force by WHO and Alzheimer’s Association reported on a protocol to systematically study the neuropsychiatric sequalae of COVID-19, and provided a comprehensive review of the evidence linking these manifestations with Alzheimer’s disease and related dementias (ADRD). In that protocol, we specifically proposed that SARS-CoV-2 would trigger ADRD-like pathology following the extended olfactory cortical network (EOCN) in older individuals with underlying genetic susceptibility (de Erausquin et al., 2021, 2022). This hypothesis emerged from the descriptions of anosmia as and early and often single symptom of SARS-CoV-2 infection, and the well-known association of the same symptom with cognitive impairment in Alzheimer’s disease (Roberts et al., 2016; Tian et al., 2022; Xiao et al., 2023).
Persistent hyposmia has been correlated with cognitive impairment in those previously exposed to COVID-19, with functional MRI (fMRI) revealing altered olfactory network connectivity in the absence of structural damage (Muccioli et al., 2023). In FDG PET studies of post-acute COVID-19 patients, hypometabolism involving areas involved in olfactory and limbic pathways has also been associated with corresponding symptoms (Verger et al., 2022). Parallels between early Alzheimer’s disease findings and COVID-19 associated neuronal damage can also be seen by the UK Biobank study, where both diffuse grey matter loss as well as focal to the EOCN and the parahippocampal/perirhinal and entorhinal cortex was identified in COVID-19 patients (Douaud et al., 2022). The implication of the EOCN and cognition is especially relevant to COVID-19, as it one sites SARS-CoV-2 either uses to port in the CNS, or cause distal inflammation communicated to proximal sites in a sterile manner. This neuroanatomical concept has been supported by emerging neuropathological studies on both animal models (Jiao et al., 2021; Philippens et al., 2022) and via human brain autopsy findings (Thakur et al., 2021).
In rhesus monkeys, SARS-CoV-2’s olfactory neuroinvasion route was shown to result gliosis and neuronal apoptosis involving the entorhinal area, hippocampus, thalamus, and midbrain in differing degrees (Jiao et al., 2021). Similarly, in a Syrian golden hamster model of SARS-CoV-2 transolfactory neuroinvasion, the hippocampi displayed decreased density of dendritic spines tandem with gliosis in the olfactory bulb (Kishimoto-Urata et al., 2022). Murine models currently indicate that both microgliosis and proteinopathy may persist beyond the acute phase of COVID-19 (Käufer et al., 2022; Kishimoto-Urata et al., 2022). As we have previously expanded on, microgliosis, tauopathy and Alzheimer’s disease-like signaling is consistently shown in brain organoids exposed to SARS-CoV-2 and mediated by IFN-I (Ramani et al., 2020; Hou et al., 2022; Kong et al., 2022, 2023), whereas this model in humans is supported by radiological evidence and neuropathology that parallels Alzheimer’s disease in its early stages, involving olfactory and limbic networks (de Erausquin et al., 2022; Douaud et al., 2022; Reiken et al., 2022; Ferrucci et al., 2023).
Similar findings have been revealed by human autopsy studies. In a study of consecutive COVID-19 patients (n = 41), microgliosis, reactive astrocytosis and neuronophagia were consistent findings, localized in the olfactory bulb, brainstem, and thalami of a subset of patients, along with ischemic / hypoxic lesions. Notably, autopsied brains contained neurofibrillary tangles, with and without amyloid plaques (17/41, 41.4%), a finding disproportionate with the age of the patients (Thakur et al., 2021). Microgliosis localized to the hippocampus and the brainstem has been consistently reported in other human neuropathological studies of COVID-19 patients, albeit in the absence of productive SARS-CoV-2 replication within the CNS (Lou et al., 2021; Poloni et al., 2021).
These observations have been reinforced by neuropathological findings from brain and brain plus fused cortical-blood vessel organoid models of SARS-CoV-2 infection, where SARS-CoV-2 infected neurons displayed aberrant tau phosphorylation and localization (Ramani et al., 2020). In the fused cortical blood vessel organoid model, tauopathy, beta amyloidosis and microglial activation was also noted as a result of SARS-CoV-2 infection (Kong et al., 2023). Notably, aberrant tau phosphorylation secondary to SARS-CoV-2 infection has also been observed in SH-SY5Y neuroblastoma cells (Di Primio et al., 2023). Type I interferon responses were elicited by SARS-CoV-2 infection of forebrain organoids, potentially in variant-dependent manner (Hou et al., 2022). IFN-I production by SARS-CoV-2-infected astrocytes in an organoid model, mediated by neuropillin-1. IFN-I signaling by infected astrocytes was shown to lead to decreased neurotransmitter levels, including choline, and neuronal destruction (Kong et al., 2022). Even in the absence of productive replication, IFN-I responses, microgliosis and a transcriptomic profile resembling neurodegenerative disease has also been shown in another brain organoid model (Suzzi et al., 2023).
We proposed that similar molecular mechanisms overlap between Alzheimer’s disease (AD) and COVID-19, with a focus on Type I interferon signaling (IFN-I) (Vavougios et al., 2022a). There are several reasons why IFN-I may account for the effects of COVID-19 on the CNS, and their overlap with Alzheimer’s disease pathogenesis. Type I interferon signaling is the premier response to viral infection, regardless of pathogens involved. This tonic innate immune signal is ubiquitous in the periphery as well as the CNS and remains in a prolonged stimulation determined by the host’s immune fitness and microbiome immunosurveillance (Abt et al., 2012; Lukhele et al., 2019; Wu et al., 2022). Peripheral infection can affect central sites such as the hippocampi in a sterile manner, and such prolonged stimulation may affect hippocampal neurogenesis and plasticity via crosstalk with the resident homeostatic IFN-I (Ekdahl et al., 2003; Michalovicz et al., 2015; Hosseini et al., 2020; Petrisko et al., 2020; Bayat et al., 2022). Type I interferon signaling has been shown to both directly affect cognition, and modulate long-term neuroinflammatory and neurotoxic effects for Alzheimer’s disease through microgliosis (Roy et al., 2020, 2022), beta-amyloidosis (Hur et al., 2020) and tauopathy (Jin et al., 2021; Udeochu et al., 2023). A growing body of human studies and disease models of Alzheimer’s disease consistently reports on dysregulated type I interferon signaling (See Supplementary Material 1 for an extended presentation of relevant studies) (Blank and Prinz, 2017; Jin et al., 2022; Sanford and McEwan, 2022; Vavougios et al., 2022b).
In COVID-19, type I interferon signaling is a primary innate immune response against SARS-CoV-2 that has been shown to be dysregulated as a result of complex host-response interactions (Lee and Shin, 2020; Shi et al., 2021; Eskandarian Boroujeni et al., 2022; Wu et al., 2022). Dysregulated type I interferon signaling has been shown to extend to sites proximal to the CNS, such as the olfactory bulb, the choroid plexus, and brain endothelial cells (reviewed in Vavougios et al., 2022a; Crook et al., 2023). Within the context of the studies presented, this peripheral activation of IFN-I due to COVID-19 can affect the CNS with a predilection for neurogenesis sites such as the hippocampus and the olfactory bulb (Ekdahl et al., 2003; Baruch et al., 2014; Bayat et al., 2022; Stępień et al., 2023), correspondingly mediate cognitive impairment (Blank et al., 2016) and anosmia (Llana et al., 2022, 2023).
Previous works from our group developed a model for the pathogenesis of CNS injury following COVID-19, and the overlap between the mechanisms that mediated with Alzheimer’s disease (Vavougios et al., 2021b, 2022a,b,c). We built this model upon in silico evidence of overlapping immune pathway dysregulation between peripheral blood and central nervous system sites in Alzheimer’s disease patients (Vavougios et al., 2020). Considering the established roles of IFN-I in cognition, immunity, and the pathogenesis of Alzheimer’s disease (Roy et al., 2020; Jin et al., 2022; Sanford and McEwan, 2022; Song et al., 2022), it is reasonable to posit that a pathogen that directly challenges IFN-I such as SARS-CoV-2 is an adequate probe into IFN-I mediated neuroinflammation and neurodegeneration.
What our model specifically describes is the transmission of an outside-in, quasi-infectious type I interferon signal from the periphery to the CNS, potentially via extracellular vesicles, which results in the disruption of tonic IFN-I centrally. Quasi-infectious here is used to describe the initial stimulation of IFN-I signaling by a pathogen, and the subsequent sterile elicitation of IFN-I activation centrally by endogenous disease associated molecular patterns (DAMPs). This transmission would require the interface between a peripheral site (e.g., the olfactory neuroepithelium, the brain vascular endothelium; Vavougios et al., 2022c) and a central site (such as the hippocampus) (Vavougios et al., 2021b). While peripheral stimulation persists, central sites would be exposed to prolonged IFN-I signaling and proinflammatory conditions which would be deleterious to neurogenesis, synaptogenesis and by extent, cognition (Vavougios et al., 2022a,b; Crook et al., 2023). Once IFN-I signaling has been localized to the CNS, it may become feed-forward due to the production of (DAMPs), a concept displayed experimentally in Alzheimer’s disease (Roy et al., 2020, 2022) with therapeutic implications for targeting both microglial responses and IFN-I (Moore et al., 2020). To probe into IFN-I dysregulation, we had focused on the interferon induced transmembrane protein 3 (IFITM3) (Vavougios et al., 2021b), an innate immunity protein effective against multiple pathogens, including SARS-CoV-2 (Prelli Bozzo et al., 2021) and a modulator of beta-amyloidogenesis, with an emerging role in the pathogenesis of Alzheimer’s disease (Hur et al., 2020). We had furthermore hypothesized that exosomal tau, shown to be closely related to beta amyloid and capable of seeding (Miyoshi et al., 2021; Polanco and Götz, 2022) would also intrinsically enhance feed-forward activation of IFN-I in the CNS. Insoluble tau uptake has been previously demonstrated in microglia (Bolós et al., 2016), and via exosome synthesis, mediate seeding (Asai et al., 2015). A subset of our hypothesis has been recently demonstrated by the pathogenic tau-mediated activation of IFN-I and cyclic GMP–AMP synthase (cGAS) in microglia, leading to diminished cognitive resilience in a murine model (Udeochu et al., 2023). Our model does neither rely on, or contradict neuroinvasion; instead, transolfactory neuroinflammation would result in, e.g., a direct elicitation of IFN-I in the hippocampus via EOCN projections (de Erausquin et al., 2022; Vavougios et al., 2022a).
Following the formulation of these works and their publication, a growing body of studies has provided robust corroboration to the mechanisms we predicted.
In this perspective, we aim to present our model within the context these studies, and the potential implications for a type I interferon-centric model for the pathogenesis of Alzheimer’s disease and post-acute COVID-19 cognitive impairment with implications for their treatment.
In the central nervous system, type I interferon responses represent an innate immune mechanism that is responsive to peripheral inflammation, and maintains the balance between danger-associated molecular pattern (DAMP) recognition and the magnitude of the inflammatory response that will sufficiently lead to the amelioration of this initial immune stimulant (Blank and Prinz, 2017). Homeostatic IFN-I responses in the human brain decrease with increased age, favoring inflammation (Baruch et al., 2014). In this setting, cognitive impairment has been shown to occur both as consequence of IFNAR activation (McNab et al., 2015), via a relatively indolent synaptopathy due to the chronic accumulation and dissemination of neurotoxic molecules and proteopathic seeds (Baruch et al., 2014; Viengkhou and Hofer, 2023).
We have previously adapted this well-observed paradigm to formulate the hypothesis of an outside-in injury of the CNS during COVID-19, where immune crosstalk results in microglial activation and immune-mediate neuronal injury (Vavougios et al., 2022a). In our model, IFN-I dysregulation occurs in the setting of peripheral infection and is communicated centrally via sites such as the olfactory bulb, the brainstem nuclei and the blood–brain barrier (Meinhardt et al., 2021; Krasemann et al., 2022). Sites specifically vulnerable to dysregulated IFN-I such as the olfactory bulb and the hippocampus (Bayat et al., 2022; Stępień et al., 2023) may furthermore readily account for both clinical (Gonzalez-Aleman et al., 2022) and neuropathological (Lou et al., 2021) manifestations of the CNS involvement in COVID-19.
The generation of amyloid-beta and hyperphosphorylated tau, molecules that we currently recognize as inducers of innate immunity (Rexach et al., 2020; Roy et al., 2020; Jin et al., 2021; Roy et al., 2022; Vavougios et al., 2022a; Udeochu et al., 2023), can in turn sustain pathogenic IFN-I in a feed-forward manner acting as danger associated molecular pattern (Asai et al., 2015; Bolós et al., 2016) in a process that has been shown to prime microglia towards phenotypes specifically associated with the earlier stages of neurodegenerative disease (Roy et al., 2020; Jin et al., 2021; Magusali et al., 2021; Kim et al., 2022; Udeochu et al., 2023). This aspect of our model, previously predicting beta amyloidosis and tauopathy based on the IFN-I response secondary to SARS-CoV-2 infection (Ramani et al., 2020; Vavougios et al., 2021a,b, 2022a,b,c; Di Primio et al., 2023) has been independently validated (Ramani et al., 2020; Green et al., 2022; Käufer et al., 2022; Ma et al., 2022; Reiken et al., 2022; Di Primio et al., 2023; Suzzi et al., 2023). A notable part of this feed-forward process is that microglial activation may be sustained via sterile DAMPs as IFN-I activators (Roy et al., 2020) such as exosomal or insoluble tau (Asai et al., 2015; Bolós et al., 2016) and errant nucleic acids (Roy et al., 2020). Molecular markers of neuronal damage have been identified in COVID-19 patients (Frontera et al., 2022; Lennol et al., 2023), albeit they are currently not routinely used along the main molecular assays used to detect COVID-19 (Habibzadeh et al., 2021).
Our model posits that the induction of peripheral IFN-I during COVID-19 may dysregulate IFN-I homeostasis in the CNS. A consequent question is whether these dysregulations are a valid biological substrate for cognitive impairment.
IFN-I represents a canonical immune surveillance response for the brain that effectively regulates pathogen dynamics and cellular permissiveness (Drokhlyansky et al., 2017; Welsch Jeremy et al., 2019). The relationship between IFN-I and cognition relies on data from several translational models that indicate its primary homeostatic role in the form of a tonic IFN-I response that oscillates between inflammation and quiescence. Homeostatic roles for type I interferon have been described in the maintenance of hippocampal synaptic plasticity by astrocytes (Hosseini et al., 2020). Tissue specific activation of the interferon alpha and beta receptor subunit 1 (IFNAR1) and the C-X-C Motif Chemokine Ligand 10 - C-X-C Motif Chemokine Receptor 3 (CXLC10-CXCR3) axis in murine brain endothelial and epithelial cells may manifest as cognitive impairment (Blank et al., 2016). Furthermore, they showed that this induction may occur in the setting of immune challenge from either viral ssRNA or type I interferons. As an ssRNA virus, SARS-CoV-2 has been shown to productively infect brain endothelial cells and alter IFN-I signaling via host-virus protein–protein interactions (Wenzel et al., 2021; Krasemann et al., 2022; Vavougios et al., 2022c; Yang et al., 2022; Suzzi et al., 2023). such as Nsp5 mediated cleavage of nuclear factor (NF)-κB essential modulator (NEMO) (Wenzel et al., 2021). Interferon signaling is also spatially resolved, however. Sites of adult neoneurogenesis have been shown to be more vulnerable to aberrant IFN-I signaling, including the subventricular zone (SVZ) and the hippocampus (Blank and Prinz, 2017; Viengkhou and Hofer, 2023) – sites furthermore important in the setting of Alzheimer’s disease (Kim et al., 2022), with the EOCN potentially affected in early or preclinical stages (Roberts et al., 2016; Yahiaoui-Doktor et al., 2019; Tian et al., 2022; Xiao et al., 2023). This concept appears to account for this specific aspect of COVID-19 neuropathology, where microgliosis (Poloni et al., 2021) and a decrease in adult human neoneurogenesis are combined (Stępień et al., 2023). Notably, aside from disease-associated microglia (DAM), IFN-I expressing microglia have also been shown to enhance neurodegeneration as well as the infiltration of the SVZ by T helper cells (Zhang et al., 2023). The interaction between microglia and T cells in the SVZ are state specific, with an apparent infiltration occurring with advanced age (Kim et al., 2022), when basal IFN-I responses are also altered (Baruch et al., 2014). Excessive exposure to the secretion of type I interferon by the microglia either in vitro or in vivo can lead to derangement of functional neuronal maturation (Hattori et al., 2020). Notably, tau and beta amyloid have been shown to stimulate microglia and upregulate IFN-I via cGAS-STING, a pathway that is furthermore linked to IFITM3 (Jin et al., 2021; Wu et al., 2023) further linking innate immunity and Alzheimer’s disease.
So far, we have established a robust body of works that validate our hypothesis of IFN-I as a central hub for cognitive impairment and neuronal damage in the setting of COVID-19. The convergence on IFN-I however constitutes a central part of the molecular, transcriptional and neuroanatomical overlap between COVID-19 and neurodegenerative disease, and specifically with Alzheimer’s disease (Magusali et al., 2021; Bayat et al., 2022; Mavrikaki et al., 2022; Vavougios et al., 2022a,b,c; Suzzi et al., 2023).
Similarly, a growing body of studies identify IFN-I responses as key molecular evens in the evolution of Alzheimer’s disease (Taylor et al., 2014; Roy et al., 2020; Sanford and McEwan, 2022; Govindarajulu et al., 2023; Sanford et al., 2023; Udeochu et al., 2023). Several works localize relationship in microglial responses to IFN-I, and their potential to drive feed-forward amyloidogenic cascades (Moore et al., 2020; Roy et al., 2020) and mediate exosomal tau seeding (Bolós et al., 2016; Wang et al., 2017; Miyoshi et al., 2021; Polanco and Götz, 2022; Udeochu et al., 2023). Aberrant IFN-I signaling in microglia has been shown to display tau-induced dystrophic and senescent phenotypes in murine models (Jin et al., 2022); recently, microglial activation in the setting of IFN-I signaling was shown to lead to synaptopathy and cognitive deficits, and conversely reversed by microglial ifnar1 deletion (Roy et al., 2022). Another notable aspect of Type I interferon signaling perturbations in Alzheimer’s disease is that they are detectable both in peripheral immune cells and the CNS in Alzheimer’s disease (Roy et al., 2020; Song et al., 2022), a concept we have previously and independently outlined (Vavougios et al., 2020).
Within the context of our model, several specific conclusions regarding IFN-I, cognition and neurodegeneration can be made: (a) Peripheral perturbations in IFN-I signalling can be communicated to the CNS via multiple sites (b) dysregulation of constitutional IFN-I responses can manifest as cognitive impairment acutely, or as a result of cognitive impairment (c) Local IFN-I dysregulation can lead to microgliosis and proteinopathy (d) once dysregulated, IFN-I requires DAMPs but not necessary a proliferating pathogen in the CNS (e) Exosomal tau, neurofibrillary tangles and errant nucleic acids may act as such DAMPs (d) this model can be activated by multiple pathogens (Dominy et al., 2019) or commensal microbiota, (Abt et al., 2012; Supplementary Figure 3) as well as other conditions resulting in IFN-I dysregulation, such as hypoxia.
In this light, COVID-19 functions as a potent perturbator of IFN-I homeostasis, and via that dysregulation molecular mechanisms associated Alzheimer’s disease may be enhanced. This proposed concept is furthermore confirmed by the overlap we and others have observed between COVID-19 and Alzheimer’s disease, and the central role of interferon stimulated genes in the development of this pathology (Mostafavi et al., 2018; Magusali et al., 2021; Poloni et al., 2021; Vavougios et al., 2021a,b; Green et al., 2022; Reiken et al., 2022; Semerdzhiev et al., 2022; Suzzi et al., 2023). Furthermore, the dysregulation of tonic IFN-I rather than the specific effect of SARS-CoV-2 can generalize this model to other pathogens and infections that have been associated with increased susceptibility to Alzheimer’s disease (Levine et al., 2023).
The interpretation of our model of cognitive impairment as well as the contribution of dysregulated tonic IFN-I and the crosstalk between peripheral and central nervous system immunity requires context and an awareness of important limitations (See Supplementary Material 2 for an extended presentation of context, strengths and limitations).
In this perspective, we presented the evolution of a model that explores how peripheral IFN-I dysregulation may affect the CNS, triggering microgliosis, proteinopathy and cognitive deficits. Furthermore, while COVID-19/PASC and Alzheimer’s disease are distinct conditions, our pathogenetic model provides insight into an implied intermediate mechanism by which infection, results in damage to structures affecting cognition and olfaction in the central nervous system, via IFN-I pathways that are also encountered in Alzheimer’s disease, and are druggable (Mavrikaki et al., 2022; Sanford and McEwan, 2022; Vavougios et al., 2022b). As innate immune- and specifically IFN-I dysregulation is increasingly recognized in Alzheimer’s disease, our model may be generalizable across pathogen and commensal microbiota-focused models where innate immunity is similarly stimulated (Dominy et al., 2019; Grabrucker et al., 2023; Levine et al., 2023).
This proposed model provides mechanistic background for the combination of olfactory dysfunction and cognitive impairment, considering the selective vulnerability of the olfactory bulb-hippocampal circuitry to IFN-I as neurogenesis sites (Ekdahl et al., 2003; Bayat et al., 2022; Stępień et al., 2023; Figure 1). As shown by a growing body of research, IFN-I can furthermore account for the effect of age on inflammation and cognition (Baruch et al., 2014), which again is a central component in the pathogenesis of ADRD. Lastly, our model supports a multi- or sequential- hit concept where multiple pathogens and immune challenges to which IFN-I is responsive may result in lasting CNS damage (Figure 2). Following an initial pathogen-induced stimulation, endogenous DAMPs (such as nucleic acids and proteopathic seeds) produced by neuronal damage would fuel second-order IFN-I dysregulation by a sterile inflammatory response (Rubartelli, 2014). Notably, interferon stimulated genes such as IFITMs and OASs, as we and other have reported (Magusali et al., 2021; Vavougios et al., 2021a,b) are linked to nucleic acid surveillance mechanisms (Magusali et al., 2021; Vavougios et al., 2022b) such as the OAS antiviral response and cGAS-STING, where Αβ and tau pathobiology converge with IFN-I dysregulation and inflammasome activation (Magusali et al., 2021; Udeochu et al., 2023; Wu et al., 2023). Focusing on IFN-I modulation and restoration of canonical signaling may thus represent an important axis of therapeutics that is currently underexplored.
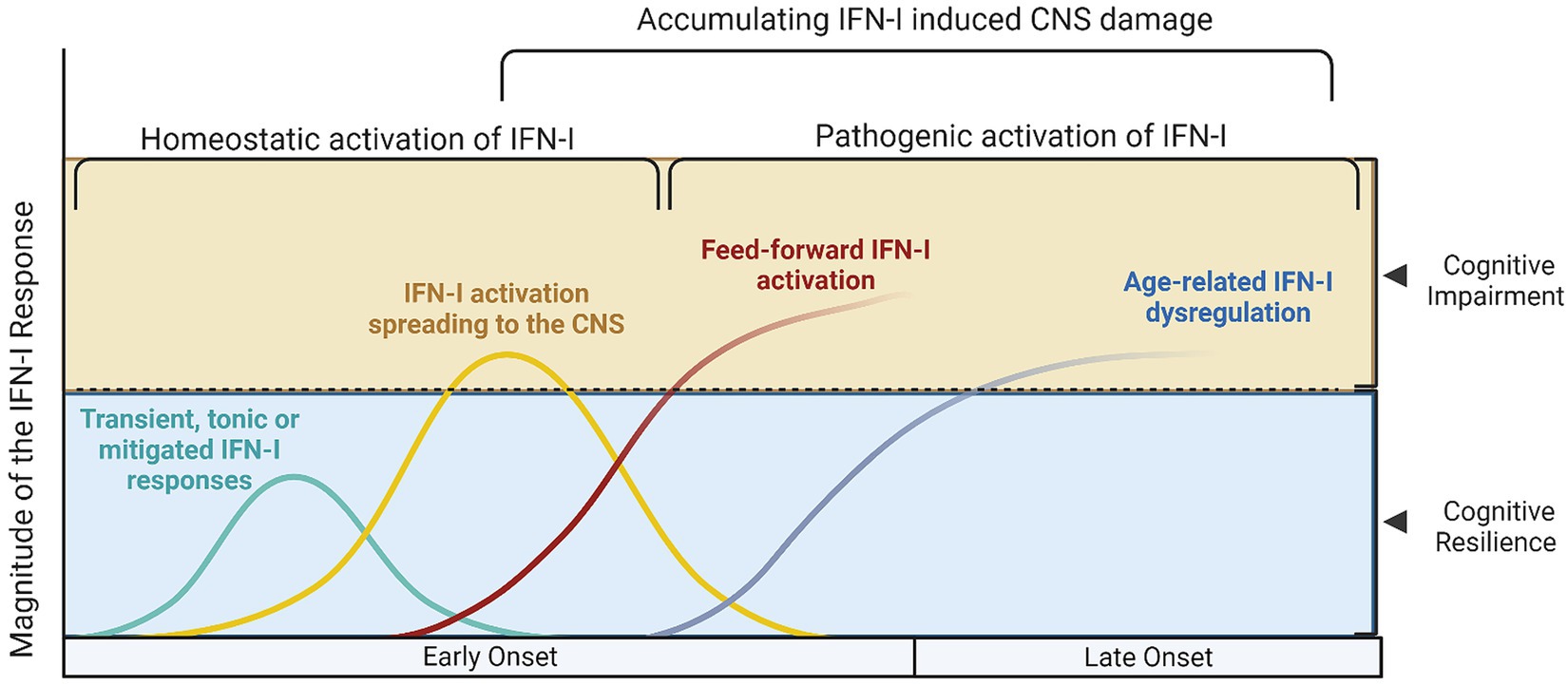
Figure 1. Different models of Type I interferon signaling in the human brain and their effects on cognition. Type I interferon signaling (IFN-I) is a primary innate immune response against pathogens and danger associated molecular patterns (DAMPs). A tonic response is maintained by commensal microbiota and transient immune challenges, whereas other, unmitigated sources of stimulation may exert a deleterious effect on the CNS. (A) The cyan curve represents a transient activation of IFN-I, where the source of DAMPs would be depleted, the offending pathogen dealt with and correspondingly, anti-inflammatory/homeostatic IFN-I responses would prevail., whereas in (B) the yellow curve represents hyperinflammatory IFN-I activation, which still however follows a canonical course and may be reversed. During this hyperactivation period, cognitive impairment may manifest, as is evidence from both murine and human studies; in this setting, acute COVID-19 and other infections would also fit. The red (C) and blue (D) lines represent cases where secondary, feed-forward activation of IFN-I responses by endogenous DAMPS exceeds the capacity of its regulatory mechanisms and may eventually establish the precipice of neurodegeneration. These curves are a function of the magnitude of IFN-I activation with age (early onset vs. late onset cognitive impairment and CNS damage, conversely).
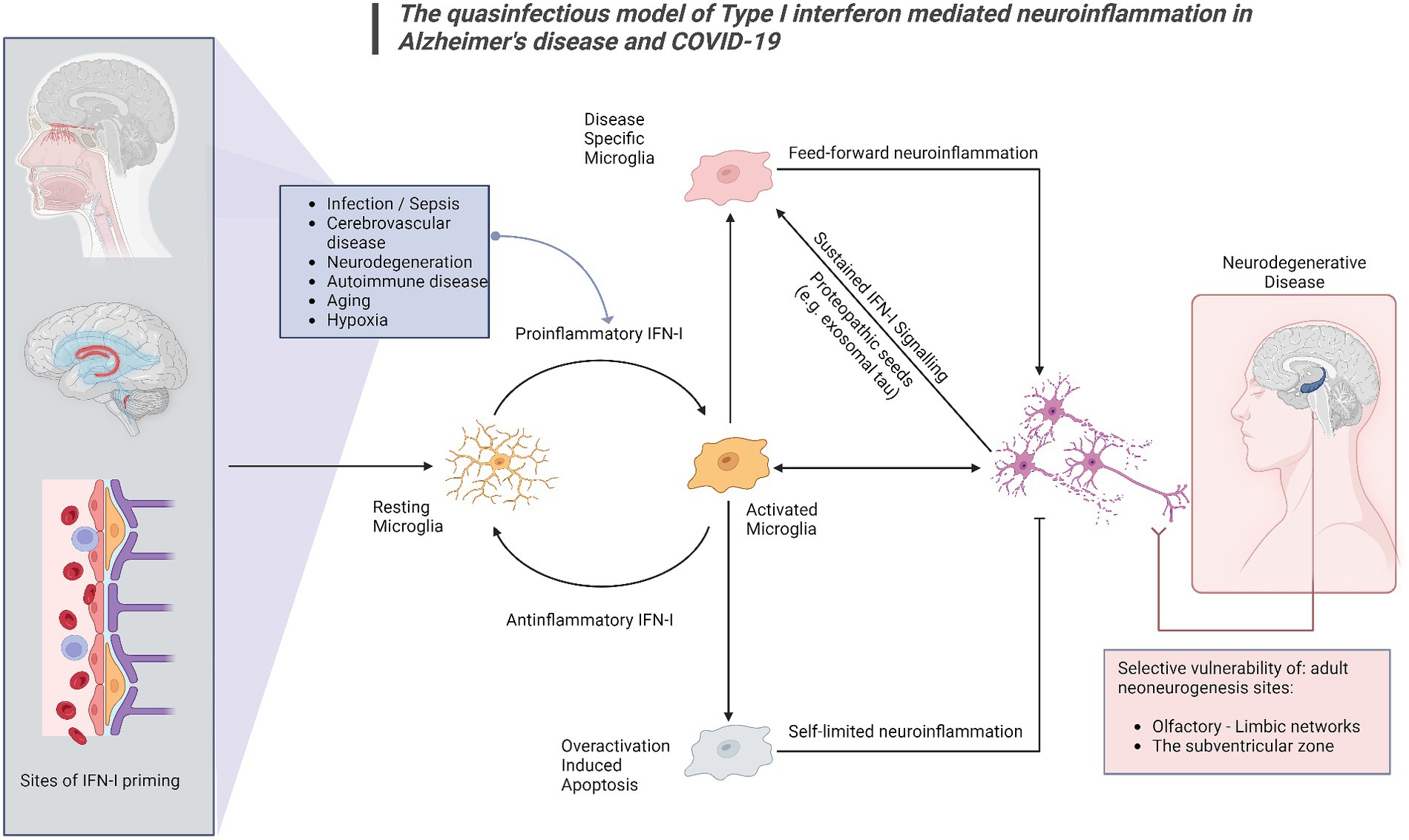
Figure 2. The quasinfectious model of Type I interferon mediated neuroinflammation in Alzheimer’s disease and COVID-19. In this model, applicable to both COVID-19-associated cognitive impairment and COVID-19, Proinflammatory IFN-I activation may occur distally via DAMPs and other IFN-I activated sites at the blood–brain barrier, the choroid plexus and the olfactory bulb. Prolonged infection, cerebrovascular disease, autoimmune disease, cancer, aging and pre-existing neurodegenerative disease may also generate endogenous DAMPs such as exosomal tau, nucleic acids and beta-amyloid. Microglia have been shown to react to these signals as IFN-I relays, switching from a resting to an activated state. A homeostatic response includes switching off proinflammatory IFN-I responses, extending to overactivation-induced apoptosis for microglia. A subsequent phenotypic switch in activated microglia, would subsequently promote neurodegeneration. Central nervous system sites adjacent to distal or peripheral inflamed sites would be primarily affected, as well as sites of adult neurogenesis, shown to be vulnerable in both COVID-19 and Alzheimer’s disease.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
GV: Conceptualization, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing. V-ST: Writing – original draft. AL: Writing – original draft. TM: Funding acquisition, Writing – original draft. GE: Conceptualization, Methodology, Validation, Writing – original draft, Writing – review & editing. GH: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. GV and GH were supported by the Cyprus Academy of Sciences, Letters and Arts on the COVALENT and COGNATE studies, on relevant projects which examine the interplay between cognition and infection. TM received funding support from the Tallacht University Hospital (TUH)/The Adelaide and Meath Hospital, Dublin, incorporating the National Children’s Hospital (AMNCH), Dublin, Ireland.
GV would like to thank Giovanni D’Avossa, Paul Edison, and Heather M. Snyder for their support in developing the concept of type I interferon-centric, quasinfectious model of CNS damage in Alzheimer’s disease and COVID-19.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnhum.2024.1352118/full#supplementary-material
Abt, M. C., Osborne, L. C., Monticelli, L. A., Doering, T. A., Alenghat, T., Sonnenberg, G. F., et al. (2012). Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170. doi: 10.1016/j.immuni.2012.04.011
Asai, H., Ikezu, S., Tsunoda, S., Medalla, M., Luebke, J., Haydar, T., et al. (2015). Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. doi: 10.1038/nn.4132
Baruch, K., Deczkowska, A., David, E., Castellano, J. M., Miller, O., Kertser, A., et al. (2014). Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93. doi: 10.1126/science.1252945
Bayat, A. H., Azimi, H., Hassani Moghaddam, M., Ebrahimi, V., Fathi, M., Vakili, K., et al. (2022). COVID-19 causes neuronal degeneration and reduces neurogenesis in human hippocampus. Apoptosis 27, 852–868. doi: 10.1007/s10495-022-01754-9
Blank, T., Detje, C. N., Spieß, A., Hagemeyer, N., Brendecke, S. M., Wolfart, J., et al. (2016). Brain endothelial- and epithelial-specific interferon receptor chain 1 drives virus-induced sickness behavior and cognitive impairment. Immunity 44, 901–912. doi: 10.1016/j.immuni.2016.04.005
Blank, T., and Prinz, M. (2017). Type I interferon pathway in CNS homeostasis and neurological disorders. Glia 65, 1397–1406. doi: 10.1002/glia.23154
Bolós, M., Llorens-Martín, M., Jurado-Arjona, J., Hernández, F., Rábano, A., and Avila, J. (2016). Direct evidence of internalization of tau by microglia in vitro and in vivo. J. Alzheimers Dis. 50, 77–87. doi: 10.3233/JAD-150704
Crook, H., Ramirez, A., Hosseini, A., Vavougyios, G., Lehmann, C., Bruchfeld, J., et al. (2023). European working group on SARS-CoV-2: current understanding, unknowns, and recommendations on the neurological complications of COVID-19. Brain Connect. 13, 178–210. doi: 10.1089/brain.2022.0058
de Erausquin, G. A., Snyder, H., Brugha, T. S., Seshadri, S., Carrillo, M., Sagar, R., et al. (2022). Chronic neuropsychiatric sequelae of SARS-CoV-2: protocol and methods from the Alzheimer's Association global consortium. Alzheimers Dement. 8:e12348. doi: 10.1002/trc2.12348
de Erausquin, G. A., Snyder, H., Carrillo, M., Hosseini, A. A., Brugha, T. S., Seshadri, S., et al. (2021). The chronic neuropsychiatric sequelae of COVID-19: the need for a prospective study of viral impact on brain functioning. Alzheimers Dement. 17, 1056–1065. doi: 10.1002/alz.12255
Deer, R. R., Rock, M. A., Vasilevsky, N., Carmody, L., Rando, H., Anzalone, A. J., et al. (2021). Characterizing Long COVID: deep phenotype of a complex condition. EBioMedicine 74:103722. doi: 10.1016/j.ebiom.2021.103722
Di Primio, C., Quaranta, P., Mignanelli, M., Siano, G., Bimbati, M., Scarlatti, A., et al. (2023). Severe acute respiratory syndrome coronavirus 2 infection leads to tau pathological signature in neurons. PNAS Nexus. 2. doi: 10.1093/pnasnexus/pgad282
Dominy, S. S., Lynch, C., Ermini, F., Benedyk, M., Marczyk, A., Konradi, A., et al. (2019). Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Advance 5:eaau3333. doi: 10.1126/sciadv.aau3333
Douaud, G., Lee, S., Alfaro-Almagro, F., Arthofer, C., Wang, C., McCarthy, P., et al. (2022). SARS-CoV-2 is associated with changes in brain structure in UK biobank. Nature 604, 697–707. doi: 10.1038/s41586-022-04569-5
Drokhlyansky, E., Göz Aytürk, D., Soh, T. K., Chrenek, R., O’Loughlin, E., Madore, C., et al. (2017). The brain parenchyma has a type I interferon response that can limit virus spread. Proc. Natl. Acad. Sci. 114, E95–E104. doi: 10.1073/pnas.1618157114
Ekdahl, C. T., Claasen, J.-H., Bonde, S., Kokaia, Z., and Lindvall, O. (2003). Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. 100, 13632–13637. doi: 10.1073/pnas.2234031100
Eskandarian Boroujeni, M., Sekrecka, A., Antonczyk, A., Hassani, S., Sekrecki, M., Nowicka, H., et al. (2022). Dysregulated interferon response and immune hyperactivation in severe COVID-19: targeting STATs as a novel therapeutic strategy. Front. Immunol. 13:888897. doi: 10.3389/fimmu.2022.888897
Ferrucci, R., Cuffaro, L., Capozza, A., Rosci, C., Maiorana, N., Groppo, E., et al. (2023). Brain positron emission tomography (PET) and cognitive abnormalities one year after COVID-19. J. Neurol. 270, 1823–1834. doi: 10.1007/s00415-022-11543-8
Frontera, J. A., Boutajangout, A., Masurkar, A. V., Betensky, R. A., Ge, Y., Vedvyas, A., et al. (2022). Comparison of serum neurodegenerative biomarkers among hospitalized COVID-19 patients versus non-COVID subjects with normal cognition, mild cognitive impairment, or Alzheimer's dementia. Alzheimers Dement. 18, 899–910. doi: 10.1002/alz.12556
Gonzalez-Aleman, G., Zamponi, H. P., Juarez-Aguaysol, L., Kukoc, G., Dominguez, M. E., Pini, B., et al. (2022). Olfactory dysfunction but not COVID-19 severity predicts severity of cognitive sequelae following SARS-CoV-2 infection in Amerindian older adults. Alzheimers Dement. 18:e066868. doi: 10.1002/alz.066868
Govindarajulu, M., Ramesh, S., Beasley, M., Lynn, G., Wallace, C., Labeau, S., et al. (2023). Role of cGAS-Sting Signaling in Alzheimer's disease. Int. J. Mol. Sci. 24. doi: 10.3390/ijms24098151
Grabrucker, S., Marizzoni, M., Silajdžić, E., Lopizzo, N., Mombelli, E., Nicolas, S., et al. (2023). Microbiota from Alzheimer’s patients induce deficits in cognition and hippocampal neurogenesis. Brain 146, 4916–4934. doi: 10.1093/brain/awad303
Green, R., Mayilsamy, K., McGill, A. R., Martinez, T. E., Chandran, B., Blair, L. J., et al. (2022). SARS-CoV-2 infection increases the gene expression profile for Alzheimer’s disease risk. Molec. Therapy Methods Clin. Develop. 27, 217–229. doi: 10.1016/j.omtm.2022.09.007
Habibzadeh, P., Mofatteh, M., Silawi, M., Ghavami, S., and Faghihi, M. A. (2021). Molecular diagnostic assays for COVID-19: an overview. Crit. Rev. Clin. Lab. Sci. 58, 385–398. doi: 10.1080/10408363.2021.1884640
Hattori, Y., Naito, Y., Tsugawa, Y., Nonaka, S., Wake, H., Nagasawa, T., et al. (2020). Transient microglial absence assists postmigratory cortical neurons in proper differentiation. Nat. Commun. 11:1631. doi: 10.1038/s41467-020-15409-3
Hosseini, S., Michaelsen-Preusse, K., Grigoryan, G., Chhatbar, C., Kalinke, U., and Korte, M. (2020). Type I interferon receptor Signaling in astrocytes regulates hippocampal synaptic plasticity and cognitive function of the healthy CNS. Cell Rep. 31:107666. doi: 10.1016/j.celrep.2020.107666
Hou, Y., Li, C., Yoon, C., Leung, O. W., You, S., Cui, X., et al. (2022). Enhanced replication of SARS-CoV-2 omicron BA.2 in human forebrain and midbrain organoids. Signal Transduct. Target. Ther. 7. doi: 10.1038/s41392-022-01241-2
Hur, J. Y., Frost, G. R., Wu, X., Crump, C., Pan, S. J., Wong, E., et al. (2020). The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer's disease. Nature 586, 735–740. doi: 10.1038/s41586-020-2681-2
Jiao, L., Yang, Y., Yu, W., Zhao, Y., Long, H., Gao, J., et al. (2021). The olfactory route is a potential way for SARS-CoV-2 to invade the central nervous system of rhesus monkeys. Signal Transduct. Target. Ther. 6:169. doi: 10.1038/s41392-021-00591-7
Jin, M., Shiwaku, H., Tanaka, H., Obita, T., Ohuchi, S., Yoshioka, Y., et al. (2021). Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 12:6565. doi: 10.1038/s41467-021-26851-2
Jin, M., Xu, R., Wang, L., Alam, M. M., Ma, Z., Zhu, S., et al. (2022). Type-I-interferon signaling drives microglial dysfunction and senescence in human iPSC models of down syndrome and Alzheimer’s disease. Cell Stem Cell 29, 1135–53.e8. doi: 10.1016/j.stem.2022.06.007
Käufer, C., Schreiber, C. S., Hartke, A.-S., Denden, I., Stanelle-Bertram, S., Beck, S., et al. (2022). Microgliosis and neuronal proteinopathy in brain persist beyond viral clearance in SARS-CoV-2 hamster model. EBioMedicine 79:103999. doi: 10.1016/j.ebiom.2022.103999
Kim, H. S., Shin, S. M., Kim, S., Nam, Y., Yoo, A., and Moon, M. (2022). Relationship between adult subventricular neurogenesis and Alzheimer’s disease: pathologic roles and therapeutic implications. Front. Ageing Neurosci. 14:1002281. doi: 10.3389/fnagi.2022.1002281
Kim, D. W., Tu, K. J., Wei, A., Lau, A. J., Gonzalez-Gil, A., Cao, T., et al. (2022). Amyloid-beta and tau pathologies act synergistically to induce novel disease stage-specific microglia subtypes. Mol. Neurodegener. 17:83. doi: 10.1186/s13024-022-00589-x
Kishimoto-Urata, M., Urata, S., Kagoya, R., Imamura, F., Nagayama, S., Reyna, R. A., et al. (2022). Prolonged and extended impacts of SARS-CoV-2 on the olfactory neurocircuit. Sci. Rep. 12:5728. doi: 10.1038/s41598-022-09731-7
Kong, W., Montano, M., Corley, M. J., Helmy, E., Kobayashi, H., Kinisu, M., et al. (2022). Neuropilin-1 mediates SARS-CoV-2 infection of astrocytes in brain organoids, inducing inflammation leading to dysfunction and death of neurons. MBio 13:e0230822. doi: 10.1128/mbio.02308-22
Kong, D., Park, K. H., Kim, D.-H., Kim, N. G., Lee, S.-E., Shin, N., et al. (2023). Cortical-blood vessel assembloids exhibit Alzheimer’s disease phenotypes by activating glia after SARS-CoV-2 infection. Cell Death Discovery 9:32. doi: 10.1038/s41420-022-01288-8
Krasemann, S., Haferkamp, U., Pfefferle, S., Woo, M. S., Heinrich, F., Schweizer, M., et al. (2022). The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Reports 17, 307–320. doi: 10.1016/j.stemcr.2021.12.011
Lee, J. S., and Shin, E.-C. (2020). The type I interferon response in COVID-19: implications for treatment. Nat. Rev. Immunol. 20, 585–586. doi: 10.1038/s41577-020-00429-3
Lennol, M. P., Ashton, N. J., Moreno-Pérez, O., García-Ayllón, M. S., Ramos-Rincon, J. M., Andrés, M., et al. (2023). Transient changes in the plasma of astrocytic and neuronal injury biomarkers in COVID-19 patients without neurological syndromes. Int. J. Mol. Sci. 24. doi: 10.3390/ijms24032715
Levine, K. S., Leonard, H. L., Blauwendraat, C., Iwaki, H., Johnson, N., Bandres-Ciga, S., et al. (2023). Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 111, 1086–93.e2. doi: 10.1016/j.neuron.2022.12.029
Llana, T., Mendez, M., Garces-Arilla, S., Hidalgo, V., Mendez-Lopez, M., and Juan, M. C. (2023). Association between olfactory dysfunction and mood disturbances with objective and subjective cognitive deficits in long-COVID. Front. Psychol. 14. doi: 10.3389/fpsyg.2023.1076743
Llana, T., Mendez, M., Zorzo, C., Fidalgo, C., Juan, M.-C., and Mendez-Lopez, M. (2022). Anosmia in COVID-19 could be associated with long-term deficits in the consolidation of procedural and verbal declarative memories. Front. Neurosci. 16. doi: 10.3389/fnins.2022.1082811
Lou, J. J., Movassaghi, M., Gordy, D., Olson, M. G., Zhang, T., Khurana, M. S., et al. (2021). Neuropathology of COVID-19 (neuro-COVID): clinicopathological update. Free Neuropathol. :2. doi: 10.17879/freeneuropathology-2021-2993
Lukhele, S., Boukhaled, G. M., and Brooks, D. G. (2019). Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin. Immunol. 43:101277. doi: 10.1016/j.smim.2019.05.001
Ma, G., Zhang, D.-F., Zou, Q.-C., Xie, X., Xu, L., Feng, X.-L., et al. (2022). SARS-CoV-2 spike protein S2 subunit modulates γ-secretase and enhances amyloid-β production in COVID-19 neuropathy. Cell Discovery 8:99. doi: 10.1038/s41421-022-00458-3
Magusali, N., Graham, A. C., Piers, T. M., Panichnantakul, P., Yaman, U., Shoai, M., et al. (2021). A genetic link between risk for Alzheimer's disease and severe COVID-19 outcomes via the OAS1 gene. Brain 144, 3727–3741. doi: 10.1093/brain/awab337
Mavrikaki, M., Lee, J. D., Solomon, I. H., and Slack, F. J. (2022). Severe COVID-19 is associated with molecular signatures of aging in the human brain. Nat. Aging 2, 1130–1137. doi: 10.1038/s43587-022-00321-w
McNab, F., Mayer-Barber, K., Sher, A., Wack, A., and O'Garra, A. (2015). Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103. doi: 10.1038/nri3787
Meinhardt, J., Radke, J., Dittmayer, C., Franz, J., Thomas, C., Mothes, R., et al. (2021). Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 24, 168–175. doi: 10.1038/s41593-020-00758-5
Michalovicz, L. T., Lally, B., and Konat, G. W. (2015). Peripheral challenge with a viral mimic upregulates expression of the complement genes in the hippocampus. J. Neuroimmunol. 285, 137–142. doi: 10.1016/j.jneuroim.2015.06.003
Miyoshi, E., Bilousova, T., Melnik, M., Fakhrutdinov, D., Poon, W. W., Vinters, H. V., et al. (2021). Exosomal tau with seeding activity is released from Alzheimer’s disease synapses, and seeding potential is associated with amyloid beta. Lab. Investig. 101, 1605–1617. doi: 10.1038/s41374-021-00644-z
Moore, Z., Mobilio, F., Walker, F. R., Taylor, J. M., and Crack, P. J. (2020). Abrogation of type-I interferon signalling alters the microglial response to Aβ(1-42). Sci. Rep. 10:3153. doi: 10.1038/s41598-020-59917-0
Mostafavi, S., Gaiteri, C., Sullivan, S. E., White, C. C., Tasaki, S., Xu, J., et al. (2018). A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 21, 811–819. doi: 10.1038/s41593-018-0154-9
Muccioli, L., Sighinolfi, G., Mitolo, M., Ferri, L., Jane Rochat, M., Pensato, U., et al. (2023). Cognitive and functional connectivity impairment in post-COVID-19 olfactory dysfunction. Neuroimage Clin. 38:103410. doi: 10.1016/j.nicl.2023.103410
Parotto, M., Gyöngyösi, M., Howe, K., Myatra, S. N., Ranzani, O., Shankar-Hari, M., et al. (2023). Post-acute sequelae of COVID-19: understanding and addressing the burden of multisystem manifestations. Lancet Respir. Med. 11, 739–754. doi: 10.1016/S2213-2600(23)00239-4
Petrisko, T. J., Bloemer, J., Pinky, P. D., Srinivas, S., Heslin, R. T., Du, Y., et al. (2020). Neuronal CXCL10/CXCR3 Axis mediates the induction of cerebral hyperexcitability by peripheral viral challenge. Front. Neurosci. 14:220. doi: 10.3389/fnins.2020.00220
Philippens, I., Böszörményi, K. P., Wubben, J. A. M., Fagrouch, Z. C., van Driel, N., Mayenburg, A. Q., et al. (2022). Brain inflammation and intracellular α-synuclein aggregates in macaques after SARS-CoV-2 infection. Viruses 14. doi: 10.3390/v14040776
Polanco, J. C., and Götz, J. (2022). Exosomal and vesicle-free tau seeds—propagation and convergence in endolysosomal permeabilization. FEBS J. 289, 6891–6907. doi: 10.1111/febs.16055
Poloni, T. E., Medici, V., Moretti, M., Visonà, S. D., Cirrincione, A., Carlos, A. F., et al. (2021). COVID-19-related neuropathology and microglial activation in elderly with and without dementia. Brain Pathol. 31:e12997. doi: 10.1111/bpa.12997
Prelli Bozzo, C., Nchioua, R., Volcic, M., Koepke, L., Krüger, J., Schütz, D., et al. (2021). IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 12:4584. doi: 10.1038/s41467-021-24817-y
Ramani, A., Müller, L., Ostermann, P. N., Gabriel, E., Abida-Islam, P., Müller-Schiffmann, A., et al. (2020). SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 39:e106230. doi: 10.15252/embj.2020106230
Reiken, S., Sittenfeld, L., Dridi, H., Liu, Y., Liu, X., and Marks, A. R. (2022). Alzheimer's-like signaling in brains of COVID-19 patients. Alzheimers Dement. 18, 955–965. doi: 10.1002/alz.12558
Rexach, J. E., Polioudakis, D., Yin, A., Swarup, V., Chang, T. S., Nguyen, T., et al. (2020). Tau pathology drives dementia risk-associated gene networks toward chronic inflammatory states and immunosuppression. Cell Rep. 33:108398. doi: 10.1016/j.celrep.2020.108398
Roberts, R. O., Christianson, T. J. H., Kremers, W. K., Mielke, M. M., Machulda, M. M., Vassilaki, M., et al. (2016). Association between olfactory dysfunction and amnestic mild cognitive impairment and Alzheimer disease dementia. JAMA Neurol. 73, 93–101. doi: 10.1001/jamaneurol.2015.2952
Roy, E. R., Chiu, G., Li, S., Propson, N. E., Kanchi, R., Wang, B., et al. (2022). Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity 55, 879–94.e6. doi: 10.1016/j.immuni.2022.03.018
Roy, E. R., Wang, B., Wan, Y. W., Chiu, G., Cole, A., Yin, Z., et al. (2020). Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 130, 1912–1930. doi: 10.1172/JCI133737
Rubartelli, A. (2014). DAMP-mediated activation of NLRP3-inflammasome in brain sterile inflammation: the fine line between healing and neurodegeneration. Front. Immunol. 5:99. doi: 10.3389/fimmu.2014.00099
Sanford, S. A. I., and McEwan, W. A. (2022). Type-I interferons in Alzheimer's disease and other tauopathies. Front. Cell. Neurosci. 16:949340. doi: 10.3389/fncel.2022.949340
Sanford, S. A. I., Miller, L. V. C., Vaysburd, M., Keeling, S., Tuck, B. J., Clark, J., et al. (2023). The type-I interferon response potentiates seeded tau aggregation and exacerbates tau pathology. Alzheimers Dement. 19. doi: 10.1002/alz.073392
Semerdzhiev, S. A., Fakhree, M. A. A., Segers-Nolten, I., Blum, C., and Claessens, M. (2022). Interactions between SARS-CoV-2 N-protein and α-synuclein accelerate amyloid formation. ACS Chem. Neurosci. 13, 143–150. doi: 10.1021/acschemneuro.1c00666
Shi, G., Kenney, A. D., Kudryashova, E., Zani, A., Zhang, L., Lai, K. K., et al. (2021). Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 40:e106501. doi: 10.15252/embj.2020106501
Song, L., Chen, J., Lo, C. Z., Guo, Q., Feng, J., and Zhao, X. M. (2022). Impaired type I interferon signaling activity implicated in the peripheral blood transcriptome of preclinical Alzheimer's disease. EBioMedicine 82:104175. doi: 10.1016/j.ebiom.2022.104175
Stępień, T., Tarka, S., Chmura, N., Grzegorczyk, M., Acewicz, A., Felczak, P., et al. (2023). Influence of SARS-CoV-2 on adult human neurogenesis. Cells 12. doi: 10.3390/cells12020244
Suzzi, S., Tsitsou-Kampeli, A., and Schwartz, M. (2023). The type I interferon antiviral response in the choroid plexus and the cognitive risk in COVID-19. Nat. Immunol. 24, 220–224. doi: 10.1038/s41590-022-01410-z
Taylor, J. M., Minter, M. R., Newman, A. G., Zhang, M., Adlard, P. A., and Crack, P. J. (2014). Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer's disease. Neurobiol. Aging 35, 1012–1023. doi: 10.1016/j.neurobiolaging.2013.10.089
Thakur, K. T., Miller, E. H., Glendinning, M. D., Al-Dalahmah, O., Banu, M. A., Boehme, A. K., et al. (2021). COVID-19 neuropathology at Columbia University Irving medical Center/New York presbyterian hospital. Brain 144, 2696–2708. doi: 10.1093/brain/awab148
Tian, Q., Bilgel, M., Moghekar, A. R., Ferrucci, L., and Resnick, S. M. (2022). Olfaction, cognitive impairment, and PET biomarkers in community-dwelling older adults. J. Alzheimers Dis. 86, 1275–1285. doi: 10.3233/JAD-210636
Udeochu, J. C., Amin, S., Huang, Y., Fan, L., Torres, E. R. S., Carling, G. K., et al. (2023). Tau activation of microglial cGAS–IFN reduces MEF2C-mediated cognitive resilience. Nat. Neurosci. 26, 737–750. doi: 10.1038/s41593-023-01315-6
Vavougios, G. D., Breza, M., Mavridis, T., and Krogfelt, K. A. (2021a). FYN, SARS-CoV-2, and IFITM3 in the neurobiology of Alzheimer's disease. Brain Disord. 3:100022. doi: 10.1016/j.dscb.2021.100022
Vavougios, G. D., de Erausquin, G. A., and Snyder, H. M. (2022a). Type I interferon signaling in SARS-CoV-2 associated neurocognitive disorder (SAND): mapping host-virus interactions to an etiopathogenesis. Front. Neurol. 13:1063298. doi: 10.3389/fneur.2022.1063298
Vavougios, G. D., Mavridis, T., Artemiadis, A., Krogfelt, K. A., and Hadjigeorgiou, G. (2022b). Trained immunity in viral infections, Alzheimer's disease and multiple sclerosis: a convergence in type I interferon signalling and IFNβ-1a. Biochim. Biophys. Acta Mol. basis Dis. 1868:166430. doi: 10.1016/j.bbadis.2022.166430
Vavougios, G. D., Nday, C., Pelidou, S. H., Gourgoulianis, K. I., Stamoulis, G., Doskas, T., et al. (2021b). Outside-in induction of the IFITM3 trafficking system by infections, including SARS-CoV-2, in the pathobiology of Alzheimer's disease. Brain Behav. Immun. Health 14:100243. doi: 10.1016/j.bbih.2021.100243
Vavougios, G. D., Nday, C., Pelidou, S. H., Zarogiannis, S. G., Gourgoulianis, K. I., Stamoulis, G., et al. (2020). Double hit viral parasitism, polymicrobial CNS residency and perturbed proteostasis in Alzheimer's disease: a data driven, in silico analysis of gene expression data. Mol. Immunol. 127, 124–135. doi: 10.1016/j.molimm.2020.08.021
Vavougios, G. D., Zarogiannis, S. G., Hadjigeorgiou, G., Krogfelt, K. A., and Gourgoulianis, K. I. (2022c). SARS-CoV-2 and type I interferon signaling in brain endothelial cells: blurring the lines between friend or foe. Stem Cell Reports 17, 1012–1013. doi: 10.1016/j.stemcr.2022.04.011
Verger, A., Barthel, H., Tolboom, N., Fraioli, F., Cecchin, D., Albert, N. L., et al. (2022). 2-[18F]-FDG PET for imaging brain involvement in patients with long COVID: perspective of the EANM neuroimaging committee. Eur. J. Nucl. Med. Mol. Imaging 49, 3599–3606. doi: 10.1007/s00259-022-05913-7
Viengkhou, B., and Hofer, M. J. (2023). Breaking down the cellular responses to type I interferon neurotoxicity in the brain. Front. Immunol. 14:1110593. doi: 10.3389/fimmu.2023.1110593
Wang, Y., Balaji, V., Kaniyappan, S., Krüger, L., Irsen, S., Tepper, K., et al. (2017). The release and trans-synaptic transmission of tau via exosomes. Mol. Neurodegener. 12:5. doi: 10.1186/s13024-016-0143-y
Welsch Jeremy, C., Charvet, B., Dussurgey, S., Allatif, O., Aurine, N., Horvat, B., et al. (2019). Type I interferon receptor Signaling drives selective permissiveness of astrocytes and microglia to measles virus during brain infection. J. Virol. 93, e00618–e00619. doi: 10.1128/JVI.00618-19
Wenzel, J., Lampe, J., Müller-Fielitz, H., Schuster, R., Zille, M., Müller, K., et al. (2021). The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 24, 1522–1533. doi: 10.1038/s41593-021-00926-1
Wu, B., Ramaiah, A., Garcia, G. Jr., Hasiakos, S., Arumugaswami, V., and Srikanth, S. (2022). ORAI1 limits SARS-CoV-2 infection by regulating tonic type I IFN Signaling. J. Immunol. 208, 74–84. doi: 10.4049/jimmunol.2100742
Wu, Z., Tang, W., Ibrahim, F., Chen, X., Yan, H., Tao, C., et al. (2023). Aβ induces neuroinflammation and microglial M1 polarization via cGAS-STING-IFITM3 Signaling pathway in BV-2 cells. Neurochem. Res. 48, 2881–2894. doi: 10.1007/s11064-023-03945-5
Xiao, Z., Wu, W., Ma, X., Wu, J., Liang, X., Cao, Y., et al. (2023). Olfactory function, neurofilament light chain, and cognitive trajectory: a 12-year follow-up of the Shanghai aging study. Alzheimers Dement. 15:e12485. doi: 10.1002/dad2.12485
Yahiaoui-Doktor, M., Luck, T., Riedel-Heller, S. G., Loeffler, M., Wirkner, K., and Engel, C. (2019). Olfactory function is associated with cognitive performance: results from the population-based LIFE-adult-study. Alzheimers Res. Ther. 11:43. doi: 10.1186/s13195-019-0494-z
Yang, R.-C., Huang, K., Zhang, H.-P., Li, L., Zhang, Y.-F., Tan, C., et al. (2022). SARS-CoV-2 productively infects human brain microvascular endothelial cells. J. Neuroinflammation 19:149. doi: 10.1186/s12974-022-02514-x
Keywords: Alzheimer’s disease, COVID-19, interferons, drug repositioning, innate immunity, neurogenesis, cognitive impairment
Citation: Vavougios GD, Tseriotis V-S, Liampas A, Mavridis T, de Erausquin GA and Hadjigeorgiou G (2024) Type I interferon signaling, cognition and neurodegeneration following COVID-19: update on a mechanistic pathogenetic model with implications for Alzheimer’s disease. Front. Hum. Neurosci. 18:1352118. doi: 10.3389/fnhum.2024.1352118
Edited by:
Kevin Nicholas Hascup, Southern Illinois University Carbondale, United StatesReviewed by:
Mohammad Mofatteh, Queen's University Belfast, United KingdomCopyright © 2024 Vavougios, Tseriotis, Liampas, Mavridis, de Erausquin and Hadjigeorgiou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George D. Vavougios, ZGFudGV2YXZvdWdpb3NAaG90bWFpbC5jb20=; dmF2b3VneWlvcy5nZW9yZ2lvc0B1Y3kuYWN5LmN5
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.