 Laetitia Francelle
Laetitia Francelle Caroline Lotz
Caroline Lotz Tiago Outeiro
Tiago Outeiro Emmanuel Brouillet
Emmanuel Brouillet Karine Merienne
Karine Merienne- 1Department of NeuroDegeneration and Restorative Research, University Medical Center Goettingen, Goettingen, Germany
- 2CNRS UMR 7364, Laboratory of Cognitive and Adaptive Neurosciences, University of Strasbourg, Strasbourg, France
- 3Commissariat à l’Energie Atomique et aux Energies Alternatives, Département de Recherche Fondamentale, Institut d’Imagerie Biomédicale, Molecular Imaging Center, Neurodegenerative diseases Laboratory, UMR 9199, CNRS Université Paris-Sud, Université Paris-Saclay, Fontenay-aux-Roses, France
Unbalanced epigenetic regulation is thought to contribute to the progression of several neurodegenerative diseases, including Huntington’s disease (HD), a genetic disorder considered as a paradigm of epigenetic dysregulation. In this review, we attempt to address open questions regarding the role of epigenetic changes in HD, in the light of recent advances in neuroepigenetics. We particularly discuss studies using genome-wide scale approaches that provide insights into the relationship between epigenetic regulations, gene expression and neuronal activity in normal and diseased neurons, including HD neurons. We propose that cell-type specific techniques and 3D-based methods will advance knowledge of epigenome in the context of brain region vulnerability in neurodegenerative diseases. A better understanding of the mechanisms underlying epigenetic changes and of their consequences in neurodegenerative diseases is required to design therapeutic strategies more effective than current strategies based on histone deacetylase (HDAC) inhibitors. Researches in HD may play a driving role in this process.
Introduction
Huntington’s disease (HD) is a genetic disease affecting preferentially medium spiny neurons (MSN) of the striatum. Increasing numbers of studies provide evidence for altered epigenetic regulations in HD. Here, after a summary of general epigenetic mechanisms in neurons, we review the state-of-the-art of epigenetic changes in HD. We discuss the mechanisms underlying these changes and their consequences, particularly on expression of neuronal identity genes. Current challenges to improve our understanding of the role of epigenetic mechanisms in HD are discussed, including the development of new technologies to profile neuronal epigenomes at single-cell or cell-type specific levels. Epigenetic modifications represent promising therapeutics. We discuss the need of identifying new epigenetic targets for therapy, representing alternatives to histone deacetylase (HDAC) inhibitors.
Epigenetic Mechanisms: General Rules
The current definition of epigenetics relates to “the study of phenomena and mechanisms that cause chromosome-bound, heritable changes to gene expression that are not dependent on changes to DNA sequence” (Deans and Maggert, 2015). Epigenetic mechanisms actually regulate several DNA/RNA-mediated processes, including transcription, DNA repair and DNA replication, through modulation of the structure of chromatin, a macromolecular complex of DNA, RNA and proteins such as histones. Two major epigenetic mechanisms influence chromatin structure: histone modifications and DNA methylation (Allis and Jenuwein, 2016) (Figure 1).
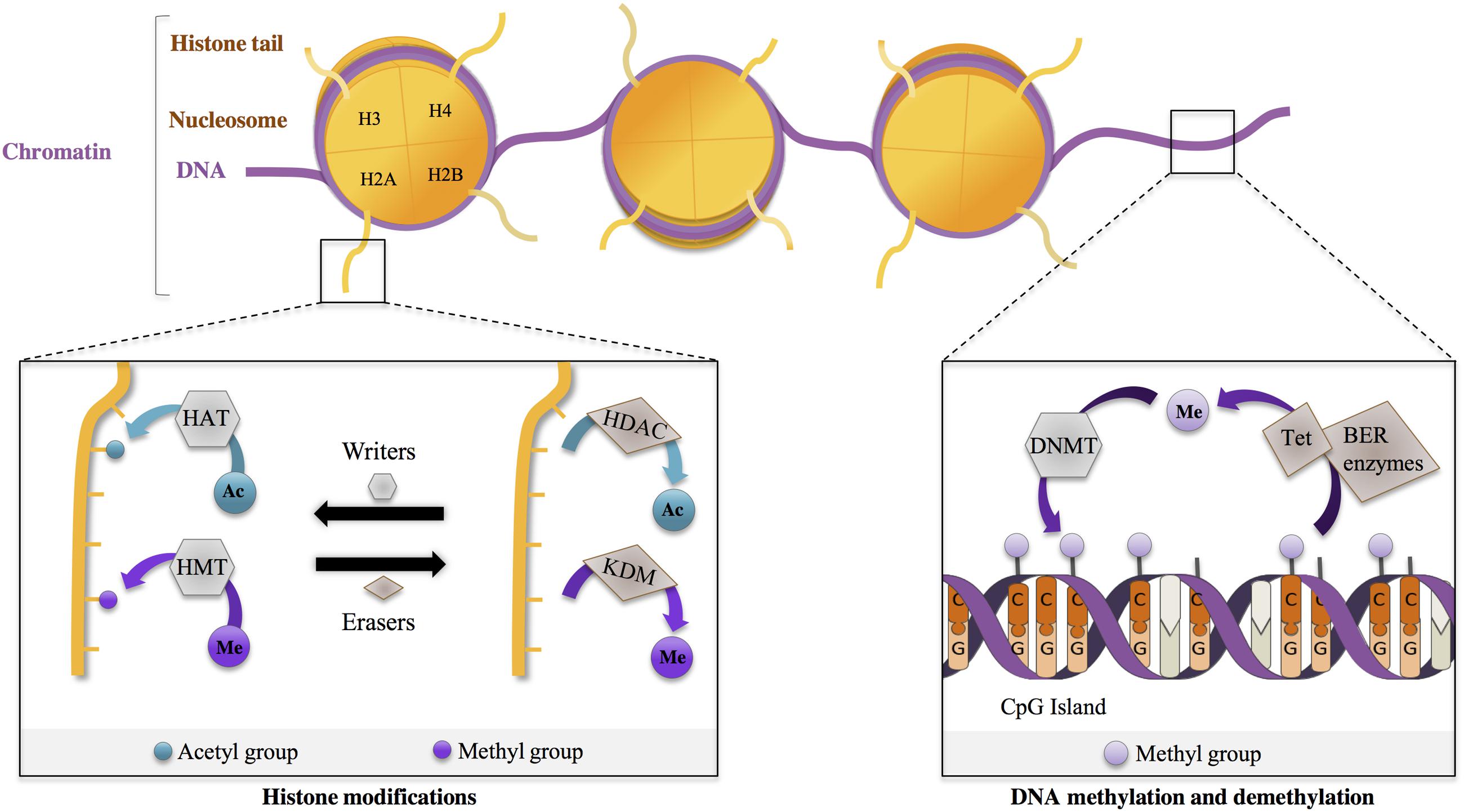
FIGURE 1. Epigenetic regulations are dynamic. Chromosomal DNA is wrapped around histone octamers (comprising dimers of H2A, H2B, H3, H4), forming nucleosomes, the core unit of chromatin. Protruding N-terminus histone tails can undergo post-translational modifications (PTM) that influence chromatin state (e.g., relaxed vs. compact state) Epigenetic writers add PTM on histone tail residues. For example, histone acetyltransferases (HAT) add acetyl- (Ac) groups, and histone methyltransferases (HMT) add methyl- (Me) groups. In a dynamic manner, these marks are removed by epigenetic erasers, such as histone deacetylases (HDAC) and lysine demethylases (KDM), removing, respectively, Ac- and Me-groups. DNA methylation at cytosines, particularly at CpG dinucleotides, is performed by DNA methyltransferases (DNMT). DNA demethylation involves hydroxylation and oxydation of methylated cytosines by Tet proteins (Tet), which are then repaired by DNA repair mechanisms, including base excision repair (BER).
In the nucleus, DNA is wrapped around core particles of chromatin, the nucleosomes, which are formed of octamers of histones, including H2A, H2B, H3, and H4. Histones are subject to various post-translational modifications (PTM) such as acetylation, methylation, phosphorylation and ubiquitylation. These histone modifications modulate the degree of compaction of nucleosomes, thereby affecting chromatin accessibility to various factors, particularly transcriptional regulators. (Jenuwein and Allis, 2001; Borrelli et al., 2008; Wang and Jin, 2010; Tsompana and Buck, 2014; Xu et al., 2014). Importantly, histone residues can be modified in a combinatorial, reversible, and targeted manner (Figure 1). For instance, histone acetyltransferases (HAT) and HDAC are involved in acetylation and deacetylation of specific histone residues, respectively (Figure 1). Similarly, methylases and demethylases regulate the addition and removal of methyl groups on histone residues (Figure 1). The combinatorial, reversible and targeted nature of histone modifications is the basis of the so-called ‘histone code’. It permits to achieve specificity in the outcome, through the action of proteins interpreting the code (e.g., “readers”) (Jenuwein and Allis, 2001; Bannister and Kouzarides, 2011; Jones et al., 2016). One major outcome is transcription. General rules of the “transcriptional” histone code are relatively well defined. For instance, histone acetylation, whatever the histone or the residue modified, promotes relaxed chromatin and is associated with transcriptional activation, whereas histone methylation, depending on the specific residue that is modified, can lead to transcriptional repression or activation. For instance, H3K9 methylation is associated with transcription repression, whereas H3K4 methylation correlates with transcription activation. Moreover, the genome comprises different regulatory regions, including promoters and enhancers (i.e., regulatory regions distal to promoters) playing specific roles in transcriptional regulation. These regions display specific histone modifications. For instance, promoters and enhancers of active genes are enriched in trimethylated H3K4 (H3K4me3) and in acetylated H3K27 (H3K27ac) respectively, further illustrating the targeting precision permitted by the histone code.
DNA methylation, another important epigenetic mechanism, consists in adding a methyl group to cytosines by DNA methyltransferases (DNMT), particularly at C5 position of cytosine in cytosine-guanine dinucleotide sequences (CpG), creating 5-methylcytosine (5-mC). DNMT1 is implicated in the maintenance of methylation patterns during DNA replication. In contrast, DNMT3a and DNMT3b have been associated with de novo DNA methylation (Jeltsch, 2006; Feng et al., 2010). Though DNA methylation was long considered as a stable process, it is now clear that post-mitotic cells can undergo active DNA demethylation, through a mechanism implicating TET proteins (Pastor et al., 2013). TET proteins induce hydroxylation of 5-mC (leading to 5-hmc) and are further involved in their oxidation. Oxidized 5-hmC are then processed by DNA repair mechanisms and converted back to their unmethylated state (Guo et al., 2011; Feng et al., 2015). 5-hmC is particularly extended in neurons, suggesting that regulation of DNA methylation is highly dynamic in these cells (Song et al., 2011; Szulwach et al., 2011). 5-mC are bound by several classes of methyl-binding proteins (such as MeCP2), which associate with other protein partners, including HDAC, forming co-repressor complexes (Urdinguio et al., 2009; Du et al., 2015). DNA methylation at gene promoters, generally enriched in CpG sites and forming so-called CpG islands, is an important mechanism involved in gene repression (Deaton and Bird, 2011) (Figure 1).
The collection of epigenetic modifications (e.g., the epigenome) can be assessed at genome-wide scale, using next generation sequencing-based techniques such as chromatin-immunoprecipitation-sequencing (ChIP-seq). Generation and integration of epigenomic data with transcriptomic and/or functional data to fully decode epigenomes at cell-type specific level is a major current challenge that requires the development of new techniques and analysis methods (Marconett et al., 2013; Shin et al., 2014). In particular, methods to process low cell numbers/single-cell need to be developed as well as powerful bioinformatics tools (Bock et al., 2016).
Neuroepigenetics: Why are Neurons Specific?
Epigenetic mechanisms, interfacing individual genomes with environmental factors, are essential to the regulation of fundamental biological processes. Historically, epigenetic mechanisms have been primarily explored in the context of development, including cellular differentiation and the establishment of stable cellular identity (Holliday, 2006; Roth and Sweatt, 2011; Boland et al., 2014). In fact, cell state transitions during development require massive epigenetic changes, whose stabilization enables the maintenance of cell-type specific identity. The mechanisms involve the establishment of defined transcriptional programs. As a result, the epigenome is a gatekeeper of cell-type specific identity. However, this view of stable epigenetics has been challenged, due to evidence showing that massive and dynamic epigenetic changes can be implicated in the regulation of cellular activity. Such “plasticity” of epigenetic regulations is particularly critical to neuronal cell activity; hence the concept of neuroepigenetics (Borrelli et al., 2008; Riccio, 2010; Sweatt, 2013; Deans and Maggert, 2015).
Neuronal excitability is one major property of neuronal cells. In response to environmental experience, including learning, drug exposure, psychological and physical stress environmental signals, neurons undergo reversible transitions from resting to active (or excited) states, which influence synaptic plasticity and promote adaptive behavior, such as learning and memory (Sultan and Day, 2011; Sweatt, 2016). These processes are highly dynamic, and can also be long-lasting. Increasing evidence indicates that epigenetic mechanisms regulate the transition from resting to active neuronal state (Korzus, 2010; Landgrave-Gomez et al., 2015; Meadows et al., 2016).
Learning and memory processes are associated with synaptic plasticity, leading to the rapid formation of new synapses, which can be strengthened or lost over time. Synaptic plasticity correlates with dynamic changes at the level of histone modifications. Specifically, changes in acetylation of histones, including H3 and H4 have been associated with early formation of new synapses (Federman et al., 2009; Chatterjee et al., 2013; Graff and Tsai, 2013; Peixoto and Abel, 2013; Benito et al., 2015). Moreover, neuronal activity can be associated with memory storage and consolidation, which may involve long-term remodeling of neuronal networks at system levels. Epigenetic regulations have also been implicated in these processes. Upon learning paradigms, the chromatin in brain tissues implicated in memory formation and/or consolidation, such as the hippocampus and the cortex, undergoes extensive modifications, including increased histone acetylation and DNA methylation changes (Day and Sweatt, 2011; Bousiges et al., 2013; Zovkic et al., 2013; Halder et al., 2016). DNA methylation and histone modifications both control memory processes, through transcriptional effects that comprise the activation of synaptic plasticity genes, including immediate early genes (IEGs) like Fos, Egr1, or Arc (Minatohara et al., 2015; Thakurela et al., 2015). Remarkably, these transcriptional effects that are experience-driven and epigenetically regulated can be long lasting. Activation of specific signaling pathways, such as the cAMP/CREB/CREB-binding protein (CBP) pathway, is involved in the coupling between epigenetic and transcriptional responses, promoting the recruitment of protein complexes at target genes, which induces chromatin remodeling and drive transcription (Cedar and Bergman, 2009; Tie et al., 2014; Alberini and Kandel, 2015; Ortega-Martinez, 2015).
However, although tight coupling between neuronal-activity-regulated epigenetic and transcriptional changes remains a favored hypothesis, recent data, integrating epigenomic, and transcriptomic data suggest that the link between both events may not be as clear as anticipated (Lopez-Atalaya and Barco, 2014; Liu et al., 2015; Valor, 2015b). Recently, contextual fear conditioning in mice was used as a learning paradigm to examine the spatiotemporal correlation between epigenetic and transcriptional modifications in neurons (Halder et al., 2016). Specifically, DNA methylation and seven histone modifications were assessed at a genome-wide scale using hippocampal and cortical neurons of mice that were subjected to contextual fear conditioning. To specify the timing of epigenetic changes in these tissues, analyses were performed at different time points with respect to contextual fear conditioning, associated with different memory processes, including memory formation and consolidation. Transcriptomic analyses were also performed on the same tissues. Generally, histone modifications were extensively modulated during memory formation, and these widespread changes weakly correlated with gene-selective transcriptional changes. In contrast, DNA methylation changes were rather locus-specific and correlated with transcriptional changes, including splicing events (Lev Maor et al., 2015; Halder et al., 2016). Thus, the coupling between epigenetic and transcriptional changes may depend on chromatin modifications. Additional studies are required to specify causal relationship between the two events. Other neuronal cells/tissues and experimental paradigms may be used, which would permit to investigate the interplay between epigenetics and transcription in brain cells/tissues associated with additional brain functions, including other cognitive functions, motor functions and functions linked to emotion and motivation regulation.
Thus, it appears that epigenetic mechanisms in neurons not only control their identity, like in other cell types, but also regulate neuronal activation, including the ability to undergo dynamic plasticity in response to environmental signals. Then, the question arising is what is going on in pathological situations, when neuronal function is impaired? Does it result from altered activation capacity of affected neurons or from a loss of neuronal identity? Are impaired epigenetic regulations implicated in pathological processes? These questions are particularly relevant to neurodegenerative diseases where specific neuronal populations (or identities) are preferentially affected. In the following sections, we have chosen to focus on one such disease, HD, playing a pioneering role in the understanding of the epigenetic regulation mechanisms in brain diseases.
Epigenetics in HD
Huntington’s disease is a neurodegenerative disease caused by an unstable expanded CAG repeats (>35–39 repeats) in the Huntingtin gene (HTT), which results in the production of mutant protein (mHtt) with a toxic polyglutamine (polyQ) tract (Landles and Bates, 2004). HD is characterized by specific symptoms, including motor impairment (e.g., chorea, bradykinesia, gait abnormalities, dystonia), cognitive deficits (motor skill learning deficits, planning, and attention troubles) and psychiatric alterations (depression, mania, apathy, suicide), usually appearing at adulthood (see as review, Roos, 2010). Since polyQ expansion toxicity is correlated with repeat size, HD patients with longer CAG expansions are more severely affected, showing earlier onset of symptoms and faster pathology progression. HD is characterized by a preferential and primary degeneration of two structures of the basal ganglia: the caudate nucleus and the putamen that form the neostriatum. However, additional brain regions, particularly the cortex, degenerate as the pathology progresses (Rosas et al., 2008). Remarkably, in HD striatum, selective neuronal populations, the GABAergic MSN, are more particularly vulnerable, whereas the large cholinergic interneurons, the medium size GABAergic interneurons and glial cells appear spared (Ferrante et al., 1987a,b; Cicchetti et al., 1996). From a biochemical point of view, polyQ-Htt presents a high propensity to misfold and aggregate, which leads to the formation of nuclear inclusions, particularly in neurons (DiFiglia et al., 1997; Li L. et al., 2016). These aggregates, a disease hallmark, recruit a number of proteins, including transcriptional regulators (Steffan et al., 2000; Arrasate and Finkbeiner, 2012).
A long period of neuronal dysfunction precedes the death of neurons sensitive to the HD mutation. Several cellular processes are believed to contribute to neuronal dysfunction, including Htt cleavage and aggregation, abnormal protein-protein interactions, dysfunctional calcium signaling, abnormal axonal transport, impaired energy metabolism, dysregulation of neuronal activity, and altered transcription (see as reviews Landles and Bates, 2004; Zuccato et al., 2010; Labbadia and Morimoto, 2013; Saudou and Humbert, 2016).
The “Neuronal” Signature of the HD Transcriptome
Transcriptional dysregulation plays a central role in HD pathogenesis (Seredenina and Luthi-Carter, 2012). Major transcriptional changes have been reported in the brains of HD patients (Hodges et al., 2006). This is also observed in HD mouse models, including transgenic and knock-in mice (Luthi-Carter et al., 2000; Luthi-Carter et al., 2002; Zucker et al., 2005; Roze et al., 2008). Transcriptomic studies using mice showed that transcriptional changes are progressive, CAG repeat-length-dependent and most extended in the striatum (Desplats et al., 2006; Kuhn et al., 2007; Becanovic et al., 2010; Langfelder et al., 2016). In the striatum of HD models, transcriptional changes occur in both directions: many genes are down- and up-regulated (Seredenina and Luthi-Carter, 2012; Francelle et al., 2014). Remarkably, down-regulated genes display a neuronal signature, since decreased genes in HD striatum are enriched in genes that define striatal neuron identity and function, such as Darpp32, Rgs9, Drd1 or Drd2 (Hodges et al., 2006; Kuhn et al., 2007; Vashishtha et al., 2013; Achour et al., 2015; Langfelder et al., 2016). In the other affected brain regions such as the cortex, fewer genes are down-regulated. However, they also present a neuronal signature, reflecting tissue identity (Vashishtha et al., 2013; Langfelder et al., 2016).
Interestingly, a recent study using RNAseq revealed a developmental signature associated with differentially expressed genes in post-mortem prefrontal cortex of HD patients. Specifically, Hox genes and additional homeobox genes were re-expressed in HD brains, suggesting that the transcriptome of HD neurons resemble that of immature neurons (Labadorf et al., 2015). Thus, these results support the view that the transcriptional program involved in the maintenance of neuronal identity is impaired in HD neurons. Up-regulated genes in the striatum or cortex of HD patients were also enriched in biological processes linked to inflammation (Hodges et al., 2006; Labadorf et al., 2015). However, up-regulation of immune response genes is not that clear in mouse models (Achour et al., 2015; Langfelder et al., 2016). Thus, down- and up-regulated genes in HD brain tissues display distinct functional signatures, suggesting that different mechanisms may operate.
The mechanisms underlying mutant Htt-induced transcriptional effects are unclear. However, they are thought to involve both altered regulation of transcription regulators and histone-modifying enzymes.
HDACi in HD: What are the Targets?
The hypothesis of an epigenetic origin of transcriptional dysregulation in HD, particularly implicating histone acetylation, has received increasing support over the years (Jaenisch and Bird, 2003; Lee et al., 2013; Glajch and Sadri-Vakili, 2015). The assumption that altered histone acetylation might contribute to HD was made in the early 2000s, when it was found that the HAT CREB-binding protein CBP is recruited into aggregates of mutant Htt, and that HDAC inhibitors (HDACi) improve phenotypes of drosophila and mouse models of HD (Nucifora et al., 2001; Steffan et al., 2001; Kazantsev et al., 2002). As a result, epigenetic strategies using HDACi to increase histone acetylation have been early considered for HD. Several preclinical studies have been performed using broad-spectrum HDACi (e.g., suberoxylanilide hydroxamic acid (SAHA), Trichostatin A (TSA), phenylbutyrate, sodium butyrate (NaB)] that target non-selectively HDAC of class I and II (Ferrante et al., 2003; Hockly et al., 2003; Gardian et al., 2005; Sadri-Vakili et al., 2007; Sharma and Taliyan, 2015b).
Histone deacetylase inhibitors improve some phenotypes of HD mice, including neuropathology and motor function. However, it is unclear whether beneficial effects require increased histone acetylation. Instead, studies suggest that the mechanism may involve acetylation of non-histone proteins. In support to this view, inactivation of a target of SAHA, Hdac 4, ameliorates neurodegeneration in HD mice through an apparently, transcription-independent mechanism, acting on mutant Htt aggregation process (Mielcarek et al., 2011). Non-histone-mediated beneficial effects of HDACi have also been documented in models of Parkinson disease (PD; Godena et al., 2014), suggesting common mechanisms between several neurodegenerative diseases.
New compounds have been developed in an attempt to generate more selective HDACi and with less toxic side effects (Herman et al., 2006; Thomas et al., 2008). The compound HDACi 4b, which was reported to ameliorate disease phenotype of HD mice, show high potency for inhibiting HDAC1 and HDAC3 (Thomas et al., 2008; Jia et al., 2012). However, physicochemical properties and metabolic profile of this compound were found suboptimal for investigation of HDAC inhibition in mice per oral administration (Beconi et al., 2012). The effect of RGFP966, an HDAC3-selective inhibitor, was recently investigated using HD mice (Jia et al., 2016). The results suggest that the compound limits glial cell response, diminishing markers of glial cell activation. Surprisingly however, a heterozygous inactivation of the Hdac3 gene in HD mice did not ameliorate disease-related phenotypes (Moumne et al., 2012), suggesting that more than 50% knock-down of the Hdac3 gene might be needed to see a beneficial effect. More recently, beneficial transgenerational effects have been reported using HDACi 4b in HD mice (Jia et al., 2015). Thus, despite some beneficial effects, the mode of action of HDACi in HD models remains elusive and may not systematically implicate histone- and transcription-dependent mechanisms.
It is still unclear whether expression of neuronal identity genes in HD brain tissues is restored upon HDACi treatment (Coppede, 2014; Wang et al., 2014; Sharma et al., 2015; Valor, 2015a). Clinical studies using HDACi are ongoing and the results are awaited. So far, Phase II studies provide indication for safety and tolerability of several compounds, including phenylbutyrate (Hogarth et al., 2007; Westerberg et al., 2015).
But what exactly is the status of the HD epigenome? Is acetylation the only histone modification impaired in HD affected tissues, and to which extent? Are acetylated histone residues all affected the same way by the HD mutation? Is DNA methylation also impaired? Is the chromatin structure globally altered and repressed or is it altered at specific genomic regions or gene loci? What is the underlying mechanism? What are the consequences of chromatin structures changes? Do they underlie transcriptional changes? Addressing these questions is certainly a prerequisite to the development of new epigenetic therapies for HD.
HD Epigenome
Many studies have already been performed that attempt to address these issues (Lee et al., 2013; Valor and Guiretti, 2014; Glajch and Sadri-Vakili, 2015). It is expected that the use of genome-scale approaches, which has remained so far rather limited in the HD field, will improve our knowledge of HD epigenome as well as provide insights into the mechanism responsible for altered epigenetic regulation in HD.
Relationship between Epigenetic and Transcriptional Changes in HD
Histone acetylation
Extensive changes in histone acetylation levels were observed in cellular systems based on mutant Htt overexpression (Steffan et al., 2001; Igarashi et al., 2003). However, global levels of H2B, H3 and H4 acetylation appeared unchanged between brain tissues of HD R6/2 and control mice (Hockly et al., 2003; Sadri-Vakili et al., 2007). Studies using the striatum of HD mice further suggested that decreased H3 acetylation occurs at selective gene loci, particularly at promoters of down-regulated genes such as Drd2, Penk1, Actb, or Grin1 (Sadri-Vakili et al., 2007). Using chromatin immunoprecipitation paired with microarray hybridization (ChIP-chip), McFarland et al. (2012) assessed histone acetylation changes at genome-wide scale in the striatum of HD R6/2 transgenic mice. H3K9 and H3K14 acetylations (H3K9,14ac) and transcriptional changes between HD and WT striatum were poorly correlated in the striatum of HD R6/2 mice, suggesting that variation in H3K9,14ac levels alone may not be sufficient to account for gene expression changes in HD mice. Valor et al. (2013) reached similar conclusions by investigating changes in H3K9,14 ac and H4K12ac using a more powerful method –ChIPseq- on hippocampus and cerebellum of the HD transgenic N171-82Q mouse model (Figure 2). However, whereas data obtained by McFarland et al. (2012) suggest broad changes in histone acetylation in HD mouse striatum when compared to WT striatum, the results reported by Valor et al. (2013) indicate that changes are restricted to few loci. The absence of bulk changes in histone acetylation is further supported by a study showing promoter deacetylation of H3 at specific loci in HD models (Guiretti et al., 2016).
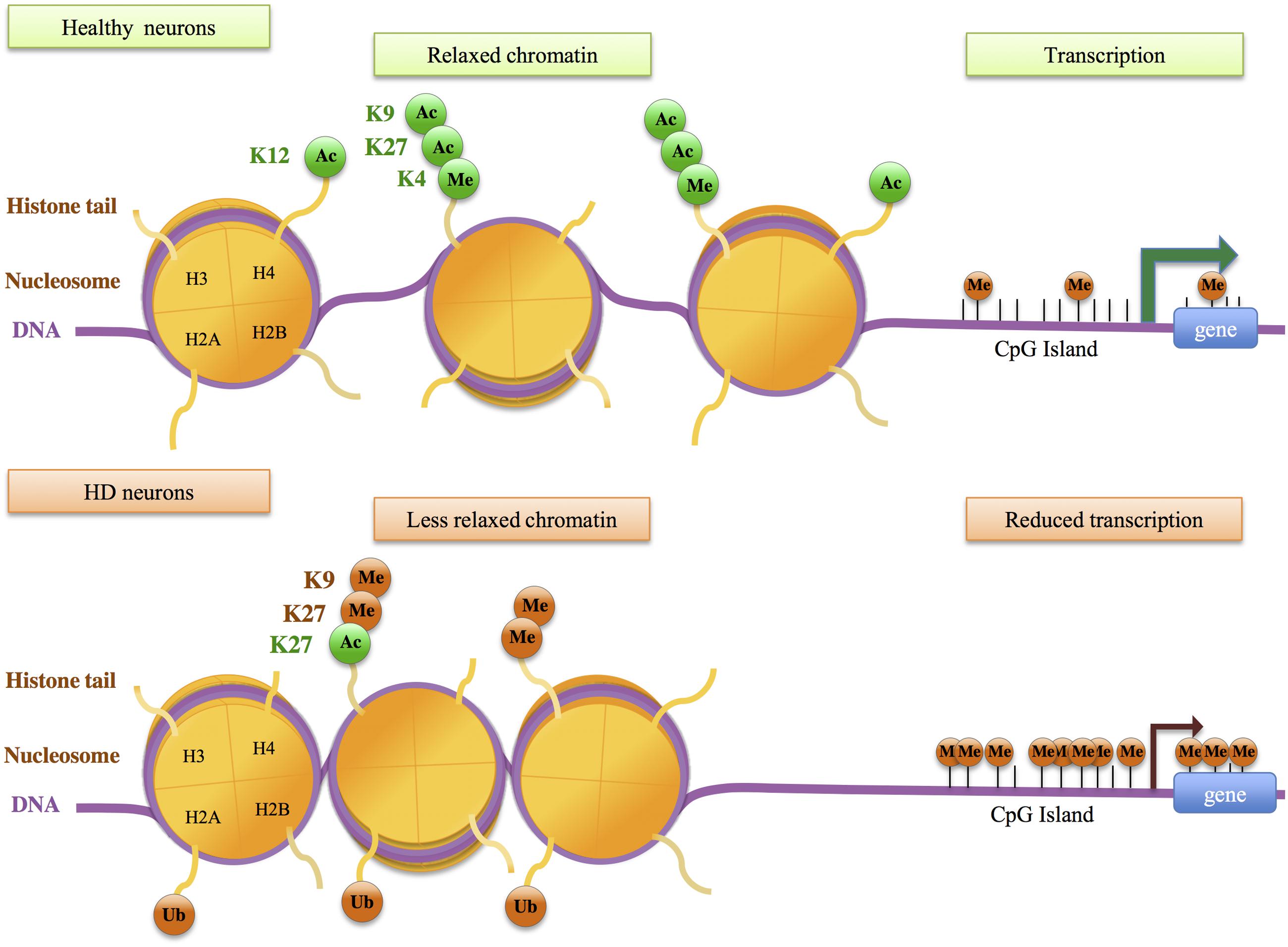
FIGURE 2. Histone modifications and DNA methylation changes in Huntington’s disease (HD) neurons. Genes in healthy neurons are enriched in transcriptionally active histone modifications, including acetylation of histone H3 at lysine 27 (H3K27ac), H3K9ac, H4K12ac, trimethylation of H3K4 (H3K4me3). Down-regulated genes in HD neurons show decreased levels of active histone modifications and increased levels of histone modifications associated with transcriptionnally repressed chromatin, including H3K9me2, H3K27me3, and ubiquitylation of H2A (H2A ub), resulting in less relaxed chromatin and decreased transcription. These events may be associated with increased DNA methylation at gene promoters.
H3K27ac, a mark of active enhancers, was also selectively decreased in the striatum of HD R6/1 mice (Achour et al., 2015) (Figure 2). Integrating H3K27ac ChIPseq with RNA polymerase II (RNAPII) ChIPseq and RNAseq data, Achour et al. (2015) showed that H3K27ac and RNAPII are decreased at regions enriched in down-regulated genes, providing evidence for a strong correlation between decreased H3K27ac, decreased RNAPII and gene down-regulation in HD mouse striatum. Moreover, enhancer regions presenting reduced H3K27ac in HD R6/1 striatum were enriched in super-enhancers, a category of broad enhancers, regulating genes that define cell type-specific identity and function (Figure 3). In fact, striatal super-enhancer genes down-regulated in HD mouse striatum were enriched in genes controlling neuronal activity, including neuronal plasticity and transmission (Achour et al., 2015). Thus, the results suggest that selective decrease in super-enhancer activity underlies HD neuronal transcriptomic signature (e.g., down-regulation of genes that define neuronal identity and function, referred to as neuronal identity genes thereafter). This supports an epigenetic origin of gene down-regulation in HD.
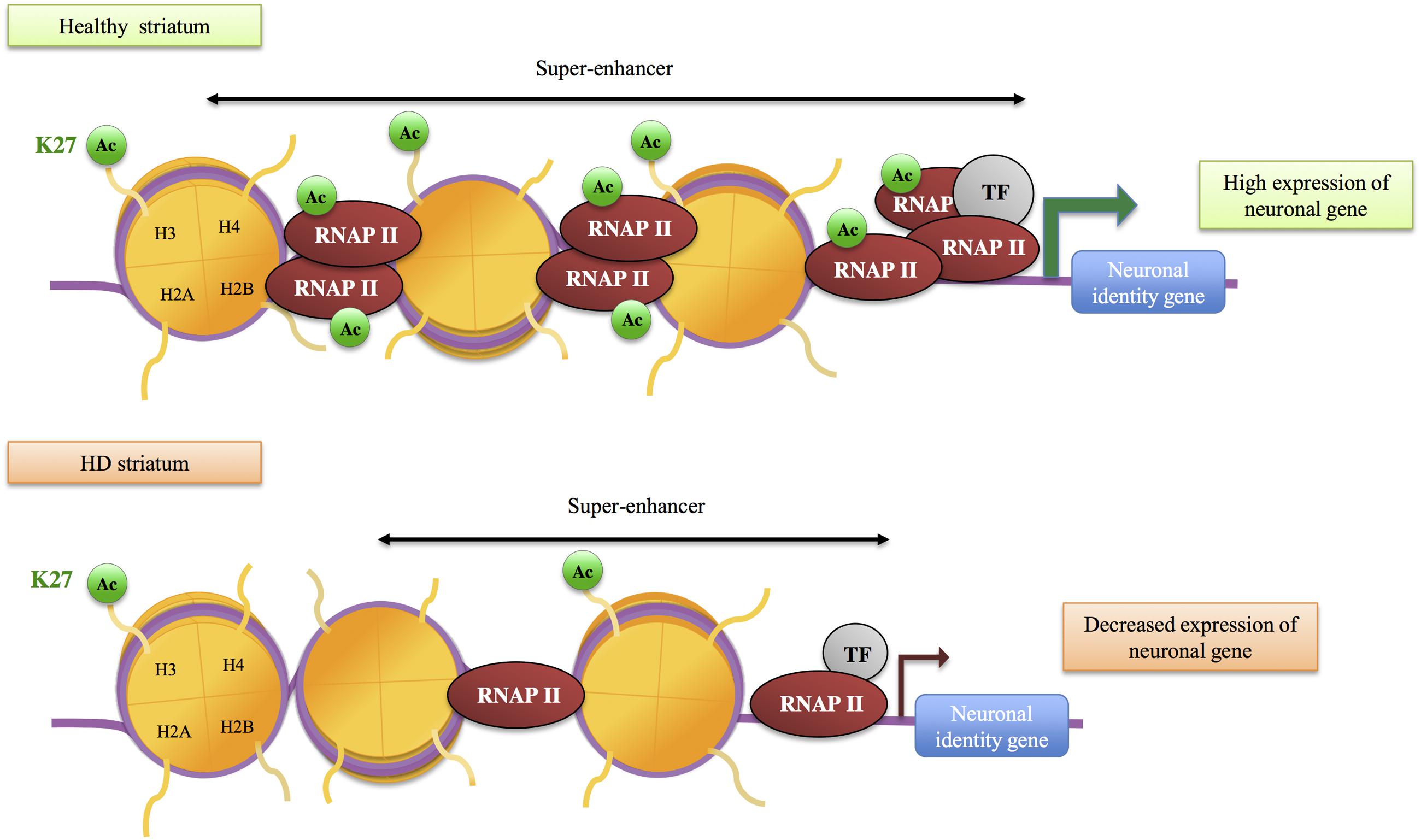
FIGURE 3. Role of super-enhancers in gene down-regulation in HD. Super-enhancers, a category of large enhancers enriched in H3K27ac, regulate transcription of neuronal identity genes in the striatum. In HD mouse striatum, H3K27ac and RNA polymerase II (RNAP II) are decreased at super-enhancers, which likely contributes to reduced transcription of neuronal identity genes. TF, transcription factor.
Other histone modifications
The above results support the view that specific regulatory elements, super-enhancers, are sensitive to the HD mutation. Recent data indicate that selective promoters are also impaired in the striatum of HD mice and patients. Investigating H3K4me3, a mark of active promoters, down-regulated genes in the striatum and cortex of HD R6/2 mice were found to preferentially associate with broad promoters, regulating genes enriched in biological processes linked to neuronal function (Vashishtha et al., 2013). Moreover, H3K4me3 appeared decreased at down-regulated genes in R6/2 vs. WT tissues (Figure 2). Thus, these results show that specific broad promoter signature associates with decreased expression of neuronal genes in the striatum and cortex of HD mice. It is very likely that target genes of broad promoters and super-enhancers in brain tissues overlap and are enriched in neuronal identity genes. Moreover, in a study using ChIPseq on embryonic stem cells (ESC) and neural progenitor cells (NPC) expressing mutant Htt with various CAG sizes, a correlation was reported between CAG-repeat-dependent changes in gene expression and in H3K4me3 levels, particularly in NSC (Biagioli et al., 2015). This raises the hypothesis that mutant Htt might alter epigenetic regulation at early stage of neuronal differentiation. Finally, H3K4me3 has been investigated in the prefrontal cortex of HD patients using ChIPseq (Bai et al., 2015; Dong et al., 2015). In contrast to mouse data, human data indicate that epigenetic and transcriptional changes in HD vs control tissues are poorly correlated (Bai et al., 2015; Dong et al., 2015). However, in human experiments, sequencing depth appears suboptimal, which may have resulted in underestimation of H3K4me3 signals. Interestingly however, the study by Dong et al. (2015) indicates that promoters differentially enriched in H3K4me3 associate with genes involved in pathways or network linked to neuronal activity and inflammation, suggesting that transcriptional changes affecting inflammatory genes, in addition to those affecting neuronal genes, involve epigenetic mechanisms (Figure 2). The hypothesis that induction of inflammatory genes in HD neuronal tissues associates with loss of identity of glial cells would need to be investigated (Gabel et al., 2016).
Additional epigenetic modifications at regulatory regions might also be impaired. Increased levels of H3K9me2, a mark associated with heterochromatin, have been reported in the striatum of HD patients and R6/2 mice, using immunohistological analyses (Ryu et al., 2006). Whether specific loci are more particularly sensitive to increased H3K9 methylation in HD models has yet to be investigated using genome-wide approach. Moreover, the level of H3K27me3, a repressive histone modification that can mark promoters and enhancers, was modulated by CAG-repeat size in both ESC and NPC (Seong et al., 2010; Biagioli et al., 2015). However, the transcriptional consequences of this effect were unclear. Finally, H2A ubiquitylation (H2Aub) was found increased at down-regulated genes in HD R6/2 mice (Kim et al., 2008), and ChIP-on-chip analysis of H2Aub changes in R6/2 striatum indicated that histone changes were not restricted to dysregulated genes (McFarland et al., 2012).
DNA methylation
Genome-wide analysis of DNA methylation was performed in HD cell models (Ng et al., 2013). Changes in DNA methylation in response to mutant Htt were observed at both promoter proximal and distal regulatory regions. Interestingly, a large fraction of the genes that changed in expression upon mutant Htt expression displayed changes in DNA methylation, suggestive of a causal relationship (Figure 2). DNA methylation was also profiled using post-mortem cortex and liver from HD patients (De Souza et al., 2016). The results revealed minimal evidence of HD-associated DNA methylation. However, the HTT gene was methylated in a tissue-specific manner, which might lead to tissue-specific regulation of HTT promoter activity. Additionally, 5-hydroxymethylcytosine (5-hmC) and 7-methyl guanine (7-MG) were globally reduced in brain tissues of HD mouse models, including YAC128 mice (5-hmC study), R6/2 and CAG140 knockin mice (7-MG study) (Thomas et al., 2013; Wang et al., 2013). While these studies further provide evidence for altered DNA methylation in HD brain tissues, transcriptional consequences of such impairments remains elusive. Analysis of DNA methylation has also been used to assess the epigenetic clock of HD patients (Horvath et al., 2016). Horvath recently developed an epigenetic measure of tissue age, so-called epigenetic age, estimated from DNA methylation levels of 353 CpG sites (Horvath, 2013). The study using brain tissues from HD patients showed accelerated epigenetic aging in HD brain, particularly in cortical tissues. However, this was not the case for striatal tissues, possibly due to excessive neuronal loss (Horvath et al., 2016). While transcriptional significance of accelerated aging in HD brain is unclear, the data might reveal an age-dependent alteration of epigenetic regulation. Finally, treatment of HD mice with the HDAC inhibitor HDACi 4b led to transgenerational effects, possibly mediated by increased DNA methylation at CpG sites associated with Kdm5d (Jia et al., 2015). Thus, DNA methylation may be a therapeutic target for HD.
Mechanisms Involved in Altered Epigenetic Regulation in HD
As mentioned above, altered histone acetylation in HD has been proposed to result from decreased activity of the HAT CBP, due to CBP recruitment into aggregates of mutant Htt, CBP depletion in neurons expressing mutant Htt and/or through an aberrant interaction of CBP with soluble mutant Htt (Steffan et al., 2000; Nucifora et al., 2001; Jiang et al., 2006; Seredenina and Luthi-Carter, 2012; Glajch and Sadri-Vakili, 2015) (Figure 4). The effect appears specific to CBP since CBP-related HAT p300 was unable to rescue cell toxicity in overexpression assays (Nucifora et al., 2001). However, although studies manipulating CBP levels in HD models support a role for CBP in pathogenesis (Steffan et al., 2000; Klevytska et al., 2010), including cognitive deficits (Giralt et al., 2012), it is still unclear whether altered CBP underlies HD neuronal transcriptional signature.
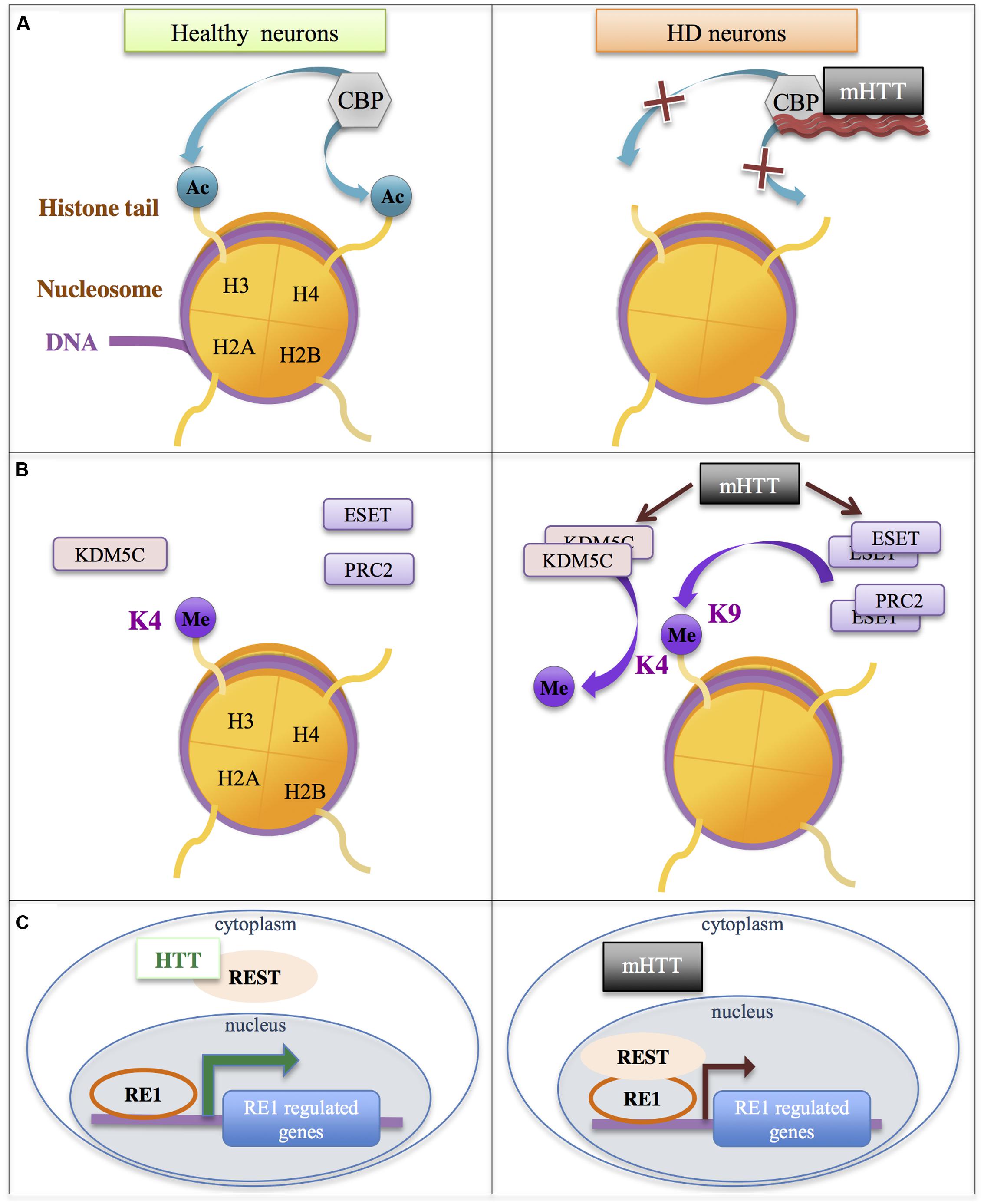
FIGURE 4. Additional mechanisms involved in gene down-regulation in HD. Mutant HTT (mHTT) leads to the modulation of the activity of chromatin modifiers/complexes (e.g., CBP, ESET, KDM5C, PRC2) and transcriptional regulators (e.g., REST). (A) The HAT CBP is sequestrated in mHTT aggregates and/or interacts with soluble mHTT, which reduces its activity, leading to reduction of histone acetylation. (B) Increased levels/activities of the H3K4me3 demethylase KDM5C, of the methyltransferase ESET and of polycomb repressive complex PRC2 lead to increased histone methylation. (C) HTT interacts with REST, limiting its availability in the nucleus. This interaction is impaired with mHTT, leading to increased binding of REST to RE1 elements and down-regulation of RE1-regulated genes. ESET, ERG-Associated Protein with SET Domain; HTT, Huntingtin; KDM5C, Lysine Demethylase 5C; PRC2, polycomb repressive complex 2; REST, RE1 Silencing Transcription Factor.
The activity of additional chromatin modulators was also found affected by Htt and/or mutant Htt. Htt was associated with polycomb repressive complex 2 (PRC2) in cell nucleus, which facilitated stimulation of PRC2 activity (Figure 4). Noticeably, CAG-expanded Htt further enhanced PRC2 activity in cell models (Seong et al., 2010). However, although H3K27me3 ChIPseq analyses revealed that Htt null mutation in ESC globally decreased H3K27me3, a result consistent with Htt-mediated stimulation of PRC2, the trend was opposite in NPC, suggesting that Htt may also be implicated in the process of H3K27me3 removal, and these different roles for Htt on H3K27me3 regulation may depend on cell differentiation state (Biagioli et al., 2015). Thus, the role of mutant Htt on genome-wide regulation of H3K27me3 needs to be further investigated, particularly in mature neurons. It is especially important to specify this issue since PRC2 deficiency in adult neurons leads to molecular, electrophysiological and behavioral phenotypes reminiscent to those seen in HD mice (von Schimmelmann et al., 2016). In fact, inactivation of PRC2 in striatal neurons resulted in re-expression of transcription factors involved in neuronal differentiation and in down-regulation of striatal identity genes, suggesting that transcriptional signatures resulting from PRC2 inactivation and from the expression of mutant Htt in the striatum share similarities (von Schimmelmann et al., 2016).
Additional enzymes modulating histone methylation were also deregulated in HD models. ESET/SETB1, a H3K9 methyltransferase, was increased in the striatum and cortex of HD R6/2 mice due to SP1/SP3-mediated transcriptional activation, which resulted in increased histone H3K9 methylation (Ryu et al., 2006) (Figure 4). Moreover, KDM5C/JARID1C, an enzyme involved in demethylation of H3K4me3, was up-regulated in the striatum and cortex of HD R6/2 mice and proposed to contribute to decreased H3K4me3 in HD brain tissues (Ng et al., 2013; Vashishtha et al., 2013) (Figure 4). Other studies further suggest that Htt and/or mutant Htt affect chromatin structure through modulation of the activity of enzymes regulating histone methylation, including H3K9 and H3K4 methylation (Ooi and Wood, 2007; Lee et al., 2013; Dietz et al., 2015).
As already mentioned, transcription factors also contribute to remodel the chromatin through the recruitment of histone modifying enzymes, including enzymes involved in histone acetylation and methylation. For instance, following binding to their cognate DNA response element, several transcription factors can interact with CBP in a stimulus-dependent manner, thereby increasing CBP concentration and histone acetylation at selective gene regulatory elements (e.g., enhancers and/or promoters) (Sterner and Berger, 2000). Thus, impaired level or activity of transcription factors in HD may also affect epigenetic regulations. Many transcription factors, including REST, SP1, TAF130, p53, were found deregulated in cells expressing the HD mutation, due to aberrant interaction with mutant Htt or altered transcriptional regulations (Nucifora et al., 2001; Bae et al., 2005; Ravache et al., 2010; Zuccato et al., 2010; Moumne et al., 2013; Langfelder et al., 2016) (Figure 3). It is tempting to speculate that some of these factors, particularly those regulating neuronal differentiation or maintenance of neuronal fate (this is for instance the case of REST), might contribute to HD epigenetic and transcriptional alterations.
Additionally, the ability of transcription factors to interact with chromatin-modifying enzymes depends in some cases on the activation of specific signaling pathways, which permits to optimize the coupling between epigenetic and transcriptional responses after stimulation. This is the case for CREB, which needs to be phosphorylated in response to activation of cAMP signaling pathway, to bind and recruit CBP on chromatin (Cardinaux et al., 2000). The Ras/MAPK/MSK1 signaling pathway is another pathway, where transcriptional and epigenetic responses are coordinated through phosphorylation events targeting transcription factors and histones (e.g., ELK1 and H3 at serine 10, respectively) (Brami-Cherrier et al., 2009; Bahrami and Drablos, 2016). These pathways, which control the dynamics of IEGs expression such as Fos, Egr1, and Arc, play a key role in the regulation of neuronal activity, including plasticity and excitability (Besnard et al., 2011; West and Greenberg, 2011; Lopez-Atalaya and Barco, 2014). Several studies suggest that these pathways are impaired in HD striatum, raising the hypothesis that the coupling between epigenetic and transcriptional mechanisms regulating neuronal activity is altered in HD (Besnard et al., 2011; Langfelder et al., 2016), which might contribute to altered epigenetic regulations (Chawla et al., 2003).
Despite these hypotheses, it remains unclear how mutant Htt leads to selective alteration of the chromatin associated with neuronal identity genes. The underlying mechanism may be the addition of direct effects of mutant Htt on the chromatin and transcription regulators described above (e.g., CBP, PRC2, ESET/SETB1, KDM5C/JARID1C, REST, SP1, CREB, TAF130, p53). Alternatively, it may be the result of an indirect mechanism caused by mutant Htt on neuronal homeostasis and activity that is yet to be identified.
Emerging Picture and Perspectives
Here, we reviewed the current understanding of the role of neuroepigenetics in HD, in an attempt to uncover the significance of epigenetic changes in HD brain, a prerequisite to the rational design of epigenetic therapies.
Three main questions underlie the review: (1) What are epigenetic changes in HD? (2) What is the consequence of epigenetic changes in HD, particularly on transcriptional regulation? (3) What mechanism(s) cause(s) epigenetic changes in HD? Although, we still lack complete answers to any of these questions, an emerging picture arises, supporting the view that maintenance and/or establishment of neuron-specific chromatin identity is altered in HD, which leads to down-regulation of neuronal identity genes. This suggests that mutant Htt might interfere with neuronal differentiation, in agreement with recent studies (Molero et al., 2016). To investigate this hypothesis, it will be crucial to assess HD epigenome and transcriptome across time, including developmental stages. If altered epigenetic regulation in HD brain results in down-regulation of neuronal identity genes, it is very likely to disturb neuronal activity, including synaptic plasticity and neuronal excitability, controlling learning and memory processes. This will need to be investigated. The issue of tissue-/cell-specificity of epigenetic alterations in HD also remains to be investigated. It is unclear whether the HD mutation induces similar epigenetic changes between cell types (e.g., neurons vs. glial cells) or between different tissues (e.g., striatum vs. cortex, hippocampus or cerebellum, neuronal tissue vs. non-neuronal tissue). It will also be crucial to investigate the timing of establishment of super-enhancer signatures relative to transcriptional changes to specify the relationship between epigenetic and transcriptional changes in HD brain tissues. Addressing these questions will certainly provide insights into the mechanism causing epigenetic and transcriptional changes in HD, which may be a key to the identification of new therapeutic targets.
Comprehensive analysis of the HD epigenome using various HD models and genome-wide techniques, including techniques that permit to investigate epigenomes at cell type-specific or single-cell scale and in 3D (using chromosome conformation capture- (3C)-based methods) is necessary to address these questions. Another challenge will be to develop bioinformatics methods to analyze the data and extract meaning. This may require the development of approaches that reduce complexity, such as network-based methods (Langfelder and Horvath, 2008; Parmentier et al., 2013).
Epigenetic therapies to treat neurodegenerative diseases have been first considered for HD, when Steffan et al. (2000) showed that HDAC inhibitors improve the phenotype of HD flies. In fact, preclinical studies to evaluate the effect of HDAC inhibitors in HD have inspired other neurodegenerative diseases, including additional trinucleotide repeat (TNR) diseases (for review see Evans-Galea et al., 2013) PD, Alzheimer disease (AD), and amyotrophic lateral sclerosis (ALS) (Xu et al., 2011; Lazo-Gomez et al., 2013; Sharma and Taliyan, 2015a). Beneficial effects have been observed in preclinical models for these different diseases, though the effects of HDAC inhibitors appear partial and underlying mechanisms are unclear (Benito et al., 2015; Harrison et al., 2015). In the specific case of TNR diseases, HDAC inhibitors may affect disease progression through two independent mechanisms: (1) through a general effect on gene expression program, (2) through modulation of the stability of TNRs, which are subject to epigenetic regulations (Goula et al., 2012; Evans-Galea et al., 2013). To overcome limitations associated with the use of HDCAi, including the lack of specificity and toxicity, HDACi-alternative epigenetic therapies are currently being developed that are based on HAT activators, histone methyltransferase inhibitors and DNMT modulators (Table 1).
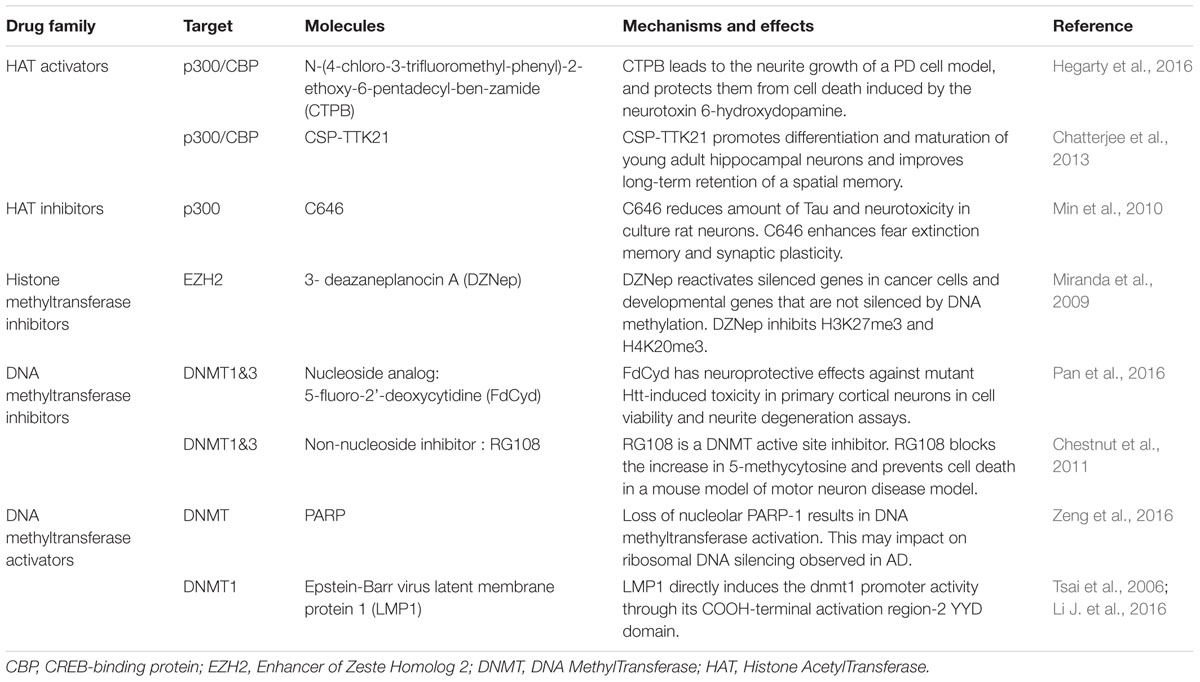
TABLE 1. Examples of histone deacetylase (HDACi)-alternative epigenetic therapies.
Epigenomic studies on HD models might benefit from studies in other neurodegenerative diseases and reciprocally. Indeed, the similarity of epigenomic and transcriptomic signatures between HD and AD models (e.g., neuronal and inflammation signatures) is intriguing and might suggest that common epigenetic mechanisms to several neurodegenerative diseases might operate (Gjoneska et al., 2015).
Author Contributions
LF and KM developed the literature review. KM, LF, and EB wrote the manuscript. LF and CL built the figures. All the authors read and commented the article throughout the writing, and collaborated in the revision of the final manuscript before submission.
Funding
This work was supported by funds from the Centre National de la Recherche Scientifique (CNRS). CL is a Ph.D. student supported by the French Research Ministry. LF is supported by a Postdoctoral Fellowship of the Alexander-von-Humboldt Foundation. TO is supported by the DFG Center for Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Achour, M., Le Gras, S., Keime, C., Parmentier, F., Lejeune, F. X., Boutillier, A. L., et al. (2015). Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington’s disease mice. Hum. Mol. Genet. 24, 3481–3496. doi: 10.1093/hmg/ddv099
Alberini, C. M., and Kandel, E. R. (2015). The regulation of transcription in memory consolidation. Cold Spring Harb. Perspect. Biol. 7:a021741. doi: 10.1101/cshperspect.a021741
Allis, C. D., and Jenuwein, T. (2016). The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500. doi: 10.1038/nrg.2016.59
Arrasate, M., and Finkbeiner, S. (2012). Protein aggregates in Huntington’s disease. Exp. Neurol. 238, 1–11. doi: 10.1016/j.expneurol.2011.12.013
Bae, B. I., Xu, H., Igarashi, S., Fujimuro, M., Agrawal, N., Taya, Y., et al. (2005). p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 47, 29–41. doi: 10.1016/j.neuron.2005.06.005
Bahrami, S., and Drablos, F. (2016). Gene regulation in the immediate-early response process. Adv. Biol. Regul. 62, 37–49. doi: 10.1016/j.jbior.2016.05.001
Bai, G., Cheung, I., Shulha, H. P., Coelho, J. E., Li, P., Dong, X., et al. (2015). Epigenetic dysregulation of hairy and enhancer of split 4 (HES4) is associated with striatal degeneration in postmortem Huntington brains. Hum. Mol. Genet. 24, 1441–1456. doi: 10.1093/hmg/ddu561
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Becanovic, K., Pouladi, M. A., Lim, R. S., Kuhn, A., Pavlidis, P., Luthi-Carter, R., et al. (2010). Transcriptional changes in Huntington disease identified using genome-wide expression profiling and cross-platform analysis. Hum. Mol. Genet. 19, 1438–1452. doi: 10.1093/hmg/ddq018
Beconi, M., Aziz, O., Matthews, K., Moumné, L., O’Connell, C., Yates, D., et al. (2012). Oral administration of the pimelic diphenylamide HDAC inhibitor HDACi 4b is unsuitable for chronic inhibition of HDAC activity in the CNS in vivo. PLoS ONE 7:e44498. doi: 10.1371/journal.pone.0044498
Benito, E., Urbanke, H., Ramachandran, B., Barth, J., Halder, R., Awasthi, A., et al. (2015). HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J. Clin. Invest. 125, 3572–3584. doi: 10.1172/JCI79942
Besnard, A., Galan-Rodriguez, B., Vanhoutte, P., and Caboche, J. (2011). Elk-1 a transcription factor with multiple facets in the brain. Front. Neurosci. 5:35. doi: 10.3389/fnins.2011.00035
Biagioli, M., Ferrari, F., Mendenhall, E. M., Zhang, Y., Erdin, S., Vijayvargia, R., et al. (2015). Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum. Mol. Genet. 24, 2442–2457. doi: 10.1093/hmg/ddv006
Bock, C., Farlik, M., and Sheffield, N. C. (2016). Multi-omics of single cells: strategies and applications. Trends Biotechnol. 34, 605–608. doi: 10.1016/j.tibtech.2016.04.004
Boland, M. J., Nazor, K. L., and Loring, J. F. (2014). Epigenetic regulation of pluripotency and differentiation. Circ. Res. 115, 311–324. doi: 10.1161/CIRCRESAHA.115.301517
Borrelli, E., Nestler, E. J., Allis, C. D., and Sassone-Corsi, P. (2008). Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974. doi: 10.1016/j.neuron.2008.10.012
Bousiges, O., Neidl, R., Majchrzak, M., Muller, M. A., Barbelivien, A., Pereira, de Vasconcelos A, et al. (2013). Detection of histone acetylation levels in the dorsal hippocampus reveals early tagging on specific residues of H2B and H4 histones in response to learning. PLoS ONE 8:e57816. doi: 10.1371/journal.pone.0057816
Brami-Cherrier, K., Roze, E., Girault, J. A., Betuing, S., and Caboche, J. (2009). Role of the ERK/MSK1 signalling pathway in chromatin remodelling and brain responses to drugs of abuse. J. Neurochem. 108, 1323–1335. doi: 10.1111/j.1471-4159.2009.05879.x
Cardinaux, J. R., Notis, J. C., Zhang, Q., Vo, N., Craig, J. C., Fass, D. M., et al. (2000). Recruitment of CREB binding protein is sufficient for CREB-mediated gene activation. Mol. Cell. Biol. 20, 1546–1552. doi: 10.1128/MCB.20.5.1546-1552.2000
Cedar, H., and Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. doi: 10.1038/nrg2540
Chatterjee, S., Mizar, P., Cassel, R., Neidl, R., Selvi, B. R., Mohankrishna, D. V., et al. (2013). A novel activator of CBP/p300 acetyltransferases promotes neurogenesis and extends memory duration in adult mice. J. Neurosci. 33, 10698–10712. doi: 10.1523/JNEUROSCI.5772-12.2013
Chawla, S., Vanhoutte, P., Arnold, F. J., Huang, C. L., and Bading, H. (2003). Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J. Neurochem. 85, 151–159. doi: 10.1046/j.1471-4159.2003.01648.x
Chestnut, B. A., Chang, Q., Price, A., Lesuisse, C., Wong, M., and Martin, L. J. (2011). Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 31, 16619–16636. doi: 10.1523/JNEUROSCI.1639-11.2011
Cicchetti, F., Gould, P. V., and Parent, A. (1996). Sparing of striatal neurons coexpressing calretinin and substance P (NK1) receptor in Huntington’s disease. Brain Res. 730, 232–237. doi: 10.1016/0006-8993(96)00307-1
Coppede, F. (2014). The potential of epigenetic therapies in neurodegenerative diseases. Front. Genet. 5:220. doi: 10.3389/fgene.2014.00220
Day, J. J., and Sweatt, J. D. (2011). Cognitive neuroepigenetics: a role for epigenetic mechanisms in learning and memory. Neurobiol. Learn. Mem. 96, 2–12. doi: 10.1016/j.nlm.2010.12.008
De Souza, R. A., Islam, S. A., McEwen, L. M., Mathelier, A., Hill, A., Mah, S. M., et al. (2016). DNA methylation profiling in human Huntington’s disease brain. Hum. Mol. Genet. 25, 2013–2030. doi: 10.1093/hmg/ddw076
Deans, C., and Maggert, K. A. (2015). What do you mean, ”epigenetic”? Genetics 199, 887–896. doi: 10.1534/genetics.114.173492
Deaton, A. M., and Bird, A. (2011). CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022. doi: 10.1101/gad.2037511
Desplats, P. A., Kass, K. E., Gilmartin, T., Stanwood, G. D., Woodward, E. L., Head, S. R., et al. (2006). Selective deficits in the expression of striatal-enriched mRNAs in Huntington’s disease. J. Neurochem. 96, 743–757. doi: 10.1111/j.1471-4159.2005.03588.x
Dietz, K. N., Di, Stefano, L., Maher, R. C., Zhu, H., Macdonald, M. E., Gusella, J. F., et al. (2015). The Drosophila Huntington’s disease gene ortholog dhtt influences chromatin regulation during development. Hum. Mol. Genet. 24, 330–345. doi: 10.1093/hmg/ddu446
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. doi: 10.1126/science.277.5334.1990
Dong, X., Tsuji, J., Labadorf, A., Roussos, P., Chen, J. F., Myers, R. H., et al. (2015). The Role of H3K4me3 in transcriptional regulation is altered in Huntington’s Disease. PLoS ONE 10:e0144398. doi: 10.1371/journal.pone.0144398
Du, Q., Luu, P. L., Stirzaker, C., and Clark, S. J. (2015). Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics 7, 1051–1073. doi: 10.2217/epi.15.39
Evans-Galea, M. V., Hannan, A. J., Carrodus, N., Delatycki, M. B., and Saffery, R. (2013). Epigenetic modifications in trinucleotide repeat diseases. Trends Mol. Med. 19, 655–663. doi: 10.1016/j.molmed.2013.07.007
Federman, N., Fustinana, M. S., and Romano, A. (2009). Histone acetylation is recruited in consolidation as a molecular feature of stronger memories. Learn. Mem. 16, 600–606. doi: 10.1101/lm.1537009
Feng, J., Shao, N., Szulwach, K. E., Vialou, V., Huynh, J., Zhong, C., et al. (2015). Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nat. Neurosci. 18, 536–544. doi: 10.1038/nn.3976
Feng, J., Zhou, Y., Campbell, S. L., Le, T., Li, E., Sweatt, J. D., et al. (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13, 423–430. doi: 10.1038/nn.2514
Ferrante, R. J., Beal, M. F., Kowall, N. W., Richardson, E. P. Jr., and Martin, J. B. (1987a). Sparing of acetylcholinesterase-containing striatal neurons in Huntington’s disease. Brain Res. 411, 162–166. doi: 10.1016/0006-8993(87)90694-9
Ferrante, R. J., Kowall, N. W., Beal, M. F., Martin, J. B., Bird, E. D., and Richardson, E. P. Jr. (1987b). Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 46, 12–27. doi: 10.1097/00005072-198701000-00002
Ferrante, R. J., Kubilus, J. K., Lee, J., Ryu, H., Beesen, A., Zucker, B., et al. (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 23, 9418–9427.
Francelle, L., Galvan, L., and Brouillet, E. (2014). Possible involvement of self-defense mechanisms in the preferential vulnerability of the striatum in Huntington’s disease. Front. Cell Neurosci. 8:295. doi: 10.3389/fncel.2014.00295
Gabel, S., Koncina, E., Dorban, G., Heurtaux, T., Birck, C., Glaab, E., et al. (2016). Inflammation Promotes a conversion of astrocytes into neural progenitor cells via NF-kappaB activation. Mol. Neurobiol. 53, 5041–5055. doi: 10.1007/s12035-015-9428-3
Gardian, G., Browne, S. E., Choi, D. K., Klivenyi, P., Gregorio, J., Kubilus, J. K., et al. (2005). Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 280, 556–563. doi: 10.1074/jbc.M410210200
Giralt, A., Puigdellívol, M., Carretón, O., Paoletti, P., Valero, J., Parra-Damas, A., et al. (2012). Long-term memory deficits in Huntington’s disease are associated with reduced CBP histone acetylase activity. Hum. Mol. Genet. 21, 1203–1216. doi: 10.1093/hmg/ddr552
Gjoneska, E., Pfenning, A. R., Mathys, H., Quon, G., Kundaje, A., Tsai, L. H., et al. (2015). Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369. doi: 10.1038/nature14252
Glajch, K. E., and Sadri-Vakili, G. (2015). Epigenetic Mechanisms Involved in Huntington’s Disease Pathogenesis. J. Huntingtons Dis. 4, 1–15. doi: 10.3233/JHD-159001
Godena, V. K., Brookes-Hocking, N., Moller, A., Shaw, G., Oswald, M., Sancho, R. M., et al. (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 5:5245. doi: 10.1038/ncomms6245
Goula, A.-V., Stys, A., Chan, J. P. K., Trottier, Y., Festenstein, R., and Merienne, K. (2012). Transcription elongation and tissue-specific somatic CAG instability. PLoS Genet. 8:e1003051. doi: 10.1371/journal.pgen.1003051
Graff, J., and Tsai, L. H. (2013). Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111. doi: 10.1038/nrn3427
Guiretti, D., Sempere, A., Lopez-Atalaya, J. P., Ferrer-Montiel, A., Barco, A., and Valor, L. M. (2016). Specific promoter deacetylation of histone H3 is conserved across mouse models of Huntington’s disease in the absence of bulk changes. Neurobiol. Dis. 89, 190–201. doi: 10.1016/j.nbd.2016.02.004
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., and Song, H. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. doi: 10.1016/j.cell.2011.03.022
Halder, R., Hennion, M., Vidal, R. O., Shomroni, O., Rahman, R. U., Rajput, A., et al. (2016). DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci. 19, 102–110. doi: 10.1038/nn.4194
Harrison, I. F., Crum, W. R., Vernon, A. C., and Dexter, D. T. (2015). Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br. J. Pharmacol. 172, 4200–4215. doi: 10.1111/bph.13208
Hegarty, S. V., O’Leary, E., Solger, F., Stanicka, J., Sullivan, A. M., and O’Keeffe, G. W. (2016). A small molecule activator of p300/CBP histone acetyltransferase promotes survival and neurite growth in a cellular model of parkinson’s disease. Neurotox. Res. 30, 510–520. doi: 10.1007/s12640-016-9636-2
Herman, D., Jenssen, K., Burnett, R., Soragni, E., Perlman, S. L., and Gottesfeld, J. M. (2006). Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2, 551–558. doi: 10.1038/nchembio815
Hockly, E., Richon, V. M., Woodman, B., Smith, D. L., Zhou, X., Rosa, E., et al. (2003). Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 100, 2041–2046. doi: 10.1073/pnas.0437870100
Hodges, A., Strand, A. D., Aragaki, A. K., Kuhn, A., Sengstag, T., Hughes, G., et al. (2006). Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 15, 965–977. doi: 10.1093/hmg/ddl013
Hogarth, P., Lovrecic, L., and Krainc, D. (2007). Sodium phenylbutyrate in Huntington’s disease: a dose-finding study. Mov. Disord. 22, 1962–1964. doi: 10.1002/mds.21632
Holliday, R. (2006). Epigenetics: a historical overview. Epigenetics 1, 76–80. doi: 10.4161/epi.1.2.2762
Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14:R115. doi: 10.1186/gb-2013-14-10-r115
Horvath, S., Langfelder, P., Kwak, S., Aaronson, J., Rosinski, J., Vogt, T. F., et al. (2016). Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging (Albany NY) 8, 1485–1512. doi: 10.18632/aging.101005
Igarashi, S., Morita, H., Bennett, K. M., Tanaka, Y., Engelender, S., Peters, M. F., et al. (2003). Inducible PC12 cell model of Huntington’s disease shows toxicity and decreased histone acetylation. Neuroreport 14, 565–568. doi: 10.1097/00001756-200303240-00007
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33(Suppl.), 245–254. doi: 10.1038/ng1089
Jeltsch, A. (2006). Molecular enzymology of mammalian DNA methyltransferases. Curr. Top. Microbiol. Immunol. 301, 203–225.
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Jia, H., Kast, R. J., Steffan, J. S., and Thomas, E. A. (2012). Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: implications for the ubiquitin-proteasomal and autophagy systems. Hum. Mol. Genet. 21, 5280–5293. doi: 10.1093/hmg/dds379
Jia, H., Morris, C. D., Williams, R. M., Loring, J. F., and Thomas, E. A. (2015). HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc. Natl. Acad. Sci. U.S.A. 112, E56–E64. doi: 10.1073/pnas.1415195112
Jia, H., Wang, Y., Morris, C. D., Jacques, V., Gottesfeld, J. M., Rusche, J. R., et al. (2016). The Effects of Pharmacological Inhibition of Histone Deacetylase 3 (HDAC3) in Huntington’s Disease Mice. PLoS ONE 11:e0152498. doi: 10.1371/journal.pone.0152498
Jiang, H., Poirier, M. A., Liang, Y., Pei, Z., Weiskittel, C. E., Smith, W. W., et al. (2006). Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 23, 543–551. doi: 10.1016/j.nbd.2006.04.011
Jones, P. A., Issa, J. P., and Baylin, S. (2016). Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17, 630–641. doi: 10.1038/nrg.2016.93
Kazantsev, A., Walker, H. A., Slepko, N., Bear, J. E., Preisinger, E., Steffan, J. S., et al. (2002). A bivalent Huntingtin binding peptide suppresses polyglutamine aggregation and pathogenesis in Drosophila. Nat. Genet. 30, 367–376. doi: 10.1038/ng864
Kim, M. O., Chawla, P., Overland, R. P., Xia, E., Sadri-Vakili, G., and Cha, J. H. (2008). Altered histone monoubiquitylation mediated by mutant huntingtin induces transcriptional dysregulation. J. Neurosci. 28, 3947–3957. doi: 10.1523/JNEUROSCI.5667-07.2008
Klevytska, A. M., Tebbenkamp, A. T., Savonenko, A. V., and Borchelt, D. R. (2010). Partial depletion of CREB-binding protein reduces life expectancy in a mouse model of Huntington disease. J. Neuropathol. Exp. Neurol. 69, 396–404. doi: 10.1097/NEN.0b013e3181d6c436
Korzus, E. (2010). Manipulating the brain with epigenetics. Nat. Neurosci. 13, 405–406. doi: 10.1038/nn0410-405
Kuhn, A., Goldstein, D. R., Hodges, A., Strand, A. D., Sengstag, T., Kooperberg, C., et al. (2007). Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 16, 1845–1861. doi: 10.1093/hmg/ddm133
Labadorf, A., Hoss, A. G., Lagomarsino, V., Latourelle, J. C., Hadzi, T. C., Bregu, J., et al. (2015). RNA sequence analysis of human huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. PLoS ONE 10:e0143563. doi: 10.1371/journal.pone.0143563
Labbadia, J., and Morimoto, R. I. (2013). Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 38, 378–385. doi: 10.1016/j.tibs.2013.05.003
Landgrave-Gomez, J., Mercado-Gomez, O., and Guevara-Guzman, R. (2015). Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell Neurosci. 9:58. doi: 10.3389/fncel.2015.00058
Landles, C., and Bates, G. P. (2004). Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in molecular medicine review series. EMBO Rep. 5, 958–963. doi: 10.1038/sj.embor.7400250
Langfelder, P., Cantle, J. P., Chatzopoulou, D., Wang, N., Gao, F., Al-Ramahi, I., et al. (2016). Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 19, 623–633. doi: 10.1038/nn.4256
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Lazo-Gomez, R., Ramirez-Jarquin, U. N., Tovar, Y. R. L. B., and Tapia, R. (2013). Histone deacetylases and their role in motor neuron degeneration. Front. Cell Neurosci. 7:243. doi: 10.3389/fncel.2013.00243
Lee, J., Hwang, Y. J., Kim, K. Y., Kowall, N. W., and Ryu, H. (2013). Epigenetic mechanisms of neurodegeneration in Huntington’s disease. Neurotherapeutics 10, 664–676. doi: 10.1007/s13311-013-0206-5
Lev Maor, G., Yearim, A., and Ast, G. (2015). The alternative role of DNA methylation in splicing regulation. Trends Genet. 31, 274–280. doi: 10.1016/j.tig.2015.03.002
Li, J., Liu, X., Liu, M., Che, K., and Luo, B. (2016). Methylation and expression of Epstein–Barr virus latent membrane protein 1, 2A and 2B in EBV-associated gastric carcinomas and cell lines. Dig. Liver Dis. 48, 673–680. doi: 10.1016/j.dld.2016.02.017
Li, L., Liu, H., Dong, P., Li, D., Legant, W. R., Grimm, J. B., et al. (2016). Real-time imaging of Huntingtin aggregates diverting target search and gene transcription. Elife 5:e17056. doi: 10.7554/eLife.17056
Liu, L., Jin, G., and Zhou, X. (2015). Modeling the relationship of epigenetic modifications to transcription factor binding. Nucleic Acids Res. 43, 3873–3885. doi: 10.1093/nar/gkv255
Lopez-Atalaya, J. P., and Barco, A. (2014). Can changes in histone acetylation contribute to memory formation? Trends Genet. 30, 529–539. doi: 10.1016/j.tig.2014.09.003
Luthi-Carter, R., Strand, A., Peters, N. L., Solano, S. M., Hollingsworth, Z. R., Menon, A. S., et al. (2000). Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 9, 1259–1271. doi: 10.1093/hmg/9.9.1259
Luthi-Carter, R., Strand, A. D., Hanson, S. A., Kooperberg, C., Schilling, G., La, Spada AR, et al. (2002). Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington’s disease mouse models reveal context-independent effects. Hum. Mol. Genet. 11, 1927–1937. doi: 10.1093/hmg/11.17.1927
Marconett, C. N., Zhou, B., Rieger, M. E., Selamat, S. A., Dubourd, M., Fang, X., et al. (2013). Integrated transcriptomic and epigenomic analysis of primary human lung epithelial cell differentiation. PLoS Genet. 9:e1003513. doi: 10.1371/journal.pgen.1003513
McFarland, K. N., Das, S., Sun, T. T., Leyfer, D., Xia, E., Sangrey, G. R., et al. (2012). Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington’s disease. PLoS ONE 7:e41423. doi: 10.1371/journal.pone.0041423
Meadows, J. P., Guzman-Karlsson, M. C., Phillips, S., Brown, J. A., Strange, S. K., Sweatt, J. D., et al. (2016). Dynamic DNA methylation regulates neuronal intrinsic membrane excitability. Sci. Signal. 9:ra83. doi: 10.1126/scisignal.aaf5642
Mielcarek, M., Benn, C. L., Franklin, S. A., Smith, D. L., Woodman, B., Marks, P. A., et al. (2011). SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS ONE 6:e27746. doi: 10.1371/journal.pone.0027746
Min, S. W., Cho, S. H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Minatohara, K., Akiyoshi, M., and Okuno, H. (2015). Role of immediate-early genes in synaptic plasticity and neuronal ensembles underlying the memory trace. Front. Mol. Neurosci. 8:78. doi: 10.3389/fnmol.2015.00078
Miranda, T. B., Cortez, C. C., Yoo, C. B., Liang, G., Abe, M., Kelly, T. K., et al. (2009). DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 8, 1579–1588. doi: 10.1158/1535-7163.MCT-09-0013
Molero, A. E., Arteaga-Bracho, E. E., Chen, C. H., Gulinello, M., Winchester, M. L., Pichamoorthy, N., et al. (2016). Selective expression of mutant huntingtin during development recapitulates characteristic features of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 113, 5736–5741. doi: 10.1073/pnas.1603871113
Moumne, L., Betuing, S., and Caboche, J. (2013). Multiple aspects of gene dysregulation in Huntington’s Disease. Front. Neurol. 4:127. doi: 10.3389/fneur.2013.00127
Moumne, L., Campbell, K., Howland, D., Ouyang, Y., and Bates, G. P. (2012). Genetic knock-down of HDAC3 does not modify disease-related phenotypes in a mouse model of Huntington’s disease. PLoS ONE 7:e31080. doi: 10.1371/journal.pone.0031080
Ng, C. W., Yildirim, F., Yap, Y. S., Dalin, S., Matthews, B. J., Velez, P. J., et al. (2013). Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. U.S.A. 110, 2354–2359. doi: 10.1073/pnas.1221292110
Nucifora, F. C. Jr., Sasaki, M., Peters, M. F., Huang, H., Cooper, J. K., Yamada, M., et al. (2001). Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291, 2423–2428. doi: 10.1126/science.1056784
Ooi, L., and Wood, I. C. (2007). Chromatin crosstalk in development and disease: lessons from REST. Nat. Rev. Genet. 8, 544–554. doi: 10.1038/nrg2100
Ortega-Martinez, S. (2015). A new perspective on the role of the CREB family of transcription factors in memory consolidation via adult hippocampal neurogenesis. Front. Mol. Neurosci. 8:46. doi: 10.3389/fnmol.2015.00046
Pan, Y., Daito, T., Sasaki, Y., Chung, Y. H., Xing, X., Pondugula, S., et al. (2016). Inhibition of DNA methyltransferases blocks mutant huntingtin-induced neurotoxicity. Sci. Rep. 6:31022. doi: 10.1038/srep31022
Parmentier, F., Lejeune, F. X., and Neri, C. (2013). Pathways to decoding the clinical potential of stress response FOXO-interaction networks for Huntington’s disease: of gene prioritization and context dependence. Front. Aging Neurosci. 5:22. doi: 10.3389/fnagi.2013.00022
Pastor, W. A., Aravind, L., and Rao, A. (2013). TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 14, 341–356. doi: 10.1038/nrm3589
Peixoto, L., and Abel, T. (2013). The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 38, 62–76. doi: 10.1038/npp.2012.86
Ravache, M., Weber, C., Merienne, K., and Trottier, Y. (2010). Transcriptional activation of REST by Sp1 in Huntington’s disease models. PLoS ONE 5:e14311. doi: 10.1371/journal.pone.0014311
Riccio, A. (2010). Dynamic epigenetic regulation in neurons: enzymes, stimuli and signaling pathways. Nat. Neurosci. 13, 1330–1337. doi: 10.1038/nn.2671
Roos, R. A. (2010). Huntington’s disease: a clinical review. Orphanet J. Rare Dis. 5:40. doi: 10.1186/1750-1172-5-40
Rosas, H. D., Salat, D. H., Lee, S. Y., Zaleta, A. K., Pappu, V., Fischl, B., et al. (2008). Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain 131, 1057–1068. doi: 10.1093/brain/awn025
Roth, T. L., and Sweatt, J. D. (2011). Annual research review: epigenetic mechanisms and environmental shaping of the brain during sensitive periods of development. J. Child Psychol. Psychiatry 52, 398–408. doi: 10.1111/j.1469-7610.2010.02282.x
Roze, E., Betuing, S., Deyts, C., Marcon, E., Brami-Cherrier, K., Pagès, C., et al. (2008). Mitogen- and stress-activated protein kinase-1 deficiency is involved in expanded-huntingtin-induced transcriptional dysregulation and striatal death. FASEB J. 22, 1083–1093. doi: 10.1096/fj.07-9814
Ryu, H., Lee, J., Hagerty, S. W., Soh, B. Y., McAlpin, S. E., Cormier, K. A., et al. (2006). ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 19176–19181. doi: 10.1073/pnas.0606373103
Sadri-Vakili, G., Bouzou, B., Benn, C. L., Kim, M. O., Chawla, P., Overland, R. P., et al. (2007). Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum. Mol. Genet. 16, 1293–1306. doi: 10.1093/hmg/ddm078
Saudou, F., and Humbert, S. (2016). The biology of huntingtin. Neuron 89, 910–926. doi: 10.1016/j.neuron.2016.02.003
Seong, I. S., Woda, J. M., Song, J. J., Lloret, A., Abeyrathne, P. D., Woo, C. J., et al. (2010). Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 19, 573–583. doi: 10.1093/hmg/ddp524
Seredenina, T., and Luthi-Carter, R. (2012). What have we learned from gene expression profiles in Huntington’s disease? Neurobiol. Dis. 45, 83–98. doi: 10.1016/j.nbd.2011.07.001
Sharma, S., and Taliyan, R. (2015a). Targeting histone deacetylases: a novel approach in Parkinson’s disease. Parkinsons Dis. 2015:303294. doi: 10.1155/2015/303294
Sharma, S., and Taliyan, R. (2015b). Transcriptional dysregulation in Huntington’s disease: the role of histone deacetylases. Pharmacol. Res. 100, 157–169. doi: 10.1016/j.phrs.2015.08.002
Sharma, S., Taliyan, R., and Singh, S. (2015). Beneficial effects of sodium butyrate in 6-OHDA induced neurotoxicity and behavioral abnormalities: Modulation of histone deacetylase activity. Behav. Brain Res. 291, 306–314. doi: 10.1016/j.bbr.2015.05.052
Shin, J., Ming, G. L., and Song, H. (2014). Decoding neural transcriptomes and epigenomes via high-throughput sequencing. Nat. Neurosci. 17, 1463–1475. doi: 10.1038/nn.3814
Song, C. X., Szulwach, K. E., Fu, Y., Dai, Q., Yi, C., Li, X., et al. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72. doi: 10.1038/nbt.1732
Steffan, J. S., Bodai, L., Pallos, J., Poelman, M., McCampbell, A., Apostol, B. L., et al. (2001). Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413, 739–743. doi: 10.1038/35099568
Steffan, J. S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y. Z., Gohler, H., et al. (2000). The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. U.S.A. 97, 6763–6768. doi: 10.1073/pnas.100110097
Sterner, D. E., and Berger, S. L. (2000). Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459. doi: 10.1128/MMBR.64.2.435-459.2000
Sultan, F. A., and Day, J. J. (2011). Epigenetic mechanisms in memory and synaptic function. Epigenomics 3, 157–181. doi: 10.2217/epi.11.6
Sweatt, J. D. (2013). The emerging field of neuroepigenetics. Neuron 80, 624–632. doi: 10.1016/j.neuron.2013.10.023
Sweatt, J. D. (2016). Neural plasticity & behavior–sixty years of conceptual advances. J. Neurochem. 139, 179–199. doi: 10.1111/jnc.13580
Szulwach, K. E., Li, X., Li, Y., Song, C. X., Wu, H., Dai, Q., et al. (2011). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616. doi: 10.1038/nn.2959
Thakurela, S., Sahu, S. K., Garding, A., and Tiwari, V. K. (2015). Dynamics and function of distal regulatory elements during neurogenesis and neuroplasticity. Genome Res. 25, 1309–1324. doi: 10.1101/gr.190926.115
Thomas, B., Matson, S., Chopra, V., Sun, L., Sharma, S., Hersch, S., et al. (2013). A novel method for detecting 7-methyl guanine reveals aberrant methylation levels in Huntington disease. Anal. Biochem. 436, 112–120. doi: 10.1016/j.ab.2013.01.035
Thomas, E. A., Coppola, G., Desplats, P. A., Tang, B., Soragni, E., Burnett, R., et al. (2008). The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 105, 15564–15569. doi: 10.1073/pnas.0804249105
Tie, F., Banerjee, R., Saiakhova, A. R., Howard, B., Monteith, K. E., Scacheri, P. C., et al. (2014). Trithorax monomethylates histone H3K4 and interacts directly with CBP to promote H3K27 acetylation and antagonize Polycomb silencing. Development 141, 1129–1139. doi: 10.1242/dev.102392
Tsai, C. L., Li, H. P., Lu, Y. J., Hsueh, C., Liang, Y., Chen, C. L., et al. (2006). Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH2-terminal kinase signaling. Cancer Res. 66, 11668–11676. doi: 10.1158/0008-5472.CAN-06-2194
Tsompana, M., and Buck, M. J. (2014). Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7:33. doi: 10.1186/1756-8935-7-33
Urdinguio, R. G., Sanchez-Mut, J. V., and Esteller, M. (2009). Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 8, 1056–1072. doi: 10.1016/S1474-4422(09)70262-5
Valor, L. M. (2015a). Epigenetic-based therapies in the preclinical and clinical treatment of Huntington’s disease. Int. J. Biochem. Cell Biol. 67, 45–48. doi: 10.1016/j.biocel.2015.04.009
Valor, L. M. (2015b). Transcription, epigenetics and ameliorative strategies in Huntington’s Disease: a genome-wide perspective. Mol. Neurobiol. 51, 406–423. doi: 10.1007/s12035-014-8715-8
Valor, L. M., and Guiretti, D. (2014). What’s wrong with epigenetics in Huntington’s disease? Neuropharmacology 80, 103–114. doi: 10.1016/j.neuropharm.2013.10.025
Valor, L. M., Guiretti, D., Lopez-Atalaya, J. P., and Barco, A. (2013). Genomic landscape of transcriptional and epigenetic dysregulation in early onset polyglutamine disease. J. Neurosci. 33, 10471–10482. doi: 10.1523/JNEUROSCI.0670-13.2013
Vashishtha, M., Ng, C. W., Yildirim, F., Gipson, T. A., Kratter, I. H., Bodai, L., et al. (2013). Targeting H3K4 trimethylation in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 110, E3027–E3036. doi: 10.1073/pnas.1311323110
von Schimmelmann M., Feinberg, P. A., Sullivan, J. M., Ku, S. M., Badimon, A., Duff, M. K., et al. (2016). Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 19, 1321–1330. doi: 10.1038/nn.4360
Wang, F., Fischhaber, P. L., Guo, C., and Tang, T. S. (2014). Epigenetic modifications as novel therapeutic targets for Huntington’s disease. Epigenomics 6, 287–297. doi: 10.2217/epi.14.19
Wang, F., Yang, Y., Lin, X., Wang, J. Q., Wu, Y. S., Xie, W., et al. (2013). Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet. 22, 3641–3653. doi: 10.1093/hmg/ddt214
Wang, X., and Jin, H. (2010). The epigenetic basis of the Warburg effect. Epigenetics 5, 566–568. doi: 10.4161/epi.5.7.12662
West, A. E., and Greenberg, M. E. (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3:a005744. doi: 10.1101/cshperspect.a005744
Westerberg, G., Chiesa, J. A., Andersen, C. A., Diamanti, D., Magnoni, L., Pollio, G., et al. (2015). Safety, pharmacokinetics, pharmacogenomics and QT concentration-effect modelling of the SirT1 inhibitor selisistat in healthy volunteers. Br. J. Clin. Pharmacol. 79, 477–491. doi: 10.1111/bcp.12513
Xu, K., Dai, X. L., Huang, H. C., and Jiang, Z. F. (2011). Targeting HDACs: a promising therapy for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2011, 143269. doi: 10.1155/2011/143269
Xu, Y. M., Du, J. Y., and Lau, A. T. (2014). Posttranslational modifications of human histone H3: an update. Proteomics 14, 2047–2060. doi: 10.1002/pmic.201300435
Zeng, J., Libien, J., Shaik, F., Wolk, J., and Hernandez, A. I. (2016). Nucleolar PARP-1 expression is decreased in alzheimer’s disease: consequences for epigenetic regulation of RDNA and cognition. Neural Plast. 2016:8987928. doi: 10.1155/2016/8987928
Zovkic, I. B., Guzman-Karlsson, M. C., and Sweatt, J. D. (2013). Epigenetic regulation of memory formation and maintenance. Learn. Mem. 20, 61–74. doi: 10.1101/lm.026575.112
Zuccato, C., Valenza, M., and Cattaneo, E. (2010). Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 90, 905–981. doi: 10.1152/physrev.00041.2009
Zucker, B., Luthi-Carter, R., Kama, J. A., Dunah, A. W., Stern, E. A., Fox, J. H., et al. (2005). Transcriptional dysregulation in striatal projection- and interneurons in a mouse model of Huntington’s disease: neuronal selectivity and potential neuroprotective role of HAP1. Hum. Mol. Genet. 14, 179–189. doi: 10.1093/hmg/ddi014
Keywords: neuroepigenetics, Huntington’s disease, epigenomics, transcriptomics, neuronal activity, HDAC inhibitors, neurodegenerative diseases
Citation: Francelle L, Lotz C, Outeiro T, Brouillet E and Merienne K (2017) Contribution of Neuroepigenetics to Huntington’s Disease. Front. Hum. Neurosci. 11:17. doi: 10.3389/fnhum.2017.00017
Received: 07 November 2016; Accepted: 10 January 2017;
Published: 30 January 2017.
Edited by:
Daniela S. Andres, ETH Zurich, SwitzerlandReviewed by:
Anthony John Hannan, Florey Institute of Neuroscience and Mental Health, AustraliaElizabeth A. Thomas, Scripps Research Institute, USA
Martin Radrizzani, National University of San Martín, Argentina
Copyright © 2017 Francelle, Lotz, Outeiro, Brouillet and Merienne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laetitia Francelle, bGFldGl0aWEuZnJhbmNlbGxlQGhvdG1haWwuZnI=