 Natalia C. Prymaczok1
Natalia C. Prymaczok1 Roland Riek
Roland Riek Juan Gerez
Juan Gerez- 1Laboratorio de Neurofisiología del Instituto Multidisciplinario de Biología Celular, Argentine Research Council (CONICET), National University of La Plata and Scientific Research Commission, Province of Buenos Aires (CIC-PBA), La Plata, Buenos Aires, Argentina
- 2Laboratory of Physical Chemistry, D-CHAB, ETH Zurich, Zurich, Switzerland
As Parkinson's disease progresses, a massive loss of dopaminergic neurons is accompanied by accumulation of alpha-Synuclein (αSyn) neuronal inclusions called Lewy bodies and Lewy neurites. Inclusions first appear in olfactory bulb and enteric neurons then in ascendant neuroanatomical interconnected areas, and finally, in late stages of the disease, Lewy bodies are observed in a substantia nigra pars compacta with clear signs of neuronal loss. It is believed that the spreading of Lewy bodies through the nervous system is a consequence of the cell-to-cell propagation of αSyn, that can occur via sequential steps of secretion and uptake. Certain pathological forms of transmitted αSyn are able to seed endogenous counterparts in healthy recipient cells, thus promoting the self-sustained cycle of inclusion formation, amplification and spreading, that ultimately underlies disease progression. Here we review the cell-to-cell propagation of αSyn focusing on its role in the progression of Parkinson's disease.
Alpha-Synuclein and Parkinon's Disease
Parkinson's disease (PD) is a complex degenerative disorder that is pathologically characterized by a massive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the progressive accumulation of Lewy bodies and Lewy neurites (LBs/LNs), two forms of inclusions rich in filaments of aggregated alpha-Synuclein (αSyn) (Spillantini et al., 1997). Although a causative role remains to be formally established, the facts that LBs/LNs are present in virtually all sporadic and familial forms of PD (Poulopoulos et al., 2012), that point mutations and multiplications of the αSyn-encoding gene, SNCA, lead to early onset PD (Polymeropoulos et al., 1997; Krüger et al., 1998; Singleton et al., 2003; Chartier-Harlin et al., 2004; Zarranz et al., 2004) and that SNCA polymorphisms positively correlate with PD risk (Satake et al., 2009; Simón-Sánchez et al., 2009; Edwards et al., 2010) attest an irrefutable link between PD and αSyn.
Since the discovery that αSyn is abundant in LBs in the late 90s, a tremendous effort has been made to determine the precise 3D conformations adopted by this protein under physiological conditions. It is clear now that in aqueous solution αSyn behaves as an intrinsically disordered protein, lacking a defined or stable structure (Uversky and Eliezer, 2009; Drescher et al., 2012). Although still a matter of extensive debate, an emerging consensus indicates that within healthy cells αSyn exists as soluble low molecular weight species that play important roles in intra and extracellular vesicle trafficking and dynamics (Burre et al., 2010; Bartels et al., 2011; Fauvet et al., 2012; Theillet et al., 2016). In disease-related contexts, however, αSyn is also found as β-sheet-enriched amyloid aggregates that reside within and constitute the building blocks of LBs/LNs (Spillantini et al., 1997; Baba et al., 1998; Conway et al., 1998, 2000). Compelling evidence indicates that the culprits of toxicity are oligomers and higher order assemblies of αSyn such as amyloid fibrils (El-Agnaf et al., 1998; Winner et al., 2011; Rockenstein et al., 2014).
The Cell-to-Cell Transmission of αSyn
Early neuroanatomical studies conducted mainly by Braak and co-workers revealed that LBs appear first in the olfactory bulb and enteric neurons and that only after several years they are found in certain areas of the midbrain such as SNpc and eventually neocortex (Wakabayashi et al., 1988; Braak et al., 2003a, 2006; Braak and Del Tredici, 2008). Thus, during the progression of the disease, LBs are found in a stereotypical and topographical distribution in the nervous system. This highly predictable pattern of LB distribution was not taken into deep consideration until the subsequent discovery (in 2008) that healthy neurons would acquire LBs when grafted into the brains of PD patients (Kordower et al., 2008a,b; Li et al., 2008, 2010). A few years later, the demonstration that αSyn is transmitted from cell-to-cell led to the unifying hypothesis that the transcellular transmission of certain forms of αSyn underlies LB pathogenesis and spreading, and by extension, PD progression (Dunning et al., 2012). This hypothesis was originally supported by clinical evidence suggesting “host-to-graft” transmission of pathological αSyn forms: when embryonic mesencephalic neurons were grafted into PD patient's brains, they developed LBs several years after grafting (Kordower et al., 2008a,b; Li et al., 2008, 2010). The in vitro evidence supporting the cell-to-cell propagation of αSyn is its release by unconventional secretion (Emmanouilidou et al., 2010) and the uptake of extracellular αSyn (both natural and recombinant forms) by active mechanisms involving endocytosis (Figure 1; Sung et al., 2001; Liu et al., 2007; Lee et al., 2008a). αSyn can also be transmitted trans-synaptically and through tunnel-like structures that connect the cytosol of neighbor cells (Danzer et al., 2011; Abounit et al., 2016). In vivo evidence includes the slow but persistent acquisition of LB-like inclusions by healthy neuronal cells that have been grafted into the brains of mice predisposed to develop LB-pathology spontaneously, such as αSyn transgenic mice (Desplats et al., 2009; Hansen et al., 2011). Similarly, an early onset and widespread LB-like pathology is observed in animals that had received an intracerebral dose of brain homogenates of diseased αSyn transgenic mice (Luk et al., 2012b). While other factors present in brain homogenates could be involved, αSyn alone is sufficient to initiate LB-like pathology and its subsequent spreading: a single intracerebral injection of synthetic αSyn preformed fibrils leads to pathogenesis and progressive accumulation LB-like inclusions in neuroanatomically-interconnected areas accompanied by pathological features of PD such as neurodegeneration, neuroinflammation and motor deficits (Luk et al., 2012a; Sacino et al., 2014). This induction of LB-like pathology by intracerebral administration of αSyn aggregates strictly depends on the presence of αSyn in the host recipient cell, as no pathology can be induced in αSyn knock out mice (Luk et al., 2012b; Mougenot et al., 2012). The requirement of endogenous αSyn on LB-like inclusion spreading is explained by the observation in cell cultures that upon uptake, preformed αSyn fibrils promote the structural corruption of endogenous αSyn and its recruitment into newly formed inclusions (Luk et al., 2009; Waxman and Giasson, 2010; Volpicelli-Daley et al., 2011, 2014). Thus, host αSyn would be essential for the amplification of inclusions, an idea that needs to be further challenged (Helwig et al., 2016). Recruitment of host αSyn by exogenous αSyn has also been demonstrated in vivo in most mouse models in which LB pathogenesis is induced by administration of exogenous αSyn seeds (Luk et al., 2012b).
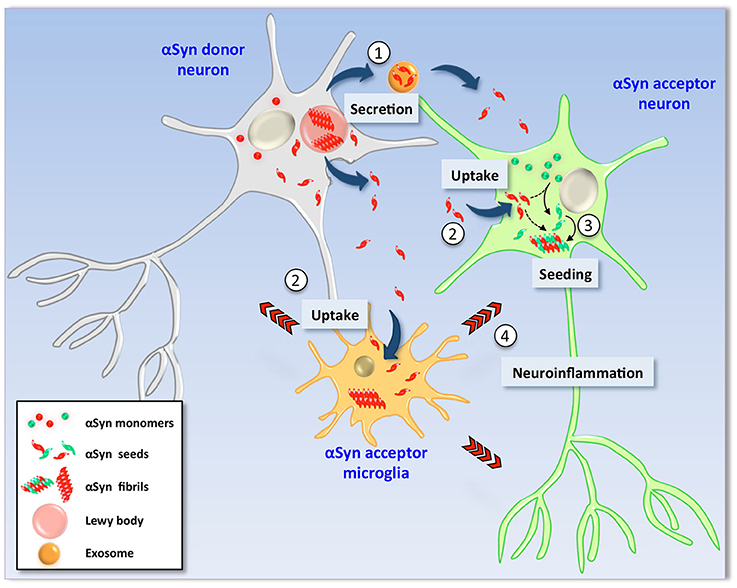
Figure 1. Hypothetical model of α-Synuclein cell-to-cell transmission. In pathological conditions, αSyn is found as β-sheet-enriched amyloid aggregates and fibrils that reside within Lewy bodies and Lewy neurites (LBs/LNs). Neurons containing LBs (left) could release αSyn aggregates and seeds into the extracellular milieu by different mechanisms such as non-classical exocytosis or via exosomes (1). Extracellular αSyn is then internalized by endocytosis by neighbor neurons as well as glial cells (2). Due to its amyloidogenic nature, uptake of exogenous seeds promotes the structural corruption of endogenous counterpart in healthy recipient neuronal cells (right). Thus, monomers of the recipient cell are converted into aggregates and fibrils by direct action of exogenous seeds (3) and new LBs are formed (not shown). Neuronal viability is severely affected by two mechanisms; (i) the intrinsic cytotoxic properties of intracellular αSyn aggregates and (ii) indirectly by action of proinflammatory molecules released by glial cells activated upon exposure to extracellular αSyn seeds (4).
Braak's Hypothesis in PD Progression
Braak and colleagues discovered that in sporadic PD, Lewy body pathology is first observed in the lower brainstem and anterior olfactory structures and that it then ascends following a caudo-rostral pattern from the dorsal motor nucleus through susceptible areas of the medulla, pontine tegmentum, midbrain, basal forebrain, reaching in some extreme cases the cerebral cortex (Braak et al., 2003a). These observations elegantly detailed LB spreading along the central nervous system (CNS), but they did not explain where and how the inclusions are originated. Thus, the provocative idea that LB-pathology begins when a neurotropic pathogen enters the nervous system and then spreads in a retrograde-axonal and transneuronal manner from one vulnerable brain region to the next was then introduced (Braak et al., 2003b; Hawkes et al., 2007). While in transit, this pathogen induces formation of αSyn-positive inclusions in these traceable CNS areas. It is now evident that certain species of αSyn can fulfill the requirements for Braak's neurotropic pathogen, and that both the olfactory and gastric tracks are largely compatible with the putative entry routes. In support of this, LB pathology is found in both the anterior olfactory nucleus as well as olfactory bulb mitral cells, the projection neurons that receive inputs from the olfactory epithelium (Daniel and Hawkes, 1992; Braak et al., 2003a). LB pathology has long been known to occur in the gastrointestinal tract of PD patients and is well documented in all the stages of PD (Braak et al., 2006; Lebouvier et al., 2008; Pouclet et al., 2012). Importantly, Braak and colleagues also provided the explanation for the observed non-random distribution of LBs along the nervous system by showing that different cell types have different susceptibilities to the development of inclusions. They concluded that neurons with a long, thin and poorly myelinated axon are highly susceptible to develop inclusions (Braak and Braak, 2000; Braak et al., 2003b). Importantly, these features are found in the enteric vagal preganglionic neurons that are susceptible to developing LB-pathology in early asymptomatic stages of the disease (Braak and Del Tredici, 2004).
Mechanisms of Toxicity
Like many aspects of its intercellular transmission, very little is known on the mechanisms of toxicity inherent to cell-to-cell transmitted αSyn. Likewise, how αSyn transmission is modulated or whether PD-related familial mutations or somatic copy number variations of the SNCA gene influence αSyn transmission remains to be determined. Taking into consideration our current knowledge on the consequences of αSyn misregulation, it is conceivable that cell-to-cell transmitted and endogenous αSyn share cytotoxic mechanisms that directly impact neuronal survival. These include, but are not limited to, the loss of function of endogenous αSyn as a consequence of its seeded aggregation, a phenomenon that massively affects neuronal physiological processes such as vesicle trafficking including neurotransmitter release and recycling (Jenco et al., 1998; Abeliovich et al., 2000; Murphy et al., 2000; Cabin et al., 2002; Chandra et al., 2005), the impairment of mitochondrial activity that perturbs not only a plethora of metabolic processes but also degradative pathways (Martin et al., 2006; Devi et al., 2008; Liu et al., 2009; Chinta et al., 2010; Loeb et al., 2010), and the disruption of vesicular transport mechanisms, in particular those that trigger endoplasmic reticulum stress (Cooper et al., 2006; Gitler et al., 2008; Thayanidhi et al., 2010). However, it is still possible that cell-to-cell transmitted αSyn has its own particular repertoire of cytotoxic properties, in addition to its probable distinct physiological functions. The fact that cell-to-cell transmitted αSyn is released to the extracellular milieu allowed neuroscientists to develop cellular and animal models based in administration of exogenous αSyn species that would recapitulate key mechanistic aspects of αSyn transmission such as its internalization and downstream events. Data obtained from glial cells might constitute the first evidence that endogenous and exogenously acquired αSyn might behave differently. It is well known that glial cells normally do not express αSyn mRNA (Reyes et al., 2014) and instead acquire the protein from the extracellular milieu (Liu et al., 2007; Lee et al., 2008b, 2010; Park et al., 2009). Thus, uptake of exogenous αSyn not only would explain the source of αSyn in glial cytoplasmic inclusions (GCIs) in multiple system atrophy (MSA), a progressive neurodegenerative disease related to PD and other synucleinopaties (Tu et al., 1998), but also would uncover a unique role for extracellular αSyn in the context of αSyn-deficient cells. Furthermore, it was shown that extracellular αSyn activates astrocytes and microglia in vitro and in vivo resulting in a neuroinflammatory response reminiscent to that observed in PD (Zhang et al., 2005; Lee et al., 2010; Alvarez-Erviti et al., 2011; Halliday and Stevens, 2011; Luk et al., 2012a). Noteworthy, neurons are highly susceptible to glial-derived proinflammatory factors, therefore representing an alternative neurotoxic process triggered specifically by cells that have acquired αSyn from the extracellular milieu. Although the neuron-glia interaction might help to elucidate specific and non-redundant roles for intracellular and extracellular cell-to-cell transmitted αSyn, the molecular and biochemical determinants that presumably make these two forms of αSyn different remain completely unexplored.
The Prion Hypothesis
As a consequence of Braak's model formulation, the idea that PD behaves as a prion disease has emerged, leading some to refer to it as a “prion-like” disorder. Of note, unlike prions, transmissibility between individuals of pathological forms of αSyn has not been demonstrated and thereby αSyn is currently considered as a non-infectious protein (Aguzzi and Rajendran, 2009; Beekes et al., 2014). The analogy to prion diseases stems for the fact that in in vivo experiments, involving mostly rodents and in some cases non-human primates, intracerebral administration of exogenous αSyn (either αSyn-containing brain material or synthetic αSyn proteins) is sufficient to trigger LB pathogenesis, amplification and spreading. These processes are usually accompanied by the phenotypic changes naturally observed in PD such as neurodegeneration, neuroinflammation, and motor deficits. Furthermore, it has been shown that in some cases the particular structural conformation of the exogenous αSyn seed, normally referred to as the “conformational strain”, is transmitted from the exogenously administered aggregates to host αSyn and thereby to the newly formed inclusions (Bousset et al., 2013; Guo et al., 2013; Peelaerts et al., 2015). This supports the idea of that the applied exogenous αSyn acts as seeds that template the aggregation of homotypic molecules of the host, a phenomenon characteristic of prions. The evidence of PD as a prion-like disorder is accumulating, however, there are still several unsolved questions that should be addressed before this terminology is broadly accepted. These questions arise from the inherent limitations of animal models for the full recapitulation of the human condition. In this sense, a constantly growing effort is being made to better characterize the brain material that contains neurotoxic αSyn species and the synthetic αSyn aggregates that are administrated intracerebrally to trigger PD-like pathology (Bousset et al., 2013; Tuttle et al., 2016). To uncover the molecular similarities and differences between the αSyn used in such in vivo experiments and those contained in Lewy bodies and Lewy neurites is critical to comprehensively understand the scopes and limitations of such animal models and the resultant hypotheses. As an example of such a gap of information, it has long been reported that certain forms of αSyn are found in the cerebrospinal fluid (CSF) of both healthy subjects and PD patients (Borghi et al., 2000; Tokuda et al., 2006; Mollenhauer et al., 2010; Parnetti et al., 2011; Foulds et al., 2012). However, it remains an enigma which structural species of αSyn are found in CSF and whether these molecules correspond to the transmitted species that mediate the pathogenic process that underlies LB pathogenesis and spreading. Elucidating this will help to reconcile clinical evidence arguing against the concept of prion-like progression in Parkinson's disease and related synucleinopaties (Hallett et al., 2014).
Concluding Remarks
The discovery of the cell-to-cell propagation of αSyn and in particular its role as mediator of disease progression has opened new therapeutic avenues for the treatment of PD and related neurological disorders, and novel therapies targeting extracellular αSyn aimed to delay or stop disease progression are currently being explored. The therapeutic potential of passive immunotherapy targeting aberrant forms of αSyn, for instance, has recently been investigated showing that it efficiently interferes with uptake of extracellular αSyn seeds preventing downstream effects such as amplification and transmission of pathological aggregates (Tran et al., 2014). Similarly, administration of rationally engineered antibodies robustly promotes degradation and neutralization of internalized αSyn preventing cell-to-cell aggregate transmission and neuronal loss (Bae et al., 2012; Spencer et al., 2014). Nevertheless, the elucidation of the mechanisms involved in αSyn transcellular transmission will be instrumental not only for the development of novel therapies for PD but also for the understanding of the “prion-like” properties of amyloid-beta (Aβ), tau and Huntingtin, all of them transmissible aggregation-prone proteins with a long history in neurodegenerative diseases such as Alzheimer and Huntington's diseases, respectively (Brettschneider et al., 2015).
Author Contributions
NP, RR, and JG carried out literature searches, assisted in generation of figures and writing of the manuscript. All authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
NP and JG are supported by a “Wilhelm Hurka” Stiftung. RR and JG are supported by a grant of the Swiss National Science Foundation (Sinergia 154461). JG is supported by an EMBO postdoctoral fellowship (ALTF-254-2012). The authors acknowledge to Jason Greenwald for critical reading of the manuscript.
References
Abeliovich, A., Schmitz, Y., Fariñas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/S0896-6273(00)80886-7
Abounit, S., Bousset, L., Loria, F., Zhu, S., de Chaumont, F., Pieri, L., et al. (2016). Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138. doi: 10.15252/embj.201593411
Aguzzi, A., and Rajendran, L. (2009). The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783–790. doi: 10.1016/j.neuron.2009.12.016
Alvarez-Erviti, L., Couch, Y., Richardson, J., Cooper, J. M., and Wood, M. J. (2011). Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 69, 337–342. doi: 10.1016/j.neures.2010.12.020
Baba, M., Nakajo, S., Tu, P. H., Tomita, T., Nakaya, K., Lee, V. M., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Bae, E. J., Lee, H. J., Rockenstein, E., Ho, D. H., Park, E. B., Yang, N. Y., et al. (2012). Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. 32, 13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Beekes, M., Thomzig, A., Schulz-Schaeffer, W. J., and Burger, R. (2014). Is there a risk of prion-like disease transmission by Alzheimer- or Parkinson-associated protein particles? Acta Neuropathol. 128, 463–476. doi: 10.1007/s00401-014-1324-9
Borghi, R., Marchese, R., Negro, A., Marinelli, L., Forloni, G., Zaccheo, D., et al. (2000). Full length α-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci. Lett. 287, 65–67. doi: 10.1016/S0304-3940(00)01153-8
Bousset, L., Pieri, L., Ruiz-Arlandis, G., Gath, J., Jensen, P. H., Habenstein, B., et al. (2013). Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 4, 2575. doi: 10.1038/ncomms3575
Braak, H., and Braak, E. (2000). Pathoanatomy of Parkinson's disease. J. Neurol. 247(Suppl. 2), II3–10. doi: 10.1007/pl00007758
Braak, H., and Del Tredici, K. (2004). Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol. Aging 25, 19–23. doi: 10.1016/j.neurobiolaging.2003.04.001
Braak, H., and Del Tredici, K. (2008). Invited article: nervous system pathology in sporadic Parkinson disease. Neurology 70, 1916–1925. doi: 10.1212/01.wnl.0000312279.49272.9f
Braak, H., Del Tredici, K., Rüb, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003a). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Braak, H., De Vos, R. A., Bohl, J., and Del Tredici, K. (2006). Gastric α-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci. Lett. 396, 67–72. doi: 10.1016/j.neulet.2005.11.012
Braak, H., Rüb, U., Gai, W. P., and Del Tredici, K. (2003b). Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. (Vienna) 110, 517–536. doi: 10.1007/s00702-002-0808-2
Brettschneider, J., Del Tredici, K., Lee, V. M., and Trojanowski, J. Q. (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120. doi: 10.1038/nrn3887
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Cabin, D. E., Shimazu, K., Murphy, D., Cole, N. B., Gottschalk, W., Mcilwain, K. L., et al. (2002). Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 22, 8797–8807.
Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schluter, O. M., and Sudhof, T. C. (2005). α-synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chartier-Harlin, M. C., Kachergus, J., Roumier, C., Mouroux, V., Douay, X., Lincoln, S., et al. (2004). α-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364, 1167–1169. doi: 10.1016/S0140-6736(04)17103-1
Chinta, S. J., Mallajosyula, J. K., Rane, A., and Andersen, J. K. (2010). Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 486, 235–239. doi: 10.1016/j.neulet.2010.09.061
Conway, K. A., Harper, J. D., and Lansbury, P. T. (1998). Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320. doi: 10.1038/3311
Conway, K. A., Harper, J. D., and Lansbury, P. T. Jr. (2000). Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry 39, 2552–2563. doi: 10.1021/bi991447r
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 313, 324–328. doi: 10.1126/science.1129462
Daniel, S. E., and Hawkes, C. H. (1992). Preliminary diagnosis of Parkinson's disease by olfactory bulb pathology. Lancet 340, 186. doi: 10.1016/0140-6736(92)93275-R
Danzer, K. M., Ruf, W. P., Putcha, P., Joyner, D., Hashimoto, T., Glabe, C., et al. (2011). Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 25, 326–336. doi: 10.1096/fj.10-164624
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Drescher, M., Huber, M., and Subramaniam, V. (2012). Hunting the chameleon: structural conformations of the intrinsically disordered protein alpha-synuclein. Chembiochem 13, 761–768. doi: 10.1002/cbic.201200059
Dunning, C. J., Reyes, J. F., Steiner, J. A., and Brundin, P. (2012). Can Parkinson's disease pathology be propagated from one neuron to another? Prog. Neurobiol. 97, 205–219. doi: 10.1016/j.pneurobio.2011.11.003
Edwards, T. L., Scott, W. K., Almonte, C., Burt, A., Powell, E. H., Beecham, G. W., et al. (2010). Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 74, 97–109. doi: 10.1111/j.1469-1809.2009.00560.x
El-Agnaf, O. M., Jakes, R., Curran, M. D., Middleton, D., Ingenito, R., Bianchi, E., et al. (1998). Aggregates from mutant and wild-type α-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of β-sheet and amyloid-like filaments. FEBS Lett. 440, 71–75. doi: 10.1016/S0014-5793(98)01418-5
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 30, 6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010
Fauvet, B., Mbefo, M. K., Fares, M. B., Desobry, C., Michael, S., Ardah, M. T., et al. (2012). α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364. doi: 10.1074/jbc.M111.318949
Foulds, P. G., Yokota, O., Thurston, A., Davidson, Y., Ahmed, Z., Holton, J., et al. (2012). Post mortem cerebrospinal fluid α-synuclein levels are raised in multiple system atrophy and distinguish this from the other α-synucleinopathies, Parkinson's disease and Dementia with Lewy bodies. Neurobiol. Dis. 45, 188–195. doi: 10.1016/j.nbd.2011.08.003
Gitler, A. D., Bevis, B. J., Shorter, J., Strathearn, K. E., Hamamichi, S., Su, L. J., et al. (2008). The Parkinson's disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 145–150. doi: 10.1073/pnas.0710685105
Guo, J. L., Covell, D. J., Daniels, J. P., Iba, M., Stieber, A., Zhang, B., et al. (2013). Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117. doi: 10.1016/j.cell.2013.05.057
Hallett, P. J., Cooper, O., Sadi, D., Robertson, H., Mendez, I., and Isacson, O. (2014). Long-term health of dopaminergic neuron transplants in Parkinson's disease patients. Cell Rep. 7, 1755–1761. doi: 10.1016/j.celrep.2014.05.027
Halliday, G. M., and Stevens, C. H. (2011). Glia: initiators and progressors of pathology in Parkinson's disease. Mov. Disord. 26, 6–17. doi: 10.1002/mds.23455
Hansen, C., Angot, E., Bergström, A. L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
Hawkes, C. H., Del Tredici, K., and Braak, H. (2007). Parkinson's disease: a dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 33, 599–614. doi: 10.1111/j.1365-2990.2007.00874.x
Helwig, M., Klinkenberg, M., Rusconi, R., Musgrove, R. E., Majbour, N. K., El-Agnaf, O. M., et al. (2016). Brain propagation of transduced alpha-synuclein involves non-fibrillar protein species and is enhanced in α-synuclein null mice. Brain 139, 856–870. doi: 10.1093/brain/awv376
Jenco, J. M., Rawlingson, A., Daniels, B., and Morris, A. J. (1998). Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by α- and β-synucleins. Biochemistry 37, 4901–4909. doi: 10.1021/bi972776r
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B., and Olanow, C. W. (2008a). Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat. Med. 14, 504–506. doi: 10.1038/nm1747
Kordower, J. H., Chu, Y., Hauser, R. A., Olanow, C. W., and Freeman, T. B. (2008b). Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov. Disord. 23, 2303–2306. doi: 10.1002/mds.22369
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., et al. (1998). Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Lebouvier, T., Chaumette, T., Damier, P., Coron, E., Touchefeu, Y., Vrignaud, S., et al. (2008). Pathological lesions in colonic biopsies during Parkinson's disease. Gut 57, 1741–1743. doi: 10.1136/gut.2008.162503
Lee, H. J., Suk, J. E., Bae, E. J., Lee, J. H., Paik, S. R., and Lee, S. J. (2008a). Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849. doi: 10.1016/j.biocel.2008.01.017
Lee, H. J., Suk, J. E., Bae, E. J., and Lee, S. J. (2008b). Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 372, 423–428. doi: 10.1016/j.bbrc.2008.05.045
Lee, H. J., Suk, J. E., Patrick, C., Bae, E. J., Cho, J. H., Rho, S., et al. (2010). Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272. doi: 10.1074/jbc.M109.081125
Li, J. Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
Li, J. Y., Englund, E., Widner, H., Rehncrona, S., Björklund, A., Lindvall, O., et al. (2010). Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson's disease. Mov. Disord. 25, 1091–1096. doi: 10.1002/mds.23012
Liu, G., Zhang, C., Yin, J., Li, X., Cheng, F., Li, Y., et al. (2009). α-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 454, 187–192. doi: 10.1016/j.neulet.2009.02.056
Liu, J., Zhou, Y., Wang, Y., Fong, H., Murray, T. M., and Zhang, J. (2007). Identification of proteins involved in microglial endocytosis of α-synuclein. J. Proteome Res. 6, 3614–3627. doi: 10.1021/pr0701512
Loeb, V., Yakunin, E., Saada, A., and Sharon, R. (2010). The transgenic overexpression of α-synuclein and not its related pathology associates with complex I inhibition. J. Biol. Chem. 285, 7334–7343. doi: 10.1074/jbc.M109.061051
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O'brien, P., Trojanowski, J. Q., et al. (2012a). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luk, K. C., Kehm, V. M., Zhang, B., O'brien, P., Trojanowski, J. Q., and Lee, V. M. (2012b). Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 209, 975–986. doi: 10.1084/jem.20112457
Luk, K. C., Song, C., O'brien, P., Stieber, A., Branch, J. R., Brunden, K. R., et al. (2009). Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 106, 20051–20056. doi: 10.1073/pnas.0908005106
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., et al. (2006). Parkinson's disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50. doi: 10.1523/JNEUROSCI.4308-05.2006
Mollenhauer, B., El-Agnaf, O. M. A., Marcus, K., Trenkwalder, C., and Schlossmacher, M. G. (2010). Quantification of α-synuclein in cerebrospinal fluid as a biomarker candidate: review of the literature and considerations for future studies. Biomark. Med. 4, 683–699. doi: 10.2217/bmm.10.90
Mougenot, A. L., Nicot, S., Bencsik, A., Morignat, E., Verchère, J., Lakhdar, L., et al. (2012). Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 33, 2225–2228. doi: 10.1016/j.neurobiolaging.2011.06.022
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., and Lee, V. M. (2000). Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220.
Park, J. Y., Kim, K. S., Lee, S. B., Ryu, J. S., Chung, K. C., Choo, Y. K., et al. (2009). On the mechanism of internalization of α-synuclein into microglia: roles of ganglioside GM1 and lipid raft. J. Neurochem. 110, 400–411. doi: 10.1111/j.1471-4159.2009.06150.x
Parnetti, L., Chiasserini, D., Bellomo, G., Giannandrea, D., De Carlo, C., Qureshi, M. M., et al. (2011). Cerebrospinal fluid Tau/α-synuclein ratio in Parkinson's disease and degenerative dementias. Mov. Disord. 26, 1428–1435. doi: 10.1002/mds.23670
Peelaerts, W., Bousset, L., Van Der Perren, A., Moskalyuk, A., Pulizzi, R., Giugliano, M., et al. (2015). α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. doi: 10.1038/nature14547
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Pouclet, H., Lebouvier, T., Coron, E., Des Varannes, S. B., Rouaud, T., Roy, M., et al. (2012). A comparison between rectal and colonic biopsies to detect Lewy pathology in Parkinson's disease. Neurobiol. Dis. 45, 305–309. doi: 10.1016/j.nbd.2011.08.014
Poulopoulos, M., Levy, O. A., and Alcalay, R. N. (2012). The neuropathology of genetic Parkinson's disease. Mov. Disord. 27, 831–842. doi: 10.1002/mds.24962
Reyes, J. F., Rey, N. L., Bousset, L., Melki, R., Brundin, P., and Angot, E. (2014). Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62, 387–398. doi: 10.1002/glia.22611
Rockenstein, E., Nuber, S., Overk, C. R., Ubhi, K., Mante, M., Patrick, C., et al. (2014). Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137, 1496–1513. doi: 10.1093/brain/awu057
Sacino, A. N., Brooks, M., Thomas, M. A., Mckinney, A. B., McGarvey, N. H., Rutherford, N. J., et al. (2014). Amyloidogenic α-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 127, 645–665. doi: 10.1007/s00401-014-1268-0
Satake, W., Nakabayashi, Y., Mizuta, I., Hirota, Y., Ito, C., Kubo, M., et al. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 41, 1303–1307. doi: 10.1038/ng.485
Simón-Sánchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41, 1308–1312. doi: 10.1038/ng.487
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). α-Synuclein locus triplication causes Parkinson's disease. Science 302, 841. doi: 10.1126/science.1090278
Spencer, B., Emadi, S., Desplats, P., Eleuteri, S., Michael, S., Kosberg, K., et al. (2014). ESCRT-mediated uptake and degradation of brain-targeted α-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol. Ther. 22, 1753–1767. doi: 10.1038/mt.2014.129
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Sung, J. Y., Kim, J., Paik, S. R., Park, J. H., Ahn, Y. S., and Chung, K. C. (2001). Induction of neuronal cell death by Rab5A-dependent endocytosis of α-synuclein. J. Biol. Chem. 276, 27441–27448. doi: 10.1074/jbc.M101318200
Thayanidhi, N., Helm, J. R., Nycz, D. C., Bentley, M., Liang, Y., and Hay, J. C. (2010). α-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 21, 1850–1863. doi: 10.1091/mbc.E09-09-0801
Theillet, F. X., Binolfi, A., Bekei, B., Martorana, A., Rose, H. M., Stuiver, M., et al. (2016). Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50. doi: 10.1038/nature16531
Tokuda, T., Salem, S. A., Allsop, D., Mizuno, T., Nakagawa, M., Qureshi, M. M., et al. (2006). Decreased α-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson's disease. Biochem. Biophys. Res. Commun. 349, 162–166. doi: 10.1016/j.bbrc.2006.08.024
Tran, H. T., Chung, C. H., Iba, M., Zhang, B., Trojanowski, J. Q., Luk, K. C., et al. (2014). α-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 7, 2054–2065. doi: 10.1016/j.celrep.2014.05.033
Tu, P. H., Galvin, J. E., Baba, M., Giasson, B., Tomita, T., Leight, S., et al. (1998). Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann. Neurol. 44, 415–422. doi: 10.1002/ana.410440324
Tuttle, M. D., Comellas, G., Nieuwkoop, A. J., Covell, D. J., Berthold, D. A., Kloepper, K. D., et al. (2016). Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 23, 409–415. doi: 10.1038/nsmb.3194
Uversky, V. N., and Eliezer, D. (2009). Biophysics of Parkinson's disease: structure and aggregation of α-synuclein. Curr. Protein Pept. Sci. 10, 483–499. doi: 10.2174/138920309789351921
Volpicelli-Daley, L. A., Luk, K. C., and Lee, V. M. (2014). Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 9, 2135–2146. doi: 10.1038/nprot.2014.143
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Wakabayashi, K., Takahashi, H., Takeda, S., Ohama, E., and Ikuta, F. (1988). Parkinson's disease: the presence of Lewy bodies in Auerbach's and Meissner's plexuses. Acta Neuropathol. 76, 217–221. doi: 10.1007/BF00687767
Waxman, E. A., and Giasson, B. I. (2010). A novel, high-efficiency cellular model of fibrillar α-synuclein inclusions and the examination of mutations that inhibit amyloid formation. J. Neurochem. 113, 374–388. doi: 10.1111/j.1471-4159.2010.06592.x
Winner, B., Jappelli, R., Maji, S. K., Desplats, P. A., Boyer, L., Aigner, S., et al. (2011). In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199. doi: 10.1073/pnas.1100976108
Zarranz, J. J., Alegre, J., Gómez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Keywords: Parkinson's disease, alpha-Synuclein, neurodegeneration, cell-to-cell propagation, Lewy bodies, prion-like diseases
Citation: Prymaczok NC, Riek R and Gerez J (2016) More than a Rumor Spreads in Parkinson's Disease. Front. Hum. Neurosci. 10:608. doi: 10.3389/fnhum.2016.00608
Received: 29 August 2016; Accepted: 14 November 2016;
Published: 02 December 2016.
Edited by:
Marcelo Merello, Fundación para la Lucha contra las Enfermedades Neurológicas de la Infancia, ArgentinaReviewed by:
Kristina Aurousseau, Centre de recherche de l'Institut universitaire de gériatrie de Montréal (CRIUGM), CanadaMartin Radrizzani, National University of San Martin, Argentina
Copyright © 2016 Prymaczok, Riek and Gerez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Gerez, anVhbi5nZXJlekBiYy5iaW9sLmV0aHouY2g=