
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Hematol., 03 April 2025
Sec. Red Cells, Iron and Erythropoiesis
Volume 4 - 2025 | https://doi.org/10.3389/frhem.2025.1549241
 Ram Prakash Thirugnanasambandam1*
Ram Prakash Thirugnanasambandam1* John Muthu2
John Muthu2Background: Pregnant individuals with sickle cell disease (SCD) face high maternal and fetal complication rates. While some studies suggest transfusions may improve outcomes, evidence is limited. This study analyzed maternal and fetal complications in pregnant SCD patients of different genotypes who received transfusions.
Methods: We performed a retrospective review of medical records at New York City Health and Hospitals/Kings County over nine years (2015–2024), including all pregnant patients with SCD (N=42). Maternal and fetal complications were analyzed based on genotype, hemoglobin levels, age, and race
Results: Patients with homozygous sickle cell disease (HbSS) comprised 69% of the cohort, with most HbSS patients over 35 years old. Baseline hemoglobin levels were 7.2 g/dl for HbSS, 9.5 g/dl for HbSC, and 7.0 g/dl for HbS Beta+ thalassemia. Transfusion needs were highest in HbSS patients (72.4%) compared to HbSC (44.4%) and HbS Beta+ thalassemia (25%) but were not statistically significant. Obstetric complications such as pre-eclampsia (20.7%) and postpartum hemorrhage (17.2%) were more common in HbSS patients but without statistical significance. Fetal complications included intrauterine growth restriction (6.9%) in HbSS patients and fetal distress (22.2%) in HbSC patients with no significant differences. Comparing transfused and non-transfused HbSS patients, transfused patients had lower hemoglobin levels and more vaso-occlusive episodes. However, obstetric and fetal complications were similar in both groups.
Conclusion: Despite transfusions, HbSS patients experienced more complications. A personalized, evidence-based approach is needed for managing SCD pregnancies, focusing on transfusion timing, comorbidities, and fetal monitoring.
Globally, the population of individuals living with sickle cell disease (SCD) has notably increased, rising by more than 41% (from 5.46 million in 2000 to 7.74 million in 2021), with many reaching reproductive age (1). Despite advancements in medical care, maternal and perinatal mortality rates among pregnant individuals with SCD remain disproportionately high (2). Optimal management of SCD during pregnancy necessitates specialized obstetric and hematology care, yet outcomes vary depending on the availability of resources and expertise (3). It is imperative to acknowledge the critical role of a team of healthcare providers, which includes experts in SCD management and experts in high-risk obstetric care, to ensure favorable pregnancy outcomes for individuals with SCD (4).
Red blood cell (RBC) transfusions are integral in managing severe anemia and acute sickle cell complications and mitigating adverse perinatal outcomes such as low birth weight and small for gestational age infants (5). However, the benefits of RBC transfusions must be carefully weighed against potential risks, including alloimmunization and iron overload (6). Previous research, including a randomized controlled trial and a retrospective study, yielded inconclusive results regarding the efficacy of prophylactic RBC transfusions in reducing maternal and perinatal mortality rates among pregnant individuals with SCD (7, 8). The need for a large-scale randomized controlled trial to assess the benefits of prophylactic transfusions in this population has been addressed by the TAPS-2 study (9).This feasibility trial assessed serial prophylactic exchange blood transfusion (SPEBT) versus standard care in pregnant women with SCD. While no statistically significant differences were observed in maternal, infant, and postnatal outcomes, trends suggested a lower incidence of vaso-occlusive crises, preterm delivery, and improved birthweight in the intervention group (9). Similarly, a systematic review and meta-analysis by AlMoshary et al. demonstrated that prophylactic blood transfusion during pregnancy in women with SCD reduced vaso-occlusive crises, pulmonary complications, perinatal mortality, and preterm births in the prophylactic transfusion group compared to controls, underscoring its potential to improve maternal and fetal outcomes (10).
While existing evidence suggests a potential association between prophylactic transfusions and improved maternal and perinatal outcomes, the limited number of studies and methodological constraints necessitate further investigation (5, 8). Considering these findings, this study attempted to retrospectively analyze the incidence of maternal and perinatal complications among both transfused and non-transfused pregnant patients of different sickle genotypes, thereby aiming to contribute to a deeper understanding of the optimal management strategies for pregnant individuals with SCD.
A retrospective analysis was conducted of medical records from NYC Health and Hospitals/Kings County to identify pregnant patients diagnosed with sickle cell disease between January 1, 2015, and January 1, 2024. Access to patient medical records was facilitated through EPIC and Quadramed software. While efforts were made to ensure the completeness of data, it is acknowledged that some women may have received care at other institutions, which could include pre-conception visits, certain aspects of antenatal care, or delivery. This limitation is inherent to retrospective analyses and underscores the potential for fragmented care outside the study center.
This study did not include a formal sample size justification or power analysis due to its retrospective nature and reliance on the available patient population over the study period. We recognize that the small sample size may limit the statistical power to detect significant differences for certain parameters. Despite these limitations, the study provides exploratory insights into maternal and perinatal complications in patients with sickle cell disease. Future prospective studies with larger cohorts and formal power analyses are warranted to validate these findings.
The analysis includes all pregnant patients diagnosed with sickle cell disease and receiving care at NYC Health and Hospitals/Kings County during the specified study period.
Demographic information was extracted, including age, ethnicity, and relevant medical history, from the medical records of eligible patients. Race was recorded based on self-reported information documented in the patient’s medical records. This approach aligns with standard practices at our institution to ensure accuracy and reflect the patient’s own identification. Additionally, data on sickle cell genotype, mean hemoglobin levels at baseline and averaged throughout pregnancy, along with the presence of comorbidities such as diabetes mellitus, hypertension, pre-eclampsia/eclampsia, and hyperlipidemia, were recorded. Sickle cell genotypes were confirmed using hemoglobin electrophoresis, the diagnostic standard at our institution. This method allowed for the identification of abnormal hemoglobin patterns and the classification of patients into HbSS, HbSC, and HbS beta+ thalassemia groups based on their electrophoretic profiles. No patients with S/beta thalassemia 0 were identified in this cohort. In this study, HbSS refers to the genotype with homozygous sickle hemoglobin, which is the underlying genetic basis for sickle cell disease. Vaso-occlusive crises (VOC) were defined as episodes of acute pain severe enough to require medical evaluation and documented as VOC in the medical record, with or without hospital admission.
Hemoglobin values were calculated using all available laboratory results documented in the medical records. For ‘baseline hemoglobin,’ values were obtained from pre-pregnancy or the first trimester. For ‘hemoglobin during pregnancy,’ values were averaged from all documented hemoglobin measurements during the second and third trimesters. Transfusion timing and frequency were analyzed by reviewing patient records for evidence of transfusion during pregnancy. However, the number of transfusions per patient, detailed categorization of transfusion timing (e.g., pre-pregnancy, during pregnancy, or postpartum), and the nature of transfusions (e.g., sporadic or scheduled) were not systematically evaluated. This limitation is due to the retrospective nature of the study and the possibility that some women may have received care at other institutions outside the NYC Health and Hospitals system, which affected our ability to collect comprehensive data beyond our system.
● Hypertension: Systolic blood pressure ≥140 mmHg and/or diastolic blood pressure ≥90 mmHg on two separate occasions at least four hours apart.
● Pre-eclampsia/Eclampsia: Pre-eclampsia was defined as hypertension with proteinuria (≥300 mg/24 hours) or evidence of end-organ dysfunction. Eclampsia was defined as the occurrence of seizures in a patient with pre-eclampsia.
● Fetal Distress: Abnormal fetal heart rate patterns requiring clinical intervention, such as late decelerations or reduced variability.
● Preterm Delivery: Delivery before 37 completed weeks of gestation.
● Postpartum Hemorrhage: Blood loss ≥500 mL after vaginal delivery or ≥1000 mL after cesarean delivery.
● Intrauterine Growth Restriction (IUGR): Fetal weight below the 10th percentile for gestational age, determined via ultrasonography.
The majority of the data are expressed as n (%). Normality testing (D’Agostino & Pearson omnibus normality test) was performed on all data sets. A comparison of groups was performed by Student’s t-test (two-tailed) when data passed the normality testing and by Mann-Whitney test if data did not pass the normality testing. Experiments with more than 2 groups were analyzed using a 1-way ANOVA or Kruskal-Wallis test with Bonferroni posttests analysis. P-values were reported to indicate the statistical significance of findings, with a threshold set at p<0.05 for determining significance. All analyses were performed using GraphPad Prism Software (Version 5.0h).
Given the relatively small sample size of this study, there is an increased risk of Type II errors, meaning that some associations may not have reached statistical significance despite potential clinical relevance. A larger dataset with greater statistical power could better differentiate significant differences among transfusion subgroups and maternal-fetal outcomes. Future studies with larger cohorts are warranted to more robustly assess these relationships and minimize the likelihood of false-negative results.
This study included a predominance of patients with homozygous sickle cell disease (HbSS), constituting 69% of the study population. The majority (66%) of these patients were above 35 years of age (Table 1). The mean baseline hemoglobin levels were consistently lower in the HbSS group at approximately 7.0 g/dl (Table 2), which remained relatively constant throughout their pregnancy for the vast majority. In comparison, patients with heterozygous sickle genotypes (HbSC and HbS Beta+ thalassemia) showed higher than mean baseline hemoglobin levels of approximately 9.5 g/dl and approximately 7.0 g/dl respectively, which averaged around 8.0g/dl during pregnancy, though the differences were not statistically significant.
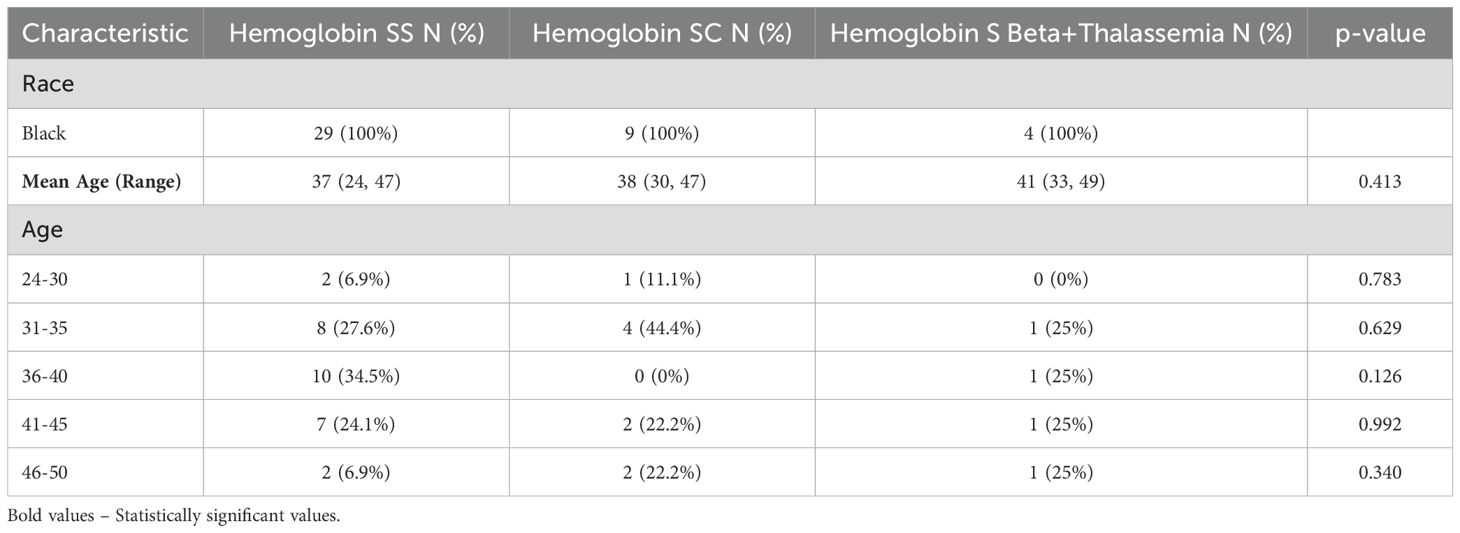
Table 1. General demographics classified by genotype.
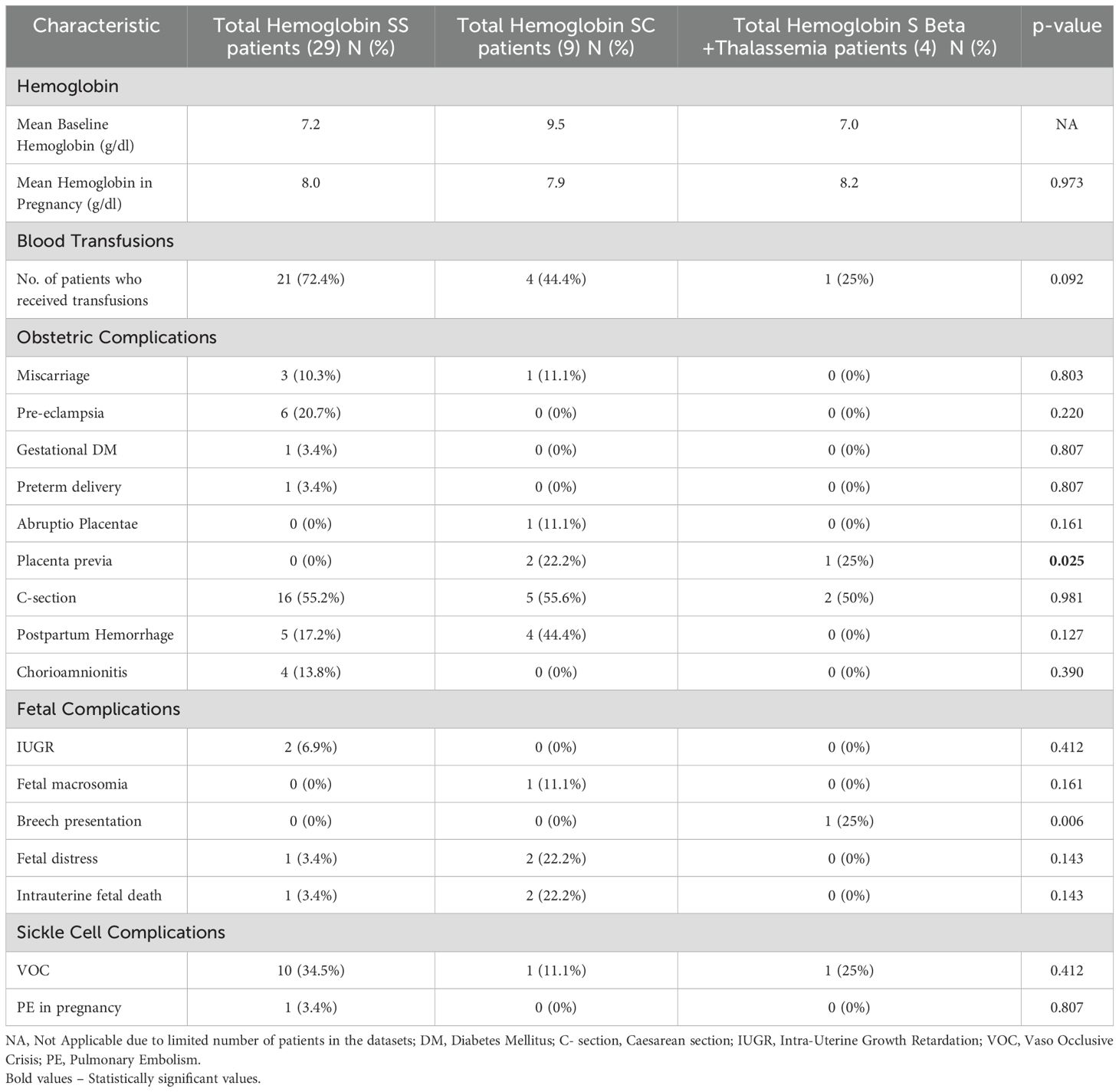
Table 2. Maternal sickle cell/obstetric and perinatal complications classified by genotype.
The need for blood transfusions during pregnancy was highest among HbSS patients, with 72.4% receiving transfusions (Table 2). This was in contrast to 44.% of HbSC patients and 25% of HbS Beta+ thalassemia patients, highlighting the varying medical needs and interventions required by different groups.These differences were not statistically significant (p = 0.092). Due to the retrospective nature of the study, we were unable to distinguish between patients receiving scheduled prophylactic exchanges and those receiving transfusions for pregnancy-related complications, limiting the interpretation of these findings.
Obstetric complications were more prevalent among HbSS patients compared to other genotypes. However, these differences did not achieve statistical significance unless otherwise noted. Pre-eclampsia was observed exclusively in the HbSS genotype (21%), while the HbSC and HbS Beta+ thalassemia groups had no cases. The incidence of postpartum hemorrhage was higher among HbSC patients (44%) compared to HbSS patients (17%), while none of the HbS Beta+ thalassemia patients experienced this complication; however, this difference was not statistically significant (p = 0.127). Cesarean section was frequent across all groups, having been conducted among 55% of HbSS, 56% of HbSC, and 50% of HbS Beta+ thalassemia patients, with no statistically significant differences. Miscarriages occurred in 10% of HbSS patients as compared to 11% of HbSC patients, while none were noted among HbS Beta+ thalassemia patients, though these differences were not statistically significant. While the majority of the obstetric complications among the three genotypes did not attain any statistical significance, placenta previa had a statistical significance with p=0.025 and was found among 22% of HbSC patients and 25%of the HbS Beta+ thalassemia patients.
Fetal complications also varied among the genotypes. The incidence of Intrauterine growth retardation (IUGR) was predominantly noted in HbSS patients (7%) These differences were not statistically significant (p = 0.412). Fetal distress was more prevalent in HbSC patients (22%) compared to HbSS patients (3%) and none in HbS Beta+ thalassemia patients, though this result did not reach statistical significance (p = 0.143). Intrauterine fetal death was more frequent among HbSC patients (22%) compared to HbSS patients (3%), while none was observed among HbS Beta+ thalassemia patients; however, this difference was not statistically significant (p = 0.143). The incidence of breech presentation was higher among HbS Beta+ thalassemia patients (25%), compared to other groups, with a p-value of 0.006, which may be interpreted as near significant.
Vaso-occlusive episodes during pregnancy occurred in 29% of all patients, with varying incidence among different genotypes. Among HbSS patients, 34% experienced vaso-occlusive episodes, compared to 11% of HbSC patients and 25% of HbS Beta+ thalassemia patients though this difference was not statistically significant (p = 0.412). The comparative analysis between HbSS patients who received transfusions and those who did not receive transfusions (Table 3) reveals several key insights. While the mean age of both cohorts was similar, transfused patients exhibited lower hemoglobin levels during pregnancy compared to non-transfused patients. Obstetric complications such as miscarriage, pre-eclampsia, and cesarean section were comparable between the two groups, with no statistically significant differences. Additionally, fetal complications, including IUGR and fetal distress, were more prevalent in the transfused groups, though not statistically significant. Notably, transfused patients have a significantly higher rate of VOC compared to non-transfused patients (p=0.015), indicating a potential association between VOC and transfusions.
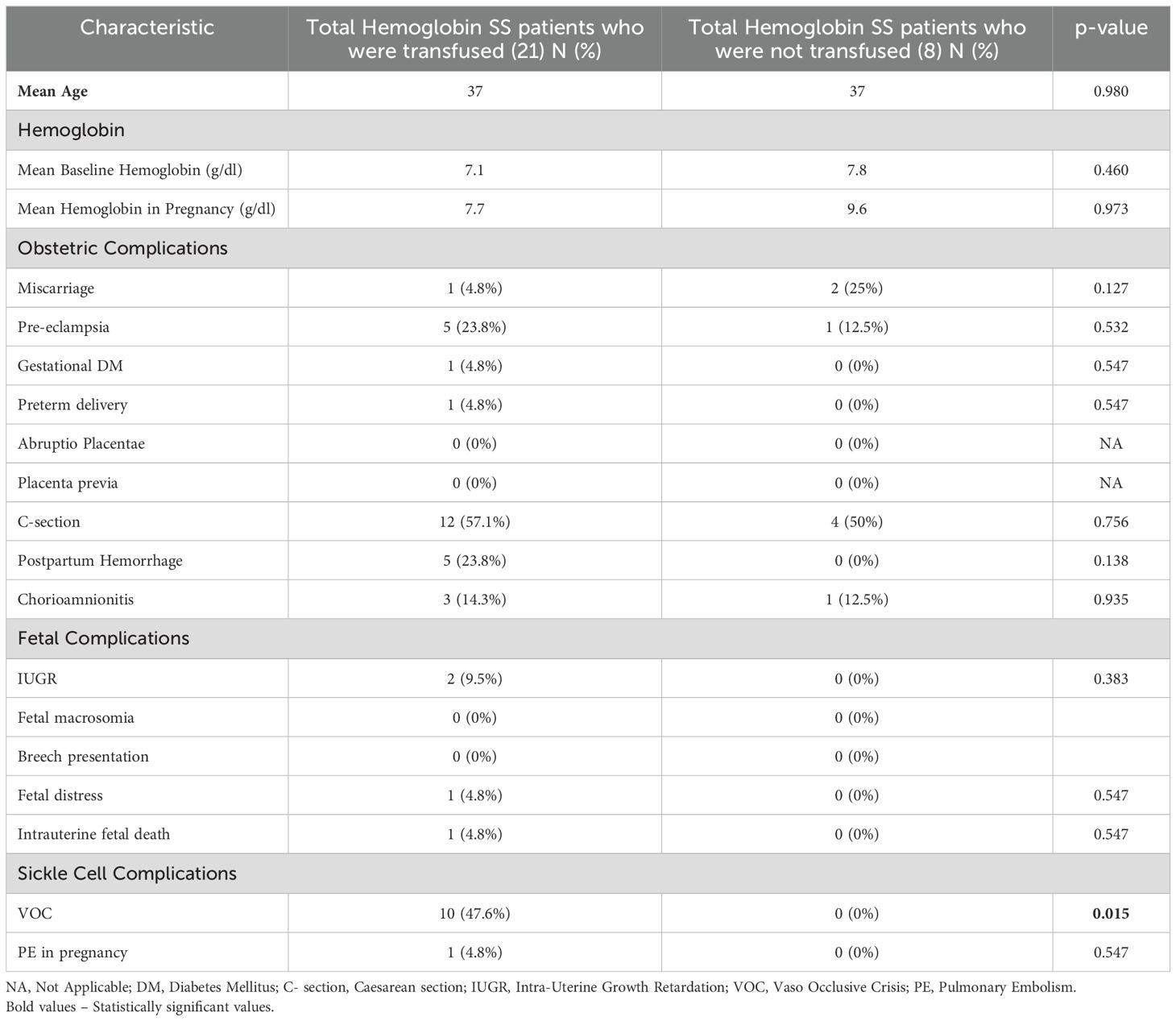
Table 3. Comparative analysis of Maternal sickle cell/obstetric and perinatal complications in Hemoglobin SS patients: Transfused versus Non-Transfused cohorts.
Sickle cell disease (SCD) affects approximately 100,000 individuals in the USA, resulting from a mutation in the βeta globin gene that leads to sickle hemoglobin production (11). In the U.S., the majority of individuals who have sickle cell disease are of black descent (12). The New York State Department of Health estimates that approximately 150-200 babies are diagnosed with sickle cell each year, with an incidence of approximately 1 in 500 live births among Black or African American infants (13). The population presented here, as depicted in Table 1, comprises exclusively women of black descent from an underserved area of Central Brooklyn. This population was further categorized into different age groups and genotypes.
This retrospective analysis highlights significant disparities in maternal and fetal outcomes among different sickle cell genotypes, emphasizing the need for tailored management strategies for each group. Patients with homozygous sickle cell disease (HbSS) exhibited the highest rates of severe obstetric complications, such as pre-eclampsia and postpartum hemorrhage, which may increase the need for blood transfusions to manage these complications. A previous study also reported that women with sickle cell disease have a higher prevalence of pregnancy-related complications, including pre-eclampsia and postpartum hemorrhage, even compared to women with other chronic conditions associated with multi-organ failure (14).In contrast, the HbSC subgroup was associated with a higher incidence of fetal complications, such as fetal distress and intrauterine fetal death, underscoring the importance of genotype-specific management strategies to optimize maternal and fetal outcomes.
Variability in mean hemoglobin levels among different sickle cell genotypes reflects the diverse pathophysiological mechanisms underlying anemia in SCD. While baseline levels differed among HbSS (7.2g/dl), HbSC (9.5g/dl), and HbS Beta+ thalassemia (7.0g/dl) patients, these differences were not statistically significant. This variability reflects the diverse pathophysiological mechanisms underlying anemia in SCD, including chronic hemolysis, hypersplenism with splenic enlargement, low erythropoietin level secondary to renal impairment, and delayed hemolytic transfusion reactions (15). During pregnancy, mean hemoglobin increased from 7.2 g/dl to 8.0 g/dl for patients with HbSS disease, decreasing to 8.0 g/dl from 9.5g/dl among HbSC and increasing to 8.0 g/dl from 7.0 g/dl in the HbS Beta+ thalassemia patient population, with no statistically significant differences. These hemoglobin changes likely reflect the severity of the disease among the different SCD genotypes, with more severe disease necessitating higher transfusion requirements, compounded by physiological anemia of pregnancy (16).
Additionally, patients with HbSS exhibited the highest number of transfusions, with 72.4% receiving transfusions throughout pregnancy, compared to 44.4% in the HbSC subgroup and 25% in the HbS Beta+ thalassemia subgroup, though these differences were not statistically significant. Prophylactic RBC transfusions are often recommended to mitigate complications of severe anemia and to reduce sickling in both maternal and placental circulation, thereby enhancing blood flow and oxygenation (17). While prophylactic transfusions have demonstrated benefits in preventing strokes and aiding preoperative preparation for a caesarian section in SCD (18), their broader use is constrained by risks such as transfusion reactions, alloimmunization, infections, and iron overload (19). A more detailed investigation into transfusion timing (pre-pregnancy versus during pregnancy) could provide critical insights into optimizing outcomes. However, our study was limited by the inability to systematically distinguish between prophylactic and reactive transfusions or to categorize them based on timing (pre-pregnancy, antenatal, peripartum, or postpartum). Future studies should aim to incorporate structured transfusion data to better assess the impact of timing and indication on maternal and fetal outcomes. Evidence suggests that transfusion timing may influence placental blood flow, oxygenation, and fetal development, but data are limited. Studies such as those by Koshy et al. (6) and Malinowski et al. (8) indicate that carefully timed transfusions could potentially reduce complications, including vaso-occlusive crises (VOC) and pre-eclampsia. Prospective, multicenter trials focusing on the temporal aspects of transfusion are essential to refine clinical guidelines and enhance care for pregnant women with SCD. Additionally, transfusion-related complications such as iron overload and alloimmunization pose significant challenges that could further exacerbate maternal and fetal outcomes. Iron overload has been associated with organ dysfunction, while alloimmunization increases the risk of hemolytic disease in future pregnancies (20, 21). Studies assessing the impact of prophylactic transfusions on pregnancy outcomes in SCD have yielded inconsistent findings, underscoring the need for further research in this area (9, 22–24).
Pregnancy in SCD is characterized by a heightened risk of obstetric complications, with pre-eclampsia, eclampsia, and Gestational Diabetes Mellitus (GDM) among the most prevalent maternal complications (25, 26). Chronic hypertension in SCD patients has been associated with higher rates of pre-eclampsia and adverse fetal outcomes (25).Similarly, diabetes can impair placental function and fetal growth, further increasing the risk of poor pregnancy outcomes (27). These studies highlight the significant interplay between these comorbidities and pregnancy outcomes, emphasizing the need for a multidisciplinary approach to care. Additionally, placental abnormalities, particularly maternal vascular malperfusion (MVM), have been reported in 40% of SCD pregnancies, with strong associations to adverse pregnancy outcomes such as preterm birth, small for gestational age infants, and stillbirth (28). These studies highlight the significant interplay between these comorbidities and pregnancy outcomes, emphasizing the need for a multidisciplinary approach to care. While comorbidities such as hypertension and diabetes likely influence maternal and fetal outcomes in our study population, we did not perform subgroup comparisons between patients with and without comorbid conditions due to the small sample size. Future studies with larger cohorts should aim to stratify patients based on comorbidities to better delineate their impact on pregnancy outcomes in individuals with SCD.
Our study, as depicted in Table 2, reveals a higher incidence of preeclampsia and GDM among HbSS patients compared to those with HbSC and HbS beta+ thalassemia genotypes, though these differences were not statistically significant. Additionally, placental complications such as abruptio placentae and placenta previa were observed in patients with HbSC and HbS beta+ thalassemia, with statistical significance seen for the incidence of placenta previa alone (p=0.025). The incorporation of placental pathology findings highlights the need for further research into therapies aimed at improving placental function and mitigating vascular complications in this population. Although the exact mechanisms by which SCD impacts placental physiology remain elusive, factors such as red cell sickling, endothelial damage, vascular occlusion, and inflammation are thought to contribute (29). Furthermore, our findings underscore the high incidence of cesarean section among all three genotypes, as reflected by several studies (27, 30), despite SCD not being a contraindication for attempting vaginal delivery (31).
Infants born to mothers with SCD are at heightened risk for intrauterine growth restriction (IUGR), low birth weight, prematurity, and perinatal mortality (25). Placental insufficiency, stemming from vascular occlusion and endothelial damage, may contribute to poor intrauterine growth in these infants (32). Additionally, women with SCD have been observed to have lower BMI compared to controls, which may contribute to adverse pregnancy outcomes (33). However, the direct impact of BMI on fetal growth restriction in this population remains unclear and warrants further investigation. In our study, we observed instances of IUGR, fetal distress, and intrauterine fetal death among both the HbSS and HbSC populations, along with a significantly higher incidence of breech presentation among the HbS Beta+ thalassemia population (25%) with a p-value of 0.006, highlighting the significant risk faced by infants born to mothers with different genotypes of SCD. While our study primarily focused on immediate perinatal outcomes such as IUGR and fetal distress, long-term follow-up studies are essential to assess neurodevelopmental outcomes, growth parameters, and potential long-term complications in infants born to mothers with SCD. For example, developmental delays and chronic health issues in children born to mothers with SCD have been documented in prior studies (34). Future research should incorporate standardized neonatal and pediatric follow-up protocols, including neurodevelopmental assessments, growth monitoring, and evaluation of cardiopulmonary function. Longitudinal cohort studies following infants into childhood and adolescence would provide deeper insights into the long-term impact of maternal SCD on offspring health.
Throughout pregnancy, individuals with SCD remain susceptible to acute SCD-related complications, including acute vaso-occlusive crises (VOC), acute chest syndrome, venous thromboembolism (VTE), and stroke. Existing studies have observed a higher frequency of acute VOC in the HbSS population than in the HbSC subtype (35). Consistent with these findings, our study population exhibited a higher incidence of VOC episodes among patients with HbSS compared to other genotypes though this difference was not statistically significant. Furthermore, one patient experienced a pulmonary embolus (PE), consistent with previously documented associations between SCD and thromboembolic events during pregnancy (36).
Furthermore, a subgroup analysis among candidates with the HbSS genotype, given their significant representation in our study and the general SCD population (37), noted that transfused patients had lower hemoglobin levels during pregnancy at baseline compared to non-transfused patients, but both subgroups exhibited comparable rates of obstetric complications, with no statistically significant differences in hemoglobin levels or obstetric outcomes. The higher mean hemoglobin value of 9.6 g/dL among non-transfused HbSS patients is unexpected and may indicate the possibility of undocumented transfusions at other institutions. Alternatively, this finding could reflect variability in disease severity, baseline hemoglobin levels, nutritional status among individuals, or the presence of concomitant alpha thalassemia trait. Alpha thalassemia trait is known to be associated with a less severe phenotype, including higher baseline hemoglobin, reduced hemolysis, and lower transfusion requirements (38). Patients with the HbSS genotype are known to have lower hemoglobin levels and higher complication rates compared to the HbSC and HbS beta+ thalassemia genotypes (39). The randomized controlled trial by Koshy et al. (22) showed no significant difference in perinatal outcomes between patients receiving prophylactic transfusions and those receiving transfusions only for medical or obstetric indications, and our findings are consistent with those of that trial. Additionally, the current study also suggests that while fetal complications were more prevalent in the transfused group, they did not reach statistical significance. Patients who were transfused had higher rates of VOC compared to non-transfused patients (p=0.015). The clinical trial by Koshy et al. (22) demonstrated that prophylactic transfusion significantly reduced VOC incidence (p<0.01). While our study did not differentiate transfusion by indication, the observed association between transfusion and VOC suggests the need for a case-by-case approach to transfusion decisions in pregnant women with SCD.
This study has several limitations. As a retrospective study, it cannot establish causation, and the data relies on records from a single center, which may not represent the broader diversity of patients with SCD. Some women may have received parts of their care, such as pre-conception visits, antenatal care, or delivery, at other institutions, leading to potential gaps in data, particularly for transfusion history and clinical events. We reviewed records for evidence of transfusions during pregnancy, but detailed information on the number, timing, or type of transfusions was not consistently available. The small sample size further limits the statistical power of our findings, particularly for detecting associations with rare maternal and fetal outcomes, which should be interpreted with caution. Additionally, the lack of detailed analysis of transfusion timing and frequency may affect the robustness of statistical interpretations, highlighting the need for future studies to address these gaps. We did not specifically examine data on hydroxyurea use or alloantibodies in this study. Finally, the wide age range in the cohort reflects improved management and longer life expectancy in SCD, allowing more women to pursue pregnancy. To overcome these limitations, a prospective study should be structured with standardized protocols for data collection. Key variables to track would include transfusion timing (pre-pregnancy, antenatal, peripartum, and postpartum), transfusion indications (prophylactic vs. reactive), hemoglobin trends, frequency of vaso-occlusive crises, maternal comorbidities, and fetal outcomes. A structured follow-up period through pregnancy and postpartum would allow for a more comprehensive assessment of maternal and neonatal health, addressing the gaps seen in retrospective data. A prospective cohort study would allow for more systematic and consistent data collection, providing a better framework for understanding the relationship between transfusion protocols, comorbidities, and pregnancy outcomes, as well as enabling stronger causal inferences.
Addressing the gaps in our current study requires future research endeavors aimed at defining optimal transfusion protocols tailored to different sickle cell genotypes during pregnancy. Larger randomized controlled trials in multiple centers are crucial for evaluating the efficacy and safety of prophylactic versus demand-driven transfusion strategies, leading to more effective treatment approaches. Furthermore, investigating the influence of confounding variables such as the comorbid history of hypertension and diabetes, which can alter pregnancy outcomes in SCD patients, is essential to provide deeper insight into managing this high-risk pregnant population subgroup. Such studies hold significant promise in advancing our understanding and may ultimately lead to the development of improved clinical guidelines for managing pregnant individuals with SCD and comorbidities.
Despite a higher percentage of HbSS patients receiving transfusions, the incidence of obstetric and sickle cell complications remained prevalent in this subgroup, though differences were not statistically significant. Our retrospective analysis underscores the complex association between pregnancy-related complications of sickle cell disease and transfusion therapy. While our findings generally indicate a correlation between mean hemoglobin levels and adverse outcomes, further research is imperative to elucidate the optimal management strategies for pregnant individuals with SCD. Randomized trials integrating comprehensive clinical and laboratory assessments are essential to delineate the role of prophylactic transfusions in mitigating maternal, fetal, and sickle cell-related complications. Notably, our subgroup analysis among patients with the HbSS genotype, consistent with the prior randomized clinical trial, emphasizes the need for a personalized approach to managing pregnant women with SCD and takes into consideration the presence of comorbid factors that can influence the outcomes.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by SUNY Downstate IRB and Privacy Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was not obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article because the IRB reviewed the application and deemed it exempt at it was a retrospective chart review.
RT: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. JM: Conceptualization, Investigation, Methodology, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Collaborators GBDSCD. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000-2021: a systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. (2023) 10:e585–e99. doi: 10.1016/S2352-3026(23)00118-7
2. Joseph L, Driessen M. A comprehensive view of pregnancy in patients with sickle cell disease in high-income countries: the need for robust data and further decline in morbidity and mortality. Lancet Haematol. (2024) 11:e75–84. doi: 10.1016/S2352-3026(23)00310-1
3. Oteng-Ntim E, Shangaris P. Evidence-based management of pregnant women with sickle cell disease in high-income countries. Hematol Am Soc Hematol Educ Program. (2022) 2022:408–13. doi: 10.1182/hematology.2022000378
4. Hassell K. Pregnancy and sickle cell disease. Hematol Oncol Clin North Am. (2005) 19:903–16,vii-viii. doi: 10.1016/j.hoc.2005.07.003
5. Okusanya BO, Oladapo OT. Prophylactic versus selective blood transfusion for sickle cell disease in pregnancy. Cochrane Database Syst Rev. (2016) 12:CD010378. doi: 10.1002/14651858.CD010378.pub3
6. Fasano RM, Leong T, Kaushal M, Sagiv E, Luban NL, Meier ER. Effectiveness of red blood cell exchange, partial manual exchange, and simple transfusion concurrently with iron chelation therapy in reducing iron overload in chronically transfused sickle cell anemia patients. Transfusion. (2016) 56:1707–15. doi: 10.1111/trf.2016.56.issue-7
7. Koshy M, Chisum D, Burd L, Orlina A, How H. Management of sickle cell anemia and pregnancy. J Clin Apher. (1991) 6:230–3. doi: 10.1002/jca.2920060412
8. Malinowski AK, Shehata N, D’Souza R, Kuo KH, Ward R, Shah PS, et al. Prophylactic transfusion for pregnant women with sickle cell disease: a systematic review and meta-analysis. Blood. (2015) 126:2424–35;quiz 37. doi: 10.1182/blood-2015-06-649319
9. Oteng-Ntim E, Oakley LL, Robinson V, Brien S, Joseph J, Sharif J, et al. Prophylactic exchange transfusion in sickle cell disease pregnancy: a TAPS2 feasibility randomized controlled trial. Blood Advances. (2024) 8:4359–4369.8. doi: 10.1182/bloodadvances.2024012923
10. AlMoshary M, Arabdin M. The role of prophylactic transfusion on the maternal and fetal outcomes in pregnant women with sickle cell disease: A systematic review and meta-analysis. Med (Baltimore). (2024) 103:e39475. doi: 10.1097/MD.0000000000039475
11. Brandow AM, Liem RI. Advances in the diagnosis and treatment of sickle cell disease. J Hematol Oncol. (2022) 15:20. doi: 10.1186/s13045-022-01237-z
12. Solovieff N, Hartley SW, Baldwin CT, Klings ES, Gladwin MT, Taylor J, et al. Ancestry of African Americans with sickle cell disease. Blood Cells Mol Dis. (2011) 47:41–5. doi: 10.1016/j.bcmd.2011.04.002
14. Boulet SL, Okoroh EM, Azonobi I, Grant A, Craig Hooper W. Sickle cell disease in pregnancy: maternal complications in a Medicaid-enrolled population. Matern Child Health J. (2013) 17:200–7. doi: 10.1007/s10995-012-1216-3
15. Xu JZ, Thein SL. Revisiting anemia in sickle cell disease and finding the balance with therapeutic approaches. Blood. (2022) 139:3030–9. doi: 10.1182/blood.2021013873
16. Jain D, Atmapoojya P, Colah R, Lodha P. Sickle cell disease and pregnancy. Mediterr J Hematol Infect Dis. (2019) 11:e2019040. doi: 10.4084/MJHID.2019.040
17. Howard RJ, Tuck SM, Pearson TC. Pregnancy in sickle cell disease in the UK: results of a multicentre survey of the effect of prophylactic blood transfusion on maternal and fetal outcome. Br J Obstet Gynaecol. (1995) 102:947–51. doi: 10.1111/j.1471-0528.1995.tb10900.x
18. Ware RE, Helms RW, Investigators SW. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood. (2012) 119:3925–32. doi: 10.1182/blood-2011-11-392340
19. Chou ST. Transfusion therapy for sickle cell disease: a balancing act. Hematol Am Soc Hematol Educ Program. (2013) 2013:439–46. doi: 10.1182/asheducation-2013.1.439
20. Porter J, Garbowski M. Consequences and management of iron overload in sickle cell disease. Hematol Am Soc Hematol Educ Program. (2013) 2013:447–56. doi: 10.1182/asheducation-2013.1.447
21. ACOG Practice Bulletin No. 192: management of alloimmunization during pregnancy. Obstet Gynecol. (2018) 131:e82–90. doi: 10.1097/AOG.0000000000002528
22. Koshy M, Burd L, Wallace D, Moawad A, Baron J. Prophylactic red-cell transfusions in pregnant patients with sickle cell disease. A randomized cooperative study. N Engl J Med. (1988) 319:1447–52. doi: 10.1056/NEJM198812013192204
23. El-Shafei AM, Kaur Dhaliwal J, Kaur Sandhu A, Rashid-Al-Sharqi M. Indications for blood transfusion in pregnancy with sickle cell disease. Aust N Z J Obstet Gynaecol. (1995) 35:405–8. doi: 10.1111/j.1479-828X.1995.tb02153.x
24. Cunningham FG, Pritchard JA, Mason R. Pregnancy and sickle cell hemoglobinopathies: results with and without prophylactic transfusions. Obstet Gynecol. (1983) 62:419–24.
25. Boafor TK, Olayemi E, Galadanci N, Hayfron-Benjamin C, Dei-Adomakoh Y, Segbefia C, et al. Pregnancy outcomes in women with sickle-cell disease in low and high income countries: a systematic review and meta-analysis. BJOG. (2016) 123:691–8. doi: 10.1111/bjo.2016.123.issue-5
26. Barfield WD, Barradas DT, Manning SE, Kotelchuck M, Shapiro-Mendoza CK. Sickle cell disease and pregnancy outcomes: women of African descent. Am J Prev Med. (2010) 38:S542–9. doi: 10.1016/j.amepre.2009.12.020
27. Adesina OO, Brunson A, Fisch SC, Yu B, Mahajan A, Willen SM, et al. Pregnancy outcomes in women with sickle cell disease in California. Am J Hematol. (2023) 98:440–8. doi: 10.1002/ajh.26818
28. Malinowski AK, Dziegielewski C, Keating S, Parks T, Kingdom J, Shehata N, et al. Placental histopathology in sickle cell disease: A descriptive and hypothesis-generating study. Placenta. (2020) 95:9–17. doi: 10.1016/j.placenta.2020.04.003
29. Baptista LC, Costa ML, Ferreira R, Albuquerque DM, Lanaro C, Fertrin KY, et al. Abnormal expression of inflammatory genes in placentas of women with sickle cell anemia and sickle hemoglobin C disease. Ann Hematol. (2016) 95:1859–67. doi: 10.1007/s00277-016-2780-1
30. Silva-Pinto AC, de Oliveira Domingues Ladeira S, Brunetta DM, De Santis GC, de Lucena Angulo I, Covas DT. Sickle cell disease and pregnancy: analysis of 34 patients followed at the Regional Blood Center of Ribeirao Preto, Brazil. Rev Bras Hematol Hemoter. (2014) 36:329–33. doi: 10.1016/j.bjhh.2014.07.002
31. Aroke D, Tchouakam DN, Kadia BM, Choukem SP. Iron supplementation in pregnant sicklers: an opinion. BMC Pregnancy Childbirth. (2018) 18:256. doi: 10.1186/s12884-018-1894-y
32. Gil GP, Ananina G, Maschietto M, Lima SCS, da Silva Costa SM, Baptista LC, et al. Epigenetic analysis in placentas from sickle cell disease patients reveals a hypermethylation profile. PloS One. (2022) 17:e0274762. doi: 10.1371/journal.pone.0274762
33. Meeks D, Robinson SE, Macleod D, Oteng-Ntim E. Birth weights in sickle cell disease pregnancies: A cohort study. PloS One. (2016) 11:e0165238. doi: 10.1371/journal.pone.0165238
34. Oteng-Ntim E, Meeks D, Seed PT, Webster L, Howard J, Doyle P, et al. Adverse maternal and perinatal outcomes in pregnant women with sickle cell disease: systematic review and meta-analysis. Blood. (2015) 125:3316–25. doi: 10.1182/blood-2014-11-607317
35. Oteng-Ntim E, Ayensah B, Knight M, Howard J. Pregnancy outcome in patients with sickle cell disease in the UK–a national cohort study comparing sickle cell anaemia (HbSS) with HbSC disease. Br J Haematol. (2015) 169:129–37. doi: 10.1111/bjh.13270
36. Agarwal S, Stanek JR, Vesely SK, Creary SE, Cronin RM, Roe AH, et al. Pregnancy-related thromboembolism in women with sickle cell disease: An analysis of National Medicaid Data. Am J Hematol. (2023) 98:1677–84. doi: 10.1002/ajh.v98.11
37. Ata F, Rahhal A, Malkawi L, Iqbal P, Khamees I, Alhiyari M, et al. Genotypic and phenotypic composition of sickle cell disease in the arab population - A systematic review. Pharmgenomics Pers Med. (2023) 16:133–44. doi: 10.2147/PGPM.S391394
38. Brewin JN, Nardo-Marino A, Stuart-Smith S, El Hoss S, Hanneman A, Strouboulis J, et al. The pleiotropic effects of α-thalassemia on HbSS and HbSC sickle cell disease: Reduced erythrocyte cation co-transport activity, serum erythropoietin, and transfusion burden, do not translate into increased survival. Am J Hematol. (2022) 97:1275–85. doi: 10.1002/ajh.v97.10
Keywords: maternal sickle cell complications, maternal obstetric and perinatal complications, HbSS, retrospective analyses, sickle cell disease
Citation: Thirugnanasambandam RP and Muthu J (2025) Assessing maternal and perinatal complication incidence in pregnant patients with sickle cell disease: a retrospective analysis of transfusion therapy at a tertiary care hospital. Front. Hematol. 4:1549241. doi: 10.3389/frhem.2025.1549241
Received: 20 December 2024; Accepted: 18 March 2025;
Published: 03 April 2025.
Edited by:
John Strouboulis, King’s College London, United KingdomReviewed by:
Panicos Shangaris, King’s College London, United KingdomCopyright © 2025 Thirugnanasambandam and Muthu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ram Prakash Thirugnanasambandam, cmFtcHJha2FzaC50aGlydWduYW5hc2FtYmFuZGFtQG91dGxvb2suY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.